
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNSPs: chromogenic linkers for fast, selective, and irreversible cysteine modification†
Yong
Hua
ab,
Zhi
Zou
ab,
Alessandro
Prescimone
 ab,
Thomas R.
Ward
abc,
Marcel
Mayor
abde and
Valentin
Köhler
*ab
ab,
Thomas R.
Ward
abc,
Marcel
Mayor
abde and
Valentin
Köhler
*ab
aDepartment of Chemistry, University of Basel, St. Johannsring 19, CH-4056 Basel, Switzerland. E-mail: valentin.koehler@unibas.ch
bDepartment of Chemistry, University of Basel, Mattenstrasse 22, CH-4058 Basel, Switzerland
cNational Center of Competence in Research (NCCR) “Molecular Systems Engineering”, 4058 Basel, Switzerland
dInstitute for Nanotechnology (INT) and Karlsruhe Nano Micro Facility (KNMFi) Karlsruhe Institute of Technology (KIT) P.O. Box 3640, DE-76021 Karlsruhe Eggenstein-Leopoldshafen, Germany
eLehn Institute of Functional Materials (LIFM), School of Chemistry, Sun Yat-Sen University (SYSU), XinGangXi Road 135, 510275 Guangzhou, P. R. China
First published on 14th June 2024
Abstract
The addition of a sulfhydryl group to water-soluble N-alkyl(o-nitrostyryl)pyridinium ions (NSPs) followed by fast and irreversible cyclization and aromatization results in a stable S–C sp2-bond. The reaction sequence, termed Click & Lock, engages accessible cysteine residues under the formation of N-hydroxy indole pyridinium ions. The accompanying red shift of >70 nm to around 385 nm enables convenient monitoring of the labeling yield by UV-vis spectroscopy at extinction coefficients of ≥2 × 104 M−1 cm−1. The versatility of the linker is demonstrated in the stapling of peptides and the derivatization of proteins, including the modification of reduced trastuzumab with Val-Cit-PAB-MMAE. The high stability of the linker in human plasma, fast reaction rates (kapp up to 4.4 M−1 s−1 at 20 °C), high selectivity for cysteine, favorable solubility of the electrophilic moiety and the bathochromic properties of the Click & Lock reaction provide an appealing alternative to existing methods for cysteine conjugation.
Protein and peptide modification by chemical means serves the study and tracking of biomolecules,1,2 the exploration of the proteome,3–6 target-selective drug delivery,7–9 the improvement of the pharmacokinetic properties of protein drugs,10,11 and the immobilization of enzymes,12 to name a few.13–23 The increasing market share of biologicals acts as an additional incentive in this industrious area of research.16,17,22,24,25
Residues targeted for protein modification are most commonly cysteines and lysines. Cysteines are particularly attractive due to their relatively low abundance, the high nucleophilicity of surface-exposed sulfhydryl groups at moderate pH values and the diverse chemistry that can be realized.13–15 Reactive cysteines can be introduced recombinantly26 or made accessible by cleavage of intramolecular disulfide bonds under mild reductive conditions. Thiols also serve as attractive nucleophiles in materials and polymer science.27–32
Traditional approaches for thiol conjugation utilize maleimides and haloacetamides as electrophilic moieties and their widespread application continues.33 For example, the majority of FDA-approved antibody-drug-conjugates are loaded with their drug cargo through a maleimide linker.34,35
Recent examples include new designs for disulfide rebridging electrophiles,36–38 tunable equilibrium constants for conjugation,28 and electrophiles that show particularly high reaction rates.39,40 Other conjugates can be cleaved41 or locked on demand,36,42 or provide additional functionality for further elaboration.43–47 High stability48 and excellent pharmacokinetic profiles49 have been demonstrated. The introduction of TM-mediated cross-coupling methods has diversified the motifs that can be directly coupled to sulfur further.22,50–52 Despite these advances, the search for valuable conjugation techniques continues since a variety of complementary properties is desired.53–56
Inspired by vinyl-pyridinium electrophiles57 and the possibility to extend the aromatic system of o-nitroarylethane derivatives and related compounds58 by intramolecular condensation,59,60 we designed N-alkyl(o-nitrostyryl)pyridinium ions 1 (Scheme 1). Here the nucleophilic attack of a thiol at an electron-deficient alkene results in an o-nitroarylethane intermediate that can subsequently form the N-hydroxyindole product under elimination of water. The reaction enables direct UV-vis spectroscopic monitoring of the conjugation progress due to the accompanying bathochromic shift. This feature can serve to conveniently estimate reaction rates, determine conjugation yields and estimate the availability of free thiols without the need for additional chromophores in the attached cargo or chromatographic analysis. In contrast to Ellman's reagent,61 the chromogenic unit doubles as the functional linker and is directly attached to the protein of interest.
