
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLeveraging phenazine and dihydrophenazine redox dynamics in conjugated microporous polymers for high-efficiency overall photosynthesis of hydrogen peroxide†
Shufan
Feng‡
a,
Lei
Wang‡
b,
Limei
Tian‡
c,
Ying
Liu‡
d,
Ke
Hu
 c,
Hangxun
Xu
*b,
Haifeng
Wang
*d and
Jianli
Hua
*a
c,
Hangxun
Xu
*b,
Haifeng
Wang
*d and
Jianli
Hua
*a
aKey Laboratory for Advanced Materials and Joint International Research Laboratory for Precision Chemistry and Molecular Engineering, Feringa Nobel Prize Scientist Joint Research Center, Frontiers Science Center for Materiobiology and Dynamic Chemistry, School of Chemistry and Molecular Engineering, East China University of Science and Technology, Shanghai, 200237, China. E-mail: jlhua@ecust.edu.cn
bKey Laboratory of Precision and Intelligent Chemistry, Department of Polymer Science and Engineering University of Science and Technology of China Hefei, Anhui 230026, China. E-mail: hxu@ustc.edu.cn
cDepartment of Chemistry Fudan University, Shanghai 200433, China
dState Key Laboratory of Green Chemical Engineering and Industrial Catalysis, Centre for Computational Chemistry and Research Institute of Industrial Catalysis East China University of Science and Technology, Shanghai 200433, China. E-mail: hfwang@ecust.edu.cn
First published on 27th June 2024
Abstract
Harnessing solar energy for hydrogen peroxide (H2O2) production from water and oxygen is crucial for sustainable solar fuel generation. Conjugated microporous polymers (CMPs), with their vast structural versatility and extended π-conjugation, are promising photocatalysts for solar-driven H2O2 generation, though enhancing their efficiency is challenging. Inspired by the crucial role of phenazine derives in biological redox cycling and electron transfer processes, the redox-active phenazine moiety is rationally integrated into a CMP framework (TPE-PNZ). By leveraging the reversible redox dynamics between phenazine and dihydrophenazine, TPE-PNZ sets a new benchmark for H2O2 production among CMP-based photocatalysts, reaching a production rate of 5142 μmol g−1 h−1 and a solar-to-chemical conversion efficiency of 0.58% without requiring sacrificial agents. This interconversion allows for the storage of photogenerated electrons by phenazine and subsequent conversion into dihydrophenazine, which then reduces O2 to H2O2 while reverting to phenazine, markedly facilitating charge transfer and mitigating charge recombination. Experimental and computational investigations further reveal that this reversible process enhances O2 adsorption and reduction, significantly lowering the energy barrier towards H2O2 formation. This study offers critical insights into designing advanced materials for sustainable energy research.
Introduction
The escalating global demand for hydrogen peroxide (H2O2), anticipated to reach 5.7 million tons by 2027, demonstrates its indispensable role across various application areas, including papermaking, mining, chemical engineering, and water treatment.1 The conventional reliance on the anthraquinone process for industrial H2O2 production, despite its high efficacy, raises significant environmental and energy consumption concerns, given its high energy inputs and hazardous waste outputs.2 These challenges have spurred the search for alternative green and sustainable production methodologies, with artificial photosynthesis at the forefront as a viable and promising solution.3 Nevertheless, this technology is still far from commercial viability for scalable H2O2 production. The primary impediment stems from the fact that the efficiency and stability of current photocatalysts fail to meet the stringent demands of industrial applications. This discrepancy highlights the urgent necessity for the innovative creation of photocatalysts that are not only cost-efficient and robust but also exhibit superior efficiency.4Conjugated microporous polymers (CMPs) emerge as a novel class of polymer photocatalysts for solar-driven H2O2 production, owing to their extensive π-electron systems, high surface areas, and flexibility in their structural modification. However, despite these fascinating characteristics, the photocatalytic efficiency of CMPs is often compromised by pronounced charge recombination and high energy barriers during H2O2 generation.5 To address these limitations, research has ventured into incorporating diverse functional groups into CMPs, such as triazines, thiophenes, and furans, aiming to enhance photocatalytic performances of CMPs.6 Unfortunately, CMP-based photocatalysts still tend to underperform when compared to other polymer photocatalysts such as resorcinol–formaldehyde resins, covalent organic frameworks, and g-C3N4 derivatives.7 Thus, enhancing the photocatalytic efficiency of CMPs through molecular engineering, particularly by introducing innovative redox-active groups that can improve charge transfer and lower the energy threshold for H2O2 production, is highly desirable.8
Phenazines are naturally abundant compounds that can be synthesized by various bacteria, including strains of Pseudomonas and Streptomyces.9 These compounds can undergo redox cycling within biological systems, oscillating between oxidized (phenazine) and reduced (dihydrophenazine) states. The interconversion between phenazine and dihydrophenazine has essential implications in biological systems, including redox reactions, electron transfer, and cellular signaling.10 In light of the exceptional ability of phenazine-based compounds to transfer electrons and their pivotal role in redox reactions within biological systems, integrating phenazine as a functional group into the structure of CMPs could potentially enhance their performances in photocatalytic H2O2 production. This makes phenazine an excellent candidate for designing CMPs with enhanced photocatalytic efficiency.
