
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnraveling mechanisms of protein encapsulation and release in coacervates via molecular dynamics and machine learning†
Yiwei
Wang‡
,
Rongrong
Zou‡
,
Yeqiang
Zhou
,
Yi
Zheng
,
Chuan
Peng
,
Yang
Liu
*,
Hong
Tan
 ,
Qiang
Fu
and
Mingming
Ding
*
,
Qiang
Fu
and
Mingming
Ding
*
College of Polymer Science and Engineering, State Key Laboratory of Polymer Materials Engineering, Sichuan University, Chengdu 610065, China. E-mail: liuyang_leon@scu.edu.cn; dmmshx@scu.edu.cn; dmmshx@163.com
First published on 29th July 2024
Abstract
Coacervates play a pivotal role in protein-based drug delivery research, yet their drug encapsulation and release mechanisms remain poorly understood. Here, we utilized the Martini model to investigate bovine serum albumin (BSA) protein encapsulation and release within polylysine/polyglutamate (PLys/PGlu) coacervates. Our findings emphasize the importance of ingredient addition sequence in coacervate formation and encapsulation rates, attributed to preference contact between oppositely charged proteins and poly(amino acid)s. Notably, coacervates composed of β-sheet poly(amino acid)s demonstrate greater BSA encapsulation efficiency due to their reduced entropy and flexibility. Furthermore, we examined the pH responsiveness of coacervates, shedding light on the dissolution process driven by Coulomb forces. By leveraging machine learning algorithms to analyze simulation results, our research advances the understanding of coacervate-based drug delivery systems, with the ultimate goal of optimizing therapeutic outcomes.
Introduction
Protein-based biologics, renowned for their low cytotoxicity, high specificity, and potent efficacy, are emerging as a rapidly growing category of therapeutic agents in medical oncology and tissue regeneration.1–4 However, these pharmaceuticals face substantial delivery challenges due to their susceptibility to biodegradation,5–14 limited natural membrane permeability and reduced circulation times following intravenous administration.15–17 To overcome these obstacles, a promising strategy involves leveraging entropy-driven18 formation of coacervate droplets. These droplets are particularly attractive for their ability to spontaneously encapsulate a variety of large molecular cargoes and engage in direct cellular interactions, serving as biomimetic microcontainers devoid of an external membrane.19–21 However, the stability of coacervates is a primary concern in their role as drug carriers, which is influenced by numerous environmental factors, including pH,22–24 salt concentration,25 and temperature.24 Achieving stable encapsulation of protein-based drugs within coacervates poses a challenge, thereby complicating their clinical utility. Furthermore, the limitations imposed by experimental accuracy make it challenging to observe the interactions between coacervates and protein-based drugs, as well as to understand the dynamics of coacervate components during protein encapsulation. Consequently, the underlying mechanisms governing the encapsulation and release of protein-based drugs are still not fully understood.Since proteins generally carry electrical charges, it is preferable to use coacervates containing charged components for encapsulating proteins and preserving their biological activity. Polylysine (PLys) and polyglutamate (PGlu) have the ability to self-assemble into coacervates through electrostatic interactions in aqueous environments.26–29 Owing to their excellent biocompatibility and susceptibility to enzymatic breakdown within organisms, these polymers are promising candidates as protein-based drug carriers, potentially circumventing undesirable accumulation in the body.30–34 This represents an excellent model for investigating the encapsulation of proteins by coacervates. Sarah, L. P. et al. have shown that both the location of charged patches and the overall net charge of a protein are crucial factors in facilitating its encapsulation within PLys/PGlu coacervates.35 Owing to the charges present in the proteins, the order in which various components are introduced to the system could potentially affect their initial contact with specific charged poly(amino acid)s, thereby influencing the encapsulation process. This implies that the system might undergo kinetic trapping, rendering it difficult to achieve the lowest thermodynamic state.36 In addition, the charged nature of the protein and the sequence in which components are added can also impact the secondary structure of the poly(amino acid)s,37 affecting the phase behavior of the coacervate. Matthew Tirrell et al. have shown that coacervates could be formed when at least one polypeptide in the PLys/PGlu mixture is racemic, disrupting hydrogen-bonding networks in the backbone and leading to the formation of a coil secondary structure. In contrast, pairs of purely chiral polypeptides, regardless of their chirality, adopt β-sheet conformation and aggregate into more compact precipitates.38 Therefore, we speculate that the sequence of ingredient addition and the secondary structures of coacervate components could have a significant impact on protein encapsulation.
In this study, we first investigated the P-L-Lys/P-D,L-Glu coacervate with a random coil secondary structure, referring to the research of Tsanai and Black et al.22,39 Martini 3.0 simulations were utilized to model the formation of these coacervates and investigate the mechanisms underlying the encapsulation and release of various proteins within these coacervates. While all-atom (AA) simulations face challenges in observing coacervation phenomena due to temporal and spatial scale limitations,40 the Martini 3.0 coarse-grained model41 offers detailed molecular insights and significantly speeds up simulations by 4–10 times. As a result, it has been increasingly applied to simulate various biomolecular systems in recent years, e.g. Lys/Glu-based coacervate,39,42–44 peptides/proteins,45 nucleic acid,46 polymers,47,48 coacervate/membrane,49etc. However, none of these simulations have specifically addressed the encapsulation and release of proteins in coacervates. The density-based spatial clustering of applications with noise (DBSCAN)50 and principal component analysis (PCA)51,52 were employed to analyze and characterize the formation of the coacervate, as well as to identify the dynamic variations in polymer chain movements across different systems. Notably, the DBSCAN machine learning method has been used to analyze the state of coacervates.53 Here, we reveal that the sequence in which ingredients are added significantly affects the encapsulation efficiency of bovine serum albumin (BSA) within the coacervates. This effect is primarily due to differences in the contact between the protein and the oppositely charged poly(amino acid). Introducing the poly(amino acid) component with an opposite charge to that of the proteins as the initial step during coacervation enhances the encapsulation efficiency of protein-based drugs within the coacervates. This result could be extended to other protein systems, such as lysozyme, saporin, actin and enhanced green fluorescent protein (EGFP). Additionally, we also used P-L-Lys/P-L-Glu coacervates with β-sheet secondary structure and conducted pioneering research on the impact of secondary structures on protein encapsulation mediated by coacervates. Coacervates with β-sheet secondary structures exhibited superior protein encapsulation efficiencies compared to those with coil systems, due to the lowered entropy and reduced flexibility of polymer chains in the β-sheet systems. Furthermore, through DBSCAN, we achieved protein release from coacervates under neutral to acidic conditions, which was driven by charge.
Results and discussion
BSA encapsulation in coacervate
We conducted a series of coarse-grained molecular dynamics simulations based on the Martini 3.0 model, and both PLys and PGlu were selected with a degree of polymerization of 30 (Fig. 1a). The simulations began with the mixing of PLys and BSA for 5 μs, followed by the introduction of PGlu for an additional 8 μs of simulation. We referred to this protocol as LG. Our simulations unveiled that the combination of PLys and PGlu, adopting coil secondary structures38 spontaneously formed coacervates (Fig. S1a†). Notably, the rate of BSA encapsulation decreased as the BSA content increased (Fig. 1b and S2a†). These findings are well supported by the experimental observations.22 To explore more molecular details, we quantified the BSA-coacervate component interactions normalized by BSA abundance. It was found that PLys established significantly more contacts with BSA than PGlu (Fig. 1c and S4†). More importantly, the normalized contact number between BSA and PLys decreased, whereas it remained relatively stable between BSA and PGlu (Fig. 1c). Thus, the decreased encapsulation rate was primarily attributable to the decreased PLys/BSA contact. Furthermore, we calculated residue-based contact maps for PLys/BSA and PGlu/BSA pairs. In addition to the higher frequency of interaction between PLys and BSA protein, PLys predominantly formed contacts with oppositely charged Glu and Asp residues in BSA (Fig. 1d and S5–S7†) because BSA possesses a total of 17 negative charges dispersed on its surface (Fig. S8†) which resulted in a heightened attraction to positively charged PLys. Therefore, a rise in BSA concentration intensified the competition among BSA molecules to interact with PLys amino acids through coulombic forces, leading to a decrease in the encapsulation rate.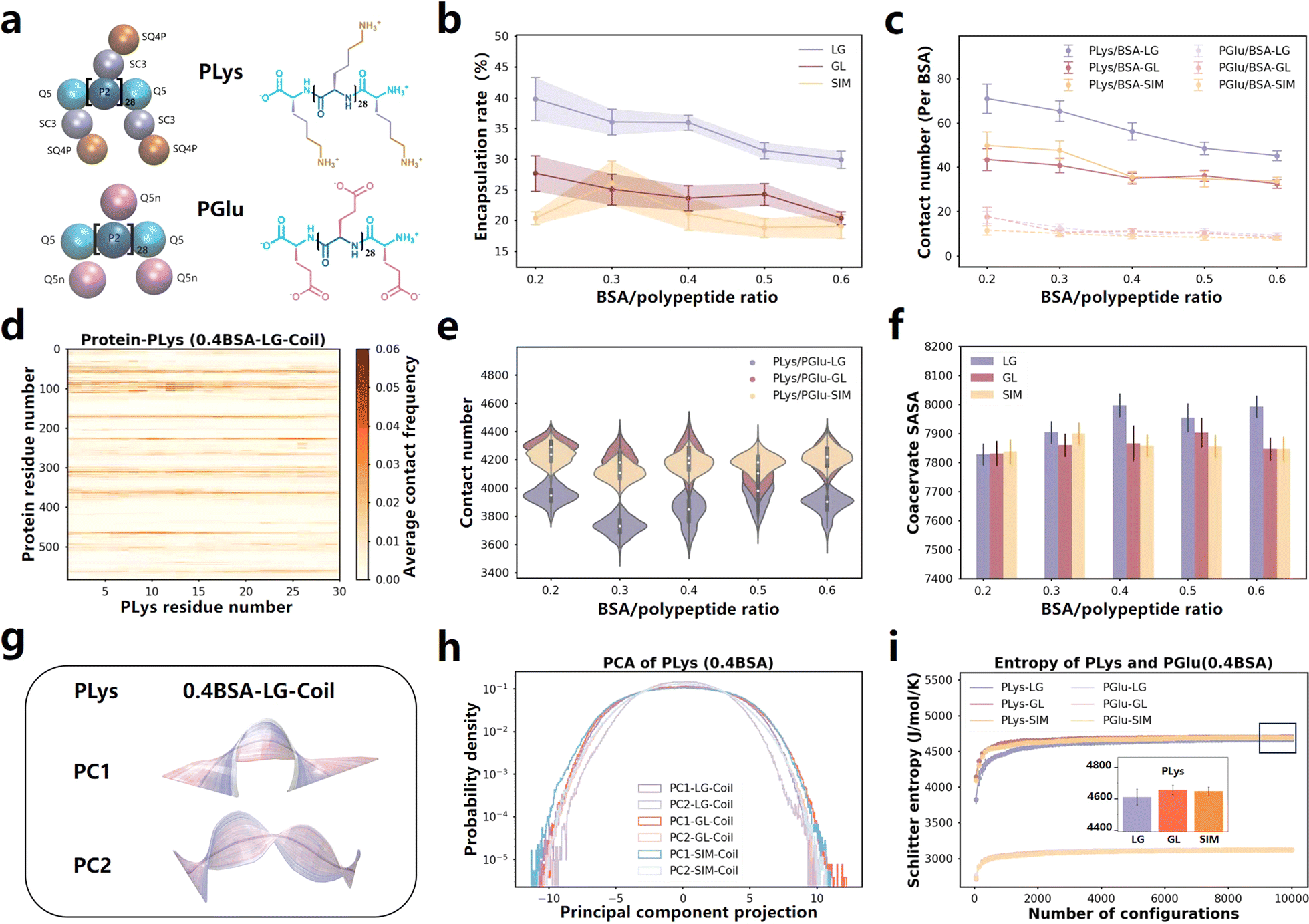 | ||
| Fig. 1 (a) Martini 3.0 CG models and chemical structures of PLys and PGlu with a degree of polymerization of 30 in simulation systems. (b) Protein encapsulation percentage by coacervates formed by different ingredient adding sequence as function of BSA content. The time-changed values are presented in Fig. S2.† (c) Contact number of PLys/BSA (solid line) and PGlu/BSA (dashed line) in various systems (normalized by BSA number). Values that change over time are displayed in Fig. S4.† (d) Contact map illustrating the residue-based contacts between PLys and BSA in 0.4BSA-LG-Coil system. The results of PLys/BSA and PGlu/BSA in other systems are presented in Fig. S5 and S6.† (e) Contact number between PLys and PGlu in various systems. Values that change over time are displayed in Fig. S9.† (f) SASA of coacervates formed by different ingredient adding sequence as BSA protein content change. The time-changed values are presented in Fig. S10.† (g) The PC1 and PC2 motion of PLys in 0.4BSA-LG-Coil system. Each illustration represents a movement of PLys from blue to white to red over time, with the mean position represented in gray. For PC1, PC2 motions of PLys and PGlu in LG, GL and SIM systems, refer to Fig. S12–S14.† (h) The PC1 and PC2 corresponding principal component projection values of PLys in various systems (0.4 BSA). The specific values of PLys and PGlu in other systems are shown in Fig. S15, S16, S18 and S19.† (i) The Schilitter entropy of PLys and PGlu in various systems with different ingredient adding sequence on coacervate formation (0.4 BSA). The numerical values of PLys in other systems and PGlu are presented in Fig. S17, S21 and S22.† | ||
Impact of ingredient adding sequence on coacervate formation and encapsulation rate
We are interested in exploring whether the ingredient adding sequence would impact the encapsulation of BSA. Apart from the LG adding sequence, we also carried out a series of simulations with different adding sequences for comparison, such as initially mixing PGlu and BSA for a period before adding PLys (GL), and simultaneously adding PLys, PGlu, and BSA (SIM). (Details can be found in Table S1.† The last frames of all the simulations are illustrated in Fig. S1b and c†). The encapsulation percentage of BSA with different adding sequences were computed (Fig. 1b, S2b and c†). The results revealed that BSA encapsulation efficiency in GL and SIM systems was lower than that in LG system. Regardless of the order of ingredient addition during coacervate formation, it was consistently observed that there were more contacts between PLys and BSA compared to PGlu and BSA. This indicated that PLys consistently played a more significant role in determining the encapsulation rate. Moreover, the number of contacts between PLys and BSA in LG systems exceeded that in other systems (see Fig. 1c). The contact map analysis (Fig. S5 and S6†) revealed that these interactions between PLys/BSA and PGlu/BSA originated from opposite charged residues. Therefore, the initial mixing of PLys and BSA in LG systems enhanced PLys/BSA contacts, leading to a higher BSA encapsulation rate compared to GL and SIM systems. These enhanced PLys/BSA contacts are primarily driven by Coulomb interactions.Moreover, we focused on the characteristics of the coacervates in LG, GL and SIM systems and found that the contact number of PLys/PGlu in all systems remained constant regardless of variations in BSA concentration (Fig. 1e, the specific values as functions of time are shown in Fig. S9†). This suggested that the stability of the coacervates was not influenced by changes in BSA concentration. In addition, the contact number between PLys and PGlu in the LG sequence were consistently lower than GL and SIM systems (see Fig. 1e). This could be attributed to the higher PLys/BSA contact in LG systems (Fig. 1c), which consequently reduced the PLys/PGlu contact due to steric hindrance. What's more, the solvent-accessible surface area (SASA) of the coacervates formed under the LG sequence was higher than those of the GL and SIM systems (Fig. 1f, S10a and c†). Analysis of the number of clusters also revealed that there are the highest number of coacervate clusters after equilibrium in LG systems (Fig. S49†).
Therefore, coacervates formed in LG systems were smaller and more dispersed than those in other systems.
To gain a comprehensive understanding of the behavior of PLys and PGlu chains during the coacervation and protein encapsulation processes in LG, GL and SIM systems, we applied the PCA machine learning algorithm and calculated the Schilitter entropy.54,55 These computations allowed us to examine the state of individual PLys and PGlu chain, classify their structures into various subcategories based on the covariance matrix, and evaluate the accessible configurational state space of amino acid chains. We investigated the effect of BSA concentration and ingredient addition sequence on coacervate formation on the motion pattern of peptide chains, respectively. In Fig. 1g, principal components (PC1, PC2) were ordered based on their eigenvalues (see Fig. S14†), with PC1 and PC2 representing the open/close and torsion motions of both PLys and PGlu in all systems (as detailed in Fig. S15–S17†). The BSA content had minimal impact on the PC1 and PC2 projection, indicating similar motion patterns of poly(amino acid)s (Fig. S11 and S12†), which aligned with the observed similarity in Schilitter entropy (Fig. S13†). When considering the ingredient adding sequence, PC1 projections showed similarities in LG, GL, and SIM systems, indicating that the sequence had limited impact on the PC1 motion of poly(amino acid) chains (Fig. 1h and S18–S19†). This observation was consistent with our discovery that end-to-end distances were similar in LG, GL, and SIM systems (see Fig. S20†). However, we detected differences in distributions of PC2 projections for PLys chains in LG, GL and SIM systems, although they were all Gaussian distributions (see Fig. 1h and S18†). In the LG system, the PC2 distribution of PLys was narrower than those in GL and SIM systems, indicating that its twisting was restricted. This was in agreement with the Schilitter entropy analysis, which showed that PLys in the LG system had lower entropy values due to its limited rotation, reducing the degree of disorder in the chain motion (Fig. 1i and S21†). This indicated that, compared to other adding sequence systems, the overall mobility of PLys around BSA was more strongly restricted, making it difficult to twist and tightly binding to the protein surface. As a result, the PLys/BSA contact and BSA encapsulation rate in LG systems were higher than the other two systems.
Non-reactive molecular dynamics we used is based on the ergodic hypothesis, which states that since there is a one-to-one correspondence between the initial conditions of a system and the state at some other time, averaging over a large number of initial conditions is equivalent to averaging over time-evolved states of the system. Given sufficient time, a system can explore all thermodynamic states located on this surface.56,57 Ideally, any initial configurations should converge to the global minimum on the free energy surface. However, in experiment, the procedure for adding ingredients affects the final results due to the constrained experimental time, leading to being captured in kinetic traps. Angela Valbuzzi found that Anti-TRAP(AT) inhibition of the RNA with trp RNA-binding attenuation protein interaction is determined kinetically. And whether AT first exists in the system will significantly affect the binding results of AT and RNA.58 The different addition sequences of I-catalysts significantly impact the rate of the ascorbic acid iodine clock reaction.59 The more limited simulation time makes it harder to sample all the state points on the free energy surface. This is equally effective for our systems. Despite being a metastable state, it plays a crucial role in fields such as catalysis and drug delivery, necessitating thorough investigation.
The impact of ingredient addition sequence on other protein systems
We conducted simulations of the coacervate encapsulation of saporin, lysozyme, actin, and EGFP proteins in the LG, GL and SIM sequence to explore the potential extension of the ingredient addition sequence effect to other proteins (details can be found in Table S2†). All simulation systems shared the same framework as BSA systems, with the only variation being the type of protein. Final simulation snapshots of all systems are shown in Fig. 2a and S23.† We calculated the SASA and the contact number between PLys and PGlu to verify the stable formation of coacervates. (Fig. S24 and S25†). Notably, the rate of protein encapsulation varied based on the specific adding sequence used. For saporin and lysozyme proteins, coacervates with the GL sequence exhibited the highest protein encapsulation rates (Fig. 2b and S27†). Conversely, for EGFP and actin proteins, coacervates with the LG sequence demonstrated the highest protein encapsulation rates (Fig. 2b and S27†). Therefore, besides BSA protein, the ingredient addition sequence could impact protein encapsulation for other proteins.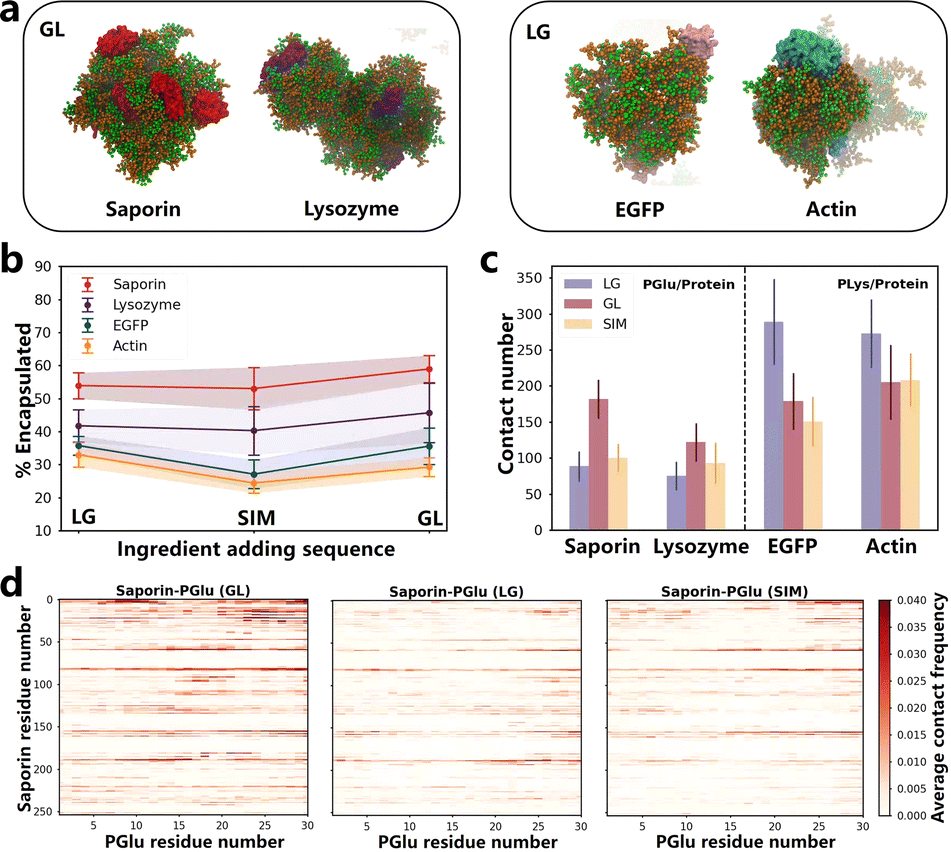 | ||
| Fig. 2 (a) The snapshots of the simulations in saporin-GL, lysozyme-GL, EGFP-LG and actin-LG systems, and the simulation snapshots of all systems are shown in Fig. S23.† Orange represents PLys, green represents PGlu, red represents saporin, purple represents lysozyme, pink represents EGFP, and light blue represents actin. Water and ions are not shown for clarity. (b) Protein encapsulation percentage in coacervates formed by different ingredient adding sequence. All P values were < 0.05, so the encapsulation rates for different ingredient adding sequence are significantly different. The time-dependent values are presented in Fig. S27.† (c) Contact number of PGlu/protein and PLys/protein in various systems. Values that change over time are displayed in Fig. S28.† (d) Contact maps illustrating the residue-based contacts between PLys and saporin in LG, GL and SIM systems. The results of PLys/protein and PGlu/protein in other systems are presented in Fig. S29–S32.† | ||
We conducted separate calculations for the number of contacts of PLys/protein and PGlu/protein within distinct coacervate systems (Fig. S28†), accompanied by the generation of their corresponding contact maps (Fig. S29–S32†). Similar to BSA protein systems, altering the adding sequence of ingredients in coacervates resulted in variations in poly(amino acid)/protein contact numbers, consequently affecting the loading efficiency.
Significant differences were noted in number of PLys/protein contacts for the negatively charged EGFP and actin proteins when exposed to different ingredient adding sequences during coacervate formation (see Fig. S28†). Specifically, the LG sequence resulted in the highest number of contacts (Fig. 2c). In contrast, for saporin and lysozyme proteins, characterized by an overall positive charge, number of PGlu/protein contacts exhibited significant differences, with the GL sequence yielding maximum contacts (Fig. 2c and S28†). These findings were consistent with outcomes of the contact maps analysis that the LG sequence for EGFP and actin proteins displayed the darkest colors in the contact map compared to the other two sequences, while the GL sequence for saporin and lysozyme proteins exhibited the deepest colors (Fig. 2d and S29–S32†). For example, in the contact maps of saporin with PGlu, it can be observed that when saporin residues are near positions 25, 60 or 80, the contact maps under the GL sequence are notably more intense (redder) compared to those under LG and SIM sequences (Fig. 2d). Consequently, in GL sequence systems involving positively charged proteins, the initial addition of PGlu resulted in strong electrostatic interactions, leading to a higher rate of protein encapsulation. Similarly, for negatively charged proteins, the initial incorporation of PLys in the LG sequence enhanced the rate of protein encapsulation within coacervates. It was worth noting that in all protein systems, the highest contact point between proteins and poly(amino acid)s was located on the charged patches of the protein surface (Fig. S8 and S28–S32†). Therefore, the adding sequence of ingredient during coacervate formation played a crucial role in influencing protein encapsulation, which was determined by the overall charge carried by the protein itself, along with patches of charged residues on the protein surface (Fig. S8†). The experiment conducted by Sarah L. Perry, et al. supported our conclusions.35 By manipulating the sequence in which poly(amino acid)s and proteins were added into systems, we could leverage electrostatic interactions to control protein encapsulation, achieving systems with high encapsulation efficiencies to enhance the therapeutic effectiveness.
Effect of secondary structure on protein encapsulation in coacervates
In the aforementioned study, we discovered that the restricted motion of poly(amino acid)s led to variations in the encapsulation efficiency of coacervates for proteins. This finding prompted us to investigate the impact of more constrained polymer chains (β-sheet secondary structure) on protein encapsulation. There are already researches to prove that the secondary structure plays a critical role in the folding and assembly behaviors of macromolecules.60–65 However, its impact on the protein encapsulation within coacervates has yet to be explored. To this end, we conducted Martini molecular dynamics simulations on coacervates exhibiting different secondary structures, comparing their ability to encapsulate BSA protein. The β-sheet and coil conformations of PLys and PGlu were controlled by altering the chirality of the monomers.61,65 As demonstrated by Matthew Tirrell, Juan J. de Pablo, et al.,38 the secondary structure formed by P-L-Lys and P-L,D-Glu are coils, while P-L-Lys and P-L-Glu form a β-sheet secondary structure. After simulations using Martini models for approximately 8 μs in LG sequence, PLys/PGlu coacervates formed and BSA proteins were encapsulated (Fig. S2a and Table S3†). Notably, β-sheet coacervate systems demonstrated higher encapsulation efficiencies compared to coil systems (Fig. 3a and S33c†). This finding was further confirmed by the radial distribution function (RDF) between poly(amino acid)s and BSA in Fig. 3b, where β-sheet systems displayed higher first and second peaks compared to coil systems.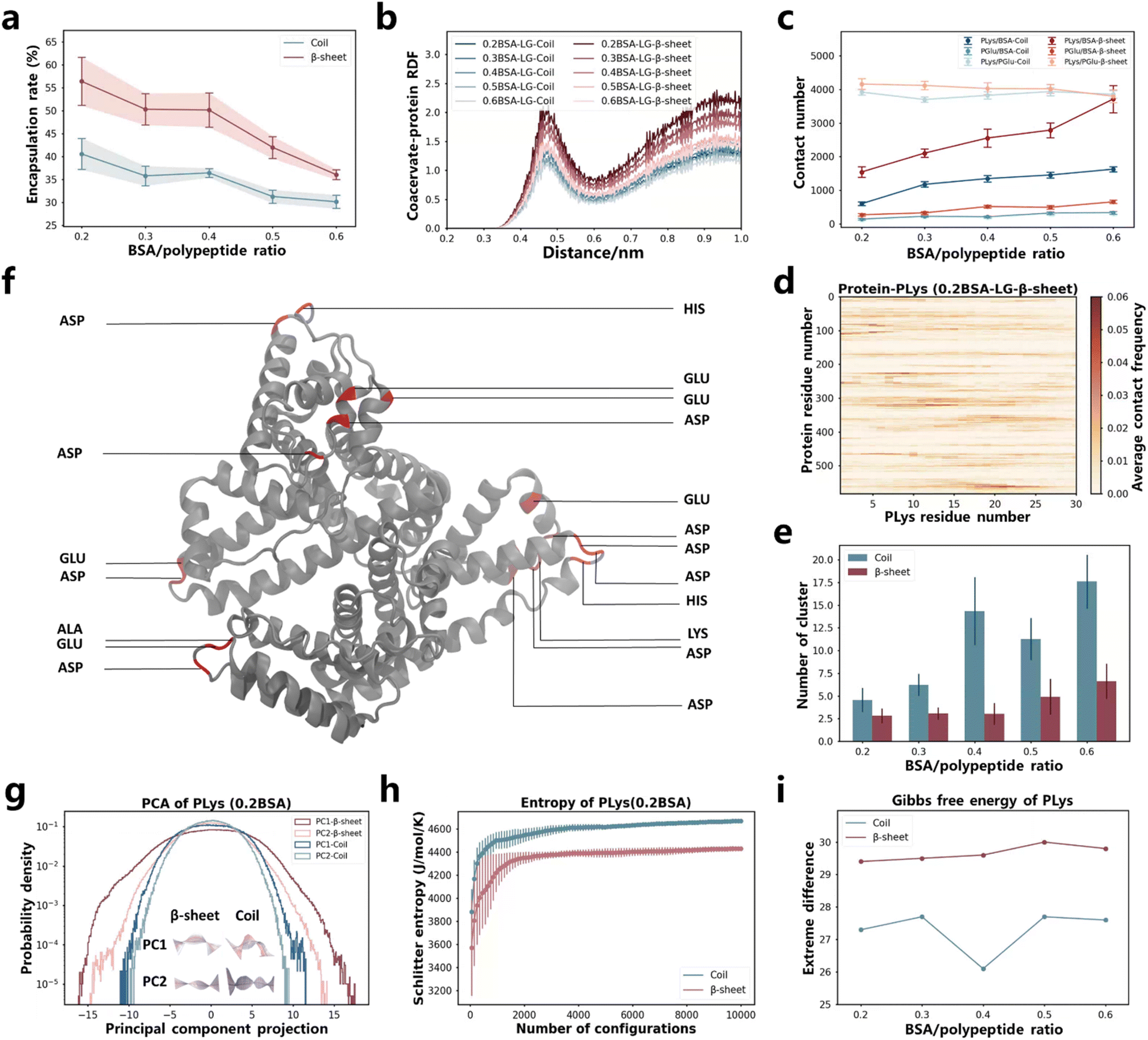 | ||
| Fig. 3 (a) Protein encapsulation percentage by coacervates as function of BSA protein content. The time-changed values are presented in Fig. S33c.† (b) RDF between coacervates (including both PLys and PGlu) and protein under different BSA content. (c) Contact number between different components in the systems. Values that change over time are displayed in Fig. S34 and S37.† (d) Contact map illustrating the residue-based contacts between BSA and PLys in the 0.2BSA-LG-β-sheet system. The results of other systems are presented in Fig. S35 and S36.† (e) NCluster of coacervates as BSA protein content change. The time-changed values are presented in Fig. S33g.† (f) The highlight of residues in BSA that have the highest contact frequency with PLys in the 0.2BSA-LG-β-sheet system. (g) The PC1 and PC2 motion of PLys and its corresponding principal component projection values in various secondary structural systems (0.2BSA). The middle illustrations represent the movement of PLys from blue to white to red over time, with the mean position represented in gray. For specific values and PC1, PC2 motion of other systems with different BSA content, refer to Fig. S38–S40.† (h) The Schilitter entropy of PLys in different secondary structural systems (0.2BSA). The numerical values of PLys in other systems and PGlu are presented in Fig. S42 and S43.† (i) Difference between extreme values of PLys Gibbs free energy. The numerical values of PLys and PGlu are presented in Fig. S44 and S45.† | ||
In order to investigate mechanism behind the encapsulation process, we computed the contact number of PLys/BSA and PGlu/BSA in both secondary structures (see Fig. 3c and S34†). Our results indicated that contact numbers between PLys and BSA, as well as between PGlu and BSA were higher in β-sheet systems than those in coil systems. This difference was particularly pronounced in the contact number between PLys and BSA due to their opposite charges. Furthermore, we generated contact maps for PLys/BSA and PGlu/BSA based on residue interaction in Fig. 3d, S35a–e and S36a–e.† We also highlighted top 20 residue pairs with the highest contact frequency in interactions between BSA protein and PLys (exemplified by the 0.2BSA-LG-β-sheet system) in Fig. 3f, and PGlu in Fig. S36f.† Apart from higher contact frequency between PLys and BSA protein, our findings indicated that most of contacts made by PLys were with Glu and Asp residues in BSA protein, while PGlu contacts were primarily made with Lys and Arg residues. These contact pairs possessed opposite charges. Therefore, the higher engulfment efficiency of β-sheet systems was primarily attributed to the higher contact between PLys and BSA, particularly involving Glu and Asp residues in the BSA protein. Moreover, we characterized the state or configuration of coacervates themselves. The number of cluster (NCluster) analysis results indicated that β-sheet systems ultimately formed fewer coacervate clusters (Fig. 3e and S33g†). And PLys/PGlu contact numbers were higher in β-sheet systems regardless of number of BSA protein numbers presented in the system (Fig. 3c and S37†). These results indicated that coacervates formed by PLys/PGlu in the β-sheet secondary structure were larger in size compared to those formed by poly(amino acid)s in coil systems (Fig. 3a and S2a†).
The behavior of PLys and PGlu during coacervation and protein encapsulation processes were analyzed using the PCA machine learning algorithm and Schilitter entropy calculations. From PCA, we obtained main motion patterns of PLys and PGlu chains during the coacervation and protein encapsulation processes, and it could be seen that PC1 and PC2 components were ranked according to their eigenvalues (Fig. S38a and S39a†), with PC1 and PC2 representing open/close and torsion motions of both PLys and PGlu in all systems (see Fig. S40†). Distributions of PC1 and PC2 projections for both PLys and PGlu chains were observed to remain unchanged regardless of the amount of BSA present, and both PC1 and PC2 adopt Gaussian distributions regardless of the secondary structure system (Fig. 3g, S38 and S39†). Moreover, our results demonstrated that the projection value distribution in β-sheet systems was considerably wider in comparison to coil systems, irrespective of whether it was PC1 or PC2, especially for PLys. This suggested that the amino acid chain within the β-sheet system exhibited a greater level of expansion and distortion. This was consistent with calculation results of the end-to-end distance analysis, where lengths of PLys and PGlu were larger in β-sheet systems (Fig. S41†). This also provided a reason for the larger SASA of whole coacervates in β-sheet systems (Fig. S33d and f†). Additionally, we calculated the Schilitter entropy for both PLys and PGlu chains, the entropy was lower in β-sheet systems than coil systems, indicating a smaller configurational space (Fig. 3h, S42 and S43†). We also computed the 2D Gibbs free energy landscape for both PLys and PGlu chains in different secondary structures as functions of PC1 and PC2, (Fig. 3i, S44 and S45†). We observed a more pronounced difference between extreme values of Gibbs free energy for both PLys and PGlu in β-sheet systems compared to coil systems. This indicated that there was greater restriction on extreme movements of poly(amino acid)s in β-sheet systems. Based on these analyses, it could be speculated that the movement of the poly(amino acid) single chain was confined to a broader structure in β-sheet systems, resulting in the formation of larger coacervates and leading to enhanced efficiency in encapsulating BSA.
pH-Induced dissolution of coacervates and protein release
One of the methods to control the formation and dissolution of coacervate droplets is to adjust the pH level.66–70 In order to investigate the state of coacervates at different pH values, we utilized the pH responsiveness of PGlu to manipulate the liquid–liquid phase separation and drove the release of BSA protein in simulations (Fig. 4b). The ratio of protonated and deprotonated PGlu species was determined at various pH levels, utilizing the pKa value as a basis for calculation. Simulation details were extracted from the experimental setting by Matthew Tirrell and his colleagues22 and could be found in Table S4.† After more than 8 μs simulations, coacervates formed and enclosed BSA when the pH level was at 5, as depicted in Fig. 4a. With an increase in acidity to pH 4 and 3, some of aggregated droplets became more dispersed. When the pH reached 2, both coacervates and BSA proteins became fully dispersed. This trend agreed well with experimental results.22 It is noteworthy that approximately after 5 μs of simulation in all systems, the contact number between poly(amino acid) and BSA remained stable over time, indicating complete dispersion of coacervates and BSA(Fig. S47d and e†).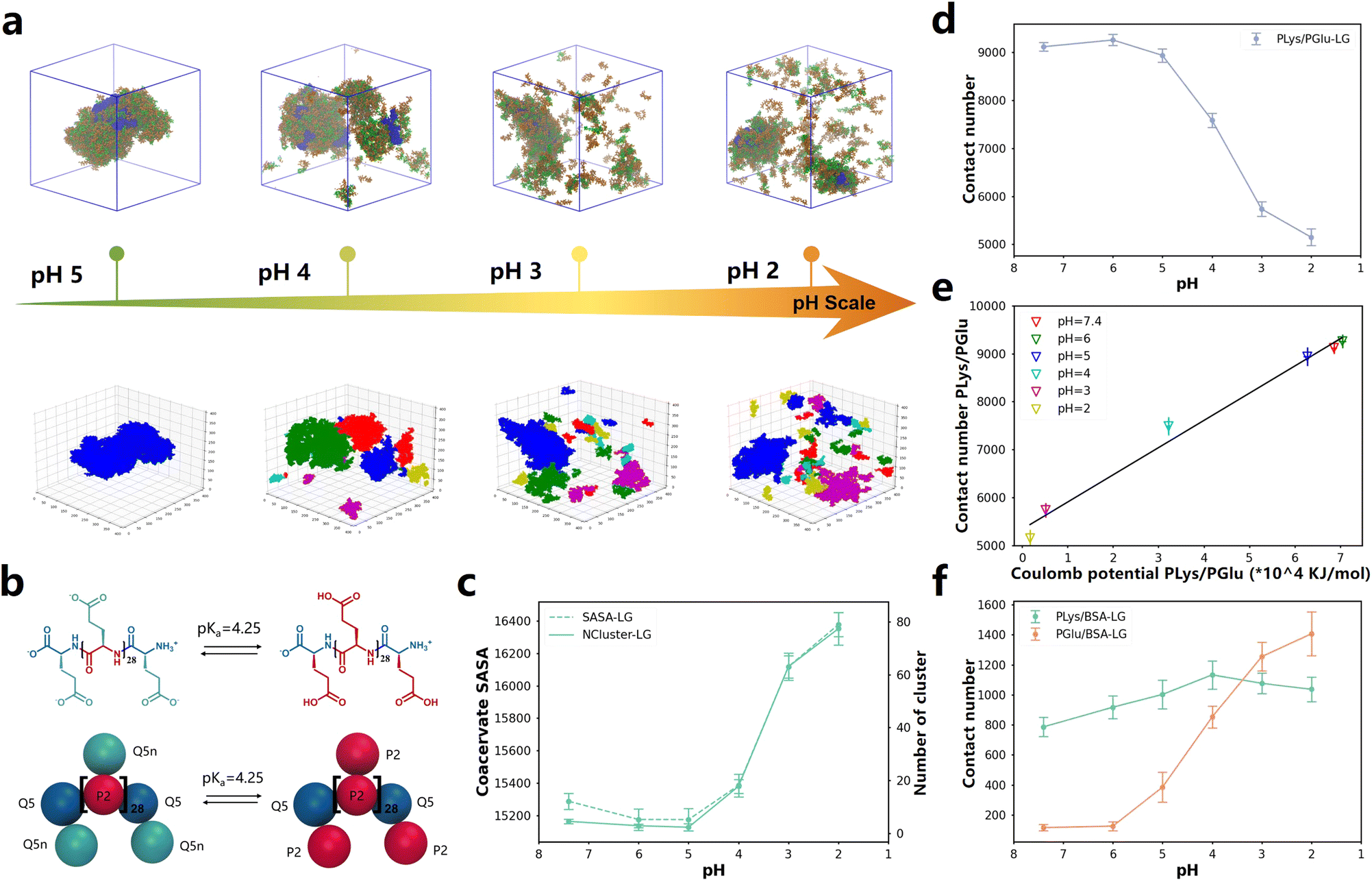 | ||
| Fig. 4 (a) Up: snapshots of the final states of 0.05BSA systems after about 8 μs of CG MD simulations at different pH values. Down: the DBSCAN algorithm clustering models. The complete snapshot is shown in Fig. S45a.† (b) Chemical structures and Martini 3.0 CG models of PGlu before and after protonation. (c) The variation curve of the NCluster and SASA with changes in pH value. The dashed line represents SASA, the solid line represents NCluster. Values that change over time are displayed in Fig. S46a and b.† (d) The contact number of PLys and PGlu after equilibrium in different pH systems. Values that change over time are displayed in Fig. S46c.† (e) Linear correlation between Coulomb potential and contact number between PLys and PGlu at different pH values. (f) The change in pH affects the quantity of contact in PLys/BSA and PGlu/BSA. Values that change over time are displayed in Fig. S46d and e.† | ||
To quantify changes in the configuration of coacervates under different pH conditions, we employed the DBSCAN machine learning algorithm to determine the phase separation level of coacervates within the system, as illustrated in Fig. 4a, S46a and ESI Movies 1–4.† Our findings revealed that both PLys and PGlu displayed notable aggregation effects at pH 5 and higher, forming continuous and dense coacervates. However, as the pH decreased, coacervates appeared fragmented, resulting in an increased number of clusters detected by DBSCAN clustering models, as shown in Fig. S46c.† Consequently, condensed droplets began to decompose as the pH decreased. This phenomenon was also confirmed in the contact number, NCluster, and SASA of different coacervate components. As the pH value decreased, there was an elevation in the NCluster and SASA of coacervate droplets (Fig. 4c and S47a and S47b†), accompanied by a reduction in the contact number between PLys and PGlu (Fig. 4d and S47c†). Furthermore, the pH value of approximately 4 exhibited the most significant changes in contact number, number of clusters, and SASA, indicating a potential phase transition point. Notably, this pH transition point between 3 and 5 aligned with experimental observations.22 To investigate the mechanism responsible for the degradation of coacervates, we quantified the Coulomb potential between PLys and PGlu in Fig. S47f.† It was observed that an increase in the Coulomb potential resulted in a linear increase in the PGlu/PLys contact number (Fig. 4e). Thus, the disintegration of coacervates was attributed to the decrease in electrostatic interaction between PLys and PGlu.
Interestingly, despite observing the disruption of coacervates as the pH value decreased, we noticed an increase in the contact between BSA and poly(amino acid)s (Fig. 4a and S47a†). To quantify this phenomenon, we computed the contact number of PGlu/BSA and PLys/BSA at different pH levels (Fig. 4f, S47d and e†). The PGlu/BSA contact increased with the disintegration of coacervate droplets (decrease of pH value), while the PLys/BSA contact remained relatively stable. This was because at high pH levels, both PGlu (prior to protonation) and BSA carried negative charges, which led to a strong repulsion between PGlu and BSA. As the pH level decreased, the protonation degree of PGlu increased, diminishing the repulsive force between PGlu and BSA, resulting in an increased contact frequency. Furthermore, within the pH range of 2–7.4, the protonation state of PLys remained constant, resulting in minimal changes in the contact number between PLys and BSA. It should be noted that at low pH levels, the BSA protein was primarily surrounded by a single layer of poly(amino acid)s, which was no longer in a coacervate state. Therefore, even though the coacervate dissolved at low pH, BSA protein was still wrapped by some PGlu and PLys chains, which could not be captured in experiments.22
Conclusions
Overall, we used the Martini 3.0 model to conduct molecular dynamics simulations, specifically focusing on coacervate drug delivery systems. Our investigation highlights the critical influence of ingredient addition sequence on coacervate formation and encapsulation rates of proteins. These effects stem from attractive forces between oppositely charged proteins and poly(amino acid)s. For instance, the positively charged saporin and lysozyme protein exhibited the highest encapsulation efficiency when first combined with PGlu, followed by the addition of PLys. Conversely, the negatively charged BSA, actin and EGFP proteins demonstrated optimal encapsulation performance when PLys was introduced prior to PGlu. Moreover, we used PLys/PGlu coacervates with different secondary structures to encapsulate BSA protein and results revealed that coacervates characterized by β-sheet configurations exhibited superior encapsulation efficiency. This enhancement was attributed to the lower entropy and reduced flexibility of PLys and PGlu in β-sheet systems.Furthermore, We investigated the pH sensitivity of coacervates and applied machine learning algorithms to analyze the dissolution process of coacervate from neutral to acidic environments. This process was found driven by Coulomb interactions between PGlu and PLys. In many disease microenvironments, such as inflammation and tumors, the pH value is lower and levels of reactive oxygen species (ROS) are higher compared to the normal cellular environment.71–73 Plys/PGlu polyelectrolytes are in a coacervate state in the cellular environment, whereas in disease microenvironments, protonated polyglutamic acid (PGlu+H) causes the dissolution of the coacervate, thereby achieving specific drug release. Therefore, our comprehensive study significantly enhances the understanding of pH sensitive coacervate mechanisms governing the encapsulation of pharmaceutical substances, and drug release within disease microenvironments. This maximizes the therapeutic effects of encapsulated agents and offers guidance for the clinical application of coacervates in drug delivery systems. However, the mechanisms by which redox-responsive coacervates form, encapsulate proteins, and penetrate the cell membrane to enter cells remain unclear, which are already included in our future research plan.
Methods
All simulations in this study were conducted using GROMACS software74 and the Martini 3.0 force field.41 A time step of 20 fs was used in constant NPT (number, pressure, and temperature) ensemble. The Lennard-Jones and Coulomb potentials were shifted to zero at a cut-off of 1.1 nm and the long-range electrostatics was treated using a reaction field (εrf = 15 beyond the cut-off). The neighbor list was updated with the Verlet neighbor search algorithm. The pressure was coupled with the Parrinello Rahman algorithm at 1 bar with a compressibility of 3 × 10−4 bar−1 and a time constant of 12 ps. The temperatures of the systems were coupled at 298 K using a v-rescale thermostat with a time constant of 1 ps. The salt concentration was 0.15 M unless otherwise stated.Principal Component Analysis (PCA) was applied to reduce dimensionality and extract motion characteristics of poly(amino acid) chains. We initiated the process by standardizing the dataset, ensuring that each feature has a mean of 0 and a standard deviation of 1. The covariance matrix of the standardized data was computed to assess the correlations between features. Following this, we determined the optimal directions for data transformation by solving for the eigenvalues and eigenvectors of the covariance matrix that we selected the eigenvectors corresponding to the top 35 largest eigenvalues to form the transformation matrix. We finally project the original motion data into a new coordinate system, completing the dimensionality reduction of the data.
Density-Based Spatial Clustering of Applications with Noise (DBSCAN) was applied in our work, we calculated the truncation radius between PLys and PGlu based on their position of the first valley in RDF and selected it as the neighborhood radius Epsilon ε = 0.6 nm (Fig. S3, S26, S33 and S48†). In order to ensure that each cluster contains at least one PLys and one PGlu, and since the content we feed is three-dimensional coordinates, we assigned a minimum point value (MinPts) of 4. Both ε and MinPts significantly influence the clustering results of DBSCAN. Subsequently, we fed the coordinates of all PLys and PGlu beads as data points and calculated the number of neighboring points within the ε neighborhood for each data point. If a data point had at least 3 neighboring points within its ε-neighborhood, we marked it as a core point. We then identified all points that were directly density-reachable, forming a cluster. This process continued until no new points could be added to any cluster. Finally, the number of clusters was calculated. We implemented this process through Python.
For more details, please refer to the ESI methods.†
Data availability
The data that support the findings of this study are available on request from the corresponding author upon reasonable request.Author contributions
Y. W. and R. Z. contributed equally. M. D., Y. L., Q. F. and H. T. supervised the project and wrote the manuscript with Y. W. and R. Z. besides, Y. W. and R. Z. performed all the experiments. Y. Z., C. P., Y. Z. was responsible for data curation and analysis, and assisted in writing. All authors discussed the results and reviewed the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support provided by the National Natural Science Foundation of China (Grant No. 52022062, 52303298, 52273140 and 51873118), the Application and Basic Research of Sichuan Department of Science and Technology and the Project of State Key Laboratory of Polymer Materials Engineering.Notes and references
- B. Leader, Q. J. Baca and D. E. Golan, Nat. Rev. Drug Discovery, 2008, 7, 21–39 CrossRef CAS PubMed.
- X. Li, W. Yang, Y. Zou, F. Meng, C. Deng and Z. Zhong, J. Contr. Release, 2015, 220, 704–714 CrossRef CAS PubMed.
- D. W. Long, N. R. Johnson, E. M. Jeffries, H. Hara and Y. Wang, J. Contr. Release, 2017, 253, 73–81 CrossRef CAS PubMed.
- A. Mullard, Nat. Rev. Drug Discovery, 2021, 20, 85–91 CrossRef CAS PubMed.
- M. Yu, J. Wu, J. Shi and O. C. Farokhzad, J. Contr. Release, 2016, 240, 24–37 CrossRef CAS PubMed.
- A. Erazo-Oliveras, K. Najjar, L. Dayani, T. Y. Wang, G. A. Johnson and J. P. Pellois, Nat. Methods, 2014, 11, 861–867 CrossRef CAS PubMed.
- J. V. Gregory, P. Kadiyala, R. Doherty, M. Cadena, S. Habeel, E. Ruoslahti, P. R. Lowenstein, M. G. Castro and J. Lahann, Nat. Commun., 2020, 11, 5687 CrossRef CAS PubMed.
- N. Yim, S. W. Ryu, K. Choi, K. R. Lee, S. Lee, H. Choi, J. Kim, M. R. Shaker, W. Sun and J. H. Park, Nat. Commun., 2016, 7, 1–9 Search PubMed.
- Y. W. Chao, Y. L. Lee, C.-S. Tseng, L. U. H. Wang, K. C. Hsia, H. Chen, J. M. Fustin, S. Azeem, T. T. Chang and C. Y. Chen, ACS Nano, 2024, 18, 4822–4839 CrossRef PubMed.
- X. Zhu, R. Tang, S. Wang, X. Chen, J. Hu, C. Lei, Y. Huang, H. Wang, Z. Nie and S. Yao, ACS Nano, 2020, 14, 2172–2182 CrossRef CAS PubMed.
- T. Wei, Q. Cheng, L. Farbiak, D. G. Anderson, R. Langer and D. J. Siegwart, ACS Nano, 2020, 14, 9243–9262 CrossRef CAS PubMed.
- T. Fang, C. H. Van Elssen, J. N. Duarte, J. S. Guzman, J. S. Chahal, J. Ling and H. L. Ploegh, Chem. Sci., 2017, 8, 5591–5597 RSC.
- S. Hyun, Y. Choi, H. N. Lee, C. Lee, D. Oh, D. K. Lee, C. Lee, Y. Lee and J. Yu, Chem. Sci., 2018, 9, 3820–3827 RSC.
- B. Kawahara, L. Gao, W. Cohn, J. P. Whitelegge, S. Sen, C. Janzen and P. K. Mascharak, Chem. Sci., 2020, 11, 467–473 RSC.
- L. R. Brown, Expet Opin. Drug Deliv., 2005, 2, 29–42 CrossRef CAS PubMed.
- S. Frokjaer and D. E. Otzen, Nat. Rev. Drug Discovery, 2005, 4, 298–306 CrossRef CAS PubMed.
- M. C. Manning, D. K. Chou, B. M. Murphy, R. W. Payne and D. S. Katayama, Pharm. Res., 2010, 27, 544–575 CrossRef PubMed.
- L. W. Chang, T. K. Lytle, M. Radhakrishna, J. J. Madinya, J. Vélez, C. E. Sing and S. L. Perry, Nat. Commun., 2017, 8, 1273 CrossRef PubMed.
- J. P. Armstrong, S. N. Olof, M. D. Jakimowicz, A. P. Hollander, S. Mann, S. A. Davis, M. J. Miles, A. J. Patil and A. W. Perriman, Chem. Sci., 2015, 6, 6106–6111 RSC.
- A. Harada and K. Kataoka, Macromolecules, 1995, 28, 5294–5299 CrossRef CAS.
- T. Trantidou, M. Friddin, Y. Elani, N. J. Brooks, R. V. Law, J. M. Seddon and O. Ces, ACS Nano, 2017, 11, 6549–6565 CrossRef CAS PubMed.
- K. A. Black, D. Priftis, S. L. Perry, J. Yip, W. Y. Byun and M. Tirrell, ACS Macro Lett., 2014, 3, 1088–1091 CrossRef CAS PubMed.
- F. P. Cakmak, S. Choi, M. O. Meyer, P. C. Bevilacqua and C. D. Keating, Nat. Commun., 2020, 11, 5949 CrossRef CAS PubMed.
- D. Priftis and M. Tirrell, Soft Matter, 2012, 8, 9396–9405 RSC.
- L. Li, S. Srivastava, M. Andreev, A. B. Marciel, J. J. de Pablo and M. V. Tirrell, Macromolecules, 2018, 51, 2988–2995 CrossRef CAS.
- K. Iwashita, A. Handa and K. Shiraki, Int. J. Biol. Macromol., 2018, 120, 10–18 CrossRef CAS PubMed.
- B. M. Johnston, C. W. Johnston, R. A. Letteri, T. K. Lytle, C. E. Sing, T. Emrick and S. L. Perry, Org. Biomol. Chem., 2017, 15, 7630–7642 RSC.
- A. C. Obermeyer, C. E. Mills, X. H. Dong, R. J. Flores and B. D. Olsen, Soft Matter, 2016, 12, 3570–3581 RSC.
- J. Wang, M. Abbas, J. Wang and E. Spruijt, Nat. Commun., 2023, 14, 8492 CrossRef CAS PubMed.
- M. I. Bajestani, S. Kader, M. Monavarian, S. M. Mousavi, E. Jabbari and A. Jafari, Int. J. Biol. Macromol., 2020, 142, 790–802 CrossRef CAS PubMed.
- O. A. Balogun-Agbaje, O. A. Odeniyi and M. A. Odeniyi, Futur. J. Pharm. Sci., 2021, 7, 125 CrossRef.
- S. Chen, S. Huang, Y. Li and C. Zhou, Front. Chem., 2021, 9, 659304 CrossRef CAS PubMed.
- Y. Meng, Q. Xue, J. Chen, Y. Li and Z. Shao, J. Dairy Sci., 2022, 105, 3746–3757 CrossRef CAS PubMed.
- Y. Zhang, W. Song, Y. Lu, Y. Xu, C. Wang, D.-G. Yu and I. Kim, Biomolecules, 2022, 12, 636 CrossRef CAS PubMed.
- W. C. B. McTigue and S. L. Perry, Soft Matter, 2019, 15, 3089–3103 RSC.
- W. C. Blocher and S. L. Perry, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2017, 9, e1442 Search PubMed.
- Z. Li, Y. Yang, C. Peng, H. Liu, R. Yang, Y. Zheng, L. Cai, H. Tan, Q. Fu and M. Ding, Chin. Chem. Lett., 2021, 32, 1563–1566 CrossRef CAS.
- S. L. Perry, L. Leon, K. Q. Hoffmann, M. J. Kade, D. Priftis, K. A. Black, D. Wong, R. A. Klein, C. F. Pierce III and K. O. Margossian, Nat. Commun., 2015, 6, 6052 CrossRef CAS PubMed.
- M. Tsanai, P. W. Frederix, C. F. Schroer, P. C. Souza and S. J. Marrink, Chem. Sci., 2021, 12, 8521–8530 RSC.
- Q. Xu, Y. Wang, Y. Zheng, Y. Zhu, Z. Li, Y. Liu and M. Ding, Biomacromolecules, 2024, 18, 382–388 Search PubMed.
- P. C. Souza, R. Alessandri, J. Barnoud, S. Thallmair, I. Faustino, F. Grünewald, I. Patmanidis, H. Abdizadeh, B. M. Bruininks and T. A. Wassenaar, Nat. Methods, 2021, 18, 382–388 CrossRef CAS PubMed.
- Y. Liu, X. Wang, Z. Wan, T. Ngai and Y. L. S. Tse, Chem. Sci., 2023, 14, 1168–1175 RSC.
- S. J. Marrink, H. J. Risselada, S. Yefimov, D. P. Tieleman and A. H. De Vries, J. Phys. Chem. B, 2007, 111, 7812–7824 CrossRef CAS PubMed.
- S. Mondal and Q. Cui, Chem. Sci., 2022, 13, 7933–7946 RSC.
- L. Monticelli, S. K. Kandasamy, X. Periole, R. G. Larson, D. P. Tieleman and S. J. Marrink, J. Chem. Theory Comput., 2008, 4, 819–834 CrossRef CAS PubMed.
- J. J. Uusitalo, H. I. Ingólfsson, P. Akhshi, D. P. Tieleman and S. J. Marrink, J. Chem. Theory Comput., 2015, 11, 3932–3945 CrossRef CAS PubMed.
- F. Grunewald, G. Rossi, A. H. De Vries, S. J. Marrink and L. Monticelli, J. Phys. Chem. B, 2018, 122, 7436–7449 CrossRef CAS PubMed.
- E. Panizon, D. Bochicchio, L. Monticelli and G. Rossi, J. Phys. Chem. B, 2015, 119, 8209–8216 CrossRef CAS PubMed.
- S. Mondal and Q. Cui, J. Phys. Chem. Lett., 2023, 14, 4532–4540 CrossRef CAS PubMed.
- M. Ester, H.-P. Kriegel, J. Sander and X. Xu, A density-based algorithm for discovering clusters in large spatial databases with noise, kdd96, 1996, pp. 226–231 Search PubMed.
- C. Labrín and F. Urdinez, in R for Political Data Science, Chapman and Hall/CRC, 2020, pp. 375–393 Search PubMed.
- S. Wold, K. Esbensen and P. Geladi, Chemometr. Intell. Lab. Syst., 1987, 2, 37–52 CrossRef CAS.
- Y. Zhang, S. Li, X. Gong and J. Chen, J. Am. Chem. Soc., 2023, 146, 342–357 CrossRef PubMed.
- I. Andricioaei and M. Karplus, J. Chem. Phys., 2001, 115, 6289–6292 CrossRef CAS.
- J. Schlitter, Chem. Phys. Lett., 1993, 215, 617–621 CrossRef CAS.
- J. v. Neumann, Proc. Natl. Acad. Sci. U. S. A., 1932, 18, 70–82 CrossRef CAS PubMed.
- M. E. Tuckerman and G. J. Martyna, J. Phys. Chem. B, 2000, 104, 159–178 CrossRef CAS.
- A. Valbuzzi, P. Gollnick, P. Babitzke and C. Yanofsky, J. Biol. Chem., 2002, 277, 10608–10613 CrossRef CAS PubMed.
- P. J. Williams, C. Killeen, I. C. Chagunda, B. Henderson, S. Donnecke, W. Munro, J. Sidhu, D. Kraft, D. A. Harrington and J. S. McIndoe, Chem. Sci., 2023, 14, 9970–9977 RSC.
- H. Liu, Y. Zhou, Y. Liu, Z. Wang, Y. Zheng, C. Peng, M. Tian, Q. Zhang, J. Li and H. Tan, Angew. Chem., Int. Ed., 2023, 135, e202213000 CrossRef.
- Y. Zheng, Y. Liu, Z. Wu, C. Peng, Z. Wang, J. Yan, Y. Yan, Z. Li, C. Liu and J. Xue, Adv. Mater., 2023, 2210986 CrossRef CAS PubMed.
- Y. Zheng, Z. Wang, Z. Li, H. Liu, J. Wei, C. Peng, Y. Zhou, J. Li, Q. Fu and H. Tan, Angew. Chem., Int. Ed., 2021, 60, 22529–22536 CrossRef CAS PubMed.
- Z. Li, Y. Zheng, J. Yan, Y. Yan, C. Peng, Z. Wang, H. Liu, Y. Liu, Y. Zhou and M. Ding, ChemBioChem, 2023, 24, e202300132 CrossRef CAS PubMed.
- H. Liu, R. Wang, J. Wei, C. Cheng, Y. Zheng, Y. Pan, X. He, M. Ding, H. Tan and Q. Fu, J. Am. Chem. Soc., 2018, 140, 6604–6610 CrossRef CAS PubMed.
- Y. Zhou, F. Fan, J. Zhao, Z. Wang, R. Wang, Y. Zheng, H. Liu, C. Peng, J. Li and H. Tan, Nat. Commun., 2022, 13, 4551 CrossRef CAS PubMed.
- D. Burgess, J. Colloid Interface Sci., 1990, 140, 227–238 CrossRef CAS.
- K. Kaibara, T. Okazaki, H. Bohidar and P. Dubin, Biomacromolecules, 2000, 1, 100–107 CrossRef CAS PubMed.
- M. G. Last, S. Deshpande and C. Dekker, ACS Nano, 2020, 14, 4487–4498 CrossRef CAS PubMed.
- C. Love, J. Steinkühler, D. T. Gonzales, N. Yandrapalli, T. Robinson, R. Dimova and T. Y. D. Tang, Angew. Chem., Int. Ed., 2020, 132, 6006–6013 CrossRef.
- W. Wei, Y. Tan, N. R. M. Rodriguez, J. Yu, J. N. Israelachvili and J. H. Waite, Acta Biomater., 2014, 10, 1663–1670 CrossRef CAS PubMed.
- Y. Sun, S. Y. Lau, Z. W. Lim, S. C. Chang, F. Ghadessy, A. Partridge and A. Miserez, Nat. Chem., 2022, 14, 274–283 CrossRef CAS PubMed.
- K. O. Margossian, M. U. Brown, T. Emrick and M. Muthukumar, Nat. Commun., 2022, 13, 2250 CrossRef CAS PubMed.
- K. Nishida, A. Tamura and N. Yui, Biomacromolecules, 2018, 19, 2238–2247 CrossRef CAS PubMed.
- D. Van Der Spoel, E. Lindahl, B. Hess, G. Groenhof, A. E. Mark and H. J. Berendsen, J. Comput. Chem., 2005, 26, 1701–1718 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc03061c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |