
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficient hydroxyl radical generation of an activatable phthalocyanine photosensitizer: oligomer higher than monomer and nanoaggregate†
Li
Li
,
Yalan
Liao
,
Shuwen
Fu
,
Zixuan
Chen
,
Tinghe
Zhao
,
Luyue
Fang
and
Xingshu
Li
 *
*
Fujian Provincial Key Laboratory of Cancer Metastasis Chemoprevention and Chemotherapy, College of Chemistry, Fuzhou University, Fuzhou 350108, China. E-mail: xingshuli@fzu.edu.cn
First published on 18th June 2024
Abstract
It remains a challenge to develop a single-component organic photosensitizer that efficiently produces hydroxyl radicals (˙OH) without oxygen involvement, especially while maintaining tumor-targeting capability. Herein, we propose an intelligent molecular design strategy whereby a tumor-targeted phthalocyanine is initially ˙OH-free and can be activated by overexpressed β-nicotinamide adenine dinucleotide sodium salt hydrate (NAD(P)H) in hypoxic tumors to efficiently produce ˙OH under light irradiation. Furthermore, the oligomer models based on the phthalocyanine molecules were constructed by a supramolecular regulation strategy, which were in an intermediate state between monomer and nanoaggregate, to achieve enhanced ˙OH generation. The level of NAD(P)H in cancer cells can be exhausted through two pathways, including spontaneous redox and the photocatalytic redox of phthalocyanines. As a result, the in vivo and in vitro assays illustrated that the oligomeric phthalocyanine containing N–O units (OligPcNOB) can specifically target cancer cells and tumor tissue with overexpressing biotin receptors. OligPcNOB exhibited significant photocytotoxicity even in an extremely low oxygen environment and successfully inhibited tumor progression.
Introduction
Photodynamic therapy (PDT) is recognized as a promising approach for cancer treatment due to its non-invasiveness, spatiotemporal controllability, and negligible systemic toxicity.1–3 Most photosensitizers (PSs), in clinical and preclinical settings, generate singlet oxygen (1O2) via the type II mechanism, which involves energy transfer between the excited triplet state (3PS*) and O2.4,5 Because of the abnormal proliferation of cancer cells, the limited O2 supply severely hinders the efficacy of these PSs for PDT.6–8 Distinguished from type II PSs, the type I PSs produce reactive oxygen species (ROS),9–11 such as superoxide (O2˙−),12–18 hydroxyl radical (˙OH),19 and hydrogen peroxide (H2O2),20,21 through an electron or hydrogen proton transfer process, with ˙OH being the most reactive.22 Using water as a reactant, ˙OH can be formed without the involvement of O2.22To date, few ˙OH-generating PSs have been reported, and the focus has mainly been on inorganic nanomaterials23–25 and organic molecule-based multicomponent systems.26–29 However, because of the low biocompatibility of inorganic materials, it is difficult for living organisms to metabolize them.30,31 In traditional organic molecule-based multicomponent systems, there is limited ˙OH-generating capacity and also increased undesired biosafety burden from the complicated components.32–34 In addition, the lack of tumor targeting results in decreased effectiveness of ˙OH-generated PSs in PDT, with potential side effects.35–37 Therefore, the development of single-component organic PSs that can efficiently oxidize water to produce ˙OH without the engagement of O2 and have precise tumor-targeting ability would further improve the overall outcome of PDT.
In this work, we have achieved efficient generation of ˙OH and precise tumor targeting through the ingenious design of the phthalocyanine molecular structure by combining the surface receptor-mediated active targeting of cancer cells and specific substance responses within the cancer cells. As shown in Fig. 1, activatable phthalocyanine with a biotin-targeting unit (PcNOB) is initially devoid of ˙OH production under light irradiation. When it is reduced to PcNB by β-nicotinamide adenine dinucleotide sodium salt hydrate (NAD(P)H), then the reduction product, PcNB, can efficiently oxidize water to produce large amounts of O2-independent ˙OH for the treatment of hypoxic tumors.
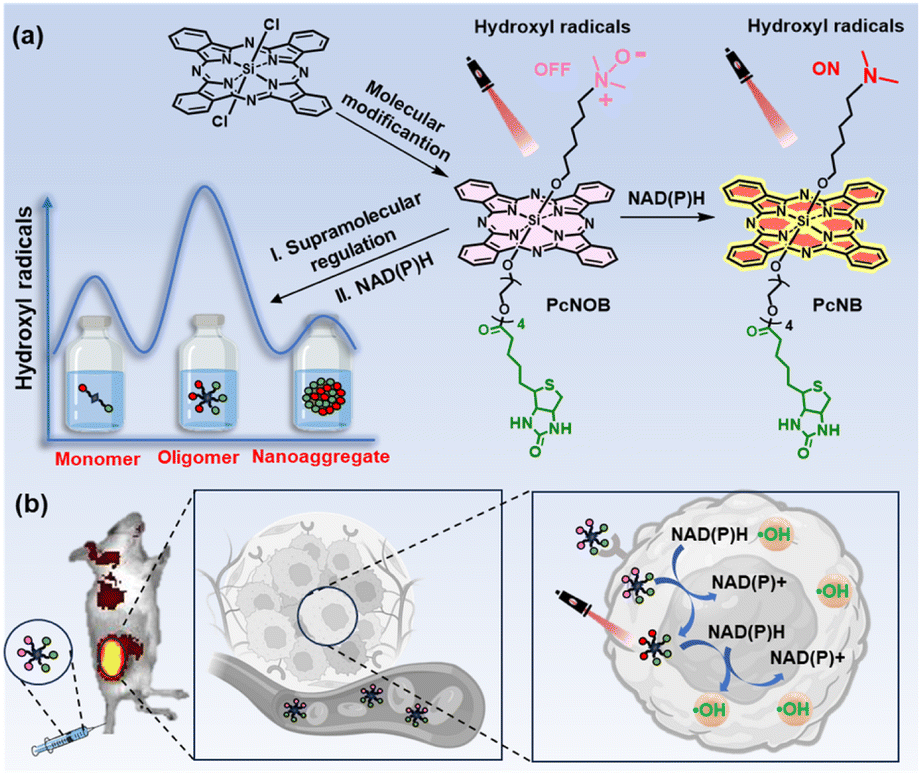 | ||
| Fig. 1 (a) Diagrammatic representation of the molecular and supramolecular strategy of an activatable phthalocyanine for highly efficient ˙OH generation and precise tumor targeting. (b) Schematic illustration of the switchable photoactivity of OligPcNOB for in vivo fluorescence imaging and tumor-targeted PDT. | ||
The level of NAD(P)H in cancer cells is exhausted through two pathways, including the spontaneous redox of PcNOB and the photocatalytic redox of PcNB. PcNOB does not require additional materials for in vivo delivery, as it is soluble in water. Compared with phthalocyanine without a biotin unit, there is a significant increase in PcNOB in tumors after tail vein injection. More importantly, we also constructed models for different aggregation states of the same phthalocyanine molecule and similar aggregation states of different phthalocyanine molecules, and comparatively studied their photosensitive properties. The first discovery of oligomeric phthalocyanine, which exists in the form between monomer and nanoaggregate, enhances ˙OH production. The oligomer phthalocyanine OligPcNOB showed 80% tumor inhibition efficiency at a dose of 0.8 nmol g−1 and a light dose of 30 J cm−2 after PDT.
Results and discussion
Synthesis and characterization
The advantages of silicon phthalocyanine are its strong absorption in the phototherapeutic window (650–850 nm), considerable ROS quantum yield, and adaptable photochemical properties. We thus selected silicon phthalocyanine as the main molecular scaffold for the design of new PSs. We attempted to introduce a reductase-responsive N–O unit to realize the activatable generation of ˙OH, as well as a tetraethyleneglycol chain modified with biotin molecules to improve biocompatibility and tumor targeting.One phthalocyanine with a biotin unit and N–O unit (PcNOB) was synthesized from dichlorosilane phthalocyanine through a three-step process of asymmetric axial substitution, esterification, and oxidation. One phthalocyanine with a biotin unit (PcNB), one phthalocyanine with an N–O unit (PcNO), and one phthalocyanine with neither biotin nor an N–O unit (PcN) were also synthesized as controls. Detailed synthesis schematics and characterization are shown in the ESI (Fig. S1–8),† and Fig. 2a shows the chemical structures of the synthesized phthalocyanines. In addition, we measured the water solubility of all the phthalocyanines, and the results are shown in Fig. S9.† PcNOB exhibits a solubility of 2.3 mg mL−1 in water, which provides a simple pharmaceutical preparation method for clinical application.
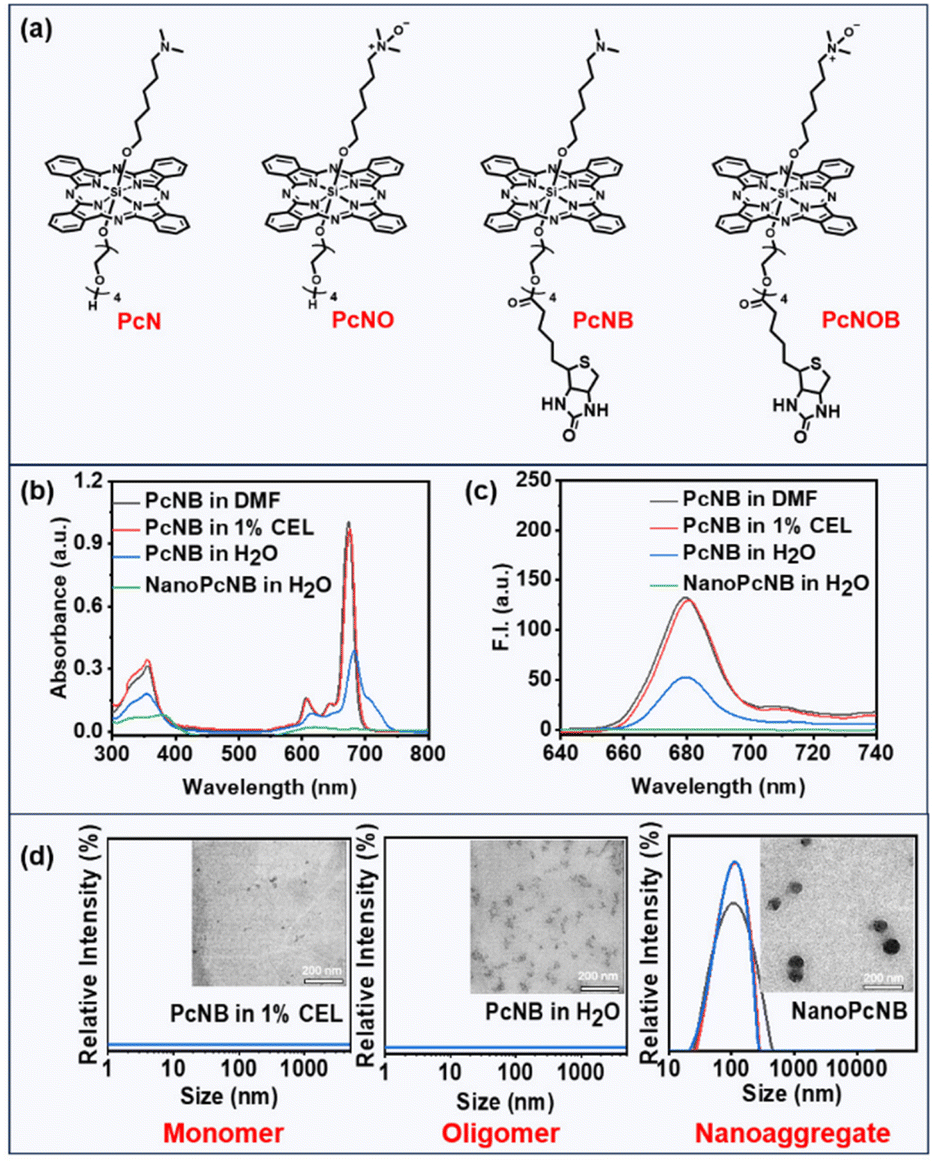 | ||
| Fig. 2 (a) Chemical structure of the synthesized phthalocyanines. (b) Electronic absorption spectra of PcNB in DMF, water containing 1% CEL, pure water, and NanoPcNB in pure water. (c) Fluorescence emission spectra of PcNB in DMF, water containing 1% CEL, pure water, and NanoPcNB in pure water. (d) TEM imaging and DLS of PcNB in water containing 1% CEL (left), pure water (middle), and NanoPcNB in pure water (right), scale bar: 200 nm. | ||
Photodynamic properties
Electronic absorption spectroscopy was used to investigate polar organic solvents and aqueous solutions. These phthalocyanine molecules (PcN, PcNO, PcNB and PcNOB) exhibited sharp Q-band absorption peaks at approximately 675 nm and superior light absorption capability in dimethylformamide (DMF) (Fig. S10 and Table S1†). In water containing 1% Cremophor EL (CEL), their absorbance spectra were similar to that in DMF, whereas in pure water, the absorbance significantly decreased, and the peak broadened (Fig. 2b and S11a†).Next, we examined the fluorescence emission spectra in polar organic solvents and aqueous solutions. As can be seen from Fig. 2c and S11b,† the fluorescence intensity of all phthalocyanine molecules in DMF remained consistent with that in water containing 1% CEL, while the fluorescence intensity in pure water was weakened. This phenomenon indicates that phthalocyanine molecules of the same structure exhibit different aggregation states in different solutions. It is well known that conventional phthalocyanines lose or reduce their photosensitizing activity due to their strong π–π stacking. However, our previous studies revealed that efficient type I photosensitizing activity was performed by the nanoaggregates formed by amine-substituted phthalocyanines.38 Thus far, the relationship between the aggregation degree of phthalocyanine molecules and photosensitizing activity has not yet been reported.
To study the relationship between supramolecular structure and photosensitizing activity, we also individually assembled phthalocyanine molecules (PcN, PcNO, PcNB, and PcNOB) into nanoparticles (NanoPcN, NanoPcNO, NanoPcNB and NanoPcNOB) under external forces. The preparation method is shown in the ESI.† All nanoparticles displayed the lowest absorbance and were able to fully quench fluorescence in pure water due to the aggregation-caused quenching (ACQ) effect. In addition, we tested the dynamic light scattering (DLS) of all phthalocyanine molecules in water containing 1% CEL, all phthalocyanine molecules in pure water, and all nanoparticles in pure water. As shown in Fig. 2d and S12,† only the nanoparticles exhibited suitable particle size and uniform distribution in pure water.
We also performed transmission electron microscopy (TEM) assays on two phthalocyanine molecules (PcNB and PcNOB) in water containing 1% CEL, two phthalocyanine molecules (PcNB and PcNOB) in pure water and two nanoparticles (NanoPcNB and NanoPcNOB) in pure water. There were tiny particles of PcNOB and PcNB in water containing 1% CEL, whereas PcNOB and PcNB in pure water behaved as multiple molecules clustered together to form irregular shapes. NanoPcNOB and NanoPcNB presented spherical nanoparticles in pure water, with particle sizes of 99 nm and 90 nm, respectively.
A comprehensive analysis of the results from absorption spectra, emission spectra and TEM imaging revealed that there were monomeric states for all phthalocyanines in water containing 1% CEL, with aggregated states for all nanoparticles in pure water. All phthalocyanines in pure water existed in a form between monomers and nanoaggregates, which are referred to as oligomeric phthalocyanines. We obtained models for different aggregation states of the same phthalocyanine molecule and similar aggregation states of different phthalocyanine molecules, including four monomeric phthalocyanines (MonoPcN, MonoPcNO, MonoPcNB, MonoPcNOB), four oligomeric phthalocyanines (OligPcN, OligPcNO, OligPcNB, OligPcNOB) and four nanoaggregated phthalocyanines (NanoPcN, NanoPcNO, NanoPcNB, NanoPcNOB).
Next, we studied the relationship between the same phthalocyanine molecule with different aggregation states and photodynamic effects. We used the indicator chemical dihydroethidium (DHE) to determine O2˙− species generation and the unique indicator aminophenyl fluorescein (APF) to determine ˙OH species creation. As shown in Fig. S13,† for the same phthalocyanine molecular structure, there was no significant difference in the ability to produce O2˙− for oligomeric phthalocyanine, monomeric phthalocyanine, or nanoaggregated phthalocyanine, and none was much higher than commercial methylene blue (MB) (Fig. S14†). However, compared with monomeric phthalocyanines and nanoaggregated phthalocyanines, oligomeric phthalocyanines produced more ˙OH (Fig. 3a and S15†).
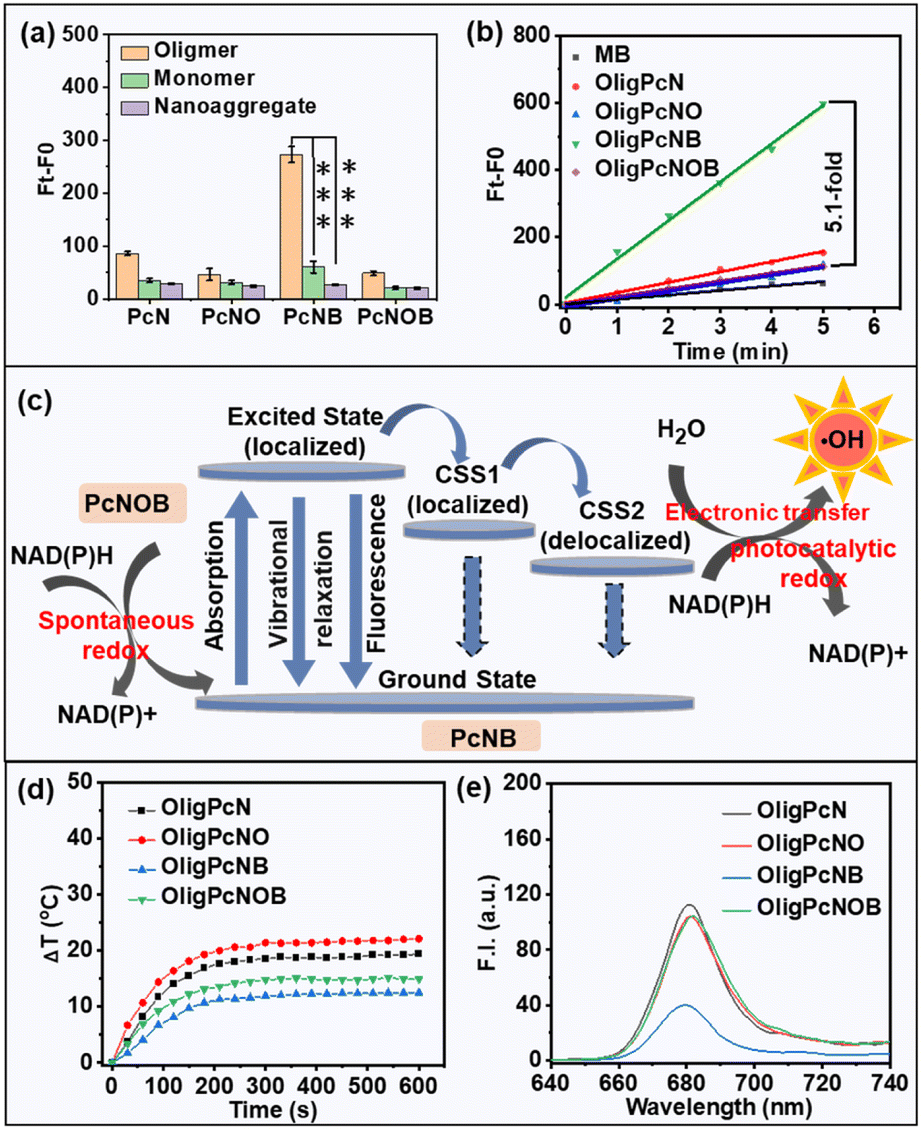 | ||
| Fig. 3 (a) ˙OH production by phthalocyanines in different aggregation states (all at 5 μM) upon red light irradiation (λ ≥ 610 nm) at the power density of 1 mW cm−2 for 5 min. Data are given as mean ± SD. **P < 0.01 and ***P < 0.001. Statistical differences were analyzed using Student's t-test. (b) Rate linear diagrams of the APF probe at 515 nm induced by oligomeric phthalocyanines (5 μM) upon red light (λ ≥ 610 nm) irradiation at a power density of 1 mW cm−2 for 5 min. MB was used as the control group. (c) Schematic illustration of the possible mechanism of ˙OH production and NAD(P)H consumption of OligPcNOB. (d) Evolution of the temperature of oligomeric phthalocyanines (5 μM) under laser exposure (680 nm, 1 W cm−2). (e) Fluorescence spectra of oligomeric phthalocyanines (2 μM) in pure water. | ||
We conjectured that oligomeric phthalocyanine may be more prone to symmetry-breaking charge separation (SBCS). As illustrated in Fig. 3c, a local excited state (S1) is quickly formed upon photon excitation of oligomeric phthalocyanine, and SBCS subsequently transpires. This process creates a localized charge-separated state (CSS1) while transferring the charge to the adjacent molecule in the ground state. The formation of a delocalized charge-separated state (CSS2) is explained by the subsequent further delocalization of the charge in the CSS1 state to the entire oligomeric phthalocyanine. There is a relatively low charge compounding rate for a molecule in the CSS2 state, which provides sufficient time for the molecule to interact with the substrate and therefore favors the production of ˙OH.
To explore the mechanism, we tested the chemical potentials of all phthalocyanines in acetonitrile and DMF using cyclic voltammetry (Fig. S16†). The results showed that the redox potential in the oligomeric phthalocyanine was more negative than that in the monomeric phthalocyanines, and thus, it is more likely that the SBCS process would occur. However, the absorption spectra of oligomeric phthalocyanines are redshifted compared to the nanoaggregated phthalocyanines. The unique slip-type stacking of J-aggregates results in less resistance to intermolecular charge transfer, which may favor the formation of the CSS2 state, and allow a greater amount of ˙OH to be produced by oligomeric phthalocyanine as compared to nanoaggregated phthalocyanine.
We also evaluated the relationship between the different structures of phthalocyanine molecules under similar aggregation states and photodynamic effects. We used the indicator APF to determine the ability of oligomeric phthalocyanines with different molecular structures to produce ˙OH. There was an improvement in fluorescence at 515 nm in the presence of the OligPcNB upon light irradiation (λ ≥ 610 nm), while no significant changes were observed for the other oligomeric phthalocyanines (Fig. S15b†). As shown in the rate linearity plot, only OligPcNB was more capable of producing ˙OH than the known MB, at 5.1-fold greater quantities than that of OligPcNOB (Fig. 3b).
We also used 5-tert-butoxycarbonyl 5-methyl-1-pyrroline N-oxide (BMPO) as a trapping agent to detect ˙OH production by means of electron spin resonance (ESR). The characteristic peak of BMPO interacting with ˙OH suggested that OligPcNB can produce ˙OH in the presence of light irradiation (Fig. S17†). The above results demonstrated that only OligPcNOB is reduced to OligPcNB, and many ˙OH moieties are generated that do not originate from OligPcNOB itself.
Next, we also investigated the impact of oxygen content on ˙OH radical production. Oxygen in water was discharged by argon bubbling (Fig. S18a†), which significantly reduced the oxygen content in water (Fig. S18b†). The results showed that OligPcNB generates a large number of ˙OH radicals under normoxic and hypoxic conditions, yet OligPcNOB failed to do so (Fig. S19†). Therefore, the ability of OligPcNB to generate ˙OH was not affected by oxygen content. More importantly, OligPcNB is effective in producing ˙OH without oxygen participation at low-dose light irradiation (0.3 J cm−2), and therefore, it is more advantageous for the treatment of hypoxic tumors. We speculated on the reason and determined from the energy level Jablonski mechanism that OligPcNOB is capable of producing the highest quantities of ˙OH.
There are three competing methods for release of photons absorbed by PSs: fluorescence emission, heat release through vibrational relaxation, and formation of ROS. As seen from Fig. 3d, the photothermal effect of OligPcNB was negligible at a power density of 1 W cm−2. The lowest fluorescence emission efficiency was for that of OligPcNB (Fig. 3e). This provides a reasonable pathway for OligPcNB to achieve the highest ˙OH-producing capacity.
Next, the valence band (VB) potential was ascertained from the ultraviolet photoelectron spectroscopy (UPS) valence band spectra. According to the current reports, the potential required (EO2/O2˙−) for O2 to gain electrons and form O2˙− is −0.33 V, and the potential required (EOH−/˙OH) to oxidise water to ˙OH is 1.99 V (Fig. S20a†).34,39 Water can be photo-oxidized to ˙OH when the VB potential of a PS is more positive than EOH−/˙OH. The VB potential of PcNOB was less than 1.99 V, which does not meet the conditions required to oxidize water to ˙OH (Fig. S20b†). Large amounts of ˙OH can be produced by PcNB, as evidenced by the fact that its calculated VB potential is 4.03 V vs. NHE (Fig. S20c†).
Interaction of phthalocyanines with NAD(P)H
Because ˙OH is the most reactive and toxic ROS in living organisms and can react with almost all endogenous biomolecules,22 we attempted to reduce the damage of ˙OH to normal tissues by means of an OFF–ON switch. In this work, our synthesized PS PcNOB initially produced no ˙OH and activation by specific substances in the hypoxic tumor tissue was required to produce large amounts of ˙OH. NAD(P)H is a typical reductase that is an ideal choice for a biomarker for hypoxic tumors.40–44 Therefore, we used NAD(P)H as an electron donor to study the bioreduction of PcNOB. The process of their interaction is shown in Fig. 3c.When the mixture of PcNOB and NAD(P)H was stirred at room temperature for 1 h, the absorption of PcNOB at 680 nm significantly increased and tended to coincide with the absorption intensity of PcNB. In addition, the color of the reaction solution deepened, and the polarity of the products increased at the end of the reaction (Fig. S21a†). These results suggested that PcNOB and NAD(P)H can spontaneously undergo redox. Next, nuclear magnetic resonance (1H NMR) hydrogen spectrum and electron spray ionization mass spectrometry (ESI-MS) were used to identify the transformation from PcNOB to PcNB. As shown in Fig. 4a and b, when PcNOB coexisted with NAD(P)H, the chemical shift of the characteristic peak of dimethylammonium migrated from 2.42 ppm to 1.81 ppm, and the relative molecular mass decreased by 15.99. These results indicate that the N–O unit can be spontaneously reduced to an amino unit under NAD(P)H catalysis.
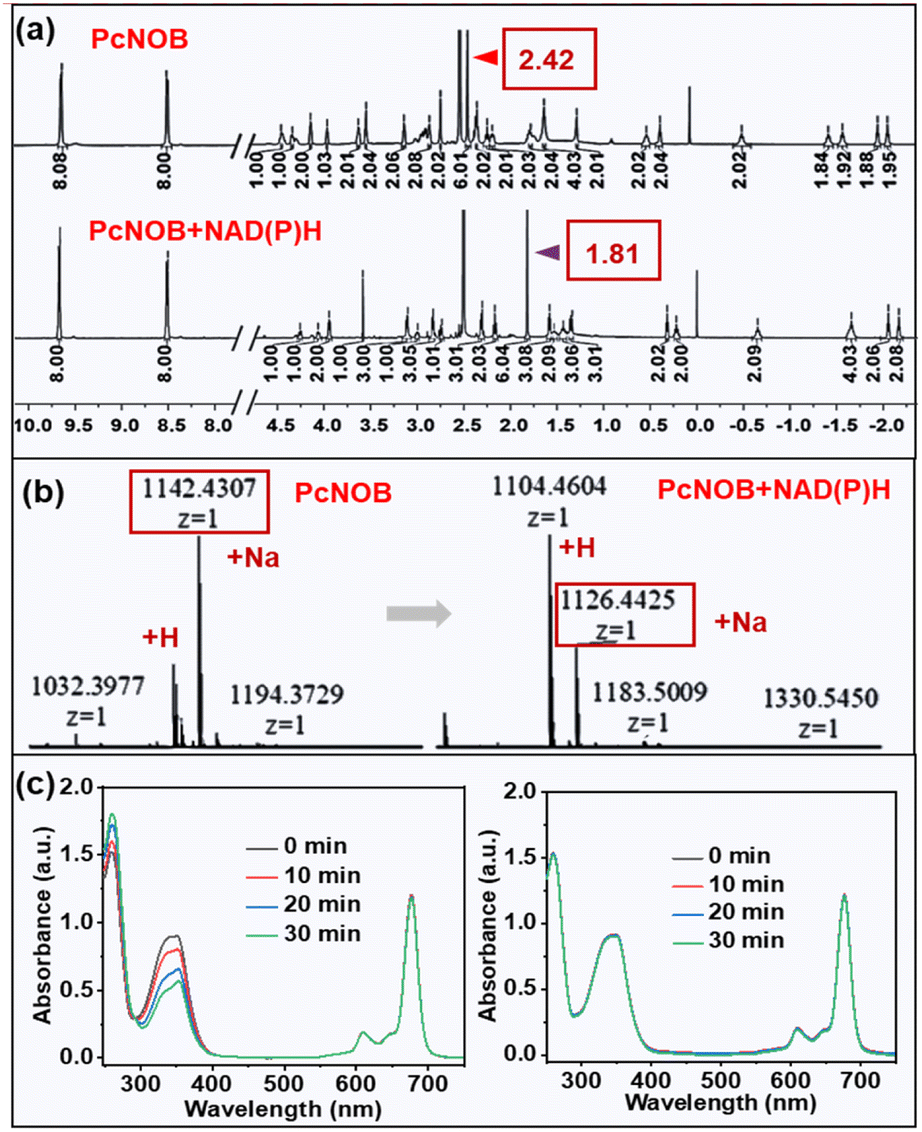 | ||
| Fig. 4 Mixed solution containing NAD(P)H and PcNOB was stirred for 1 h and later purified for 1H NMR (a) and ESI-MS (b). (c) Electronic absorption spectra of PcNB coexistence with NAD(P)H under light irradiation with a power density of 5.4 mW cm−2 (left) or absence of light irradiation (right). | ||
Herein, we propose a mechanism for the redox that may occur in organisms. When the oxidizing agent PcNOB and the reducing agent NAD(P)H coexist, the lone pair of electrons in the reducing agent is transferred from the 4-position of the dihydropyridine ring to the N–O unit in the oxidizing agent, which causes the N–O unit to shed its oxygen atom to produce the amino unit (Fig. S21b†). PcNOB allows it to remain OFF even under light illumination, and it will be switched ON by NAD(P)H. Because of this switchable strategy, it is possible to minimize off-target side effects to obtain precise anti-tumor therapy.
Next, we tested the photocatalytic oxidation of PcNB with NAD(P)H using electronic absorption spectroscopy. It has been well documented that when NAD(P)H is oxidized to NAD(P)+, the absorption decreases at 339 nm and increases at 260 nm. As demonstrated in Fig. 4c, when light irradiates an aqueous solution containing PcNB and NAD(P)H, the absorption at 339 nm clearly decreases, while it remained stable when there was no light or no PcNB (Fig. S22†). This result indicated that photocatalytic redox can be performed when PcNB and NAD(P)H coexist.
In addition, 1H NMR spectroscopy was applied to detect the conversion from NAD(P)H to NAD(P)+. The emerging peaks of NAD(P)+ were observed in the presence of PcNB under the light, including the peaks at 6.83, 8.08, 8.33, 8.72, 9.05, and 9.23 ppm. This result confirmed the successful progress of NAD(P)H to NAD(P)+ by photocatalytic oxidation. There were no new detected chemical shift peaks in the absence of light (Fig. S23†). This result suggests that PcNOB can be activated by NAD(P)H and also can efficiently oxidize NAD(P)H once more, exacerbating cancer cell death in a manner that is independent of O2.
In vitro selectivity evaluation and PDT efficacy
The above studies demonstrated that PcNOB can be reduced by NAD(P)H. In addition, the reduction product PcNB can produce a large amount of ˙OH via an O2-independent photosensitizing pathway. Motivated by this, we explored the cellular uptake and photosensitizing activity. First, cellular uptake was studied in human hepatocarcinoma (HepG2) cells, mouse breast cancer (4T1) cells, and normal human liver (L02) cells by confocal laser scanning microscopy (CLSM). 4T1 and HepG2 cells are biotin receptor-positive, and L02 cells are biotin receptor-negative. Compared with L02 cells, more significant fluorescent signals were observed in HepG2 and 4T1 cells after incubating the OligPcNOB (Fig. 5a). In contrast, the fluorescence signal of OligPcNO without a biotin unit was not significantly different in HepG2, 4T1, or L02 cells (Fig. S24a†).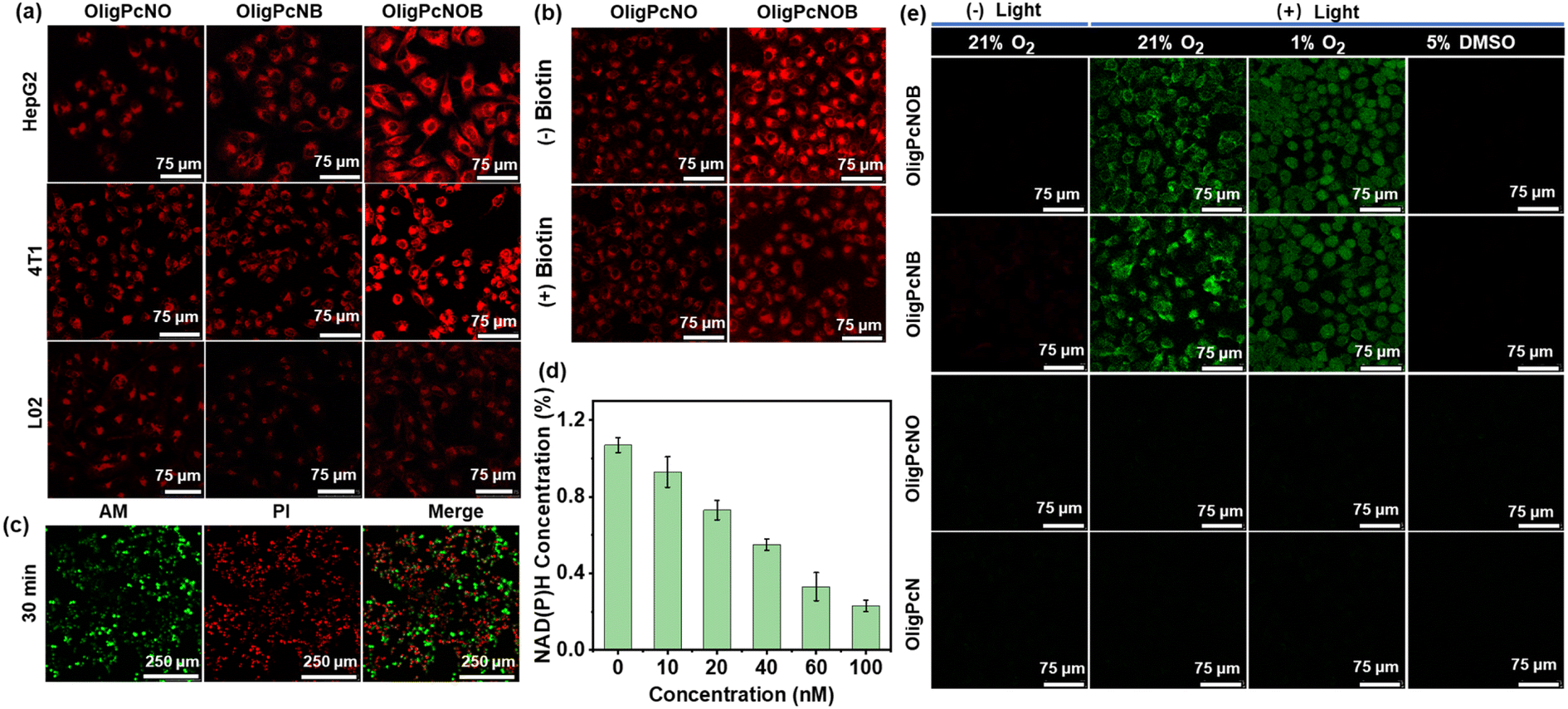 | ||
| Fig. 5 (a) CLSM imaging of cellular uptake of oligomeric phthalocyanines in HepG2, B16 and L02 cells. Cells were incubated with oligomer phthalocyanine for 2 h and later washed 3 times to remove excess phthalocyanine before continuing incubation for 6 h. The scale bar represents 75 μm. (b) Intracellular fluorescence images of HepG2 cells after incubation with OligPcNO or OligPcNOB (100 nM) for 8 h in the presence and absence of extra biotin (100 nM). The scale bar represents 75 μm. (c) CLSM imaging of HepG2 cells incubated with OligPcNOB (50 nM) for 8 h under red light (λ ≥ 610 nm, 15 mW cm−2) irradiation for 30 min using Calcein acetoxymethyl ester (AM)/propidium iodide (PI) staining. The scale bar represents 250 μm. (d) Measurement of intracellular NAD(P)H levels in HepG2 cells exposed to various concentrations of OligPcNOB after light irradiation (λ ≥ 610 nm, 15 mW cm−2). Data are displayed as mean ± SD produced using n = 5 biological samples. (e) CLSM images of intracellular ˙OH generated by oligomer phthalocyanine (300 nM) using APF as the fluorescent probe in HepG2 cells under normoxia (21% O2) without light irradiation, normoxia with light irradiation, hypoxia (1% O2) with light irradiation, and normoxia with light irradiation and in the presence of 5% dimethyl sulfoxide (DMSO). Light condition: λ ≥ 610 nm, 15 mW cm−2, 5 min. The scale bar represents 75 μm. | ||
These results suggest that there is a high binding affinity by OligPcNOB for cancer cells with overexpressing biotin receptors. OligPcNB containing biotin units exhibited a lower fluorescence signal than OligPcNOB in 4T1 and HepG2 cells, which was attributed to the decreased fluorescence emission ability of OligPcNB (Fig. 3e). To confirm the cellular uptake of biotin receptor mediation, OligPcNO or OligPcNOB was co-incubated with extra biotin in a competitive inhibition assay. As illustrated in Fig. 5b, after co-incubation, the intracellular fluorescence of OligPcNOB decreased by approximately 40%, whereas there was no significant alteration in OligPcNO (Fig. S24b†). The above results suggest that there is specific selectivity for cancer cells with overexpressed biotin receptors by oligomeric phthalocyanines with biotin units.
Then, the 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) assay was utilized to evaluate the PDT efficacy of OligPcNOB through the cell viability of HepG2 cells. It demonstrated that there was nearly no dark toxicity by OligPcNOB in normoxic and hypoxic environments (Fig. S25a†). Following a 30 min exposure to light (15 mW cm−2), OligPcNOB demonstrated notable cytotoxicity against HepG2 cells, with a half-maximal inhibitory concentration (IC50) of 25.0 ± 4.3 nM under normoxia and 30.0 ± 5.1 nM under hypoxia (Fig. S25b†). The cytotoxicity of OligPcNOB was largely unaffected by the decrease in O2 concentration, which further indicated that OligPcNOB exhibited excellent PDT efficacy in an O2-independent manner. Similarly, the live/dead assay was used to verify the proliferation inhibition of HepG2 cells by OligPcNOB under light irradiation. The live cells were labeled with Calcein AM to emit green fluorescence, and the dying or dead cells were labeled with propidium iodide (PI) to emit red fluorescence. After light irradiation, obvious red fluorescence signals were observed in OligPcNOB-treated cells (Fig. S26†). Additionally, the duration of light increased the red fluorescence signal (Fig. 5c).
Motivated by the above mechanisms, we investigated NAD(P)H depletion in HepG2 cells with a NAD(P)+/NAD(P)H assay kit with water-soluble tetrazolium salt (WST)-8. As seen in Fig. 5d, under light irradiation, the intracellular NAD(P)H levels clearly decreased as the OligPcNOB concentration increased. Then, the fluorescent probe APF was used to assess the intracellular ˙OH during PDT under normoxic (21% O2) and hypoxic (1% O2) conditions. After illumination, strong fluorescence signals were observed by CLSM in OligPcNOB-treated HepG2 cells due to the reaction of APF with ˙OH (Fig. 5e). The fluorescence intensity was not affected by the O2 concentration, which suggested that ˙OH generation was O2-independent (Fig. S27†).
Subsequently, 5% dimethyl sulfoxide (DMSO) was chosen as a quencher for ˙OH. In the presence of DMSO, the signal of fluorescence was extinguished, which further indicated the production of ˙OH in cancer cells. Notably, as shown in Fig. 3b, OligPcNOB initially failed to produce a large amount of ˙OH in pure water. However, after co-incubating it with HepG2 cells for 8 h, intracellular ˙OH production was detected using the APF probe. This proved that OligPcNOB can be reduced by NAD(P)H in cancer cells. Thus, the OligPcNOB can be activated by NAD(P)H and produces plentiful ˙OH, which is independent of O2 concentration.
In vivo tumor-targeting evaluation and PDT efficacy
We proceeded to study the tumor targeting and PDT efficacy of phthalocyanines by constructing subcutaneous hepatocarcinoma (H22) tumor-bearing mice, which were selected because of the relative simplicity in tumor model construction. First, the in vivo biodistribution of OligPcNOB was investigated by intravenous injection, and OligPcNO was used as a control. Furthermore, to compare the PDT effects of phthalocyanine under different aggregation states, we simultaneously investigated the in vivo biodistribution of NanoPcNOB.The results showed that OligPcNOB and NanoPcNOB were significantly and efficiently enriched in the tumor region at 36 h after intravenous injection. In contrast, OligPcNO was distributed in the periphery of tumor tissue (Fig. 6a and b). Consequently, the tumor accumulation capacity of OligPcNOB was 2.1-fold higher than that of OligPcNO. Second, an evaluation of the distribution of phthalocyanines in the resected tumor and primary organs was conducted by taking ex vivo fluorescence images. The findings demonstrated that there was greater uptake of OligPcNOB and NanoPcNOB in tumor tissues as compared to normal tissues, and their fluorescence signals in the tumor tissue were 2.2-fold and 4.3-fold higher than those in the liver, respectively (Fig. 6c and S28†).
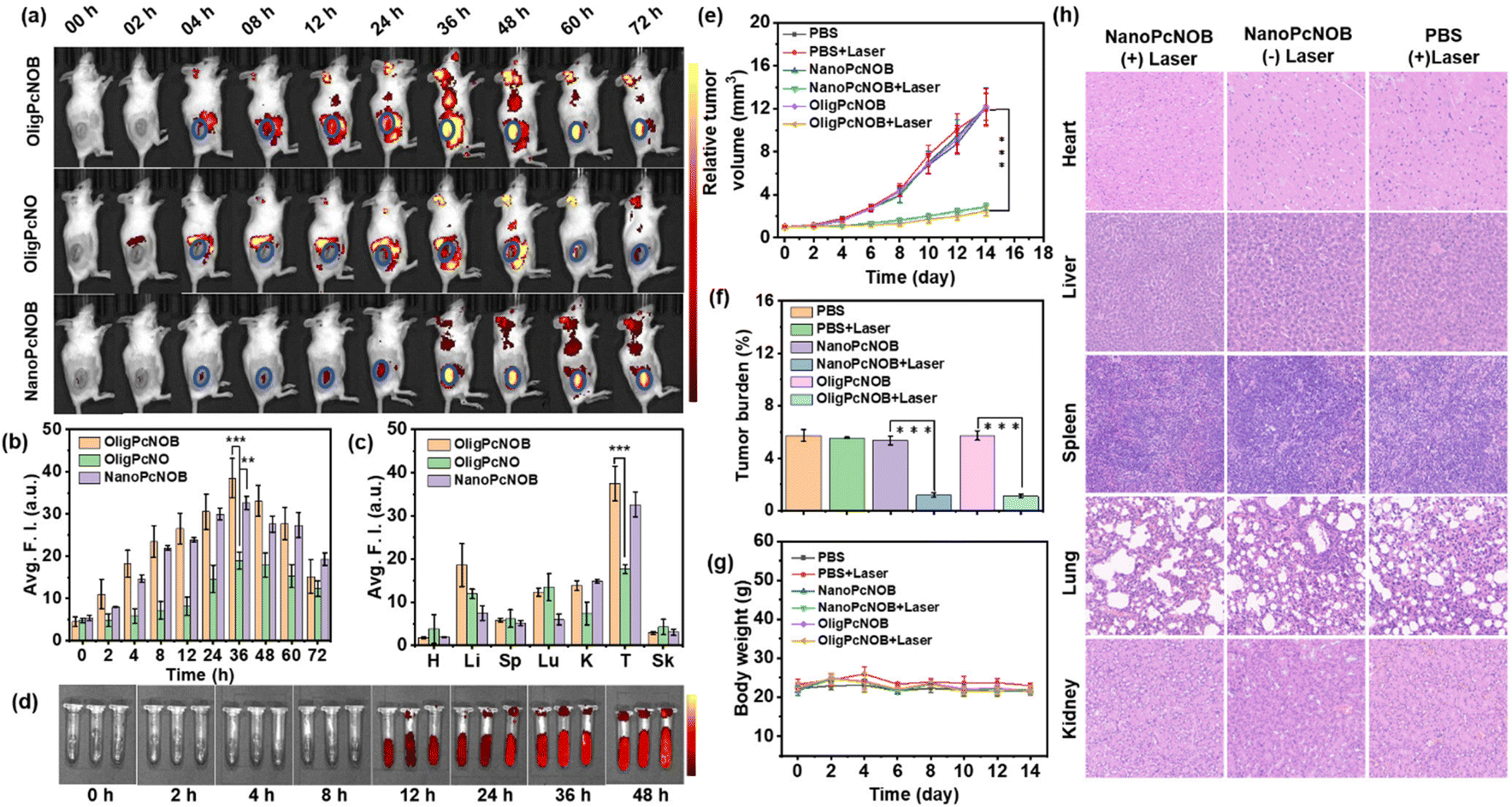 | ||
| Fig. 6 (a) Fluorescence images were captured from H22 tumor-bearing mice before and after intravenous injections of OligPcNO, OligPcNOB, and NanoPcNOB (the units of fluorescence intensity: 108 ps−1 cm−2 sr−1 μW−1 cm2). (b) Quantification of the fluorescence intensity of phthalocyanine accumulation in tumor tissue at various intervals after intravenous injection of phthalocyanine at a concentration of 200 μM (100 μL). (c) Quantification of the fluorescence intensity in the tumor tissue and various organs of mice harboring H22 tumors at 48 h following intravenous injection. H, heart; Li, liver; Sp, spleen; Lu, lung; K, kidney; T, tumor; and Sk, skin. (d) Representative photos of NanoPcNOB in mouse blood at different times. (e) Growth curves of tumors in H22-bearing mice following various treatments (n = 5). On the zeroth day, PDT treatments were conducted using a 680 nm laser (100 mW cm−2) for a duration of 5 min. (f) Tumor weight averages following 14 days of treatment as prescribed. (g) Changes in the average body weight of mice after receiving recommended treatments. (h) Images of various organs in mice subjected to different treatments and stained using the hematoxylin and eosin (H&E) stain. Three separate independent runs of the experiment produced comparable outcomes. Data are given as mean ± SD, with n = 5. It was found that **P < 0.01 and ***P < 0.001. Statistical differences were analyzed by Student's t-test. | ||
NanoPcNOB completely quenched the fluorescence in pure water, whereas it emitted a fluorescence signal in the tumor tissue of mice, probably because of nanoparticles that can depolymerize in blood. We added NanoPcNOB to the blood of mice and monitored the fluorescence intensity for 48 h. It was found that the fluorescence signal of NanoPcNOB gradually increased (Fig. 6d). All these results indicate that phthalocyanines containing biotin units either as oligomers or nanoaggregates possess the ability to precisely target tumor tissue, which provides a new means of precision cancer treatment.
Finally, the in vivo PDT efficacy of NanoPcNOB and OligPcNOB under laser irradiation (labeled as NanoPcNOB + Laser and OligPcNOB + Laser) was investigated in H22 tumor-bearing mice. Four other groups of mice treated with phosphate-buffered saline (PBS), PBS with laser irradiation (PBS + Laser), and NanoPcNOB and OligPcNOB without laser irradiation (NanoPcNOB and OligPcNOB) were used as controls. As depicted in Fig. 6e, the tumors of the control mice continued to grow for 14 days, while photodynamic treatment with NanoPcNOB and OligPcNOB effectively inhibited tumor growth, and the efficiency of both treatments remained consistent, with an inhibition rate of approximately 80%.
The exceptional anti-tumor efficaciousness of NanoPcNOB and OligPcNOB under light irradiation was further validated by the average tumor weights (Fig. 6f). The average body weight of each group did not decrease (Fig. 6g), indicating that there was little systemic toxicity to these treatments. To further demonstrate biosafety, histological measurements of the vital organs were conducted by H&E staining, which showed that there were no obvious histopathological lesions in all three groups of organs (Fig. 6h).
Biosafety was also evidenced by hemolysis experiments. When NanoPcNOB was added to PBS solution, all erythrocytes underwent sedimentation, and there was no absorption at 400–600 nm for the supernatant, indicating that no hemolysis occurred (Fig. S29†). The aforementioned results highlight the similar tumor accumulation capacity and PDT effects of NanoPcNOB and OligPcNOB. Additionally, a very simple pharmaceutical preparation method was used for OligPcNOB that ensures facile and effective clinical application.
Conclusions
We report for the first time an activatable PDT agent that can be reduced by NAD(P)H, is abundant in hypoxic tumors and whose reduction product oxidizes water to efficiently produce ˙OH under light irradiation for hypoxic tumor treatment. Notably, this activatable PS can reduce intracellular NAD(P)H levels via two methods. Moreover, we reveal for the first time that oligomeric phthalocyanine, which is a derivative of silicon phthalocyanine with an enzyme-activated N–O unit and a biotin unit for targeting, can enhance ˙OH-generation capacity. In vitro assays demonstrated that oligomeric phthalocyanine OligPcNOB exhibited considerable phototoxicity to cancer cells, and the generation of ˙OH was nearly unaffected by intracellular O2 partial pressure. Furthermore, in vivo assays revealed that OligPcNOB actively accumulated in the lesion, accounting for distinct solid tumor eradication. This work will inspire passion for future exploration of novel O2-independent activatable PSs for cancer therapy.Data availability
All experimentally relevant data can be obtained from the manuscript and ESI.†Author contributions
All authors contributed to the manuscript, and all authors have given their approval for the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the National Natural Science Foundation of China (Grant No. T2322004 and 22078066).References
- D. W. Felsher, Cancer revoked: oncogenes as therapeutic targets, Nat. Rev. Cancer, 2003, 3(5), 375–379 CrossRef CAS PubMed.
- N. Lu, Z. Deng, J. Gao, C. Liang, H. Xia and P. Zhang, An osmium-peroxo complex for photoactive therapy of hypoxic tumors, Nat. Commun., 2022, 13(1), 2245–2256 CrossRef CAS PubMed.
- X. Li, J. Kim, J. Yoon and X. Chen, Cancer-associated, stimuli-driven, turn on theranostics for multimodality imaging and therapy, Adv. Mater., 2017, 29(23), 1606857–1606881 CrossRef PubMed.
- X. Li, S. Yu, Y. Lee, T. Guo, N. Kwon, D. Lee, S. C. Yeom, Y. Cho, G. Kim, J.-D. Huang, S. Choi, K. T. Nam and J. Yoon, In vivo albumin traps photosensitizer monomers from self-assembled phthalocyanine nanovesicles: a facile and switchable theranostic approach, J. Am. Chem. Soc., 2018, 141(3), 1366–1372 CrossRef.
- C. Wang, Y. Sun, S. Huang, Z. Wei, J. Tan, C. Wu, Q. Chen and X. Zhang, Self-immolative photosensitizers for self-reported cancer phototheranostics, J. Am. Chem. Soc., 2023, 145(24), 13099–13113 CrossRef CAS PubMed.
- X. Li, N. Kwon, T. Guo, Z. Liu and J. Yoon, Innovative strategies for hypoxic-tumor photodynamic therapy, Angew. Chem., Int. Ed., 2018, 57(36), 11522–11531 CrossRef CAS PubMed.
- X. Li, S. Lee and J. Yoon, Supramolecular photosensitizers rejuvenate photodynamic therapy, Chem. Soc. Rev., 2018, 47(4), 1174–1188 RSC.
- J. Tian, B. Li, F. Zhang, Z. Yao, W. Song, Y. Tang, Y. Ping and B. Liu, Pctivatable type I photosensitizer with quenched photo-sensitization pre and post photodynamic therapy, Angew. Chem., Int. Ed., 2023, 62(46), e202307288–e202307299 CrossRef CAS PubMed.
- D. Chen, Q. Xu, W. Wang, J. Shao, W. Huang and X. Dong, Type I photosensitizers revitalizing photodynamic oncotherapy, Small, 2021, 17(31), 2006742–2006763 CrossRef CAS PubMed.
- Q. Yao, J. Fan, S. Long, X. Zhao, H. Li, J. Du, K. Shao and X. Peng, The concept and examples of type-III photosensitizers for cancer photodynamic therapy, Chem, 2022, 8(1), 197–209 CAS.
- X. Li, C. Y. Kim, S. Lee, D. Lee, H.-M. Chung, G. Kim, S.-H. Heo, C. Kim, K.-S. Hong and J. Yoon, Nanostructured phthalocyanine assemblies with protein-driven switchable photoactivities for bio-photonic imaging and therapy, J. Am. Chem. Soc., 2017, 139(31), 10880–10886 CrossRef CAS PubMed.
- Y.-Y. Zhao, L. Zhang, Z. Chen, B.-Y. Zheng, M. Ke, X. Li and J.-D. Huang, Nanostructured phthalocyanine assemblies with efficient synergistic effect of type I photoreaction and photothermal action to overcome tumor hypoxia in photodynamic therapy, J. Am. Chem. Soc., 2021, 143(34), 13980–13989 CrossRef CAS PubMed.
- W. Chen, Z. Wang, M. Tian, G. Hong, Y. Wu, M. Sui, M. Chen, J. An, F. Song and X. Peng, Integration of TADF photosensitizer as “electron pump” and BSA as “electron reservoir” for boosting type I photodynamic therapy, J. Am. Chem. Soc., 2023, 145(14), 8130–8140 CrossRef CAS PubMed.
- J. An, S. Tang, G. Hong, W. Chen, M. Chen, J. Song, Z. Li, X. Peng, F. Song and W.-H. Zheng, An unexpected strategy to alleviate hypoxia limitation of photodynamic therapy by biotinylation of photosensitizers, Nat. Commun., 2022, 13(1), 2225–2235 CrossRef CAS PubMed.
- K. X. Teng, W. K. Chen, L. Y. Niu, W. H. Fang, G. Cui and Q. Z. Yang, BODIPY-based photodynamic agents for exclusively generating superoxide radical over singlet oxygen, Angew. Chem., Int. Ed., 2021, 60(36), 19912–19920 CrossRef CAS PubMed.
- L. Yu, Y. Xu, Z. Pu, H. Kang, M. Li, J. L. Sessler and J. S. Kim, Photocatalytic superoxide radical generator that induces pyroptosis in cancer cells, J. Am. Chem. Soc., 2022, 144(25), 11326–11337 CrossRef CAS PubMed.
- V.-N. Nguyen, S. Qi, S. Kim, N. Kwon, G. Kim, Y. Yim, S. Park and J. Yoon, An emerging molecular design approach to heavy-atom-Free photosensitizers for enhanced photodynamic therapy under hypoxia, J. Am. Chem. Soc., 2019, 141(41), 16243–16248 CrossRef CAS PubMed.
- D. Zhang, K. X. Teng, L. Zhao, L. Y. Niu and Q. Z. Yang, Ultra-small nano-assemblies as tumor-targeted and Renal Clearable theranostic agent for photodynamic therapy, Adv. Mater., 2023, 35(19), 2209789–2209801 CrossRef CAS PubMed.
- C. Li, J. Ye, X. Yang, S. Liu, Z. Zhang, J. Wang, K. Zhang, J. Xu, Y. Fu and P. Yang, Fe/Mn Bimetal-doped ZIF-8-coated lumines-cent nanoparticles with up/down conversion dual-mode emission for tumor self-enhanced NIR-II imaging and catalytic therapy, ACS Nano, 2022, 16(11), 18143–18156 CrossRef CAS PubMed.
- J. Ye, W. Lv, C. Li, S. Liu, X. Yang, J. Zhang, C. Wang, J. Xu, G. Jin, B. Li, Y. Fu and X. Liang, tumor response and NIR-II photonic thermal co-enhanced catalytic therapy based on single-atom manganese nanozyme, Adv. Funct. Mater., 2022, 32(47), 2206157–2206171 CrossRef CAS.
- L. Wu, W. Zeng, Y. Ishigaki, J. Zhang, H. Bai, T. Harimoto, T. Suzuki and D. Ye, A ratiometric photoacoustic probe with a reversible response to hydrogen sulfide and hydroxyl radicals for dynamic imaging of liver inflammation, Angew. Chem., Int. Ed., 2022, 61(37), e202209248–e202209255 CrossRef CAS PubMed.
- B. Halliwell, A. Adhikary, M. Dingfelder and M. Dizdaroglu, Hydroxyl radical is a significant player in oxidative DNA damage-in vivo, Chem. Soc. Rev., 2021, 50(15), 8355–8360 RSC.
- X. Zeng, Y. Ruan, Q. Chen, S. Yan and W. Huang, Biocatalytic cascade in tumor microenvironment with a Fe2O3/Au hybrid nanozyme for synergistic treatment of triple negative breast cancer, Chem. Eng. J., 2023, 452(2), 138422–138434 CrossRef CAS.
- W. Xie, G. Zhang, Z. Guo, J. Lu, J. Ye, W. Xu, X. Gao, K. Yue, Y. Wei and L. Zhao, Ultra-sensitive iron-doped palladium nano-crystals with enhanced hydroxyl radical generation for chemo/chemodynamic nanotherapy, Adv. Funct. Mater., 2021, 32(12), 2107518–2107528 CrossRef.
- T. Jia, J. Du, J. Yang, F. Li, X. Fang and G. Chen, Near-infrared upconversion photosensitizer enabling photodynamic production and in situ dynamic monitoring of hydroxyl radical in live mice, Nano Today, 2023, 51(31), 101932–101946 CrossRef CAS.
- L. Liang, L. Wen, Y. Weng, J. Song, H. Li, Y. Zhang, X. He, W. Zhao, M. Zhan, Y. Li, L. Lu, Y. Xin and C. Lu, Homologous-targeted and tumor microenvironment-activated hydroxyl radical nano-generator for enhanced chemoimmunotherapy of non-small cell lung cancer, Chem. Eng. J., 2021, 425(89), 131451–131462 CrossRef CAS.
- R. Wang, Y. Yu, M. Gai, A. Mateos-Maroto, S. Morsbach, X. Xia, M. He, J. Fan, X. Peng, K. Landfester, S. Jiang and W. Sun, Lipo-somal enzyme nanoreactors based on nanoconfinement for efficient antitumor therapy, Angew. Chem., Int. Ed., 2023, 62(44), e202308761–e202308770 CrossRef CAS PubMed.
- W. Xie, J. Ye, Z. Guo, J. Lu, W. Xu, X. Gao, H. Huang, R. Hu, L. Mao, Y. Wei and L. Zhao, TME-responded Full-biodegradable nanocatalyst for mitochondrial calcium Overload-induced hydroxyl radical bursting cancer treatment, Chem. Eng. J., 2022, 438(91), 135372–135384 CrossRef CAS.
- P. Das, G. Chakraborty, J. Roeser, S. Vogl, J. Rabeah and A. Thomas, Integrating bifunctionality and chemical stability in covalent organic frameworks via one-pot multicomponent reactions for solar-driven H2O2 production, J. Am. Chem. Soc., 2023, 145(5), 2975–2984 CrossRef CAS PubMed.
- T. Luo, K. Ni, A. Culbert, G. Lan, Z. Li, X. Jiang, M. Kauf-mann and W. Lin, Nanoscale Metal–Organic Frameworks Stabilize Bacteriochlorins for Type I and Type II Photodynamic Therapy, J. Am. Chem. Soc., 2020, 142(16), 7334–7339 CrossRef CAS PubMed.
- T. Zhang, Z. Liu, W. Tang, D. Zhu, M. Lyu, J. W. Y. Lam, Q. Huang and B. Z. Tang, Mitochondria-targeting type I AIE photosensitizer combined with H2S therapy: uninterrupted hydroxyl radical generation for enhancing tumor therapy, Nano Today, 2022, 46(8), 101620–101629 CrossRef CAS.
- H. Wang, T. Qin, W. Wang, X. Zhou, F. Lin, G. Liang, Z. Yang, Z. Chi and B. Z. Tang, Selenium-containing type-I organic photosensitizers with dual reactive oxygen species of superoxide and hydroxyl radicals as switch-hitter for photodynamic therapy, Adv. Sci., 2023, 10(24), 2301902–2301913 CrossRef CAS PubMed.
- X. Zhao, S. He, J. Wang, J. Ding, S. Zong, G. Li, W. Sun, J. Du, J. Fan and X. Peng, Near-infrared self-assembled hydroxyl radical generator based on photoinduced cascade electron transfer for hypoxic tumor phototherapy, Adv. Mater., 2023, 35(44), 2305163–2305172 CrossRef CAS PubMed.
- K.-X. Teng, L.-Y. Niu and Q.-Z. Yang, Supramolecular photo-sensitizer enables oxygen-independent generation of hydroxyl radicals for photodynamic therapy, J. Am. Chem. Soc., 2023, 145(7), 4081–4087 CrossRef CAS PubMed.
- Y. Li, D. Zhang, Y. Yu, L. Zhang, L. Li, L. Shi, G. Feng and B. Z. Tang, A cascade strategy boosting hydroxyl radical generation with aggregation-induced emission photosensitizers-albumin complex for photodynamic therapy, ACS Nano, 2023, 17(17), 16993–17003 CrossRef CAS PubMed.
- R. Lin, J. Liu, W. Xu, Z. Liu, X. He, C. Zheng, M. Kang, X. Li, Z. Zhang, H. T. Feng, J. W. Y. Lam, D. Wang, M. Chen and B. Z. Tang, Type I photosensitization with strong hydroxyl radical generation in NIR dye boosted by vigorous intramolecular motions for synergistic therapy, Adv. Mater., 2023, 35(33), 2303212–2303224 CrossRef CAS PubMed.
- S. Wang, M. Rong, H. Li, T. Xu, Y. Bu, L. Chen, X. Chen, Z. P. Yu, X. Zhu, Z. Lu and H. Zhou, Unveiling mechanism of organic photogenerator for hydroxyl radicals generation by molecular modulation, Small, 2021, 18(6), 2104857–2104867 CrossRef PubMed.
- X. Li, D. Lee, J. D. Huang and J. Yoon, Phthalocyanine-assembled Nanodots as photosensitizers for highly efficient type I photoreactions in photodynamic therapy, Angew. Chem., Int. Ed., 2018, 57(31), 9885–9890 CrossRef CAS PubMed.
- L. Yang, G. Dong, D. L. Jacobs, Y. Wang, L. Zang and C. Wang, Two-channel photocatalytic production of H2O2 over g-C3N4 nanosheets modified with perylene imides, J. Catal., 2017, 352(8), 274–281 CrossRef CAS.
- Y. Chen, J. Shi, Y. Wu, Z. Guo, S. Li, W. Li, Z. Wu, H. Wang, H. Jiang and Z. Jiang, NADH photosynthesis system with affordable electron supply and inhibited NADH oxidation, Angew. Chem., Int. Ed., 2023, 62(42), e202310238–e202310246 CrossRef CAS PubMed.
- E. King, S. Maxel, Y. Zhang, K. C. Kenney, Y. Cui, E. Luu, J. B. Siegel, G. A. Weiss, R. Luo and H. Li, Orthogonal glycolytic pathway enables directed evolution of noncanonical cofactor oxidase, Nat. Commun., 2022, 13(1), 7282–7296 CrossRef CAS.
- N. Zhang, S. Trépout, H. Chen and M.-H. Li, AIE polymer micelle/vesicle photocatalysts combined with native enzymes for aerobic photobiocatalysis, J. Am. Chem. Soc., 2022, 145(1), 288–299 CrossRef PubMed.
- K.-X. Teng, L.-Y. Niu, N. Xie and Q.-Z. Yang, Supramolecular photodynamic agents for simultaneous oxidation of NADH and generation of superoxide radical, Nat. Commun., 2022, 13(1), 6179–6191 CrossRef CAS PubMed.
- W. Wei, F. Mazzotta, I. Lieberwirth, K. Landfester, C. T. J. Ferguson and K. a. I. Zhang, Aerobic photobiocatalysis enabled by combining core–shell nanophotoreactors and native enzymes, J. Am. Chem. Soc., 2022, 144(16), 7320–7326 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc02179g |
| This journal is © The Royal Society of Chemistry 2024 |