In this study, we show that the unique redox-active property of phenazine can be leveraged to develop a phenazine-incorporated CMP (TPE-PNZ) to enhance the photocatalytic production of H2O2 from H2O and O2. The integration of phenazine into TPE-PNZ allows it to serve as an effective electron transfer mediator, reflecting its role in natural microbial redox processes. This capability enables the transition of phenazine to dihydrophenazine through the photoreduction process, facilitating the reduction of O2 to H2O2, while dihydrophenazine can be oxidized by oxygen to regenerate phenazine. As a result, TPE-PNZ achieves an impressive H2O2 production rate of 5142 μmol g−1 h−1 and a remarkable solar-to-chemical conversion (SCC) efficiency of 0.58%. This is in stark contrast to a similarly designed CMP without the phenazine component, TPE-AC, which exhibits a much reduced H2O2 production rate, highlighting the critical role of the reversible phenazine/dihydrophenazine conversion in enhancing photocatalytic efficiency. Further studies indicate that the strategically incorporated phenazine enhances both the adsorption and reduction of O2, effectively lowering the energy barrier for H2O2 production. This study not only introduces a valuable functional moiety for the design of CMPs but also offers substantial insights into optimizing charge dynamics and active sites within CMPs for future solar fuel production.
Results and discussion
As shown in Fig. 1a, TPE-PNZ and TPE-AC were synthesized via Sonogashira coupling reactions using tetrakis(4-ethynylphenyl)ethane (TEPE) with 2,7-dibromophenazine (PNZ-Br) and 2,6-dibromoanthracene (AC-Br), respectively. The detailed synthetic routes are illustrated in Fig. S1 and S2.† In this manner, the phenazine unit was rationally integrated into TPE-PNZ, creating a donor–acceptor (D–A) structure through the interaction of the electron-deficient phenazine and the electron-rich TEPE. The Fourier transform infrared (FTIR) spectra, shown in Fig. 1b, were employed to confirm the chemical structures of TPE-PNZ and TPE-AC. The stretching vibration peaks around 3275 cm−1 can be attributed to the ≡C–H bonds of the terminal alkynes in TEPE, while the peaks at 1042 cm−1 in PNZ-Br and AC-Br correspond to the C–Br bond. Remarkably, the characteristic stretching vibration peaks observed in the precursors are absent in the spectra of TPE-PNZ and TPE-AC, suggesting the complete polymerization of TEPE with PNZ-Br and AC-Br, respectively.11 In addition, the high-resolution C 1s X-ray photoelectron spectroscopy (XPS) spectra of TPE-PNZ display peaks at approximately 282.0, 284.0, and 286.0 eV (Fig. 1c), which can be assigned to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, C
C, C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C, and CN bond, respectively.12 In contrast, the peak associated with the CN bond cannot be found in the XPS spectra of TPE-AC. The chemical structures of both CMPs were further characterized by solid-state 13C NMR spectroscopy (Fig. 1d). For TPE-PNZ, peaks in the range of 120–140 ppm are ascribed to aromatic carbons, and peaks at 90 ppm are associated with carbon atoms in the alkyne groups. Moreover, the peaks at 170 ppm can be attributed to the carbon atoms in phenazine moieties, which are not observed from the spectrum of TPE-AC.13 These characterizations conclusively confirmed the successful synthesis of designed CMPs.
C, and CN bond, respectively.12 In contrast, the peak associated with the CN bond cannot be found in the XPS spectra of TPE-AC. The chemical structures of both CMPs were further characterized by solid-state 13C NMR spectroscopy (Fig. 1d). For TPE-PNZ, peaks in the range of 120–140 ppm are ascribed to aromatic carbons, and peaks at 90 ppm are associated with carbon atoms in the alkyne groups. Moreover, the peaks at 170 ppm can be attributed to the carbon atoms in phenazine moieties, which are not observed from the spectrum of TPE-AC.13 These characterizations conclusively confirmed the successful synthesis of designed CMPs.
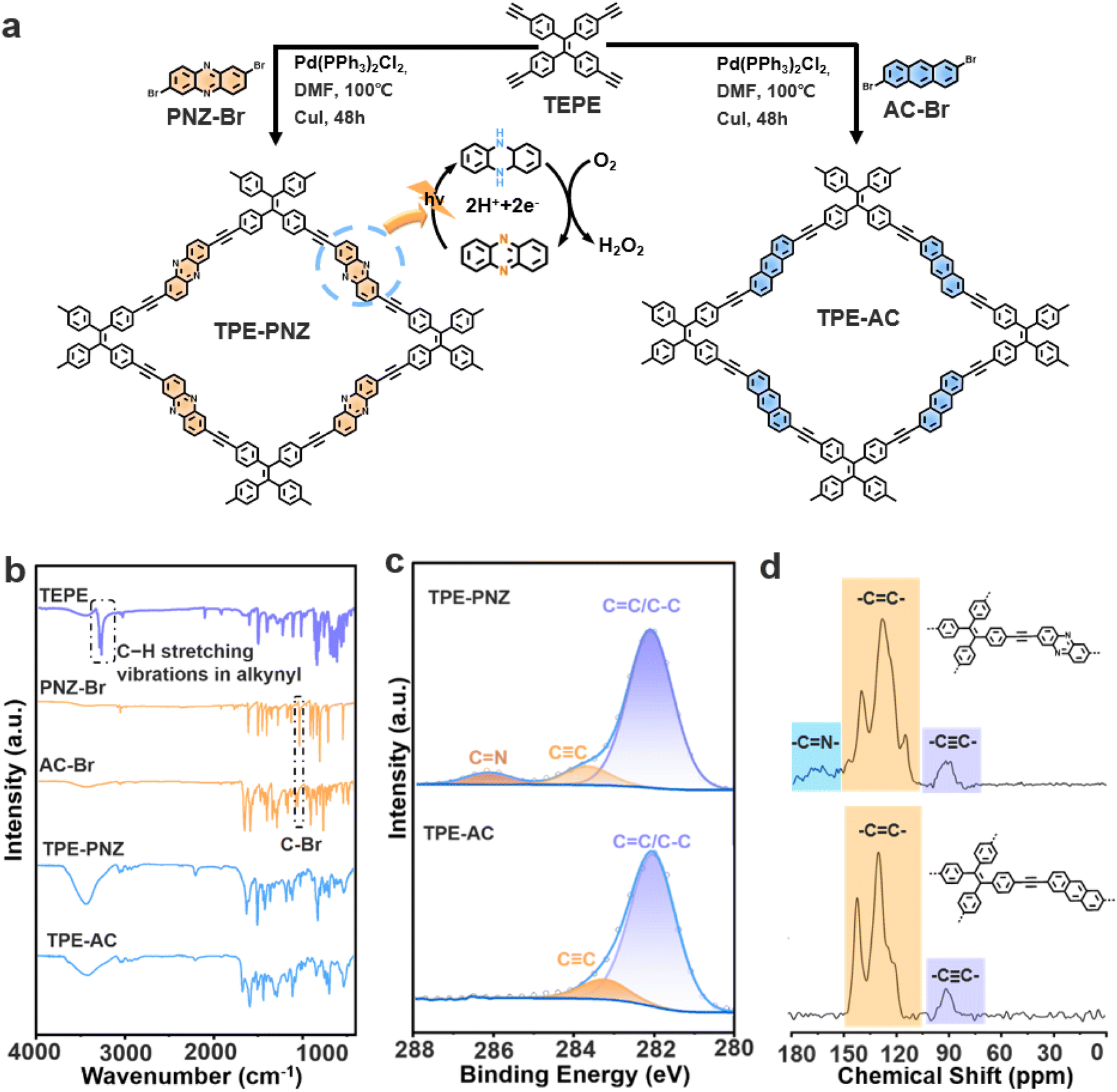 | ||
| Fig. 1 (a) Schematic illustration of the synthetic routes towards TPE-PNZ and TPE-AC from their corresponding precursors. (b) FTIR spectra of TPE-PNZ, TPE-AC, and their corresponding precursors. (c) High-resolution C 1s XPS spectra of TPE-PNZ and TPE-AC. (d) Solid-state 13C NMR spectra of TPE-PNZ and TPE-AC. | ||