 | ||
| Scheme 1 The reaction of a cysteine sulhydrylgroup with a nitrostyryl electrophile results in an irreversible bionconjugation that is accompanied by a large bathochromic shift. | ||
Results and discussion
In the initial set of experiments we employed glutathione as the nucleophile and a simple methyl group as the ‘cargo’ in electrophile 1a. Glutathione conjugates were formed quantitatively within 1 hour as revealed by LC-MS (2 mM 1a, 1.2 eq. of GSH, 37 °C, 100 mM [NH4][HCO3] (pH 8.0), buffer:DMF = 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). The reaction proceeded cleanly and was accompanied by a red-shift of 75 nm. A range of substituents at the nitroaryl moiety were tolerated; while electron-withdrawing substituents in the 4-position (1b–1f), accelerated the reaction, it was slowed down by electron-donating substituents (1g, 1j and 1l, Table 1). The products showed in all cases a strong red shift (>70 nm) relative to the N-alkyl(o-nitrostyryl)pyridinium ions allowing product formation to be detected by eye as a yellow hue of the solution.
:1). The reaction proceeded cleanly and was accompanied by a red-shift of 75 nm. A range of substituents at the nitroaryl moiety were tolerated; while electron-withdrawing substituents in the 4-position (1b–1f), accelerated the reaction, it was slowed down by electron-donating substituents (1g, 1j and 1l, Table 1). The products showed in all cases a strong red shift (>70 nm) relative to the N-alkyl(o-nitrostyryl)pyridinium ions allowing product formation to be detected by eye as a yellow hue of the solution.
|
|
|||||
|---|---|---|---|---|---|
| ID | Conversionb | λ max (nm) Substrate (1)c | λ max (nm) Product (3)c | ε (×104 M−1 cm−1) Product (3)c | k app (M−1 s−1) |
|
a 0.40 mM (1a–1n), 1.2 equiv. glutathione, 0.1 M NH4HCO3 buffer (pH 8.0)/DMF = 9:1, 1 h, 37 °C.
b Determined by LC-MS and LC-UV. Consistent with UV-vis conversions.
c In 0.1 M NH4HCO3 buffer (pH 8.0).
d
k
app was determined at 40 μM 1a–1i, 1k–1m in the presence of 4.0 mM glutathione at 20 °C in 0.1 M NH4HCO3 buffer (pH 8.0) containing 1% DMF.
e 0.20 mM 1g, 1l, 1m, 2.4 equiv. glutathione.
f Conversion determined by UV-vis.
g Isolated yield; DMF was replaced by MeOH, bodipy is the dominant chromophore. Isolated yields for all compounds (except 3j) are reported in the ESI. Preparative reactions were carried at room temperature, at higher concentrations (2–4 mM) in buffer/DMF = 5:1 (except 3n, see ESI for further details).
|
|||||
| 3a | >99% | 310 | 385 | 2.2 | 2.2 |
| 3b | >99% | 309 | 385 | 2.2 | 2.6 |
| 3c | >99% | 311 | 387 | 2.2 | 3.1 |
| 3d | >99% | 313 | 387 | 2.2 | 3.1 |
| 3e | >99% | 308 | 379 | 2.1 | 3.9 |
| 3f | >99% | 299 | 374 | 2.0 | 4.2 |
| 3g | 90% (4 h)ef | 312 | 422 | 2.2 | — |
| 3h | >99% | 316 | 391 | 2.0 | 4.4 |
| 3i | >99% | 324 | 395 | 2.3 | 4.1 |
| 3j | Trace (24 h) | — | — | — | — |
| 3k | >99e% | 320 | 405 | 2.3 | — |
| 3l | 90% (4 h)ef | 332 | 413 | 2.1 | — |
| 3m | >99e% | 309 | 387 | 2.3 | 1.1 |
| 3n | 53% (4 h)g | 525 | 524 | 6.1 | — |

Cargo can be attached to the pyridine ring directly by alkylation as shown for the bodipy chromophore in 3n or via additional linker moieties that have alternative functional groups, such as an alkyne in 3h or a primary amine displayed by a hydrophilic PEG-linker in 3i. Intriguingly, the nature of the linker can also affect reaction rates: i.e., the reaction of 1i to 3i carrying a triazole-PEG-amine cargo was significantly faster than the reaction of 1m to 3m having a lipophilic hydrocarbon chain and a terminal carboxylate. Possibly, limited solubility and mass transfer play a role for the lower reaction rate observed with 3m.
We next probed the bathochromic properties of the reaction for the estimation of reaction rate constants under pseudo-first-order conditions. The UV-spectrum of a solution containing initial concentrations of 1a (40 μM) and GSH (4.0 mM) in 100 mM NH4HCO3 buffer (pH 8.0) was recorded in the wavelength range from 270–470 nm every 3 seconds at 20 °C (Fig. 1). The decreasing absorption of 1a and the increasing absorption of 3a in the course of the reaction resulted in a well-defined isosbestic point at 355 nm. Its sharp definition indicates that the cyclization (the ‘lock’) is indeed very fast since the presence of underlying intermediates in significant concentrations can be excluded. The lower limits for the apparent second order rate constants obtained by the method of initial rates range from 1.1 M−1 s−1 for 3m to 4.4 M−1 s−1 for 3i (Table 1 and Fig. 1).
 | ||
| Fig. 1 (a) Electrophiles of type 1 react with thiol nucleophiles under fast addition and even faster cyclization (b) time-resolved UV-vis of the reaction mixture: 1a (40 μM) and glutathione (4.0 mM), 0.1 M NH4HCO3 buffer (pH 8.0), scan from 270 nm to 470 nm every 3 s; (c) apparent second order rate constants for transformation of 1a–1f, 1h, 1i, 1m (40 μM) with glutathione (4.0 mM), 20 °C obtained by a linear fit of the early data-points of the UV-vis data under pseudo-first order conditions; the lines show a first order fit to guide the eye. (d) Reversibility of thiol-Michael addition for slow reacting electrophile 1m; 2m was isolated and re-exposed to the reaction buffer with or without cysteine (e) LC-MS (m/z = 100–1000) of 2m (0.40 mM) in NH4HCO3 buffer (100 mM, pH 8.0), 37 °C, 2 h incubation; (f) LC-MS (m/z = 100–1000) of a mixture of 2m (0.40 mM) and cysteine (4.0 mM) in NH4HCO3 buffer (100 mM, pH 8.0), 37 °C, 4 h incubation. | ||
No sharp isosbestic point was observed for the reactions of electrophiles 1g–1l to products 3g–3l, respectively, possibly due to a reduced electrophilicity of the nitro group. When the reactions for slow reacting substrates 1g and 1m with GSH (1.2 eq.) were performed at higher substrate concentrations (4.0 mM) for preparative purposes, the intermediates 2g and 2m, respectively, could be detected by LC-MS. A larger scale reaction under these conditions allowed the isolation of intermediate 2m by preparative HPLC in the presence of TFA. When 2m was exposed to the reaction buffer, a mixture of 1m, 2m and 3m was observed by LC-MS after 2 hours (Fig. 1e). The addition of cysteine to 2m led to competitive formation of the cysteine addition and cyclization product, 2m(cys) and 3m(cys), respectively (Fig. 1f), confirming that the aromatization is indeed necessary to render the reaction irreversible.
The X-ray structure of the cyclized conjugate 6g revealed the presence of a water molecule associated with the N-hydroxy group, hinting at a potential acidity (Fig. 2).62,63 Since the OH-group is part of the chromophore, the absorption spectrum of the cyclized conjugates was expected to be pH-dependent. A red-shift of 24 nm was observed for 6g between pH 2 and 12 and the pKa-value (pKa = 6.71) could be conveniently determined by UV-vis-titration (see ESI†). In the reaction buffer (pH 8.0) 95% of the formed hydroxyindole 6g is deprotonated.
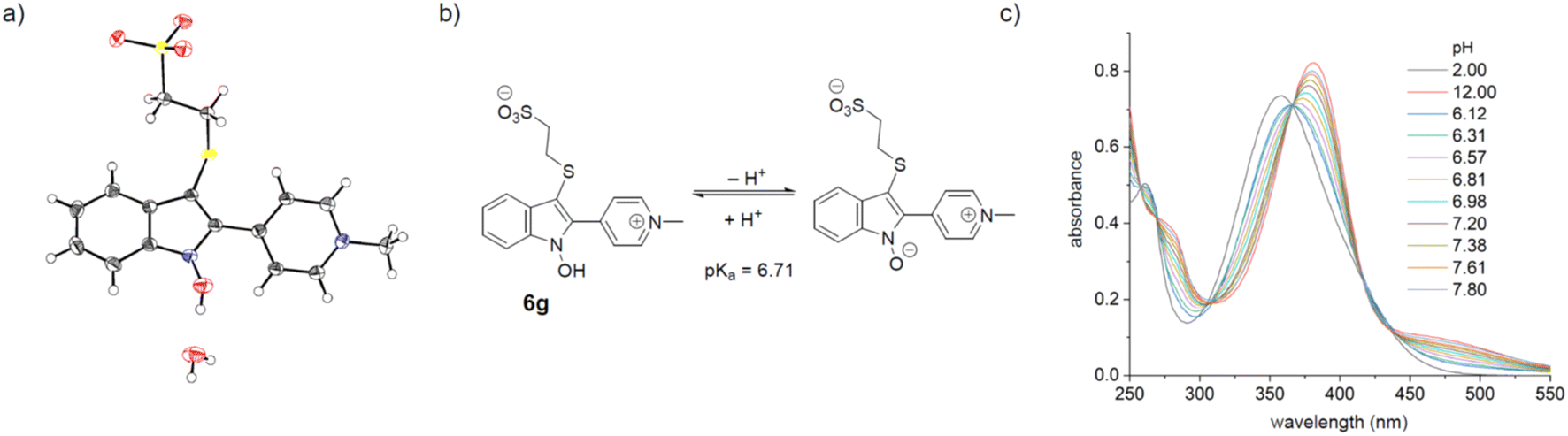 | ||
| Fig. 2 (a) The X-ray structure of conjugate 6g reveals an associated water molecule; CCDC ID 2278235 (b) protonation equilibrium of conjugate 6g (c) pH-dependence of the absorption spectrum of conjugate 6g. | ||
To test the applicability of the NSP-motif, first a small set of peptides (4–16 AA) with a single cysteine residue were reacted with electrophile 1a (Fig. 3). The conjugates were isolated in good yields after preparative HPLC (75–80%) and had formed quantitatively after 30 minutes (6c, 4 amino acids; 6d, 8 amino acids) or after 2 hours for longer peptides (6e, 6f, 12 and 16 amino acids respectively). Importantly, only singly modified products were observed, confirming that a potential cross-reactivity with lysine is of no concern under the conditions employed. We further scrutinized this conclusion by an attempted reaction of 3a with lysine itself where no reaction was observed (see ESI†).
 | ||
| Fig. 3 Selective modification of one, two or three sulfhydryl groups in peptides with mono-, bi- and trifunctional 4-(2-nitrostyryl)pyridinium linkers. Conditions for the coupling reactions: peptides with a single cysteine residue: 2.0 mM 1a, 1.5 equiv. peptide 5a–5f, 100 mM NH4HCO3 buffer (pH 8.0), 37 °C, 0.5–2 h; peptides with two cysteine residues: 0.50 mM 4a, 1.0 equiv. peptide 5g, 5h, 100 mM NH4HCO3 buffer (pH 8.0), rt, 1 hour; peptides with three cysteine residues: 0.20 mM 4b, 1.0 equiv. peptide 5i, 5j, 100 mM NH4HCO3 buffer (pH 8.0), 0 °C→rt, 1 hour. Isolated yields; products purified by prep. HPLC. | ||
Next, bi- and trifunctional linkers (4a, 4b) for peptide stapling were prepared by reacting dibromopropane or 1,3,5-tri(bromomethyl)benzene with (E)-4-(2-nitrostyryl)pyridine, respectively. Peptides with two or three cysteine residues could be conveniently constrained into cyclic or bicyclic structures under moderately dilute conditions. The monocyclic constructs with ring sizes of 7 and 8 amino acids were isolated in good yields (≥60%). Bicyclic structures were prepared with 6,7- and 7,8-membered rings respectively in ≥25% yield.
We further tested the modification of bovine aprotinin (58 AA) where the three disulfide bonds had been reduced by addition of 6 eq. of TCEP. Direct addition of 20 eq. of 1a to the reduced mixture without workup resulted in the hexa-modified polypeptide (see ESI†). The excess of 1a served to compensate for competing addition of TCEP to 1a.
Next, to probe NSPs for the selective modification of an unpaired cysteine on a protein, β-lactoglobulin A (BLG-A, 162 AA) that contains two disulfide bridges and an unpaired buried cysteine was selected. Despite being buried, this single cysteine can be modified under mild conditions. Reagent 1a, alkynyl-functionalized 5 and bifunctional 4a were tested for modification at 37 °C and at pH 8.0: all reactions resulted in singly modified BLG-A within 2 h (Fig. 4).
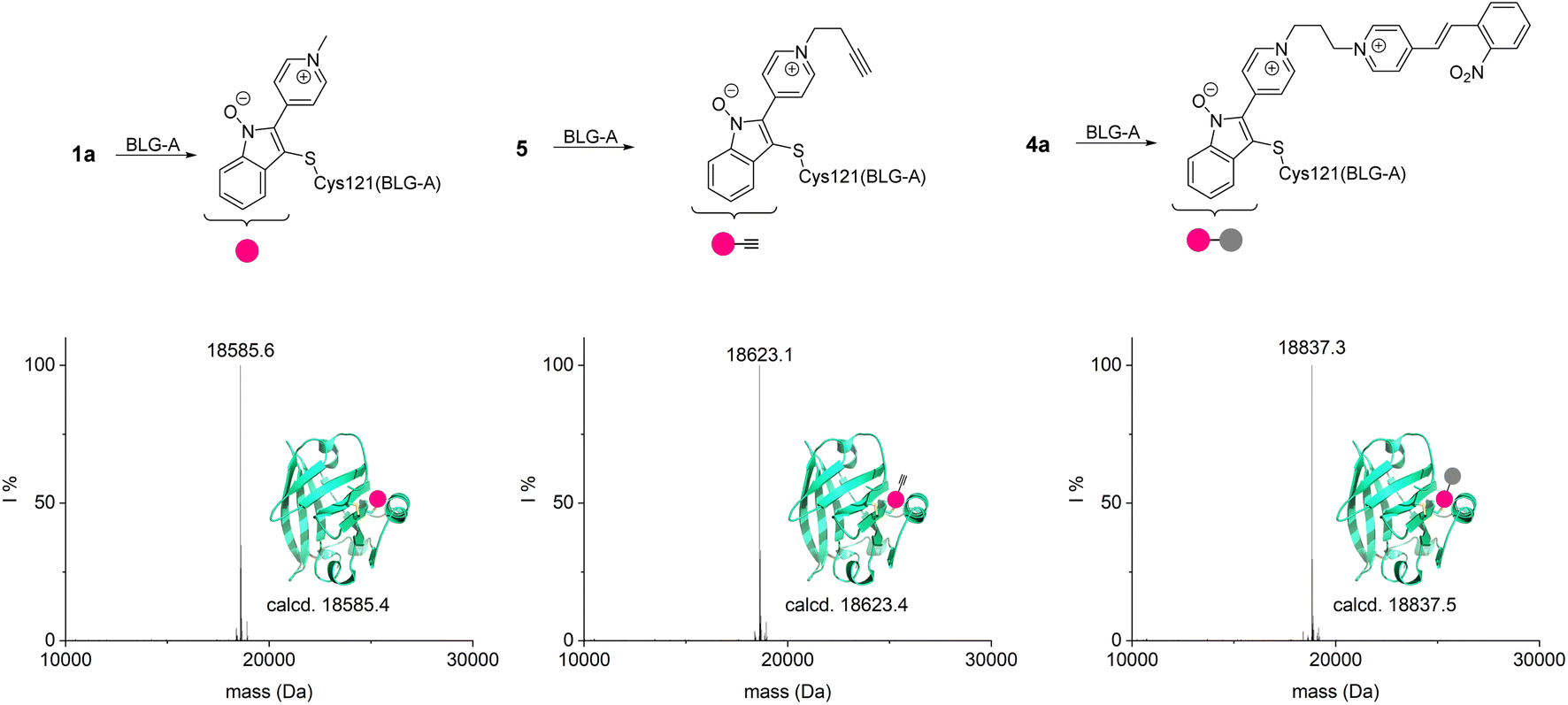 | ||
| Fig. 4 Modification of BLG-A with different NSPs (20 μM BLG-A, 10 eq. 1a and 5, 5 eq. 4a, respectively). Deconvoluted mass spectra are shown. | ||
Toward a pharmacologically relevant application, we equipped the cytotoxic Val-Cit-PAB-MMAE with the electrophilic NSP moiety. The resulting reactive drug-linker NSP-Val-Cit-PAB-MMAE was first tested in the reaction with glutathione. A similar reaction rate to 1a (kapp = 1.9 M−1 s−1) indicates that the larger payload does not impede the reaction rate (Val-Cit-PAB-MMAE vs. methyl in 1a). Importantly the GSH conjugate (0.20 mM) showed only minimal decomposition after 7 days of incubation at 37 °C and pH 8.0 in the presence of 2.0 mM cysteine according to LC-MS and LC-UV analysis (Fig. 5b). This contrasts with observations made with the commercial maleimide drug-linker MC-Val-Cit-PAB-MMAE (see ESI†).64
 | ||
| Fig. 5 (a) The GSH conjugate of the NSP-MMAE linker does not exchange with cysteine and shows superior stability. (b) Stability test of incubated GS-NHI-MMAE by LC-MS. (c) Conjugation of NSP-MMAE with trastuzumab. (d) Determination of DAR by UV-vis. (e) Deconvoluted HRMS of the trastuzumab NSP-MMAE conjugates. (f) SDS-PAGE of reduced trastuzumab after reaction with NSP-MMAE, A: no NSP-MMAE; B: 20 eq. NSP-MMAE; 3 h incubation; 3 days of dialysis at room temperature; samples were heated to 95 °C for 10 minutes before they were applied to the gel. | ||
We further tested the stability of the glutathione conjugates 3a, 3f, 3g in human plasma (pooled human plasma (blood-derived) K2 EDTA, Loxo GmbH). Within 48 hours more than half of the species with an intact conjugated-N-hydroxy indole pyridinium unit remained. Identified decomposition products stemed from cleavage of the glutamate–cysteine amide bond in the glutathione moiety (49% for 3a, 56% for 3f, and 12% for 3g, see ESI†).
The high stability of the conjugated N-hydroxy indole pyridinium unit against thiol exchange and in human plasma illustrates the superior applicability of the conjugation method.
Encouraged by these results, we modified the widely applied anti-HER2 antibody trastuzumab. Interchain disulfide bonds were reduced with TCEP and the mixture subsequently treated with 2.5 eq. of NSP-MMAE per free thiol (4 μM trastuzumab, 40 μM TCEP, 37°, 90 min, followed by 80 μM NSP-MMAE, 3 h; 3 days dialysis at room temperature). HRMS showed, as expected, the singly modified light chain and the triply modified heavy chain. Additional signals of the heavy chain are due to heterogeneous glycosylation commonly observed in commercial trastuzumab.65
The red-shifted absorption spectrum of the conjugate allows for a simple estimation of the drug to antibody ratio by comparing the absorption at 280 nm and 392 nm resulting in a value of DAR = 7.3 under not further optimized conditions. A small signal for the doubly modified half-antibody indicates that the DAR < 8.0 might be caused by incomplete TCEP reduction (Fig. 5e).
Conclusion
In summary we present a versatile and readily accessible electrophilic moiety for aqueous cysteine conjugation that allows facile monitoring of the reaction progress by virtue of its bathochromic properties and whose conjugates show superior stability to thiol exchange. The thiol N-hydroxy indole pyridinium unit furthermore showed half-lifes of >48 hours in human plasma. The chromogenic reaction proved to be an effective method for the determination of the reaction rate constants, the concentration of free sulfhydryl residues in the reaction mixture and the conjugation yield as exemplified in the drug-to-antibody ratio (DAR) of an antibody-drug conjugate (ADC). Moreover, a synthesized drug conjugate (GS-NHI-MMAE) displayed excellent stability, even when confronted with an excess of free thiol groups. These features collectively make Click & Lock an appealing alternative to existing methods for cysteine conjugation and open new possibilities in the field of protein modification and bioconjugation strategies.Data availability
The data is included in the ESI.†Author contributions
Conceptualization: Y. Hua; formal analysis: Y. Hua, V. Köhler; funding acquisition: M. Mayor, V. Köhler, T. R. Ward; investigation: Y. Hua, Z. Zou, A. Prescimone; methodology: Y. Hua; project administration: M. Mayor, V. Köhler; resources: M. Mayor, T. R. Ward, V. Köhler; supervision: V. Köhler, M. Mayor, T. R. Ward; validation: Y. Hua, V. Köhler; visualization: Y. Hua, V. Köhler, Z. Zou, A. Prescimone; writing – orginal draft: Y. Hua, V. Köhler; writing – review & editing: Y. Hua, V. Köhler, T. R. Ward, M. Mayor.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This project has received funding from the European Union's Horizon 2020 research and innovation programme under grant agreements #860713. We acknowledge funding by the Gordon & Betty Moore foundation under grant agreement #10771 and the Swiss National Science Foundation #200020_207744. We thank Michael Pfeffer for the HRMS analysis and Daniel Häussinger for advice on NMR spectroscopy.References
- B. N. G. Giepmans, S. R. Adams, M. H. Ellisman and R. Y. Tsien, Science, 2006, 312, 217–224 CrossRef CAS.
- L. Wang, M. S. Frei, A. Salim and K. Johnsson, J. Am. Chem. Soc., 2019, 141, 2770–2781 CrossRef CAS.
- E. Weerapana, C. Wang, G. M. Simon, F. Richter, S. Khare, M. B. Dillon, D. A. Bachovchin, K. Mowen, D. Baker and B. F. Cravatt, Nature, 2010, 468, 790–795 CrossRef CAS.
- K. M. Backus, B. E. Correia, K. M. Lum, S. Forli, B. D. Horning, G. E. González-Páez, S. Chatterjee, B. R. Lanning, J. R. Teijaro, A. J. Olson, D. W. Wolan and B. F. Cravatt, Nature, 2016, 534, 570–574 CrossRef CAS.
- Y. Liao, S. Chin Chan, E. A. Welsh, B. Fang, L. Sun, E. Schonbrunn, J. M. Koomen, D. R. Duckett, E. B. Haura, A. Monastyrskyi and U. Rix, ACS Chem. Biol., 2023, 18, 251–264 CrossRef CAS.
- K. C. Tang, S. M. Maddox, K. M. Backus and M. Raj, Chem. Sci., 2022, 13, 763–774 RSC.
- A. Beck, L. Goetsch, C. Dumontet and N. Corvaia, Nat. Rev. Drug Discovery, 2017, 16, 315–337 CrossRef CAS.
- M. Fani, H. R. Maecke and S. M. Okarvi, Theranostics, 2012, 2, 481–501 CrossRef CAS.
- S. J. Walsh, J. D. Bargh, F. M. Dannheim, A. R. Hanby, H. Seki, A. J. Counsell, X. Ou, E. Fowler, N. Ashman, Y. Takada, A. Isidro-Llobet, J. S. Parker, J. S. Carroll and D. R. Spring, Chem. Soc. Rev., 2021, 50, 1305–1353 RSC.
- J. M. Horn and A. C. Obermeyer, Biomacromolecules, 2021, 22, 4883–4904 CrossRef CAS.
- P. Bailon and C.-Y. Won, Expert Opin. Drug Delivery, 2009, 6, 1–16 CrossRef CAS.
- C. Mateo, J. M. Palomo, G. Fernandez-Lorente, J. M. Guisan and R. Fernandez-Lafuente, Enzyme Microb. Technol., 2007, 40, 1451–1463 CrossRef CAS.
- J. M. Chalker, G. J. Bernardes, Y. A. Lin and B. G. Davis, Chem.–Asian J., 2009, 4, 630–640 CrossRef CAS.
- S. B. Gunnoo and A. Madder, ChemBioChem, 2016, 17, 529–553 CrossRef CAS.
- P. Ochtrop and C. P. R. Hackenberger, Curr. Opin. Chem. Biol., 2020, 58, 28–36 CrossRef CAS.
- O. Boutureira and G. J. L. Bernardes, Chem. Rev., 2015, 115, 2174–2195 CrossRef CAS.
- N. Krall, F. P. da Cruz, O. Boutureira and G. J. L. Bernardes, Nat. Chem., 2016, 8, 103–113 CrossRef CAS.
- N. Stephanopoulos and M. B. Francis, Nat. Chem. Biol., 2011, 7, 876–884 CrossRef CAS.
- L. Taiariol, C. Chaix, C. Farre and E. Moreau, Chem. Rev., 2022, 122, 340–384 CrossRef CAS.
- E. D. Holz, M. Zou, D. B. Tesar and W. Shatz-Binder, Pharmaceutics, 2023, 15, 1–54 CrossRef.
- P. G. Isenegger and B. G. Davis, J. Am. Chem. Soc., 2019, 141, 8005–8013 CrossRef CAS.
- J. Ohata, S. C. Martin and Z. T. Ball, Angew. Chem., Int. Ed., 2019, 58, 6176–6199 CrossRef CAS.
- C. S. Wu and L. Cheng, ChemBioChem, 2023, 24, e202200468 CrossRef CAS.
- N. L. Kjaersgaard, T. B. Nielsen and K. V. Gothelf, ChemBioChem, 2022, 23, e202200245 CrossRef CAS.
- C. Sornay, V. Vaur, A. Wagner and G. Chaubet, R. Soc. Open Sci., 2022, 9, 211563 CrossRef CAS.
- R. Ohri, S. Bhakta, A. Fourie-O'Donohue, J. dela Cruz-Chuh, S. P. Tsai, R. Cook, B. Wei, C. Ng, A. W. Wong, A. B. Bos, F. Farahi, J. Bhakta, T. H. Pillow, H. Raab, R. Vandlen, P. Polakis, Y. Liu, H. Erickson, J. R. Junutula and K. R. Kozak, Bioconjugate Chem., 2018, 29, 473–485 CrossRef CAS.
- C. Bednarek, U. Schepers, F. Thomas and S. Bräse, Adv. Funct. Mater., 2024, 34, 2303613 CrossRef CAS.
- A. E. Crolais, N. D. Dolinski, N. R. Boynton, J. M. Radhakrishnan, S. A. Snyder and S. J. Rowan, J. Am. Chem. Soc., 2023, 145, 14427–14434 CrossRef CAS.
- A. B. Lowe, Polym. Chem., 2010, 1, 17–36 RSC.
- M. H. Stenzel, ACS Macro Lett., 2013, 2, 14–18 CrossRef CAS.
- D. P. Nair, M. Podgórski, S. Chatani, T. Gong, W. Xi, C. R. Fenoli and C. N. Bowman, Chem. Mater., 2014, 26, 724–744 CrossRef CAS.
- C. Bahou, R. J. Spears, A. M. Ramírez Rosales, L. N. C. Rochet, L. J. Barber, K. S. Stankevich, J. F. Miranda, T. C. Butcher, A. M. Kerrigan, V. K. Lazarov, W. Grey, V. Chudasama and C. D. Spicer, Biomacromolecules, 2023, 24, 4646–4652 CrossRef CAS.
- G. T. Hermanson, in Bioconjugate Techniques, ed. G. T. Hermanson, Academic Press, Boston, 3rd edn, 2013, pp. 229–258 Search PubMed.
- C. S. B. Chia, ChemistryOpen, 2022, 17, e202200032 CAS.
- J. You, J. Zhang, J. Wang and M. Jin, Bioconjugate Chem., 2021, 32, 1525–1534 CrossRef CAS.
- A. M. Tallon, Y. Xu, G. M. West, C. W. am Ende and J. M. Fox, J. Am. Chem. Soc., 2023, 145, 16069–16080 CrossRef CAS.
- F. M. Dannheim, S. J. Walsh, C. T. Orozco, A. H. Hansen, J. D. Bargh, S. E. Jackson, N. J. Bond, J. S. Parker, J. S. Carroll and D. R. Spring, Chem. Sci., 2022, 13, 8781–8790 RSC.
- L. Xu, M. J. S. A. Silva, P. M. P. Gois, S. L. Kuan and T. Weil, Chem. Sci., 2021, 12, 13321–13330 RSC.
- B. M. Lipka, D. S. Honeycutt, G. M. Bassett, T. N. Kowal, M. Adamczyk, Z. C. Cartnick, V. M. Betti, J. M. Goldberg and F. Wang, J. Am. Chem. Soc., 2023, 145, 23427–23432 CrossRef CAS.
- F. J. Chen and J. Gao, Chem.–Eur. J., 2022, 28, e202201843 CrossRef CAS.
- S. Maity, C. Bingham, W. Sheng, N. Ehyaei, D. Chakraborty, S. Tahmasebi-Nick, T. E. Kimmel, C. Vasileiou, J. H. Geiger and B. Borhan, Analyst, 2023, 148, 1085–1092 RSC.
- K. Gavriel, D. C. A. van Doeselaar, D. W. T. Geers and K. Neumann, RSC Chem. Biol., 2023, 4, 685–691 RSC.
- I. Koutsopetras, A. K. Mishra, R. Benazza, O. Hernandez-Alba, S. Cianférani, G. Chaubet, S. Nicolai and J. Waser, Chem.–Eur. J., 2023, 29, e202302689 CrossRef CAS.
- M. Ahangarpour, I. Kavianinia, P. A. Hume, P. W. R. Harris and M. A. Brimble, J. Am. Chem. Soc., 2022, 144, 13652–13662 CrossRef CAS.
- C. F. Afonso, M. C. Marques, J. P. M. Antonio, C. Cordeiro, P. M. P. Gois, P. M. S. D. Cal and G. J. L. Bernardes, Angew. Chem., Int. Ed., 2022, 61, e202208543 CrossRef CAS.
- L. Xu, S. L. Kuan and T. Weil, Angew. Chem., Int. Ed., 2021, 60, 13757–13777 CrossRef CAS.
- S. Teng, Z. Zhang, B. Li, L. Li, M. C. L. Tan, Z. Jia and T.-P. Loh, Angew. Chem., Int. Ed., 2023, 62, e202311906 CrossRef CAS.
- I. N. Gober, R. Sharan and M. Villain, J. Pept. Sci., 2023, 29, e3495 CrossRef CAS.
- P. Ochtrop, J. Jahzerah, P. Machui, I. Mai, D. Schumacher, J. Helma, M.-A. Kasper and C. P. R. Hackenberger, Chem. Sci., 2023, 14, 2259–2266 RSC.
- A. J. Rojas, J. M. Wolfe, H. H. Dhanjee, I. Buslov, N. L. Truex, R. Y. Liu, W. Massefski, B. L. Pentelute and S. L. Buchwald, Chem. Sci., 2022, 13, 11891–11895 RSC.
- H. R. Montgomery, M. S. Messina, E. A. Doud, A. M. Spokoyny and H. D. Maynard, Bioconjugate Chem., 2022, 33, 1536–1542 CrossRef CAS.
- J. Rodriguez, H. H. Dhanjee, B. L. Pentelute and S. L. Buchwald, J. Am. Chem. Soc., 2022, 144, 11706–11712 CrossRef CAS.
- Z. Zhang, L. Li, H. Xu, C. K. Lee, Z. Jia and T. P. Loh, J. Am. Chem. Soc., 2024, 146, 1776–1782 CrossRef CAS.
- R. J. Spears and V. Chudasama, Curr. Opin. Chem. Biol., 2023, 75, 102306 CrossRef CAS.
- W.-Y. O, J.-F. Cui, Q. Yu, K. K.-Y. Kung, S.-F. Chung, Y.-C. Leung and M.-K. Wong, Angew. Chem., Int. Ed., 2023, 62, e202218038 CrossRef CAS.
- H. Seki, S. J. Walsh, J. D. Bargh, J. S. Parker, J. Carroll and D. R. Spring, Chem. Sci., 2021, 12, 9060–9068 RSC.
- M. J. Matos, C. D. Navo, T. Hakala, X. Ferhati, A. Guerreiro, D. Hartmann, B. Bernardim, K. L. Saar, I. Compañón, F. Corzana, T. P. J. Knowles, G. Jiménez-Osés and G. J. L. Bernardes, Angew. Chem., Int. Ed., 2019, 58, 6640–6644 CrossRef CAS.
- J. Hanusek and V. Machacek, Collect. Czech. Chem. Commun., 2009, 74, 811–833 CrossRef CAS.
- Z. Wróbel and M. Mąkosza, Synlett, 1993, 597–598 CrossRef.
- Z. Wrobel and M. Makosza, Tetrahedron, 1997, 53, 5501–5514 CrossRef CAS.
- G. L. Ellman, Arch. Biochem. Biophys., 1959, 82, 70–77 CrossRef CAS.
- M. Somei, in Advances in Heterocyclic Chemistry, Academic Press, 2002, vol. 82, pp. 101–155 Search PubMed.
- E. E. Bykov, S. N. Lavrenov and M. N. Preobrazhenskaya, Chem. Heterocycl. Compd., 2006, 42, 42–44 CrossRef CAS.
- R. P. Lyon, J. R. Setter, T. D. Bovee, S. O. Doronina, J. H. Hunter, M. E. Anderson, C. L. Balasubramanian, S. M. Duniho, C. I. Leiske, F. Li and P. D. Senter, Nat. Biotechnol., 2014, 32, 1059–1062 CrossRef CAS.
- T. Li, X. Tong, Q. Yang, J. P. Giddens and L.-X. Wang, J. Biol. Chem., 2016, 291, 16508–16518 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2278235. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc01710b |
| This journal is © The Royal Society of Chemistry 2024 |