
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA dual heterostructure enables the stabilization of 1T-rich MoSe2 for enhanced storage of sodium ions†
Yunfeng
Chao
a,
Shenghui
Jia
a,
Jinzhao
Li
a,
Guohui
Chen
a,
Lu
Liu
a,
Fei
Tang
a,
Jianhua
Zhu
*a,
Caiyun
Wang
*b and
Xinwei
Cui
 a
a
aHenan Institute of Advanced Technology, Zhengzhou University, Zhengzhou 450052, China. E-mail: jianhuazhu@zzu.edu.cn
bIntelligent Polymer Research Institute, Faculty of Engineering and Information Sciences, University of Wollongong, Innovation Campus, North Wollongong, NSW 2500, Australia. E-mail: caiyun@uow.edu.au
First published on 6th June 2024
Abstract
Electron injection effectively induces the formation of a 1T-rich phase to address the low conductivity of MoSe2. Nevertheless, overcoming the inherent metastability of the 1T phase (particularly during the conversion reactions that entail the decomposition–reconstruction of MoSe2 and volume expansion) remains a challenge. Guided by DFT results, we designed a composite with bimetal selenides-based heterostructures anchored on reduced graphene oxide (rGO) nanosheets (G-Cu2Se@MoSe2) to obtain stabilized 1T-rich MoSe2 and enhanced ion transfer. The construction of 1T-rich MoSe2 and built-in electric fields (BiEF) through electron transfer at the heterointerfaces were realized. Moreover, the rGO-metal selenides heterostructures with in situ-formed interfacial bonds could facilitate the reconstruction of the 1T-rich MoSe2-involved heterostructure and interfacial BiEF. Such a dual heterostructure endowed G-Cu2Se@MoSe2 with an excellent rate capability with a capacity of 288 mA h g−1 at 50 A g−1 and superior cycling stability with a capacity retention ratio of 89.6% (291 mA h g−1) after 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles at 10 A g−1. Insights into the functional mechanism and structural evolution of the 1T MoSe2-involved dual heterostructure from this work may provide guidelines for the development of MoSe2 and phase-engineering strategies for other polymorphistic materials.
000 cycles at 10 A g−1. Insights into the functional mechanism and structural evolution of the 1T MoSe2-involved dual heterostructure from this work may provide guidelines for the development of MoSe2 and phase-engineering strategies for other polymorphistic materials.
1 Introduction
The increasing demand for high-mileage electric vehicles and large-scale energy-storage systems has raised concerns about the impact on natural reserves and cost of lithium metal, a key component in lithium-ion batteries (LIBs). Sodium-ion batteries (SIBs) have emerged as an alternative to LIBs owing to the abundance and low cost of sodium, high safety, and impressive electrochemical performance over a wide temperature range.1,2 Despite sharing a similar working mechanism with LIBs, SIBs cannot directly utilize the commercial graphite anode of LIBs.3 The development of sodium-storage anode materials is also hindered by a low rate of ion diffusion and huge volume expansion resulting from the inherently larger size of Na+ (1.02 Å vs. 0.76 Å for Li+).4,5 Transition-metal selenides (TMS) are promising conversion-type anode materials that offer good electrochemical activity and high capacity.2,6 Compared with transition–metal oxides or transition–metal sulfides, the larger size of Se atoms and weaker transition metal–Se bonds endow TMS with higher mobility of electrons, better ion diffusion and lower rate of volume change.3,7 Among TMS, two-dimensional (2D)-layered MoSe2 has been attracting extensive attention due to its high interlayer distance of 0.65 nm, large surface area, small bandgap of 1.1 eV and high theoretical sodium storage of 422 mA h g−1.8 However, its development is constrained by the common issues for TMS, such as low conductivity, low rate of ion diffusion, and large volume changes. These challenges affect the reaction kinetics and reversibility.The poor electron transfer of MoSe2 can be solved by phase engineering.9 The polymorphism feature enables the transformation from a semiconductive 2H-phase to metallic-like 1T state for promoting electronic conductivity, rate of ion diffusion and electrochemical activity.10–12 However, the Mo 4d-orbitals of 1T-MoSe2 exhibit lower energy levels that are not fully filled, so the metastable 1T phase is commonly observed. Strategies such as introducing heteroatoms,8 single atoms13 and an intercalator14 have been developed to realize a 1T structure in MoSe2 or the analogous 2D material MoS2 for enhanced sodium storage. All those studies have highlighted the importance and efficacy of electron injection from a donator species (P, Fe or PO43−) to Mo atoms to achieve a balanced electron occupation in Mo 4d orbitals and promote a high content of the 1T phase. On the other hand, the formation of a composite with a second material having different redox potential from MoSe2 can prevent the aggregation of conversion products and alleviate the mechanical stress derived from volume changes, thus benefiting the structural stability.15,16 Metallic selenides with various bandgaps can induce spontaneous electron redistribution at heterointerfaces to generate a built-in electric field (BiEF) that enhances ion transfer.16–18 Analogically, designing a heterostructure involving MoSe2 and another selenide could be promising to overcome aforementioned drawbacks, thereby achieving stabilized 1T-MoSe2 and a BiEF for promoting the transfer of electrons and ions. Such a structure has not been reported for battery materials.
Cu2Se is an anode material with excellent sodium storage kinetics.19,20 In particular, the β-phase Cu2Se, with a face-centered cubic structure, possesses liquid-like Cu ions within the crystals. This feature endows it with good ionic and electronic conductivity while acting as an electron donor.21,22 Benefiting from this unique property, β-Cu2Se was selected to construct a bimetal selenides-based heterostructure to achieve the construction of interfacial BiEF and 1T-rich MoSe2 in this work. Nevertheless, the stability of an engineered electron-injection heterostructure faces challenges due to structural evolution during the conversion reactions, which involve volume changes and decomposition–reconstruction processes.13,23 The migration of the conversion products must be prevented during volume expansion. From this perspective, anchoring the metal selenides-based heterostructure on a suitable substrate may enhance the structural stability further by strengthening the interfacial bonds and guiding their reconstruction.24,25 The 2D material graphene oxide (GO), with large surface area and abundant surface groups, is an ideal substrate to load metal selenides and construct interfacial bonds.4,25 Considering all these advantages, it is reasonable to anticipate that a graphene-supported β-Cu2Se@MoSe2 heterostructure could exhibit superior sodium storage kinetics.
Herein, β-Cu2Se was chosen as the coupling material to construct a heterostructure with MoSe2. GO with abundant functional groups was introduced as the substrate. The formed GO-Cu2+-MoO42− complex in the precursor solution could guide the growth of bimetallic selenides during the hydrothermal selenylation process to form heterogenous Cu2Se@MoSe2 anchored on rGO nanosheets. DFT calculations and XPS data demonstrated the formation of 1T-rich MoSe2 with improved conductivity. The improved adsorption of sodium ions and construction of BiEF at the Cu2Se@MoSe2 interface promoted the diffusion rate of Na+. We also demonstrated that the rGO-metal selenides heterointerface played a critical part in facilitating the cyclic reconstruction of Cu2Se@MoSe2 heterostructures. As a result, an excellent sodium storage kinetic was realized in this dual-heterostructured G-Cu2Se@MoSe2 composite, as reflected by a high capacity of 288 mA h g−1 at 50 A g−1 and a high retention ratio of 89.6% after 15000 cycles at 10 A g−1.
2 Results and discussion
2.1 Density functional theory
Density functional theory (DFT) calculations were initially employed to explore the influence of β-Cu2Se on MoSe2 and to understand the possible functional mechanism of Cu2Se@MoSe2 heterostructures. As depicted in Fig. S1,† the computation model was optimized using β-Cu2Se with an exposed (111) facet and MoSe2 with an (002) facet. The selection of these exposed facets was based on previous reports.20,26,27 The bandgap near the Fermi level in the electronic density of states (DOS) revealed the semiconductive nature of MoSe2 (Fig. 1a) and the conductive feature of Cu2Se (Fig. 1b). For Cu2Se@MoSe2 (Fig. 1c), the Mo-d was activated through the interfacial d–p–d hybridization between Cu, Se and Mo atoms. As a result, the total DOS of Cu2Se@MoSe2 at the Fermi level was much higher than that of Cu2Se and MoSe2, implying a metallic feature with excellent mobility of electrons (Fig. 1d).18 More importantly, compared with that in Cu2Se and MoSe2, the Cu-d band and Mo-d band in Cu2Se@MoSe2 approached each other and sat closer to the Fermi energy, revealing that an electronic interaction between Cu-d to Mo-d. As displayed in the differential charge density images of Cu2Se@MoSe2 (Fig. 1e), depletion of electrons at interfacial Cu atoms and accumulation of electrons at Mo atoms were observed. Such interfacial electron transfer should enable the reorganization of the Mo 4d orbitals of MoSe2 to facilitate the formation of the 1T phase and construction of BiEF.28 The energy difference between the 2H and 1T phases decreased to 0.086 eV per atom after constructing heterostructures with Cu2Se (Fig. S1†), which is significantly lower than the reported value for neat MoSe2 (0.237 eV per atom).14,29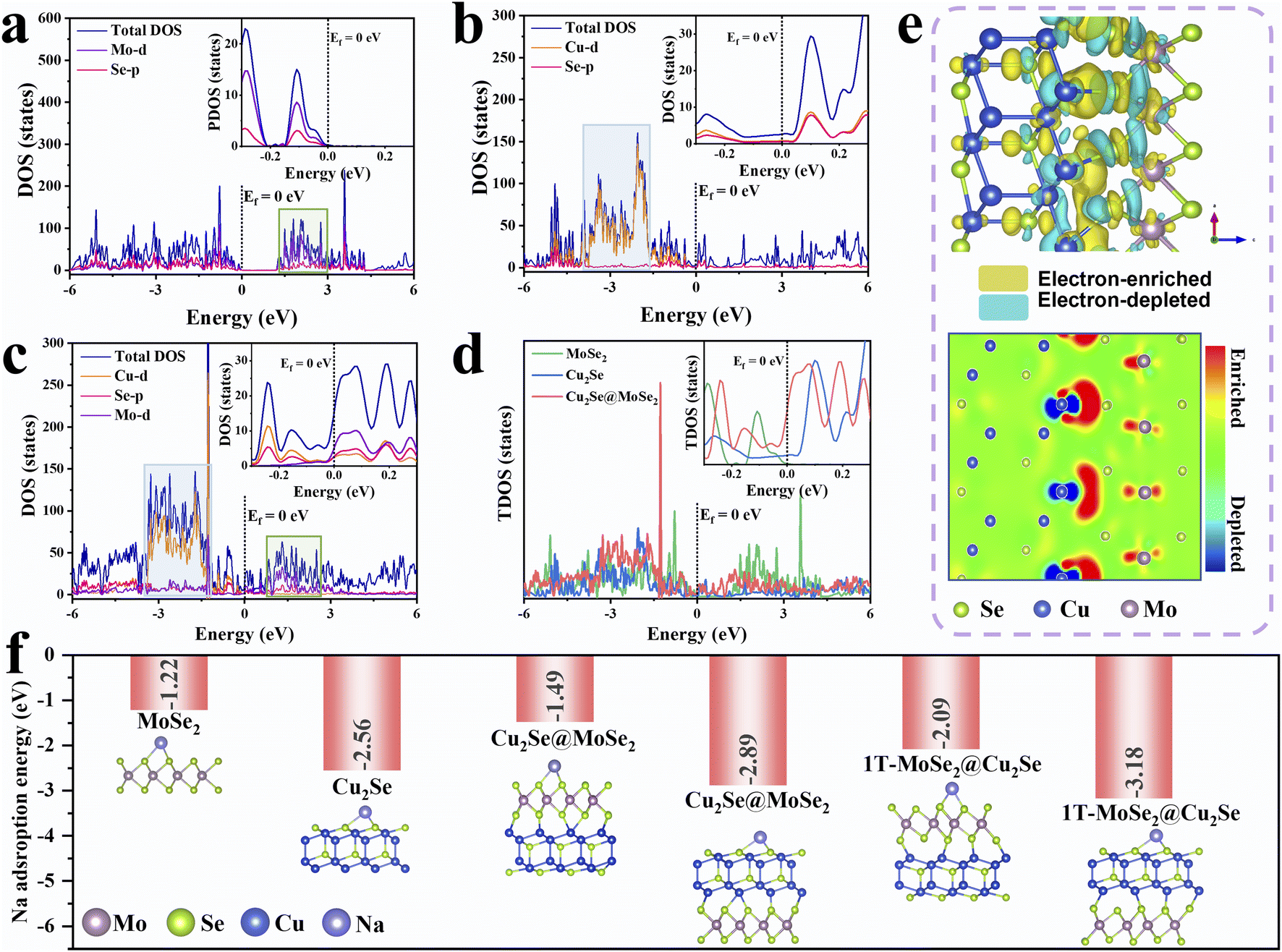 | ||
| Fig. 1 Density functional theory (DFT) calculations. The partial density of states (PDOS) of (a) MoSe2, (b) β-Cu2Se and (c) Cu2Se@MoSe2. (d) The total density of states (TDOS) of MoSe2, Cu2Se and Cu2Se@MoSe2. (e) The 3D isosurface and 2D slice images showing the differential charge density of Cu2Se@MoSe2. (f) Sodium adsorption energy of the optimized models of MoSe2, Cu2Se, Cu2Se@MoSe2 and Cu2Se@1T-MoSe2. | ||
Based on analyses of the Bader charge and difference in charge density (Fig. 1e), the electron-rich Cu was capable of offering a partial electron of 0.124 e− to Mo, resulting in the phase transition from 2H to 1T. The sodium adsorption energy was calculated based on the computational models in Fig. 1f, and was −1.22 eV for MoSe2, −2.56 eV for Cu2Se, and −1.49 eV for the MoSe2 side of the Cu2Se@MoSe2 heterostructure along with −2.89 eV for the Cu2Se side. With regard to Cu2Se@1T-MoSe2, the sodium adsorption energy was increased further to −2.09 and −3.18 eV for the 1T-MoSe2 side and Cu2Se side, respectively. Clearly, this bimetal selenides-based heterostructures could enhance the ability to capture sodium ions of Cu2Se and 1T-rich MoSe2. The enhanced adsorption energies, coupled with the BiEF generated by re-distributed electrons at the heterointerface, significantly accelerated the transfer kinetics of Na+.30 This theoretical analysis supports the hypothesis that the Cu2Se@MoSe2 heterostructure exhibits superior electronic conductivity and ion adsorption, indicating its potential to enhance sodium-storage performance.
2.2 Synthesis and characterization of G-Cu2Se@MoSe2
The preparation process and structural evolution of heterogeneous G-Cu2Se@MoSe2 composite are shown in Fig. 2a. In the precursor solution, the electrostatic attraction between the negatively charged surface groups on the GO surface, copper ions, and MoO42− induced the formation of a GO-Cu2+-MoO42− complex, which facilitated the subsequent hydrothermal selenylation process for the formation of dual heterostructures. Cu2Se@MoSe2 heterostructures were directly grown on reduced graphene oxide (rGO) nanosheets to form in situ-generated interfacial bonds. In contrast, the Cu2Se/MoSe2 sample obtained without the use of GO (Fig. S2†) displayed large polygonal Cu2Se nanosheets and small flocculent MoSe2 with loose contact, suggesting less of a heterostructure. This observation further supported the important role of GO on guiding the growth of metal selenides to construct a Cu2Se@MoSe2 heterostructure by forming a GO-Cu2+-MoO42− complex in the precursor solution. The G-MoSe2 sample displayed a hierarchical morphology with flower-like MoSe2 clusters anchored on the surface of 2D rGO nanosheets (Fig. S3a and b†). As shown in the SEM images of G-Cu2Se@MoSe2 (Fig. 2b and c), the microsized rGO plane was fully covered with hierarchical layers. The TEM image in Fig. 2d demonstrated the uniform coverage of MoSe2 layers and the existence of polygonal nanosheets. HRTEM images in Fig. 2e displays two lattice fringes with a distance of 3.3 nm and 0.67 nm, which match the (111) planes of Cu2Se and (002) planes of MoSe2 nanosheets, respectively. This result also certified the flatly decorated Cu2Se and vertically anchored MoSe2 nanosheets. Fig. 2f displays the co-existence of 1T-MoSe2 and 2H-MoSe2, with their positions supporting the important role of heterostructures in the formation of the 1T phase. The uniform distribution of elements C, O, Mo, Cu and Se (EDS mapping images, Fig. 2g) further evidenced the homogenous decoration of Cu2Se and MoSe2 on the rGO nanosheets.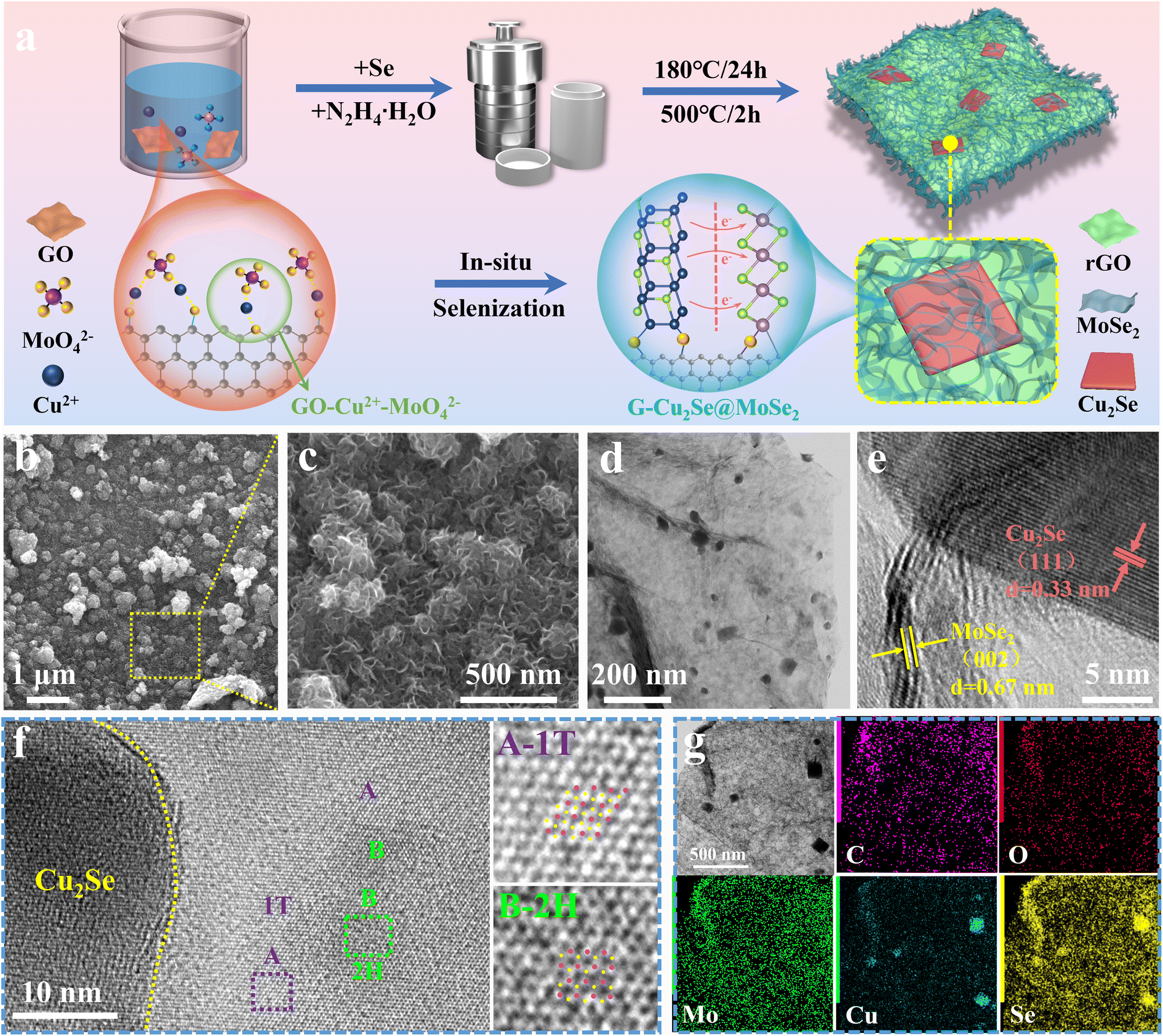 | ||
| Fig. 2 Morphology of the heterogeneous G-Cu2Se@MoSe2 composite. (a) Preparation procedures of the G-Cu2Se@MoSe2 heterogeneous composite (schematic). (b and c) SEM images showing surface morphologies. TEM (d) and HRTEM (e and f) images. (g) EDS mapping images displaying the distribution of C, O, Mo, Cu and Se elements. | ||
The distinct peaks of the G-Cu2Se@MoSe2 composite in the XRD pattern (Fig. 3a) matched well with the diffraction peaks of MoSe2 (JCPDS Card No. 29-0914) and β-Cu2Se (JCPDS Card No. 65-2982). The XRD pattern of G-MoSe2 in Fig. S3c† also evidenced the existence of MoSe2. In the Raman spectra of Cu2Se/MoSe2 and G-MoSe2 (Fig. 3b and S3d†), the main peak centered at 241 cm−1 was assigned to the A1g peaks of 2H MoSe2 and Cu2Se.31 With respect to G-Cu2Se@MoSe2, the representative peaks of the 1T phase emerged as J1, J2, E1g, J3, and E12g, thereby confirming the formation of 1T-rich MoSe2.8,32 Besides, the weak peak at around 256 cm−1 could be attributed to the A1g peak of the Cu2Se nanosheets underneath.26 The appearance of the D band (1342.2 cm−1) and G band (1593.1 cm−1) in G-Cu2Se@MoSe2 verified the presence of disordered carbon and graphite carbon. The higher intensity ratio of the D band to the G band (ID/IG = 1.65) than that for G-MoSe2 (1.4) illustrated the abundance of defects on the rGO planes, possibly resulting from the formation of more interfacial C–Mo and C–O–M (M: Cu or Mo) bonds. The BET surface area and distribution of pore widths were characterized by N2 adsorption–desorption isotherms (Fig. 3c). G-Cu2Se@MoSe2 and Cu2Se/MoSe2 exhibited similar type-IV isotherm plots, delivering a comparable surface area of 45.8 and 50.1 m2 g−1, respectively. The slightly higher surface area of Cu2Se/MoSe2 could be attributed to the presence of flocculent MoSe2 with a large exposed surface and fewer heterostructures. In contrast, G-Cu2Se@MoSe2 possessed more covalently bonded interfaces, leading to the formation of rich heterostructures and a decrease in the exposed surface area. From this perspective, the comparable specific surface area exhibited by the heterostructure-rich G-Cu2Se@MoSe2 should be attributed to the rGO nanosheets that prevented a significant decrease in the specific surface area. The majority of pores in Cu2Se/MoSe2 had a diameter of about 2.57 nm. G-Cu2Se@MoSe2 exhibited three main pores with a diameter of 2.07, 2.57 and 7.61 nm. The hierarchical morphology and dual heterostructure of G-Cu2Se@MoSe2 may lead to the formation of multiscale pores and channels to facilitate electrolyte penetration and ion transfer.33
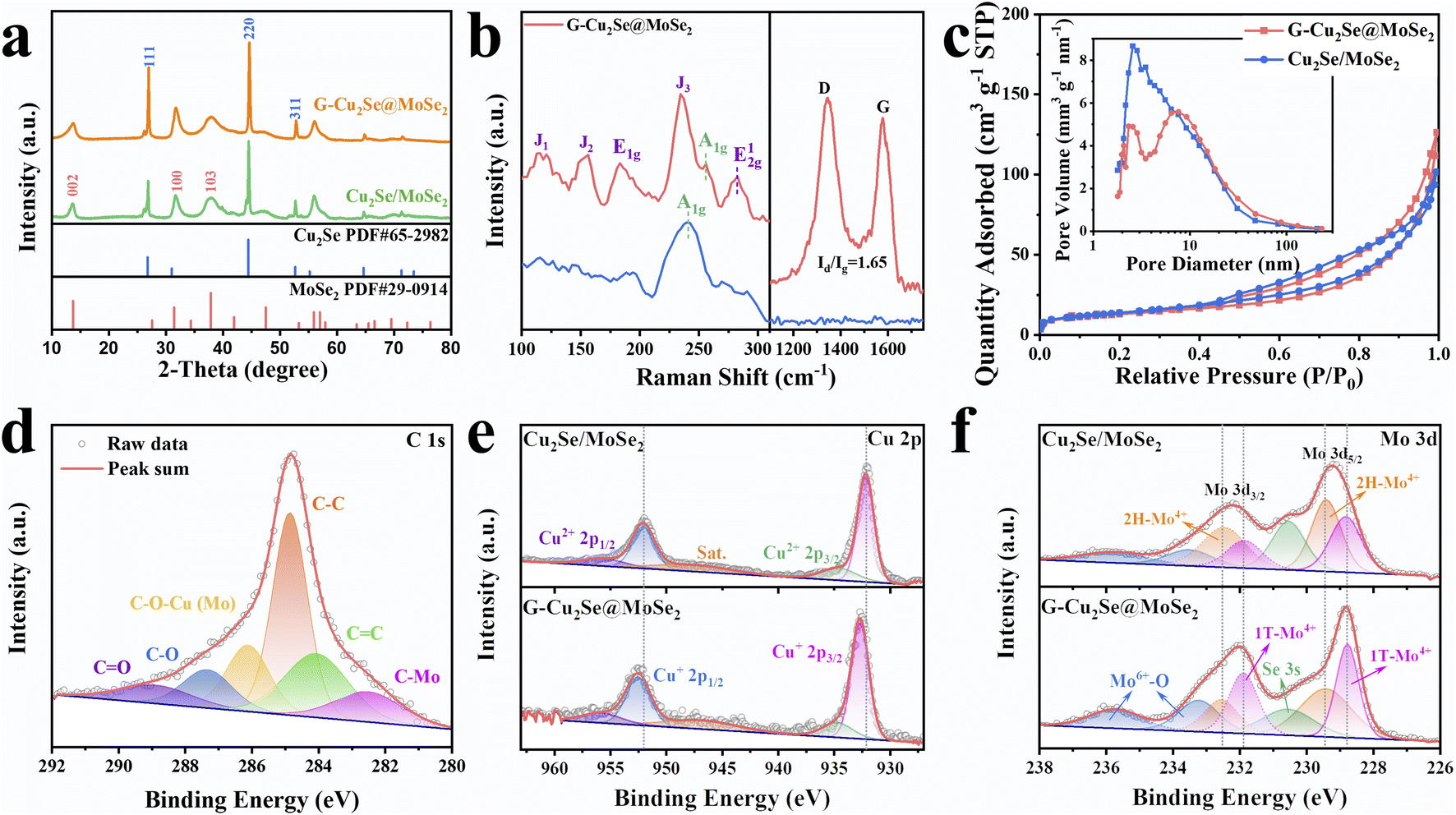 | ||
| Fig. 3 Characterizations of the microstructures in Cu2Se/MoSe2 and G-Cu2Se@MoSe2 composite. (a) XRD pattern of the G-Cu2Se@MoSe2 composite. Raman spectra (b) and N2 adsorption–desorption isotherm plots (c) of Cu2Se/MoSe2 and G-Cu2Se@MoSe2 composites. The insert in (c) is the distribution of pore sizes. (d) XPS C 1s of the G-Cu2Se@MoSe2 composite. Cu 2p (e) and Mo 3d (f) spectra of Cu2Se/MoSe2 and G-Cu2Se@MoSe2 composites. | ||
These two composites presented similar signals in the XPS spectra (Fig. S4a†). The main signals of Cu, C, Mo and Se were deconvoluted to analyze their elemental valence and chemical states. In the high-resolution C 1s spectra of G-Cu2Se@MoSe2 and G-MoSe2 (Fig. 3d and S3e†), the peaks at 282.5, 284, 284.8, 286.1, 287.3, and 288.9 eV could be indexed to C–Mo, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, C–C, C–O–M (M: Cu or Mo), C–O and CO bonds, respectively.34,35 The CC and C–C arose from rGO basal planes, while the C–O and CO bonds were attributed to residual functional groups on the rGO surface. The presence of the C–O–M bond and C–Mo bond revealed the covalently bridged interfaces between rGO and metal atoms in selenides, which suggested the formation of rGO-metal selenides heterostructures. In the Cu 2p spectra (Fig. 3e), two weak peaks at 934.8 and 955.8 eV belonged to the Cu 2p1/2 and Cu 2p3/2 of Cu2+, which could be ascribed to the interfacial C–O–Cu bond derived from the GO-Cu2+-MoO42− structure.36 Two strong peaks centered at 932.7 and 952.6 eV contributed to the Cu 2p1/2 and Cu 2p3/2 of Cu+ in Cu2Se, respectively.26 The Mo 3d spectra in Fig. 3f and S3f† showed the peaks of Mo4+, Mo6+ and Se 3s. The peaks at 228.8 and 231.7 eV referred to the Mo4+ in the 1T phase, while another two peaks at 229.4 and 233 eV represented the Mo4+ in the 2H state, thereby confirming the existence of 1T/2H MoSe2 in these two samples.37,38 Based on the integral area of 1T-Mo4+ peaks, G-Cu2Se@MoSe2 demonstrated a higher 1T-MoSe2 content of 52.9%, compared with 24.7% in Cu2Se/MoSe2 and 22.1% in G-MoSe2. The peak of Mo6+ at 232.4 and 235.6 eV could be ascribed to the partial oxidation of surface Mo atoms.4 Compared with Cu2Se/MoSe2, the Cu+ peaks of G-Cu2Se@MoSe2 shifted to higher binding energies, while the Mo4+ peaks shifted to lower binding energies, confirming electronic transfer from Cu atoms to Mo atoms at the heterointerfaces. This observation was in good agreement with the DFT results, confirming the modulation of MoSe2 to achieve a higher content of the 1T phase. C 1s, Cu 2p and Mo 3d spectra also supported construction of a dual heterostructure in G-Cu2Se@MoSe2. With regard to Se 3d spectra (Fig. S4b and c†), G-Cu2Se@MoSe2 and Cu2Se/MoSe2 displayed two similar peaks of Se 3d5/2 and Se 3d3/2, which corresponded to Se2− in metal selenides.39
C, C–C, C–O–M (M: Cu or Mo), C–O and CO bonds, respectively.34,35 The CC and C–C arose from rGO basal planes, while the C–O and CO bonds were attributed to residual functional groups on the rGO surface. The presence of the C–O–M bond and C–Mo bond revealed the covalently bridged interfaces between rGO and metal atoms in selenides, which suggested the formation of rGO-metal selenides heterostructures. In the Cu 2p spectra (Fig. 3e), two weak peaks at 934.8 and 955.8 eV belonged to the Cu 2p1/2 and Cu 2p3/2 of Cu2+, which could be ascribed to the interfacial C–O–Cu bond derived from the GO-Cu2+-MoO42− structure.36 Two strong peaks centered at 932.7 and 952.6 eV contributed to the Cu 2p1/2 and Cu 2p3/2 of Cu+ in Cu2Se, respectively.26 The Mo 3d spectra in Fig. 3f and S3f† showed the peaks of Mo4+, Mo6+ and Se 3s. The peaks at 228.8 and 231.7 eV referred to the Mo4+ in the 1T phase, while another two peaks at 229.4 and 233 eV represented the Mo4+ in the 2H state, thereby confirming the existence of 1T/2H MoSe2 in these two samples.37,38 Based on the integral area of 1T-Mo4+ peaks, G-Cu2Se@MoSe2 demonstrated a higher 1T-MoSe2 content of 52.9%, compared with 24.7% in Cu2Se/MoSe2 and 22.1% in G-MoSe2. The peak of Mo6+ at 232.4 and 235.6 eV could be ascribed to the partial oxidation of surface Mo atoms.4 Compared with Cu2Se/MoSe2, the Cu+ peaks of G-Cu2Se@MoSe2 shifted to higher binding energies, while the Mo4+ peaks shifted to lower binding energies, confirming electronic transfer from Cu atoms to Mo atoms at the heterointerfaces. This observation was in good agreement with the DFT results, confirming the modulation of MoSe2 to achieve a higher content of the 1T phase. C 1s, Cu 2p and Mo 3d spectra also supported construction of a dual heterostructure in G-Cu2Se@MoSe2. With regard to Se 3d spectra (Fig. S4b and c†), G-Cu2Se@MoSe2 and Cu2Se/MoSe2 displayed two similar peaks of Se 3d5/2 and Se 3d3/2, which corresponded to Se2− in metal selenides.39
2.3 Sodium storage of G-Cu2Se@MoSe2
The sodium-storage performances of G-Cu2Se@MoSe2 and Cu2Se/MoSe2 composites were first checked in half cells over a potential range of 0.1–2.5 V at a scan rate of 0.1 mV s−1 (Fig. 4a and S5a†). Their first three CV curves displayed three cathodic peaks and three anodic peaks at similar potential in the first scan. The peaks at 1.93 V and 1.3 V were derived from the insertion of Na+ into Cu2Se and MoSe2 to form NaxCu2Se and NayMoSe2, respectively.4 The conversion from NaxCu2Se to metal Cu and Na2Se contributed to the peak at ∼0.92 V.36 The peak at 0.31 V in the first scan could be assigned as the conversion of NayMoSe2 and the formation of a solid electrolyte interphase (SEI).35 It became weaker in subsequent cycles due to the irreversibility of SEI formation. The peaks at 1.5 V in the anodic sweeps resulted from the conversion reaction to form NaxCu2Se and NayMoSe2.26,34 Another two peaks at 1.74 and 2.05 V were related to the extraction of Na+ to form Cu2Se and MoSe2, respectively.40 The positions of CV peaks also matched well with the plateaus in their galvanostatic charge–discharge (GCD) curves (Fig. S5b and c†). At a current density of 0.1 A g−1, the heterogeneous G-Cu2Se@MoSe2 composite delivered a higher initial discharge capacity of 600 mA h g−1 and a reversible capacity of 417 mA h g−1, compared with 556 and 378 mA h g−1 for Cu2Se/MoSe2. The loss of initial capacity resulted mainly from the generation of a SEI. The higher initial coulombic efficiency for G-Cu2Se@MoSe2 (70%) compared with that of Cu2Se/MoSe2 (68%) may have been due to its smaller surface area, which consumed less Na+ to form the SEI. It was also worthy to note that G-Cu2Se@MoSe2 possessed a smaller voltage hysteresis between charge and discharge curves, indicative of its low electrode polarization, which benefits the reversibility of the reaction.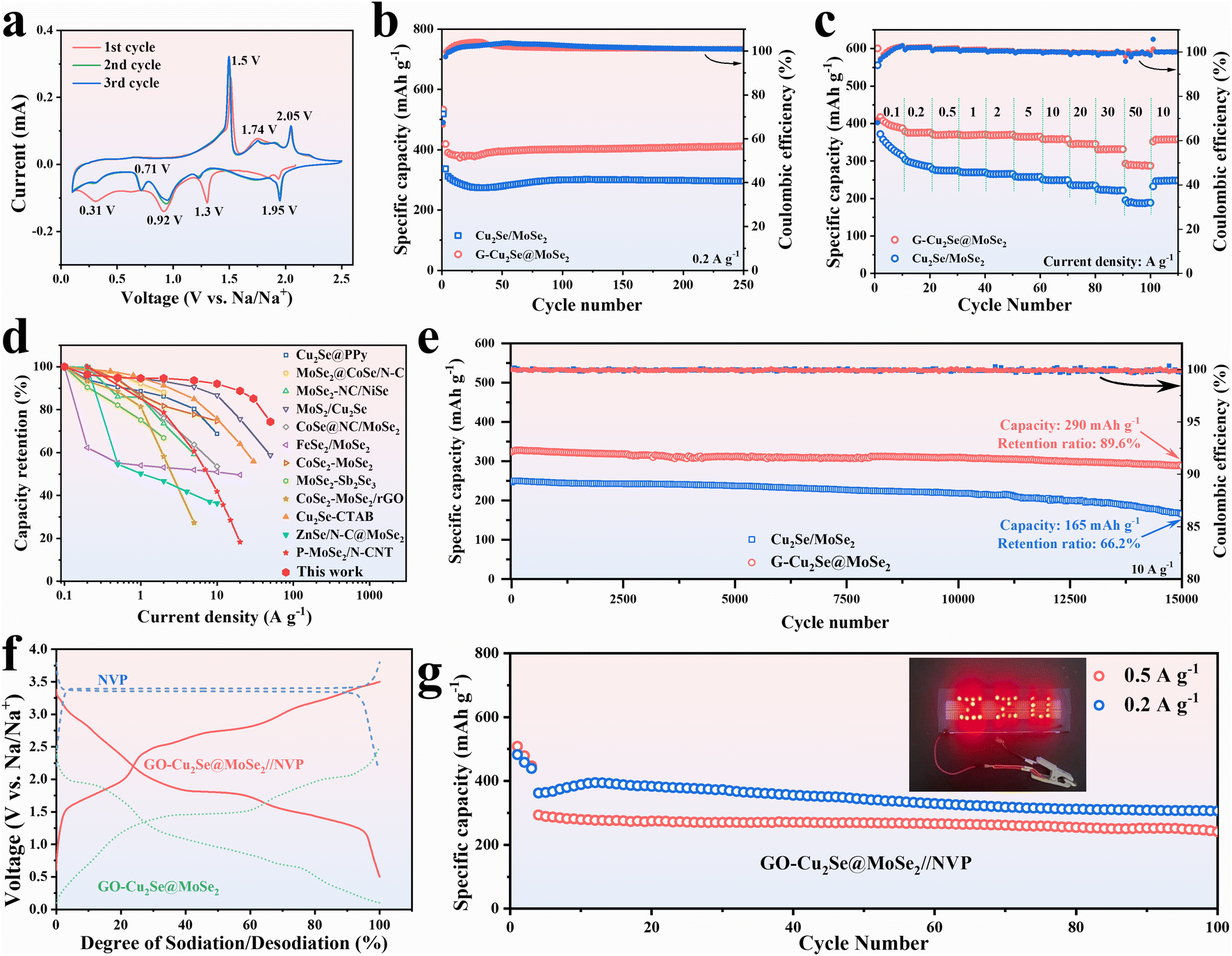 | ||
| Fig. 4 Electrochemical performance in sodium ion batteries. (a) The first three cyclic voltammograms of the G-Cu2Se@MoSe2 composite at a scan rate of 0.1 mV s−1. (b) Cycling performance of Cu2Se/MoSe2 and G-Cu2Se@MoSe2 at 0.2 A g−1. (c) Rate capability of Cu2Se/MoSe2 and G-Cu2Se@MoSe2 composites. (d) Comparison of rate performance between G-Cu2Se@MoSe2 and reported Cu2Se- or MoSe2-based anodes. (e) Long-term cycling stability of Cu2Se/MoSe2 and G-Cu2Se@MoSe2 at a current density of 10 A g−1. (f) Charge–discharge profiles of the Na3V2(PO4)3 (NVP) cathode, G-Cu2Se@MoSe2 anode and G-Cu2Se@MoSe2//NVP full cell. (g) Cycling performance of the G-Cu2Se@MoSe2//NVP full cell. | ||
As displayed in Fig. 4b, the sodium-storage performance was evaluated at 0.2 A g−1 after being activated at 0.1 A g−1 for three cycles. The capacity of G-Cu2Se@MoSe2 reached as high as 412 mA h g−1 (103.5% compared with the capacity at fourth cycle) after 250 cycles, which could be ascribed to the activation of electrodes upon cycling.41 In contrast, it was only 295 mA h g−1 (93%) for Cu2Se/MoSe2. The capacity decreased slightly then increased at the initial stage, which could be attributed to electrode activation. Rate performance was investigated from 0.1 A g−1 to 50 A g−1 (Fig. 4c). Heterogeneous G-Cu2Se@MoSe2 displayed much higher capacities at all currents, which were 390, 376, 370, 369, 369, 365, 359, 346, and 332 mA h g−1 at a current density of 0.1, 0.2, 0.5, 1, 2, 5, 10, 20 and 30 A g−1, respectively. Even at a high current density of 50 A g−1, a high capacity of 288 mA h g−1 was obtained, much higher than that obtained for Cu2Se/MoSe2 (188 mA h g−1) and for G-MoSe2 (93 mA h g−1) (Fig. S3g†). After reverting to 10 A g−1, the G-Cu2Se@MoSe2 composite maintained a higher capacity of 358 mA h g−1 than the 246 mA h g−1 for Cu2Se@MoSe2. The GCD profiles at different current densities of G-Cu2Se@MoSe2 are presented in Fig. S6.† The gradually weakened plateaus with an increase in current density revealed the domination of the capacitive process. The lower voltage hysteresis of G-Cu2Se@MoSe2 than that of Cu2Se/MoSe2 was also displayed at all current densities, evidencing its excellent rate capability. The rate performance of G-Cu2Se@MoSe2 was compared with that of other reported Cu2Se- or MoSe2-based anodes (Fig. 4d), which clearly supported the superiority of G-Cu2Se@MoSe2.4,19,26,42–50 Such excellent rate performance could be attributed to the dual heterostructure, which increased the proportion of 1T-phase MoSe2 and established interfacial BiEF for enhanced electron mobility, improved sodium adsorption, and promotion of ion transfer.
Long-term cycling stability tests were performed at 10 A g−1 after the rate tests (Fig. 4e). The G-Cu2Se@MoSe2 composite displayed excellent stability during the whole test, with a retention ratio of 89.6% (291 mA h g−1) after 15000 cycles. However, the Cu2Se/MoSe2 anode displayed a faster capacity fading with a retention ratio of only 66.2%. The large fluctuation in the coulombic efficiency of Cu2Se/MoSe2 also indicated its unstable conversion reactions. G-MoSe2 delivered a capacity of only 194 mA h g−1 after 8600 cycles at 10 A g−1 (Fig. S3h†). The cycling performance of G-Cu2Se@MoSe2 was tested further using fresh cells (Fig. S7†), eliciting a capacity of 331 mA h g−1 with a retention ratio of 95.1% after 3300 cycles at 1 A g−1 and 323 mA h g−1 with a retention ratio of 94.7% after 10000 cycles at 10 A g−1. Based on the ESI in Fig. S8,† the rGO content in G-Cu2Se@MoSe2 was estimated to be 12%, while the capacity of pure rGO anodes was only 110 mA h g−1 at 0.1 A g−1, 32 mA h g−1 at 50 A g−1, and 42 mA h g−1 after 15000 cycles at 10 A g−1. These findings strongly suggested that the role of the rGO substrate lay in facilitating rapid reaction kinetics and mitigating volume changes rather than directly enhancing the capacity. Unequivocally, the outstanding sodium-storage performance of the G-Cu2Se@MoSe2 composite could be attributed to the construction of dual heterostructures. The bimetal selenides-based heterostructure limited the aggregation of conversion products and accommodated the expansion stress, while the rGO-metal selenides heterostructure facilitated the reconstruction of a bimetal selenides-based heterostructure, leading to stabilization of 1T-rich MoSe2 and BiEF.
To evaluate the practical application of the G-Cu2Se@MoSe2 composite further, full SIB batteries coupled with Na3V2(PO4)3 (NVP) cathodes were assembled. NVP cathodes exhibited a plateau of about 3.35 V in half cells with metal sodium as counter electrodes (Fig. 4f) and delivered a high specific capacity of 89.3 mA h g−1 after 100 cycles at 1 A g−1 (Fig. S9a†). With regard to G-Cu2Se@MoSe2//NVP full cells, a voltage window of 0.5–3.5 V was chosen to perform electrochemical tests. Based on the weight of active materials (G-Cu2Se@MoSe2) in anodes, a high specific capacity of 335, 283 and 190 mA h g−1 was obtained at 0.5, 1 and 2 A g−1, respectively (Fig. S9b†). The calculated energy density of the full cell could reach as high as 144.7 W h kg−1. After 100 cycles (Fig. 4g), the full cell could deliver a reversible capacity of 306 mA h g−1 (84.5%) at 0.2 A g−1 and at 241 mA h g−1 (82.2%) at 0.5 A g−1. The inset image in Fig. 4g demonstrates a “ZZU” pattern composed by 29 LED bulbs that had been lit-up by a fully charged G-Cu2Se@MoSe2//NVP cell, thereby proving its practical potential.
2.4 Reaction kinetics of G-Cu2Se@MoSe2
The electrochemical reaction kinetics of the G-Cu2Se@MoSe2 composite were investigated by analyzing the capacitive contribution, impedance properties and ion diffusivity. The CV curves of G-Cu2Se@MoSe2 and Cu2Se@MoSe2 composites at various scan rates were collected and are shown in Fig. 5a and S10a,† respectively. The marked five peaks were selected to analyze the capacitive contributions on the basis of the power-law relationship (i = aνb) between peak current (i) and scan rate (ν).47 A b value of 1 indicates a surface-capacitive dominated electrochemical process. A b value of 0.5 denotes a diffusion-controlled process. By fitting the linear slopes of log (i) versus log (v) (Fig. 5b), all values of b were determined in the range of 0.77–1, suggesting that the discharge/charge processes of G-Cu2Se@MoSe2 was mainly controlled by surface-capacitive behavior.34 In contrast, the slightly lower b values for Cu2Se/MoSe2 (Fig. S10b†) indicated a lower capacitive contribution. Then, the contribution of surface-capacitive processes at each scan rate (ν) was calculated through the equation i = k1ν + k2ν0.5, where k1ν and k2ν0.5 are the sections of capacitive and diffusion processes, respectively.51 Unsurprisingly, G-Cu2Se@MoSe2 displayed a higher capacitive contribution at all scan rates (Fig. 5c and S10c†). For example, the capacitive contribution for G-Cu2Se@MoSe2 and Cu2Se/MoSe2 (Fig. S11†) was 92.1% and 89.6% at 0.3 mV s−1, respectively. The higher contribution of the capacitive process in G-Cu2Se@MoSe2 could enable fast reaction kinetics and, thus, excellent rate performance.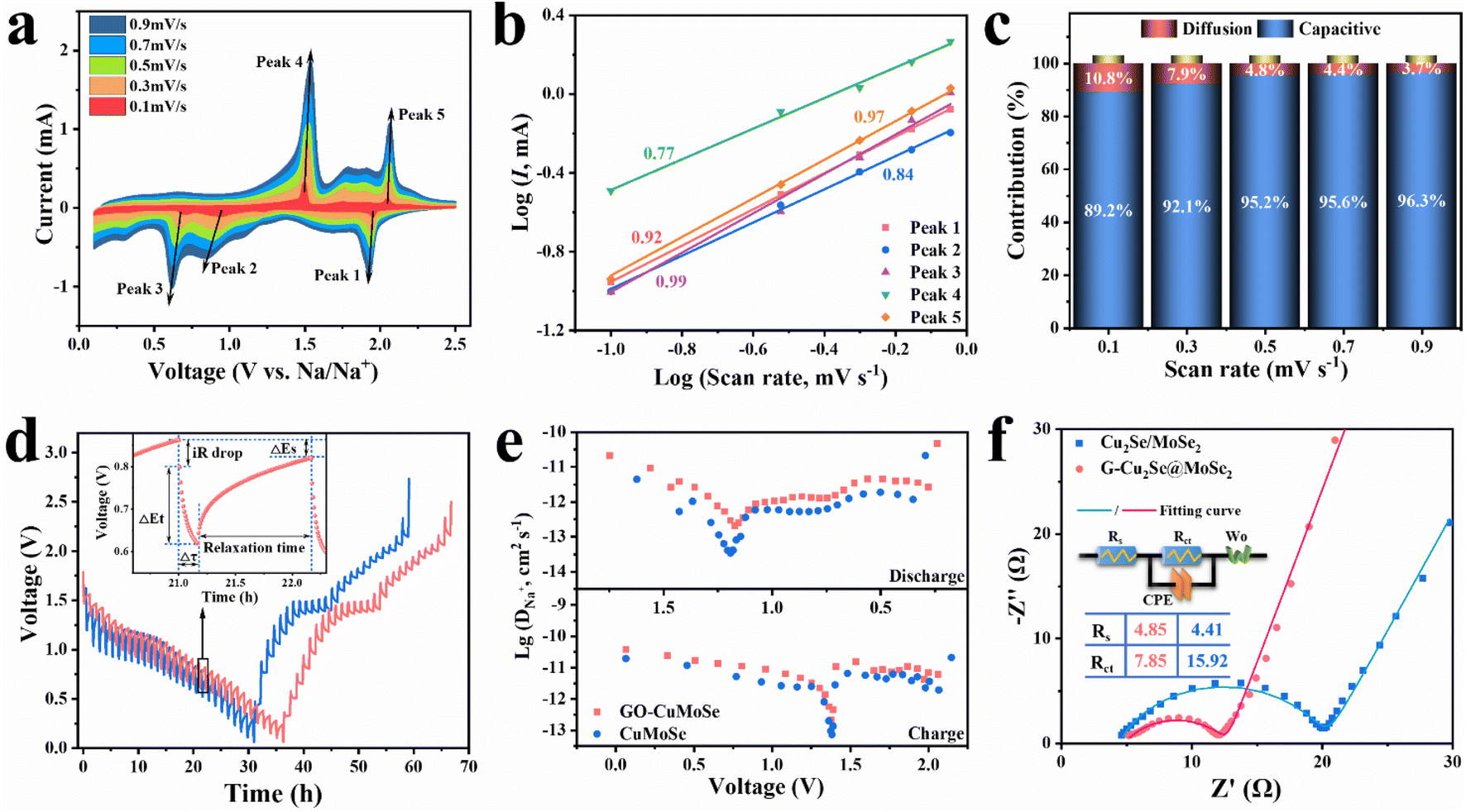 | ||
| Fig. 5 Electrochemical reaction kinetics of the G-Cu2Se@MoSe2 composite. (a) CV of the G-Cu2Se@MoSe2 electrode recorded at scan rates from 0.1 to 0.9 mV s−1. (b) b values of the marked peaks in (a) fitted from the logarithmic values of their peak currents and scan rates. (c) Contribution of a surface-capacitive process at different scan rates of the G-Cu2Se@MoSe2 electrode. (d) GITT curves of G-Cu2Se@MoSe2 and Cu2Se/MoSe2 composites (inset is an enlarged cycle). (e) Diffusion coefficient of Na+ during charge and discharge processes. (f) Nyquist plots of G-Cu2Se@MoSe2 and G-Cu2Se/MoSe2 composites (inset is the model of an equivalent circuit and fitted values of Rs and Rct). | ||
Ion diffusion coefficients (D) were studied by the galvanostatic intermittent titration technique (GITT) as presented in Fig. 5d. According to eqn (S1†),52 plots of D values during charge and discharge processes are given in Fig. 5e. Heterogenous G-Cu2Se@MoSe2 possessed a higher diffusion coefficient of Na+ during the entire electrochemical reaction. This excellent Na+ transfer kinetics could have mainly resulted from the enhanced adsorption energy of Na+ and the BiEF at the heterointerfaces of Cu2Se@MoSe2. The kinetic data of G-MoSe2 were also collected (Fig. S12†) and showed a low contribution of surface capacitive process at all scans and poor ion diffusivity. These sluggish reaction kinetics were in good accordance with the poor rate performance of G-MoSe2 (Fig. S3g†), further emphasizing the crucial role of heterostructures. Nyquist plots, an equivalent circuit and fitted results of fresh cells are presented in Fig. 5f. The labels of Rs, Rct, CPE and Wo used in the equivalent circuit represent contact resistance, charge transfer resistance, constant phase element and Warburg impedance, respectively.53 The G-Cu2Se@MoSe2 composite displayed a smaller semicircle in the high-frequency region with a lower charge transfer resistance (Rct) of 7.85 Ω than that for Cu2Se/MoSe2 (15.92 Ω). Conductive rGO nanosheets, 1T-phase rich MoSe2 and interfacial bonds were likely the main reasons. Warburg factors were acquired by fitting the linear slopes of Z′ versus ω−1/2 in Fig. S13a.†43 According to eqn (S2),† the smaller Warburg factor (σ) for G-Cu2Se@MoSe2 indicated a faster diffusion rate of Na+ than that for the control sample. These results confirmed the exceptional Na+ transfer kinetics of G-Cu2Se@MoSe2, which could be ascribed to improved sodium adsorption and interfacial BiEF. The impendence property of G-Cu2Se@MoSe2 upon cycling was also investigated (Fig. S13b†). The Rct decreased to about 4.07 after 20 cycles and retained almost no change at 50 cycles. On the other hand, the Warburg factor decreased to 25.7 at 20 cycles and 18.8 at 50 cycles, indicating the continuous increase of ion diffusivity along cycling, which could be attributed to electrode activation creating more ion-diffusion channels.54 This activation process also reconstructed the electrode structure to increase the amount of ion-accessible active materials, thereby enhancing the capacity during the initial cycling stage of fresh G-Cu2Se@MoSe2 anodes, as observed in Fig. S7.†
2.5 Structural evolution and reaction mechanism
The stability of dual heterostructure and 1T-rich MoSe2 during cycling tests were critical for the cycling performance of G-Cu2Se@MoSe2. To investigate the structural evolution, cycled cells were disassembled to examine the active materials by TEM and XPS. HRTEM images of G-Cu2Se@MoSe2 with the corresponding selected area electron diffraction (SAED) pattens at 0.1 V and 2.5 V are presented in Fig. 6a and b, respectively. The lattices observed at 0.1 V were the (311) and (200) planes of Na2Se. They matched well with the SAED patterns, indicating completion of the conversion reaction. At 2.5 V, the lattice distance of 0.215 and 0.24 nm belonged to the 006 and 103 planes of MoSe2, respectively. Another crystal lattice with a d-spacing of 0.33 nm was the (111) plane of Cu2Se. They were also in good agreement with SAED patterns, demonstrating good recovery of metal selenides after desodiation. EDS mapping images in Fig. S14† confirmed the uniform distribution of Cu, Mo and Se elements in the cycled G-Cu2Se@MoSe2 composite, evidencing the reconstruction of the Cu2Se@MoSe2 heterostructure. In addition, HRTEM images in Fig. 6c demonstrated the co-existence of 1T-MoSe2 and 2H MoSe2. The Raman spectrum in Fig. S15† also displays the representative peaks of 1T MoSe2, revealing the well-preserved 1T phase in the cycled G-Cu2Se@MoSe2 anode. The Cu 2p spectra in Fig. 6d and Mo 3d patterns in Fig. 6e were similar to their initial states. The shifting of Mo4+ peaks to lower energies and Cu+ to higher energies could be detected in the G-Cu2Se@MoSe2 composite, illustrating the preservation of 1T-rich MoSe2. Interestingly, the portion of 1T-MoSe2 in G-Cu2Se@MoSe2 was increased slightly to 62.5%, while the control sample remained unchanged. Moreover, the presence of C–O–M (M: Cu or Mo) and C–Mo bonds in the C 1s spectra of G-Cu2Se@MoSe2 at 0.1 V and 2.5 V (Fig. 6f) confirmed their stability during the conversion reaction, which facilitated the reconstruction of heterostructures. This finding could also explain the absence of Cu and Mo in the TEM image of G-Cu2Se@MoSe2 at 0.1 V. The generated Cu and Mo should be fixed on the surface of rGO and coverage by Na2Se. This result, combined with TEM data, could strongly support that the dual heterostructure ensured the excellent stability of the Cu2Se@MoSe2 heterostructure to facilitate the reconstruction of 1T-MoSe2 upon cycling.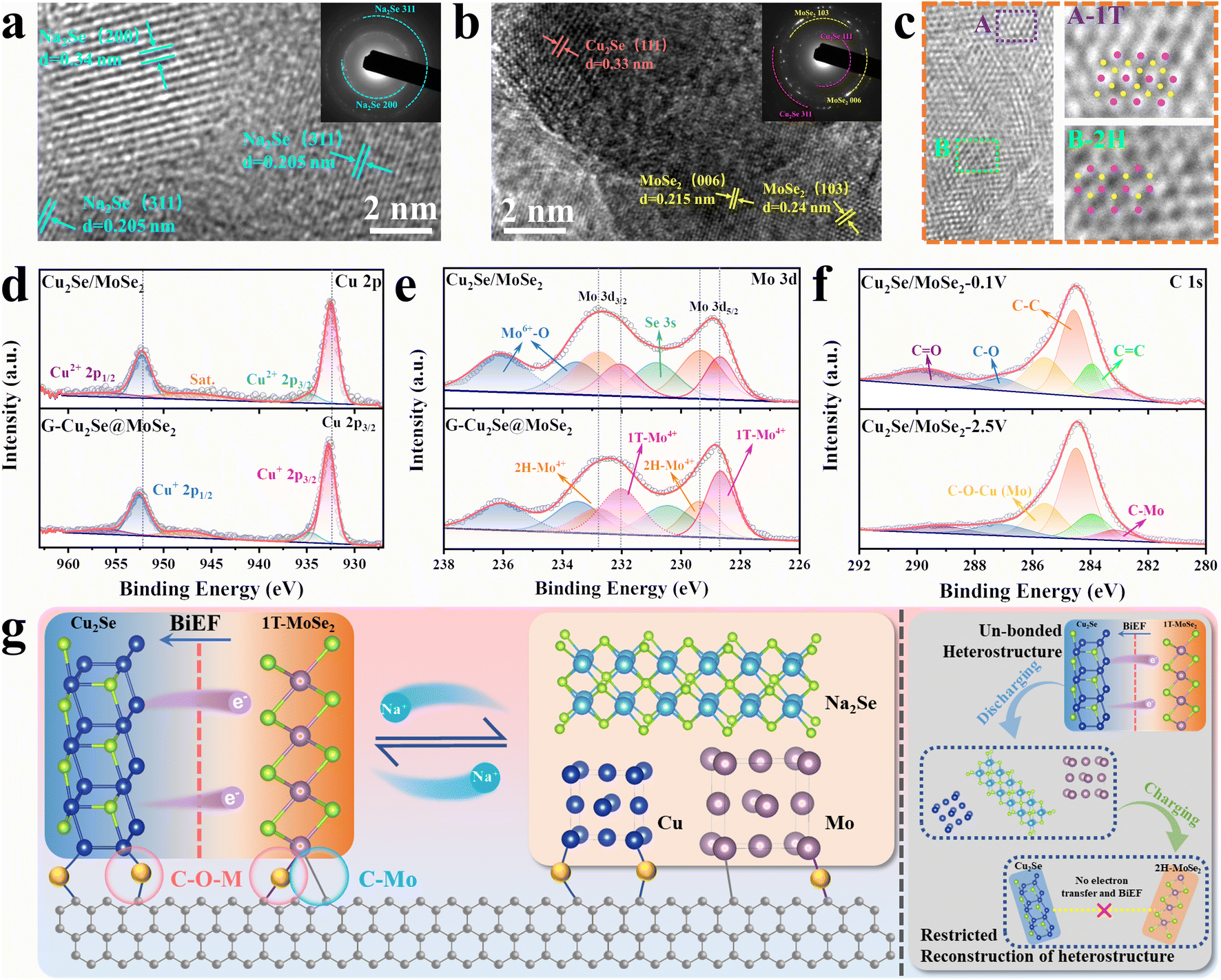 | ||
| Fig. 6 Structural evolution of G-Cu2Se@MoSe2. TEM images and corresponding SAED patterns of cycled G-Cu2Se@MoSe2 anodes that disassembled at 0.01 V (a) and 2.5 V (b). HRTEM images of cycled G-Cu2Se@MoSe2 showing the presence of 1T/2H MoSe2. Cu 2p (d) and Mo 3d (e) spectra of cycled Cu2Se/MoSe2 and G-Cu2Se@MoSe2 anodes. (f) C 1s spectra of cycled G-Cu2Se@MoSe2 anodes at 0.01 V and 2.5 V. (g) Reaction mechanism of G-Cu2Se@MoSe2 for stabilizing the reconstruction of the Cu2Se@MoSe2 heterostructure and 1T-rich MoSe2. | ||
The reaction mechanism and structural evolution of dual heterostructures in G-Cu2Se@MoSe2 is summarized and illustrated in Fig. 6g. The electron transfer generated at the heterointerfaces contributes to the formation of BiEF and a high content of 1T phase in MoSe2. Crucially, the stability of interfacial C–O–M (M: Cu or Mo) and C–Mo bonds at the rGO-metal selenides heterointerfaces is essential during the conversion process. This stability can effectively hinder the unrestrained growth and migration of the formed metal agglomerations, thereby promoting the subsequent reconstruction of Cu2Se@MoSe2 heterostructures throughout the charging process. This action further stabilizes the cyclic formation of interfacial BiEF and 1T-rich MoSe2. In contrast, the un-bonded Cu2Se@MoSe2 heterostructures experience unrestricted growth of conversion products, ultimately leading to the degradation of heterostructures and a decreased content of 1T phased MoSe2.55–57 Consequently, it can be inferred that the interfacial bonds between rGO and metal selenides have a pivotal role in facilitating the transfer of electrons and ions during long-term charging/discharging cycles. This functional mechanism highlights the crucial interactions between the dual heterostructures in the G-Cu2Se@MoSe2 composite, which holds pivotal importance for the prospective design and development of bimetal selenides-based heterostructures.
3 Conclusions
To summarize, a dual-heterostructured G-Cu2Se@MoSe2 composite was developed through a facile hydrothermal process by leveraging the GO-Cu2+-MoO42− complexes in the precursor. Electron transfer at the Cu2Se@MoSe2 interface, confirmed by DFT and XPS, induces the formation of the 1T phase in MoSe2, improving conductivity and creating a built-in electric field to enhance ion transfer. Moreover, the stable interfacial bonds between rGO and metal selenides immobilize the conversion products, thereby facilitating the reconstruction of heterostructures during cyclic conversion reactions. Consequently, the 1T-rich MoSe2 and BiEF are maintained, enabling the continuous enhancement of electron and ion transfer during long-term cycling processes. As a result, the G-Cu2Se@MoSe2 composite delivered excellent electrochemical performance, such as a high capacity of 390 mA h g−1 at 0.1 A g−1, excellent rate performance of 288 mA h g−1 at 50 A g−1, and long-term cycling stability with a capacity retention ratio of 89.6% after 15000 cycles at 10 A g−1. We also revealed the working mechanism of the 1T MoSe2-involved heterostructure and the structural evolution during conversion reactions. The insights from this work may offer valuable guidelines for the development of conversion materials for sodium ion batteries.
Data availability
The data underlying this study are available in the ESI.†Author contributions
Y. C.: conceptualization, investigation, experimental methodology, and writing – original draft. S. J.: investigation, synthetic methodology, and writing – original draft (synthesis). J. L.: validation, data curation, and visualization. G. C., L. L. and F. T.: investigation. J. Z.: conceptualization, supervision, and writing – review and editing. N. H.: conceptualization and supervision. C. W.: conceptualization and supervision.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors gratefully acknowledge the financial support from National Natural Science Foundation of China (No. 52202119 and 52272242), Natural Science Foundation of Henan Province (No. 242300421432) and fellowship of China Postdoctoral Science Foundation (No. 2022TQ0283).Notes and references
- P. K. Nayak, L. Yang, W. Brehm and P. Adelhelm, Angew. Chem., Int. Ed., 2018, 57, 102–120 CrossRef CAS PubMed.
- H. Kim, H. Kim, Z. Ding, M. H. Lee, K. Lim, G. Yoon and K. Kang, Adv. Energy Mater., 2016, 6, 201600943 Search PubMed.
- Y. Gong, Y. Li, Y. Li, M. Liu, Y. Bai and C. Wu, Small, 2023, 19, 2206194 CrossRef CAS PubMed.
- Y. Xu, X. Liu, H. Su, S. Jiang, J. Zhang and D. Li, Energy Environ. Mater., 2022, 5, 627–636 CrossRef CAS.
- J. Y. Hwang, S. T. Myung and Y. K. Sun, Chem. Soc. Rev., 2017, 46, 3529–3614 RSC.
- M. Luo, H. Yu, F. Hu, T. Liu, X. Cheng, R. Zheng, Y. Bai, M. Shui and J. Shu, Chem. Eng. J., 2020, 380, 122557 CrossRef CAS.
- Z. Hao, X. Shi, Z. Yang, L. Li and S.-L. Chou, Adv. Funct. Mater., 2022, 32, 2208093 CrossRef CAS.
- H. He, H. Zhang, D. Huang, W. Kuang, X. Li, J. Hao, Z. Guo and C. Zhang, Adv. Mater., 2022, 34, 2200397 CrossRef CAS PubMed.
- Y. Chao, Y. Han, Z. Chen, D. Chu, Q. Xu, G. Wallace and C. Wang, Adv. Sci., 2023, 2305558 Search PubMed.
- S. Li, C. Huang, L. Gao, Q. Shen, P. Li, X. Qu, L. Jiao, Y. Liu, S. Li, C. Huang, L. Gao, Q. Shen, P. Li, X. Qu, Y. Liu and L. Jiao, Angew. Chem., Int. Ed., 2022, 61, e202211478 CrossRef CAS PubMed.
- S. Li, X. Zhao, T. Wang, J. Wu, X. Xu, P. Li, X. Ji, H. Hou, X. Qu, L. Jiao and Y. Liu, Angew. Chem., Int. Ed., 2024, 63, e202320075 CrossRef CAS PubMed.
- G. Lin, Q. Ju, X. Guo, W. Zhao, S. Adimi, J. Ye, Q. Bi, J. Wang, M. Yang and F. Huang, Adv. Mater., 2021, 33, 2007509 CrossRef CAS PubMed.
- H. Xia, L. Zan, P. Yuan, G. Qu, H. Dong, Y. Wei, Y. Yu, Z. Wei, W. Yan, J. Hu, D. Deng and J. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202218282 CrossRef CAS PubMed.
- Y. Zhang, J. Wang, L. Shan, B. Han, Q. Gao, Z. Cai, C. Zhou, X. Tian, R. Sun and L. Mai, Adv. Energy Mater., 2023, 2303464 Search PubMed.
- G. Fang, Q. Wang, J. Zhou, Y. Lei, Z. Chen, Z. Wang, A. Pan and S. Liang, ACS Nano, 2019, 13, 5635–5645 CrossRef CAS PubMed.
- J. Cheng, Z. Niu, Z. Zhao, X. Pei, S. Zhang, H. Wang, D. Li and Z. Guo, Adv. Energy Mater., 2023, 13, 2203248 CrossRef CAS.
- X. Xie, X. Ma, Z. Yin, H. Tong, H. Jiang, Z. Ding and L. Zhou, Chem. Eng. J., 2022, 446, 137366 CrossRef CAS.
- Y. Li, Z. Yuan, D. Li, J. Li, Y. Zhang, M. Wang, G. Li, L. Wang and W. Han, ACS Nano, 2024, 18, 4733–4745 CrossRef CAS PubMed.
- Y. Xiao, X. Zhao, X. Wang, D. Su, S. Bai, W. Chen, S. Fang, L. Zhou, H.-M. Cheng and F. Li, Adv. Energy Mater., 2020, 10, 2000666 CrossRef CAS.
- H. Peng, W. Miao, S. Cui, Z. Liu, X. Wang, B. Tao, W. Hou, Z. Zhang and G. Ma, Chem. Eng. J., 2024, 487, 150701 CrossRef CAS.
- P. Y. Deng, K. K. Wang, H. Y. Sung, W. W. Wu and H. J. Wu, Cell Rep. Phys. Sci., 2023, 4, 101413 CrossRef CAS.
- F. Jia, S. Zhang, X. Zhang, X. Peng, H. Zhang and Y. Xiang, Chem.–Eur. J., 2014, 20, 15941–15946 CrossRef CAS PubMed.
- Y. Fang, D. Luan, X. Wen, D. Lou, Y. J. Fang, D. Y. Luan and X. W. Lou, Adv. Mater., 2020, 32, 2002976 CrossRef CAS PubMed.
- C. Wang, J. Lu, H. Tong, S. Wu, D. Wang, B. Liu, L. Cheng, Z. Lin, L. Hu, H. Wang, W. Zhang and Q. Chen, Nano Res., 2021, 14, 3545–3551 CrossRef CAS.
- S. Li, Y. Liu, X. Zhao, Q. Shen, W. Zhao, Q. Tan, N. Zhang, P. Li, L. Jiao and X. Qu, Adv. Mater., 2021, 33, 2007480 CrossRef CAS PubMed.
- L. Yue, D. Wang, Z. Wu, W. Zhao, Y. Ren, L. Zhang, B. Zhong, N. Li, B. Tang, Q. Liu, Y. Luo, A. M. Asiri, X. Guo and X. Sun, Chem. Eng. J., 2022, 433, 134477 CrossRef CAS.
- Y. Yin, Y. Zhang, T. Gao, T. Yao, X. Zhang, J. Han, X. Wang, Z. Zhang, P. Xu, P. Zhang, X. Cao, B. Song, S. Jin, Y. Yin, Y. Zhang, X. Zhang, J. Han, T. Yao, X. Wang, B. Song, Z. Zhang, P. Xu, P. Zhang, X. Cao and S. Jin, Adv. Mater., 2017, 29, 1700311 CrossRef PubMed.
- Z. Liu, Y. Tian, S. Li, L. Wang, B. Han, X. Cui and Q. Xu, Adv. Funct. Mater., 2023, 33, 2301994 CrossRef CAS.
- K.-A. N. Duerloo, Y. Li and E. J. Reed, Nat. Commun., 2014, 5, 4214 CrossRef CAS PubMed.
- P. Yan, L. Ji, X. Liu, Q. Guan, J. Guo, Y. Shen, H. Zhang, W. Wei, X. Cui and Q. Xu, Nano Energy, 2021, 86, 106139 CrossRef CAS.
- R. Zhou, Y. Huang, J. Zhou, H. Niu, L. Wan, Y. Li, J. Xu and J. Xu, Dalton Trans., 2018, 47, 16587–16595 RSC.
- Y. Yu, G.-H. Nam, Q. He, X.-J. Wu, K. Zhang, Z. Yang, J. Chen, Q. Ma, M. Zhao, Z. Liu, F.-R. Ran, X. Wang, H. Li, X. Huang, B. Li, Q. Xiong, Q. Zhang, Z. Liu, L. Gu, Y. Du, W. Huang and H. Zhang, Nat. Chem., 2018, 10, 638–643 CrossRef CAS PubMed.
- G. Zhu, D. Luo, X. Chen, J. Yang and H. Zhang, ACS Nano, 2023, 17, 20850–20874 CrossRef PubMed.
- Z. Li, L. Yu, X. Tao, Y. Li, L. Zhang, X. He, Y. Chen, S. Xiong, W. Hu, J. Li, J. Wang, H. Jin and S. Wang, Small, 2023, 2304124 Search PubMed.
- S. Chong, X. Wei, Y. Wu, L. Sun, C. Shu, Q. Lu, Y. Hu, G. Cao and W. Huang, ACS Appl. Mater. Interfaces, 2021, 13, 13158–13169 CrossRef CAS PubMed.
- X. Liu, Z. Niu, Y. Xu, Z. Zhao, C. Li, Y. Yi, H. Guan, S. Zhang, X. Pei and D. Li, Chem. Eng. J., 2022, 430, 133176 CrossRef CAS.
- X. Zhang, Y.-Y. Zhang, Y. Zhang, W.-J. Jiang, Q.-H. Zhang, Y.-G. Yang, L. Gu, J.-S. Hu and L.-J. Wan, Small Methods, 2019, 3, 1800317 CrossRef.
- Y. Zhang, S. Deng, Y. Shen, B. Liu, G. Pan, Q. Liu, X. Wang, Y. Wang, X. Xia and J. Tu, ChemSusChem, 2020, 13, 1575–1581 CrossRef CAS PubMed.
- Y. R. Pei, H. Y. Zhou, M. Zhao, J. C. Li, X. Ge, W. Zhang, C. C. Yang and Q. Jiang, Carbon Energy, 2023, e374 Search PubMed.
- R. Jin, X. Liu, L. Yang, G. Li and S. Gao, Electrochim. Acta, 2018, 259, 841–849 CrossRef CAS.
- Y. Chao, R. Jalili, Y. Ge, C. Wang, T. Zheng, K. Shu and G. G. Wallace, Adv. Funct. Mater., 2017, 27, 1700234 CrossRef.
- Y. Hoe Seon, Y. Chan Kang and J. S. Cho, Chem. Eng. J., 2021, 425, 129051 CrossRef CAS.
- N. Shi, Y. Chu, B. Xi, M. Huang, W. Chen, B. Duan, C. Zhang, J. Feng and S. Xiong, Adv. Energy Mater., 2020, 10, 2002298 CrossRef CAS.
- Y. Liu, Y. Yi, Z. Niu, S. Wei, X. Pei, Y. Fu, J. Wang, M. Ge, Z. Liu and D. Li, ACS Appl. Mater. Interfaces, 2022, 14, 6926–6936 CrossRef CAS PubMed.
- L. Zhang, H. Dong, H. Wei, E. H. Ang, J. Yang, X. Miao, H. Geng and X. Zuo, J. Power Sources, 2021, 506, 230216 CrossRef CAS.
- S. Li, H. Zhang, Y. Cao, S. Zhang, Z. Liu, C. Yang, Y. Wang and B. Wan, Nanoscale, 2023, 15, 5655–5664 RSC.
- J. Feng, S. hua Luo, P. wei Li, Y. cheng Lin, L. Zhang, Q. Wang and Y. hui Zhang, Appl. Surf. Sci., 2023, 619, 156775 CrossRef CAS.
- B. Wang, J. Y. Xue, F. L. Li, H. Geng and J. P. Lang, ChemSusChem, 2021, 14, 5304–5310 CrossRef CAS PubMed.
- Q. Sun, Y. Wen, S. Jiang, X. Li, Z. Yao, T. Liu, S. Shen and Y. Yang, J. Alloys Compd., 2023, 944, 169157 CrossRef CAS.
- J. Chen, A. Pan, Y. Wang, X. Cao, W. Zhang, X. Kong, Q. Su, J. Lin, G. Cao and S. Liang, Energy Storage Materials, 2019, 21, 97–106 CrossRef.
- Z. Yan, L. Zhao, Y. Liang, L. Zhang, H. Liu, Z. Zhu, Y. Wang and S. L. Chou, Carbon Energy, 2022, 4, 586–597 CrossRef CAS.
- Y. Liang, Z. Wang, Z. Xu, S. Li, H. Luo, C. Xu and X. Cui, Appl. Surf. Sci., 2024, 651, 159234 CrossRef CAS.
- M. Yousaf, Y. Wang, Y. Chen, Z. Wang, A. Firdous, Z. Ali, N. Mahmood, R. Zou, S. Guo and R. P. S. Han, Adv. Energy Mater., 2019, 9, 1900567 CrossRef.
- F. Zhou, S. Xin, H. W. Liang, L. T. Song and S. H. Yu, Angew. Chem., Int. Ed., 2014, 53, 11552–11556 CrossRef CAS PubMed.
- L. Wang, J. Światowska, S. Dai, M. Cao, Z. Zhong, Y. Shen and M. Wang, Mater. Today Energy, 2019, 11, 46–60 CrossRef CAS.
- X. Y. Yu and X. W. (David) Lou, Adv. Energy Mater., 2018, 8, 1701592 CrossRef.
- J. B. Cook, H.-S. Kim, Y. Yan, J. S. Ko, S. Robbennolt, B. Dunn and S. H. Tolbert, Adv. Energy Mater., 2016, 6, 1501937 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Additional information about experimental and computational details, SEM and TEM images, XPS spectra, more electrochemical results. See DOI: https://doi.org/10.1039/d4sc02400a |
| This journal is © The Royal Society of Chemistry 2024 |