The powder X-ray diffraction (PXRD) patterns, as shown in Fig. S3,† reveal that both TPE-PNZ and TPE-AC polymers possess amorphous structures. Both TPE-PNZ and TPE-AC exhibit irregularly aggregated particle structures as observed from scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images (Fig. S4 and S5†). This observation is consistent for both TPE-PNZ and TPE-AC, highlighting a common structural feature despite they incorporate different functional moieties. Meanwhile, energy-dispersive X-ray spectroscopy (EDS) mapping confirms the homogeneous distribution of carbon (C) and nitrogen (N) elements across the surface of TPE-PNZ (Fig. S6†). The uniform distribution of elements further suggests that the phenazine moieties are well-integrated throughout the polymer framework.14 The N2 sorption measurements were conducted at 77 K to assess the permanent porosity of both CMPs (Fig. S7†). The measured Brunauer–Emmett–Teller (BET) surface areas of TPE-PNZ and TPE-AC are 722.4 and 759.6 m2 g−1, respectively. Notably, the primary pore sizes were found to be below 2 nm, indicating predominantly microporous structures for both TPE-PNZ and TPE-AC. The microporosity of TPE-PNZ and TPE-AC can be particularly advantageous for photocatalytic applications, providing abundant active sites for photocatalytic reactions and facilitating efficient mass transfer.15 Furthermore, it is noteworthy that the N2 adsorption/desorption isotherms of TPE-PNZ and TPE-AC do not exhibit closure. The phenomenon may result from a variety of reasons related to the physical and chemical environments experienced by N2 molecules during the adsorption/desorption process. During adsorption, N2 molecules can penetrate deeply into the micropores, causing the pores to swell and the polymer structure to reorganize. This reorganization can create new pathways and barriers that alter the adsorption dynamics. During desorption, N2 molecules encounter different kinetic barriers compared to adsorption. These barriers can include narrow pore openings, irregular pore shapes, and variations in pore connectivity, which can slow down or hinder the release of N2 molecules. The process of adsorption and desorption may follow different thermodynamic pathways. This can result in the isotherms not overlapping perfectly, especially at lower pressures where the effects of pore structure and molecular interactions are more pronounced.16,17
Fig. 2a presents the UV-vis diffuse reflectance spectroscopy spectra of both CMPs, which exhibit pronounced absorption within the visible light range, indicating their potential for solar-driven photocatalytic applications. Based on the Tauc plots, the optical bandgaps were determined to be approximately 2.19 eV for TPE-PNZ and 2.01 eV for TPE-AC (Fig. S8†).18 Meanwhile, the positive slope observed in the Mott–Schottky plots for the CMPs implied that both CMPs were n-type semiconductors (Fig. S9†). This characteristic is crucial for their photocatalytic applications, indicating efficient electron conduction upon photoexcitation.19 The flat band potentials of TPE-PNZ and TPE-AC, when measured against the saturated calomel electrode (SCE) at a neutral pH of 7, were identified to be −0.63 and −0.56 V, respectively. Consequently, the conduction band (CB) levels were calculated to be −0.59 V for TPE-PNZ and −0.52 V for TPE-AC relative to the normal hydrogen electrode (NHE).20 Therefore, the band structures of both TPE-PNZ and TPE-AC are well-positioned for photocatalytic H2O2 production from pure water without using any sacrificial agents (Fig. 2b).3
 | ||
| Fig. 2 (a) UV/vis DRS spectra and (b) band structures of TPE-PNZ and TPE-AC. (c) Typical time course of H2O2 production in pure water using TPE-PNZ and TPE-AC, error bars are derived from standard errors from three tests. (d) Summary of H2O2 production rates from recently reported CMP-based photocatalysts for H2O2 production. (e) Wavelength-dependent AQEs of TPE-PNZ, error bars are derived from standard errors from three tests. | ||
The photocatalytic performances of TPE-PNZ and TPE-AC were assessed in pure water under visible-light irradiation (λ > 420 nm). As shown in Fig. 2c, TPE-PNZ demonstrates a remarkable H2O2 production rate of 5142 μmol g−1 h−1, surpassing all previously reported CMP-based photocatalysts (Fig. 2d).5,11,21–34 Meanwhile, TPE-PNZ achieved an impressive SCC efficiency of 0.58%, surpassing most CMP-based photocatalysts reported to date (Table S1†). In contrast, TPE-AC, designed as a comparative benchmark by replacing phenothiazine with anthracene in TPE-PNZ, only exhibited a much lower H2O2 production rate of 2183 μmol g−1 h−1. This observation indicates that the phenazine moiety plays a crucial role in enhancing the photocatalytic activity towards H2O2 generation. The apparent quantum efficiency (AQE) of TPE-PNZ at a wavelength of 420 nm was calculated to be 9.37%. The AQE values across different wavelengths correlate well with the absorption spectrum of TPE-PNZ, as illustrated in Fig. 2e. Notably, TPE-PNZ also exhibits a high photocatalytic H2O2 production rate in seawater (5250 μmol g−1 h−1), indicating that its photocatalytic activity would not be influenced by the high concentration of ions in seawater (Fig. S10†). This observation indicates that the as-synthesized TPE-PNZ holds great potential for solar-driven H2O2 production from seawater.35
The stability of TPE-PNZ, an essential aspect of performance evaluation, was confirmed through cyclic tests showing that TPE-PNZ retains its prominent H2O2 production rate after five rounds of photoirradiation (Fig. S11†). In addition, FTIR and XPS measurements revealed no substantial changes in the chemical structure of TPE-PNZ, evidencing that CMPs with fully π conjugated structures are highly robust photocatalysts for the photosynthesis of H2O2 (Fig. S12†). Both TPE-PNZ and TPE-AC exhibited minimal H2O2 decomposition activity, suggesting that the variance in H2O2 production rates between them is not due to their H2O2 decomposition capabilities (Fig. S13†).3 Furthermore, inductively coupled plasma (ICP) analyses found no obvious correlation between the photocatalytic performances of TPE-PNZ and TPE-AC and their residual Pd contents (Table S2†). This finding rules out the possibility that the performance difference between the two CMPs is caused by different Pd content levels.36
In biological systems, phenazine derivatives are produced by certain microorganisms and play a crucial role in electron transfer processes.9 TPE-PNZ exhibits notably enhanced photocatalytic performance compared to TEP-AC, implying that the redox-active phenazine moiety has a significant impact on the charge transfer process in CMPs. The comparative analysis of photocurrent responses and electrochemical impedance spectroscopy (EIS) measurements for TPE-PNZ and TPE-AC are presented in Fig. 3a and b, respectively. Here, TPE-PNZ demonstrates a substantially enhanced photocurrent with lower electrochemical impedance, implying that the phenazine moiety can facilitate charge separation.37 Additionally, we also evaluated the internal electric fields within CMPs by measuring their surface voltages and surface charge densities (Fig. 3c and S14†). The results reveal that TPE-PNZ exhibits an enhanced built-in electric field intensity (3.94) compared to TPE-AC (1.00), providing further evidence that the phenazine component can effectively promote charge separation.38 Density functional theory (DFT) calculations were then utilized to elucidate the differences in charge transfer dynamics between TPE-PNZ and TPE-AC (Fig. 3d). For TPE-PNZ, photoexcitation prompts a transition from the highest occupied molecular orbital (HOMO) to the lowest unoccupied molecular orbital (LUMO), resulting in an electron density shift from the TPE segments to the phenazine units. This redistribution facilitates effective intramolecular charge transfer within the π-conjugated framework. In contrast, for TPE-AC, DFT calculations show that both HOMO and LUMO levels are uniformly distributed throughout the entire polymer framework. The uniform distribution of HOMO and LUMO levels is unfavorable for charge separation within the polymer framework due to the lack of a preferential direction or region for the dynamic movement of photogenerated electrons and holes.39 We also utilized nanosecond transient absorption (ns-TA) spectroscopy to probe the dynamics of photogenerated charge carriers within CMPs. Based on the results of the spectroelectrochemical test (Fig. S15†), the absorption peak at 500 nm in the TA spectra of both TPE-PNZ and TPE-AC corresponds to their photoinduced charge-separated states.40 As shown in Fig. 3e and f, the average relaxation lifetime of photoinduced charge-separated states in TPE-PNZ under an Ar atmosphere is 3.09 ms, which is about 5060 times the lifetime observed for TPE-AC (610.30 ns). This stark contrast substantiates the vital role of the phenazine moiety in enhancing charge transfer efficiency and inhibiting charge recombination.12 Under an ambient air environment, the charge separation lifetimes for TPE-PNZ and TPE-AC decrease markedly to 1.96 μs and 115.71 ns, respectively (Fig. 3g and h). Our calculations reveal that the quenching effect of O2 on the charge separation lifetime of TPE-PNZ reaches an efficiency of 99.98%, notably higher than that for TPE-AC, which is only 81.01%. This demonstrates a faster electron transfer from TPE-PNZ to O2 as compared to TPE-AC, highlighting the significance of phenazine moiety in enhancing the interaction with O2 and improving the efficiency of O2 reduction to H2O2.41
 | ||
| Fig. 3 (a) Transient photocurrents, (b) Nyquist plots, and (c) relative internal electric field intensities of TPE-PNZ and TPE-AC. (d) The calculated distribution of the HOMO and LUMO of TPE-PNZ and TPE-AC. The TA kinetics trace of (e) TPE-PNZ and (f) TPE-AC probed at 500 nm under Ar. The TA kinetics trace of (g) TPE-PNZ and (h) TPE-AC probed at 500 nm under air. | ||
A series of control experiments were conducted to elucidate the mechanism responsible for photocatalytic H2O2 production. Fig. S16† demonstrates that no H2O2 production was observed under the N2 atmosphere, confirming that H2O2 is generated through the reduction of O2. We also performed steady-state fluorescence tests on TPE-PNZ and TPE-AC under Ar and O2 conditions, respectively. The experimental results showed that the fluorescence of both catalysts could be quenched by O2, confirming the efficient transfer of photo-generated electrons to O2 for the production of H2O2 (Fig. S17†).42,43 To further unravel reactive species during H2O2 formation, radical trapping experiments were carried out. These experiments revealed that the introduction of AgNO3 led to the absence of H2O2 detection while adding 10% benzyl alcohol notably enhanced H2O2 production, reflecting that H2O2 synthesis in both CMPs is a result of the O2 reduction process.44 The introduction of para-benzoquinone (p-BQ) into the reaction system inhibited H2O2 production, further suggesting that superoxide radicals (˙O2−) act as the primary intermediates in H2O2 formation, through a mechanism involving stepwise single-electron transfer process (O2 → ˙O2− → H2O2).45 This mechanism is corroborated by the results obtained from electron paramagnetic resonance (EPR) spectroscopic measurements, with the EPR spectra of TPE-PNZ under visible light showing distinctive peaks for the DMPO-˙O2− adducts (Fig. S18a†). On the other hand, the absence of DMPO-˙OH spin adduct peaks in the EPR spectra of TPE-PNZ when AgNO3 was employed as an electron scavenger under light irradiation (Fig. S18b†) rules out the possibility of hydroxyl radicals (˙OH) formation through water oxidation. Moreover, only O2 can be detected when AgNO3 is used as an electron scavenger, implying that photogenerated holes in TPE-PNZ oxidize water to produce O2 through a four-electron process (Fig. S19†).46 The average electron transfer number for the O2 reduction reactions, as determined from the RDE tests, is calculated to be 2.05 and 1.66 for TPE-PNZ and TPE-AC, respectively (Fig. S20†). This indicates that the incorporation of the phenazine moiety into the CMP framework is beneficial for the conversion of O2 into H2O2via the two-electron reduction pathway.14,45 We also monitored the changes in the photocatalytic system's pH value over time. As shown in Fig. S21,† no detectable change in the pH value of the solution can be observed over time.47,48
In situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) measurements were carried out to further explore the mechanism for H2O2 production from TPE-PNZ. As illustrated in Fig. 4a, spectral features appearing at 1384 and 1260 cm−1 can be attributed to the stretching vibrations of C–N and CN bonds in TPE-PNZ, respectively.49,50 Upon exposure to visible light under the Ar atmosphere, there was a noticeable decrease in the intensity of the CN bond peak within the phenazine units, along with an increase in the C–N bond peak intensity. This observation provides strong evidence that the phenazine moiety in TPE-PNZ can be photochemically reduced to dihydrophenazine under photoirradiation. To better illustrate the conversion process from phenazine to dihydrophenazine, we overlaid the curves in the spectrum (Fig. S22†). It's note that in spectroscopic analyses, changes in peak intensity are more pronounced during the initial stages of the reaction. This phenomenon can be attributed to the system approaching equilibrium between the interconversion of phenazine and dihydrophenazine under the given reaction conditions. Once equilibrium is established, the rate at which peak intensities change diminishes, reflecting the characteristic behavior of dynamic equilibrium systems where the rates of forward and reverse reactions balance over time.23,51–53 Meanwhile, the continuous production of H2O2, even in the absence of light, further indicates that the phenazine moiety is capable of storing electrons (Fig. S23†).54 In contrast, when O2 was purged under dark conditions, the peak intensity of the C–N bond gradually decreased, whereas the intensity of the peak corresponding to the CN band gradually increased (Fig. 4b), suggesting that O2 can oxidize dihydrophenazine back to phenazine. Notably, no significant changes in the intensities of C–N and CN bands were observed when TPE-PNZ was irradiated under light with O2 purging, as shown in Fig. 4c. This suggests the existence of a dynamic equilibrium between phenazine and dihydrophenazine in the presence of O2 under photoirradiation. Additionally, it suggests that during the photocatalytic production of H2O2, the rate of water oxidation matches the rate of O2 reduction.25,51,52 The insights from the in situ DRIFTS measurements conclusively demonstrate the reversible interconversion between phenazine and dihydrophenazine during the photocatalytic generation of H2O2 by TPE-PNZ.51
 | ||
| Fig. 4 (a) In situ DRIFT spectra of TPE-PNZ saturated with Ar under visible light irradiation. (b) In situ DRIFT spectra of TPE-PNZ saturated with O2 under dark. (c) In situ DRIFT spectra of TPE-PNZ saturated with O2 under visible light irradiation. (d) Free energy diagrams of TPE-PNZ and TPE-AC for the photosynthesis of H2O2via the oxygen reduction process. (e) Comparison of the Gibbs free energy for H2O2 production with and without the interconversion of phenazine and dihydrophenazine in TPE-PNZ. | ||
DFT calculations were performed to thoroughly understand how the rationally embedded phenazine moiety in TPE-PNZ can facilitate H2O2 production. These calculations pinpointed the rate-determining step of the stepwise single-electron O2 reduction reaction in both TPE-PNZ and TPE-AC as the formation of the *OOH intermediate.45 For TPE-PNZ, electrons generated upon photoexcitation initially reduce phenazine to dihydrophenazine (Fig. 4d). This is followed by the adsorption of O2 onto dihydrophenazine, where it gains an electron and a proton to form *OOH. For TPE-AC, anthracene acts as the adsorption site for both *O2 and *OOH. The Gibbs free energy for *OOH formation on TPE-AC (1.69 eV) is much higher than that on TPE-PNZ (−0.24 eV). This disparity indicates that the phenazine moiety can drastically lower the energy requirement for *OOH formation, thereby enhancing the efficiency of H2O2 production.1 To further validate the impact of the reversible interconversion between phenazine and dihydrophenazine on H2O2 generation, we also simulated the photocatalytic pathways for H2O2 production in TPE-PNZ by excluding the reversible interconversion process. In this model, *O2 is protonated by the surrounding medium to yield *OOH,14 with the *OOH adsorption occurring at the N atom of the phenazine unit (Fig. 4e). The Gibbs free energy associated with *OOH formation in this scenario is −0.08 eV, a value markedly higher than that obtained during the reversible phenazine–dihydrophenazine conversion (−0.24 eV). This finding highlights the crucial role of phenazine–dihydrophenazine interconversion in reducing the energy barrier for O2 reduction to facilitate H2O2 production.
Combining both experimental evidence and computational results, we have elucidated a comprehensive reaction mechanism for high-efficiency H2O2 production by TPE-PNZ. Initially, upon photoirradiation, the phenazine moiety in TPE-PNZ undergoes a redox process, wherein it captures two photogenerated electrons along with two protons, resulting in its conversion to dihydrophenazine. Following this reduction, O2 is adsorbed onto the dihydrophenazine unit, where it engages in a sequential, single-electron transfer process, receiving two electrons and two protons from dihydrophenazine. This reaction leads to the direct synthesis of H2O2, concurrently regenerating the phenazine unit by oxidizing dihydrophenazine back to its original state, as illustrated in Fig. 5. This intricate interplay of redox reactions and electron–proton transfers points out a clear pathway for the efficient production of H2O2, highlighting the pivotal role of the phenazine moiety within the TPE-PNZ framework.
 | ||
| Fig. 5 Schematic illustration of the reversible transformation between phenazine and dihydrophenazine in TPE-PNZ during H2O2 production. (Color denotation: gray: C; blue: N; red: O; white: H in benzene; green: H in dihydrophenazine). | ||
Conclusions
In summary, our study unambiguously indicates that embedding the redox-active phenazine moiety within CMPs can markedly enhance the efficiency of photocatalytic H2O2 generation. This enhancement is primarily attributed to the phenazine units acting as critical sites for the reduction of O2 and promoting effective charge separation within the polymer framework. More importantly, our study sheds light on the fundamental principles guiding the molecular design of metal-free polymers for photocatalytic applications. This approach not only exemplifies the strategic incorporation of functional moieties to enhance photocatalytic efficiency but also offers a blueprint for the development of advanced materials aimed at harnessing solar energy for chemical synthesis.Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.Author contributions
S. Feng, L. Wang and L. Tian synthesized the materials and performed the experiments. Y. Liu and H. Wang performed the DFT calculations. J. Hua and K. Hu wrote the manuscript. H. Xu modified the figures and the manuscript. H. Wang, H. Xu and J. Hua supervised and supervised the project. All authors discussed the results and contributed to the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
For financial support of this research, the authors thank the projects supported by the National Natural Science Foundation of China (21788102, 22271093, 21971064, 22109151, and 52225307), Shanghai Municipal Science and Technology Major Project (Grant No. 2018SHZDZX03), the National Key Research and Development Program of China (2021YFA1500800), the Fundamental Research Funds for the Central Universities (222201717003 and 50321101918001) and the Programme of Introducing Talents of Discipline to Universities (B16017).Notes and references
- J. Yue, L. Song, Y. Fan, Z. Pan, P. Yang, Y. Ma, Q. Xu and B. Tang, Angew. Chem., Int. Ed., 2023, 62, e202309624 CrossRef CAS PubMed.
- Y. Hou, P. Zhou, F. Liu, Y. Lu, H. Tan, Z. Li, M. Tong and J. Ni, Angew. Chem., Int. Ed., 2024, 63, e202318562 CrossRef CAS PubMed.
- Y. Luo, B. Zhang, C. Liu, D. Xia, X. Ou, Y. Cai, Y. Zhou, J. Jiang and B. Han, Angew. Chem., Int. Ed., 2023, 62, e202305355 CrossRef CAS PubMed.
- D. Chen, W. Chen, Y. Wu, L. Wang, X. Wu, H. Xu and L. Chen, Angew. Chem., Int. Ed., 2023, 62, e202217479 CrossRef CAS PubMed.
- T. Gao, C. Yang, Y. Zheng, Y. Shen, Q. Wang, L. Wang, R. Xu, M. Lv, M. Li, Z. Shi, L. Wang, X. Kong and L. Ye, Appl. Catal., B, 2024, 346, 123761 CrossRef CAS.
- L. Wang, L. Liu, Y. Li, Y. Xu, W. Nie, Z. Cheng, Q. Zhou, L. Wang and Z. Fan, Adv. Energy Mater., 2023, 14, 2303346 CrossRef.
- X. Xu, Y. Sui, W. Chen, W. Huang, X. Li, Y. Li, D. Liu, S. Gao, W. Wu, C. Pan, H. Zhong, H. Wen and M. Wen, Appl. Catal., B, 2024, 341, 123271 CrossRef CAS.
- Z. Yong and T. Ma, Angew. Chem., Int. Ed., 2023, 62, e202308980 CrossRef CAS PubMed.
- J. B. Laursen and J. Nielsen, Chem. Rev., 2004, 104, 1663–1686 CrossRef CAS PubMed.
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed.
- X. Xu, R. Sa, W. Huang, Y. Sui, W. Chen, G. Zhou, X. Li, Y. Li and H. Zhong, ACS Catal., 2022, 12, 12954–12963 CrossRef CAS.
- H. Cheng, H. Lv, J. Cheng, L. Wang, X. Wu and H. Xu, Adv. Mater., 2022, 34, 2107480 CrossRef CAS PubMed.
- L. Chen, L. Wang, Y. Wan, Y. Zhang, Z. Qi, X. Wu and H. Xu, Adv. Mater., 2020, 32, e1904433 CrossRef PubMed.
- Q. Zhi, W. P. Liu, R. Jiang, X. Zhan, Y. Jin, X. Chen, X. Yang, K. Wang, W. Cao, D. Qi and J. Jiang, J. Am. Chem. Soc., 2022, 144, 21328–21336 CrossRef CAS PubMed.
- J. M. Lee and A. I. Cooper, Chem. Rev., 2020, 120, 2171–2214 CrossRef CAS PubMed.
- J. Jeromenok and J. Weber, Langmuir, 2013, 29, 12982–12989 CrossRef CAS PubMed.
- B. Ghanem, N. McKeown, P. Budd, J. Selbie and D. Fritsch, Adv. Mater., 2008, 20, 2766–2771 CrossRef CAS PubMed.
- J. Chang, Q. Li, J. Shi, M. Zhang, L. Zhang, S. Li, Y. Chen, S. Li and Y. Lan, Angew. Chem., Int. Ed., 2023, 62, e202218868 CrossRef CAS PubMed.
- J. Zhou, Y. Wang, Z. Cui, Y. Hu, X. Hao, Y. Wang and Z. Zou, Appl. Catal., B, 2020, 277, 119228 CrossRef CAS.
- J. Wang, W. Shi, D. Liu, Z. Zhang, Y. Zhu and D. Wang, Appl. Catal., B, 2017, 202, 289–297 CrossRef CAS.
- C. Wu, Z. Teng, C. Yang, F. Chen, H. Yang, L. Wang, H. Xu, B. Liu, G. Zheng and Q. Han, Adv. Mater., 2022, 34, 2110266 CrossRef CAS PubMed.
- L. Cao, C. Wang, H. Wang, X. Xu, X. Tao, H. Tan and G. Zhu, Angew. Chem., Int. Ed., 2024, 63, e2024020 Search PubMed.
- H. Yan, M. Shen, Y. Shen, X. Wang, W. Lin, J. Pan, J. He, Y. Ye, X. Yang, F. Zhu, J. Xu, J. He and G. Ouyang, Proc. Natl. Acad. Sci. U.S.A., 2022, 119, e2202913119 CrossRef CAS PubMed.
- Y. Guo, Y. Dong, B. Liu, B. Ni, C. Pan, J. Zhang, H. Zhao, G. Wang and Y. Zhu, Adv. Funct. Mater., 2024, 2402920 CrossRef.
- Y. Ye, J. Pan, Y. Shen, M. Shen, H. Yan, J. He, X. Yang, F. Zhu, J. Xu, J. He and G. Ouyang, Proc. Natl. Acad. Sci. U.S.A., 2021, 118, e2115666118 CrossRef CAS PubMed.
- X. Li, G. Zhang, N. Li, Q. Xu, H. Li, J. Lu and D. Chen, Adv. Funct. Mater., 2024, 2316773 CrossRef.
- X. Xia, J. Feng, Z. Zhong, X. Yang, N. Li, D. Chen, Y. Li, Q. Xu and J. Lu, Adv. Funct. Mater., 2023, 34, 2311987 CrossRef.
- T. Yang, Y. Jin, Y. Wang, A. Kong, Y. Chen, Y. Zou, C. Liu, G. Wei and C. Yu, Adv. Funct. Mater., 2023, 33, 2300714 CrossRef CAS.
- Y. Yang, Q. Guo, Q. Li, L. Guo, H. Chu, L. Liao, X. Wang, Z. Li and W. Zhou, Adv. Funct. Mater., 2024, 2400612 CrossRef.
- Q. Zhang, J. Zhou, H. Zhang, C. Qi, Q. Zhou, R. Guo, H. Yang, T. Xing, M. Wang, M. Wu and W. Wu, Adv. Funct. Mater., 2024, 2401579 CrossRef.
- Z. Luo, X. Chen, Y. Hu, X. Chen, W. Lin, X. Wu and X. Wang, Angew. Chem., Int. Ed., 2023, 62, e202304875 CrossRef CAS PubMed.
- P. Gao, C. Wu, S. Wang, G. Zheng and Q. Han, J. Colloid Interface Sci., 2023, 650, 40–46 CrossRef CAS PubMed.
- R. Zhang, H. Zhao, C. Pan, J. Zhang, L. Jian, X. Sun, R. Ji, J. Li, Y. Dong and Y. Zhu, New J. Chem., 2024, 48, 3316–3324 RSC.
- X. Xu, Y. Sui, W. Chen, G. Zhou, Y. Li, H. Zhong and H. Wen, ACS Appl. Polym. Mater., 2023, 5, 7571–7580 CrossRef CAS.
- C. Chu, D. Yao, Z. Chen, X. Liu, Q. Huang, Q. Li and S. Mao, Small, 2023, 19, 2303796 CrossRef CAS PubMed.
- L. Li, Z. Cai, Q. Wu, W. Lo, N. Zhang, L. X. Chen and L. Yu, J. Am. Chem. Soc., 2016, 138, 7681–7686 CrossRef CAS PubMed.
- J. Xiong, X. Li, J. Huang, X. Gao, Z. Chen, J. Liu, H. Li, B. Kang, W. Yao and Y. Zhu, Appl. Catal., B, 2020, 266, 118602 CrossRef CAS.
- H. Zhang, X. Chen, Z. Zhang, K. Yu, W. Zhu and Y. Zhu, Appl. Catal., B, 2021, 287, 119957 CrossRef CAS.
- L. Yang, J. Yuan, G. Wang, Q. Cao, C. Zhang, M. Li, J. Shao, Y. Xu, H. Li and J. Lu, Adv. Funct. Mater., 2023, 33, 2300954 CrossRef CAS.
- B. Cai, H. Song, A. Brnovic, M. V. Pavliuk, L. Hammarström and H. Tian, J. Am. Chem. Soc., 2023, 145, 18687–18692 CrossRef CAS PubMed.
- C. Zhao, X. Wang, Y. Yin, W. Tian, G. Zeng, H. Li, S. Ye, L. Wu and J. Liu, Angew. Chem., Int. Ed., 2023, 62, e202218318 CrossRef CAS PubMed.
- C. Ye, J. Li, Z. Li, X. Li, X. Fan, L. Zhang, B. Chen, C. Tung and L. Wu, ACS Catal., 2015, 5, 6973–6979 CrossRef CAS.
- Y. Negishi, Y. Matsuura, R. Tomizawa, W. Kurashige, Y. Niihori, T. Takayama, A. Iwase and A. Kudo, J. Phys. Chem. C, 2015, 119, 11224–11232 CrossRef CAS.
- Z. Wei, M. Liu, Z. Zhang, W. Yao, H. Tan and Y. Zhu, Energy Environ. Sci., 2018, 11, 2581–2589 RSC.
- P. Li, F. Ge, Y. Yang, T. Wang, X. Zhang, K. Zhang and J. Shen, Angew. Chem., Int. Ed., 2024, e202319885 CAS.
- D. Chen, W. Chen, Y. Wu, L. Wang, X. Wu, H. Xu and L. Chen, Angew. Chem., Int. Ed., 2023, 62, 202217479 CrossRef PubMed.
- D. Tan, R. Zhuang, R. Chen, M. Ban, W. Feng, F. Xu, X. Chen and Q. Wang, Adv. Funct. Mater., 2024, 34, 2311655 CrossRef CAS.
- X. Zeng, Y. Liu, X. Hu and X. Zhang, Green Chem., 2021, 23, 1466–1494 RSC.
- Y. Kou, Y. Xu, Z. Guo and D. Jiang, Angew. Chem., Int. Ed., 2011, 50, 8753–8757 CrossRef CAS PubMed.
- X. Gao, Y. Chen, C. Gu, J. Wen, X. Peng, J. Liu, Z. Zhang, Q. Zhang, Z. Liu and C. Wang, J. Mater. Chem. A, 2020, 8, 19283–19289 RSC.
- Z. Wei, S. Zhao, W. Li, X. Zhao, C. Chen, D. L. Phillips, Y. Zhu and W. Choi, ACS Catal., 2022, 12, 11436–11443 CrossRef CAS.
- D. Lee, M. Park, N. Kim, M. Gu, H. Kim and B. Kim, J. Catal., 2022, 411, 235–244 CrossRef CAS.
- L. Zhang, Y. Huang, H. Yan, Y. Cheng, Y. X. Ye, F. Zhu and G. Ouyang, Adv. Mater., 2024, 2401162 CrossRef PubMed.
- Q. Ruan, X. Xi, B. Yan, L. Kong, C. Jiang, J. Tang and Z. Sun, Chem, 2023, 9, 1850–1864 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc02832e |
| ‡ Contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |