DOI:
10.1039/D4SC03553D
(Edge Article)
Chem. Sci., 2024,
15, 12856-12860
Asymmetric total syntheses of sarglamides A, C, D, E, and F†
Received
30th May 2024
, Accepted 8th July 2024
First published on 9th July 2024
Abstract
Sarglamides A–E were identified as a structurally new class of alkaloids with potential application for inflammation-associated diseases. Reported is the first asymmetric total synthesis of sarglamides A, C, D, E, and F within 7 steps, featuring an intermolecular Diels–Alder cycloaddition of (S)-phellandrene and 1,4-benzoquinone and intramolecular (aza-)Michael addition to construct the tetracyclic core of sarglamides. Importantly, our work demonstrated that the hypothetic Diels–Alder reaction of α-phellandrene with dienophile toussaintine C (or analogues) originally proposed as a biosynthetic pathway was not viable under non-enzymatic conditions. Additionally, we discovered novel and efficient double cyclization (cycloetherification and oxa-Michael cyclization) to construct the core framework of sarglamides E and D. Our concise synthetic strategy might allow rapid access to a library of sarglamide analogues for further evaluation of their bioactivity and mode of action.
Introduction
Sarglamides A–E1 (Fig. 1a) were identified in 2023 by Yue et al. from the plants of genus Sarcandra, which have been used as traditional Chinese medicines for treatments of inflammation and physical injuries. Sarglamides C–E were found to inhibit NO production against lipopolysaccharide-induced inflammation in BV-2 microglial cells at 10–20 μM concentrations without obvious cytotoxicity. Structurally, sarglamides are the first examples of natural products derived from heterodimerization of α-phellandrene and trans-N-cinnamoylindolinoids, which was suggested to be the biosynthetic pathway for the formation of sarglamides. The endo-selective intermolecular Diels–Alder (DA) reaction of α-phellandrene and toussaintine C2 well rationalizes the tetracyclic core and relative stereochemistry of sarglamides (Fig. 1b). Sarglamides D and E might be biosynthetically derived from 1′′,7- or 1′′,4-cycloetherification of sarglamide C. The unprecedented caged pentacyclic skeleton of sarglamide E along with the inspirational biosynthetic hypothesis of sarglamides A–C prompted our interest in their total synthesis.
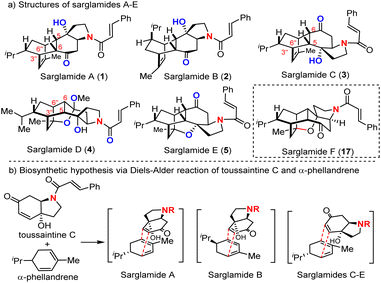 |
| | Fig. 1 (a) Molecular structures of sarglamides A–E and (b) their biosynthetic hypothesis involving the Diels–Alder reaction (transition states are provided). | |
Inspired by the proposed biosynthetic hypothesis, we set out to investigate the “straightforward” intermolecular Diels–Alder reaction of α-phellandrene (6a) and N-tosyl-5,6-dehydro-4-hydroxyindolidin-7-one (7f, the analogue of toussaintine C) (Scheme 1a). Surprisingly, the Diels–Alder reaction did not occur under either classical Lewis acid (Et2AlCl or BF3-Et2O) or thermal conditions (200 °C). We suspected that the tertiary hydroxy group from 7f presented a significant steric interaction with the diene moiety of α-phellandrene (6a) as suggested in the transition state in Fig. 1b. Under harsh conditions, appreciable decomposition of 7f was observed. This unexpected failure drove us to re-examine this type of DA reaction more systematically. Literature survey revealed that only a few reports3–5 documented the intermolecular Diels–Alder reactions of cyclohexa-1,3-dienes and cyclohexenone derivatives (Scheme 1b) and the inherent challenge was uncovered with poor yield under thermal conditions (30% yield)4 or protic acid catalysis (0% yield).3 It was recognized that the reactivity of cyclopentadiene as a diene for the DA reaction was much higher than that of cyclohexadiene: TfOH promoted the reaction of cyclopentadiene and cyclohexanone in 65% yield while no reaction occurred under identical conditions for cyclohexadiene.3 Since our targets (sarglamides) required the use of cyclohexadiene (α-phellandrene) as the diene for the DA reaction, we sought appropriate cyclohexene-containing dienophiles. We performed a study of the DA reaction of cyclohexadiene 6a–c and dienophiles 7a–7g under either Lewis acid or heating conditions (Scheme 1b). It was found that only 7a (cyclohexenone) and 7g (p-benzoquinone) could react with cyclohexadienes 6a–c. This success might be attributed to an effective minimization of the steric repulsion between R2 and diene in the transition state when R2 was hydrogen (7a) or R2/R3 were ketonic oxygen (7g). In the case of R2 being a methyl (7b–d) or hydroxyl (7c, 7e and 7f) group, the severe steric repulsion between R2 and diene prevented the Diels–Alder reaction. Notably, p-benzoquinone (7g) as the dienophile for the Diels–Alder reaction was historically significant as the very first example investigated by Diels and Alder.6,7 On the basis of these preliminary results, we re-designed our synthetic strategy towards sarglamides and focused on the most structurally complex E by exploiting the Diels–Alder reaction of α-phellandrene (6a)8 and the structurally planar p-benzoquinone (7g) as depicted in Scheme 1c. Sarglamide E could be synthesized from pentacyclic compound 9b. Transamination9 or aminolysis10–16 of lactone 9b followed through a late-stage amidation with cinnamoyl chloride. We envisioned that acid promoted cycloetherification17–23 and cis-selective oxa-Michael cyclization24–28 of 10b might occur to provide the key pentacyclic lactone 9b. The substrate for such cascade double cyclization could be obtained by the acetate aldol reaction of 8 (path b) that was readily available from the Diels–Alder reaction of α-phellandrene (6a) and p-benzoquinone (7g). We also recognized that the structurally less complex sarglamide C could be obtained from 10b if the alcohol was protected to thwart cycloetherification. Reduction of the ester 10b into aldehyde followed by reductive amination29–32 might trigger the intramolecular aza-Michael addition33–37 to afford the tetracyclic framework of sarglamide C. If the acetate aldol reaction of 8 proceeded through pathway a or in a nonselective fashion, 10a might be obtained and elaborated to sarglamide A through similar reductive amination (11a), aza-Michael addition, and amidation with cinnamoyl chloride. Our synthesis started with the intermolecular Diels–Alder reaction of enantio-enriched (S)-(+)-α-phellandrene (6a) and 1,4-benzoquinone (Scheme 2). Since (S)-(+)-α-phellandrene (6a) was expected to deliver the corresponding correct stereochemistry of sarglamides but was not commercially available, it was prepared from (R)-(−)-carvone through selective hydrogenation and the Shapiro reaction38,39 (see the ESI† for details). The Diels–Alder reaction of sublimated 1,4-benzoquinone occurred smoothly under solvent-free conditions at room temperature to provide the desired cyclization adduct 8 in 60% yield (gram scale, >4.9 g obtained) with exclusive endo selectivity. For the reaction at a larger scale (200 mmol), the reaction needed to be carried out in reflux EtOH/H2O to afford comparable yields. The aldol reaction of 8 and tbutyl acetate/LiHMDS was achieved with 70% yield in a non-regioselective manner with the assistance of CeCl3, which was believed to improve the nucleophilicity of lithium enolate.40,41 Due to the poor solubility of CeCl3 in THF, the maximum scale for this aldol reaction was 12.2 g of 8. The resulting stereoisomers 10a and 10b were separated by column chromatography on silica gel, determined at the late stage, and used for the synthesis of sarglamides A/F and C/E, respectively. The stereoisomer 10b was subjected to the conditions optimized for double cyclization (cycloetherification42,43 and oxa-Michael cyclization44). Examination of different acid promoters led to the identification of iron(III) chloride (FeCl3) as the optimal acid for the double cyclization to provide pentacyclic 9b in 77% yield. Other acid promoters including Amberlyst-15, BF3-Et2O, TMSOTf, and TBSOTf gave similar results (see the ESI† for details). The relative and absolute configuration of pentacyclic 9b was confirmed by X-ray diffraction analysis (CCDC 2358359). Transamination of lactone 9b: replacement of “O” with “NH” was successfully achieved with 89% yield by using ammonium hydroxide (NH4OH) at room temperature, which was believed to involve aminolysis45,46 of lactone (13a), dehydration (14) and intramolecular cis-selective aza-Michael addition (15b). Reductive deoxygenation of lactam 15b with silane could be achieved with rhodium catalysis47–49 when the reaction time was well controlled to minimize the competing reduction of the ketonic group into alcohol (16a). Subsequent
amidation completed the total synthesis of sarglamide E in only 6 steps with 14.38% overall yield. The spectroscopic data of our synthetic sample were well in agreement with those reported for the natural sarglamide E.1 The isomer 10a was found to undergo similar cycloetherification/oxa-Michael cyclization under protic acid conditions (Amberlyst-15) to provide pentacyclic 9a in 53% yield, which was elaborated in an identical 3-step sequence to sarglamide F (17), a compound that has not yet been identified from the natural source but shares the pentacyclic structural framework of sarglamide E. Next, we set out to elaborate the acetate aldol product 10a/b into sarglamides A/C, respectively. Our strategy was exploiting cascade reductive amination and intramolecular aza-Michael addition50 to construct the pyrrolidine moiety (18a/b → 19a/b). To this end, the tertiary alcohol of 10a/b was protected as trimethylsilyl ether (without the protection, the retro-aldol reaction occurred during the reduction) and DIBAL-H reduction and Dess–Martin periodinane51 (DMP) oxidation delivered the desired aldehyde 18a/b. The cascade reductive amination/intramolecular aza-Michael addition50 was successful but low yields were obtained (45% and 22% yield, respectively). Attempts to improve the yield of this cascade reaction proved fruitless. A major side-product was isolated, but its structure could not be fully determined. We suspected that the ammonia mediated (aza-)Morita–Baylis–Hillman reaction52–54 might effectively compete with reductive amination/aza-Michael addition. The resulting pyrrolidine was unstable and used without purification for subsequent amidation with cinnamoyl chloride (18a/b → 19a/b). Desilylation of 19a/b could afford sarglamides A and C, respectively. All spectroscopic data of our synthetic samples were in good agreement with those of natural sarglamides A and C.1
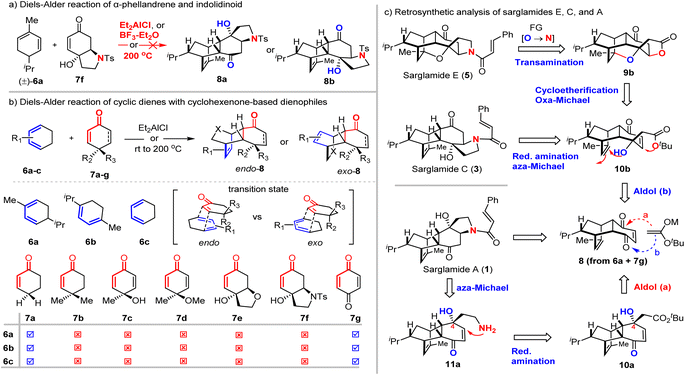 |
| | Scheme 1 (a) Diels–Alder reaction of α-phellandrene (6a) and N-tosyl-5,6-dehydro-4-hydroxyindolidin-7-one (7f), (b) Diels–Alder reaction of cyclohexadienes and cyclic dienophiles, and (c) our synthetic strategy towards sarglamides A, C and E. | |
 |
| | Scheme 2 Asymmetric total syntheses of sarglamides A, C, E, and F. | |
It was noted that sarglamide C could be converted into sarglamides E and D under acidic conditions through cycloetherification,1 which implied their possible biosynthetic relationship or artifact of sarglamides E/D. We were intrigued by such transformation and performed the cycloetherification of sarglamide C with p-TsOH and isolated sarglamide E (35% yield) and compound 20 (35% yield), which upon treatment with HCl/MeOH resulted in production of sarglamide D (90% yield) (Scheme 3). Similarly, p-TsOH triggered the cycloetherification of sarglamide A to furnish sarglamide F (67% yield). When 10c was treated with FeCl3, cycloetherification occurred to provide 9c in 60% yield. These findings unveiled that the pyrrolidine moiety as a cis-fused ring was not essential for the cycloetherification and the caged conformation of the tricyclic framework might be responsible for the high efficiency of the cycloetherification.17–23
 |
| | Scheme 3 Cycloetherification of sarglamides A and C and compound 10c. | |
Conclusion
In summary, we have accomplished the first asymmetric total syntheses of sarglamides A, C, D, E, and F in 6 or 7 steps. Our study revealed that the straightforward biosynthetic hypothesis involving the Diels–Alder reaction of α-phellandrene and toussaintine C (N-cinnamoyl-5,6-dehydro-4-hydroxyindolidin-7-one) did not occur under either acid catalysis or thermal conditions. We strategically designed the Diels–Alder reaction of α-phellandrene and planar 1,4-benzoquinone and one-pot sequential reductive amination/aza-Michael cyclization/amidation to construct the tetracyclic core of sarglamides. Importantly, new cascade cyclization involving cycloetherification and oxa-Michael cyclization is developed, which enables the 6-step total synthesis of sarglamides E and F. The biomimetic cycloetherification of sarglamide C to sarglamides D and E as well as sarglamide A to F is successfully achieved with biosynthetic implications. We believe that our concise synthetic strategy might be applicable to the facile synthesis of a library of structurally related compounds for bioactivity evaluation.
Data availability
Experimental procedures and characterization data are available within this article and its ESI.† Data are also available from the corresponding author on request.
Author contributions
R. K. and Y. W. performed the synthetic experiments. R. T. conceptualized and directed the project and drafted the manuscript with the assistance from all co-authors.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research was financially supported by the Research Grants Council of Hong Kong (C6022-22W, 16300921, 16308922, and 16304023).
Notes and references
- B. Zhou, Q. Gong, Y. Fu, J.-S. Zhou, H.-Y. Zhang and J.-M. Yue, Org. Lett., 2023, 25, 1464–1469 CrossRef CAS PubMed.
- S. S. Nyandoro, J. Ndanu, J. J. E. Munissi, A. Gruhonjic, P. A. Fitzpatrick, G. Landberg, Y. Lu, B. Wang, F. F. Pan, K. Rissanen and M. Erdelyi, J. Nat. Prod., 2015, 78, 2045–2050 CrossRef CAS.
- R. K. Schmidt, K. Müther, C. Mück-Lichtenfeld, S. Grimme and M. Oestreich, J. Am. Chem. Soc., 2012, 134, 4421–4428 CrossRef CAS.
- R. A. Valiulin, T. M. Arisco and A. G. Kutateladze, Org. Lett., 2010, 12, 3398–3401 CrossRef CAS.
- F. Fringuelli, M. Guo, L. Minuti, F. Pizzo, A. Taticchi and E. Wenkert, J. Org. Chem., 1989, 54, 710–712 CrossRef CAS.
- C. C. Nawrat and C. J. Moody, Angew. Chem., Int. Ed., 2014, 53, 2056–2077 CrossRef CAS PubMed.
- Y. Shi, X. Liu, Y. Han, P. Yan, F. Bie and H. Cao, RSC Adv., 2020, 10, 739–745 RSC.
- T. Sunakawa and C. Kuroda, Molecules, 2005, 10, 244–250 CrossRef CAS PubMed.
- K. Kim and S. H. Hong, J. Org. Chem., 2015, 80, 4152–4156 CrossRef CAS PubMed.
- W. Liu, D. D. Xu, O. Repič and T. J. Blacklock, Tetrahedron Lett., 2001, 42, 2439–2441 CrossRef CAS.
- C. B. Lan and K. Auclair, J. Org. Chem., 2023, 88, 10086–10095 CrossRef CAS.
- J. Wang, M. Rosingana, R. P. Discordia, N. Soundararajan and R. Polniaszek, Synlett, 2001, 2001, 1485–1487 CrossRef.
- C. Lalli, A. Trabocchi, G. Menchi and A. Guarna, Synlett, 2008, 2008, 189–192 CrossRef.
- W. A. Jacobs and M. Heidelberger, Org. Synth., 1927, 7, 16 CrossRef CAS.
- E. V. Mercado-Marin, P. R. Chheda, A. Faulkner and D. Carrera, Tetrahedron Lett., 2020, 61, 151552 CrossRef CAS.
- W. C. Fernelius and G. B. Bowman, Chem. Rev., 1940, 26, 3–48 CrossRef CAS.
- X. Wan, J. Hu, D. Xu, Y. Shang, Y. Zhen, C. Hu, F. Xiao, Y.-P. He, Y. Lai and W. Xie, Tetrahedron Lett., 2017, 58, 1090–1093 CrossRef CAS.
- X.-Z. Liao, R. Wang, X. Wang and G. Li, Nat. Commun., 2024, 15, 2647 CrossRef CAS PubMed.
- H. Qian, X. Han and R. A. Widenhoefer, J. Am. Chem. Soc., 2004, 126, 9536–9537 CrossRef CAS PubMed.
- A. Dzudza and T. J. Marks, Org. Lett., 2009, 11, 1523–1526 CrossRef CAS.
- H. Shigehisa, M. Hayashi, H. Ohkawa, T. Suzuki, H. Okayasu, M. Mukai, A. Yamazaki, R. Kawai, H. Kikuchi, Y. Satoh, A. Fukuyama and K. Hiroya, J. Am. Chem. Soc., 2016, 138, 10597–10604 CrossRef CAS.
- C.-G. Yang, N. W. Reich, Z. Shi and C. He, Org.
Lett., 2005, 7, 4553–4556 CrossRef CAS.
- H. Murayama, K. Nagao, H. Ohmiya and M. Sawamura, Org. Lett., 2015, 17, 2039–2041 CrossRef CAS.
- C. F. Nising and S. Bräse, Chem. Soc. Rev., 2008, 37, 1218–1228 RSC.
- G. Su, M. Formica, K. Yamazaki, T. A. Hamlin and D. J. Dixon, J. Am. Chem. Soc., 2023, 145, 12771–12782 CrossRef CAS.
- C. F. Nising and S. Bräse, Chem. Soc. Rev., 2012, 41, 988–999 RSC.
- T. Ahmad and N. Ullah, Org. Chem. Front., 2021, 8, 1329–1344 RSC.
- J. Hu, M. Bian and H. Ding, Tetrahedron Lett., 2016, 57, 5519–5539 CrossRef CAS.
- D. Enders, J. X. Liebich and G. Raabe, Chem.–Eur. J., 2010, 16, 9763–9766 CrossRef CAS.
- O. I. Afanasyev, E. Kuchuk, D. L. Usanov and D. Chusov, Chem. Rev., 2019, 119, 11857–11911 CrossRef CAS.
- T. Irrgang and R. Kempe, Chem. Rev., 2020, 120, 9583–9674 CrossRef CAS.
- K. Murugesan, T. Senthamarai, V. G. Chandrashekhar, K. Natte, P. C. J. Kamer, M. Beller and R. V. Jagadeesh, Chem. Soc. Rev., 2020, 49, 6273–6328 RSC.
- A. Yu. Rulev, Eur. J. Org Chem., 2023, 26, e202300451 CrossRef CAS.
- M. Sánchez-Roselló, J. L. Aceña, A. Simón-Fuentes and C. del Pozo, Chem. Soc. Rev., 2014, 43, 7430–7453 RSC.
- M. Sánchez-Roselló, M. Escolano, D. Gaviña and C. del Pozo, Chem. Rec., 2022, 22, e202100161 CrossRef.
- G. Pandey, M. Balakrishnan and P. S. Swaroop, Eur. J. Org Chem., 2008, 2008, 5839–5847 CrossRef.
- P. R. Krishna, A. Sreeshailam and R. Srinivas, Tetrahedron, 2009, 65, 9657–9672 CrossRef CAS.
- W. Zhang, H. Yao, J. Yu, Z. Zhang and R. Tong, Angew. Chem., Int. Ed., 2017, 56, 4787–4791 CrossRef CAS PubMed.
- S. J. Miller, H. Ishitani, Y. Furiya and S. Kobayashi, Org. Process Res. Dev., 2021, 25, 192–198 CrossRef CAS.
- I. I. R. A. Craig, J. L. Roizen, R. C. Smith, A. C. Jones, S. C. Virgil and B. M. Stoltz, Chem. Sci., 2017, 8, 507–514 RSC.
- T. Imamoto, Pure Appl. Chem., 1990, 62, 747–752 CrossRef CAS.
- G. Majetich and Y. Zhang, J. Am. Chem. Soc., 1994, 116, 4979–4980 CrossRef CAS.
- H. Kim, H. Lee, J. Kim, S. Kim and D. Kim, J. Am. Chem. Soc., 2006, 128, 15851–15855 CrossRef CAS.
- D. Gawas and U. Kazmaier, Org. Biomol. Chem., 2010, 8, 457–462 RSC.
- Y. Xu, M. McLaughlin, E. N. Bolton and R. A. Reamer, J. Org. Chem., 2010, 75, 8666–8669 CrossRef CAS.
- L. Furiassi, E. J. Tonogai and P. J. Hergenrother, Angew. Chem., Int. Ed., 2021, 60, 16119–16128 CrossRef CAS.
- S. Das, Y. Li, L.-Q. Lu, K. Junge and M. Beller, Chem.–Eur. J., 2016, 22, 7050–7053 CrossRef CAS PubMed.
- S. Das, Y. Li, C. Bornschein, S. Pisiewicz, K. Kiersch, D. Michalik, F. Gallou, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2015, 54, 12389–12393 CrossRef CAS.
- R. Kuwano, M. Takahashi and Y. Ito, Tetrahedron Lett., 1998, 39, 1017–1020 CrossRef CAS.
- W. Wang, J. Dai, Q. Yang, Y.-H. Deng, F. Peng and Z. Shao, Org. Lett., 2021, 23, 920–924 CrossRef CAS PubMed.
- D. B. Dess and J. C. Martin, J. Org. Chem., 1983, 48, 4155–4156 CrossRef CAS.
- D. Basavaiah and R. T. Naganaboina, New J. Chem., 2018, 42, 14036–14066 RSC.
- D. Basavaiah, A. J. Rao and T. Satyanarayana, Chem. Rev., 2003, 103, 811–892 CrossRef CAS PubMed.
- V. Declerck, J. Martinez and F. Lamaty, Chem. Rev., 2009, 109, 1–48 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*



