
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reversible C–C bond formation in group 4 metal complexes: nitrile extrusion via β-aryl elimination†
Pavel S.
Kulyabin‡
 a,
Georgy P.
Goryunov
a,
Andrei N.
Iashin
a,
Dmitry Y.
Mladentsev
a,
Dmitry V.
Uborsky
a,
Christian
Ehm
b,
Jo Ann M.
Canich
c,
John R.
Hagadorn
c and
Alexander Z.
Voskoboynikov
*a
a,
Georgy P.
Goryunov
a,
Andrei N.
Iashin
a,
Dmitry Y.
Mladentsev
a,
Dmitry V.
Uborsky
a,
Christian
Ehm
b,
Jo Ann M.
Canich
c,
John R.
Hagadorn
c and
Alexander Z.
Voskoboynikov
*a
aDepartment of Chemistry, M. V. Lomonosov Moscow State University, Leninskie Gory, 1/3, Moscow 119991, Russian Federation
bDipartimento di Scienze Chimiche, Università di Napoli Federico II, Via Cintia, Napoli 80126, Italy
cBaytown Technology and Engineering Complex, ExxonMobil Technology and Engineering Company, Baytown, Texas 77520, USA
First published on 27th August 2024
Abstract
Pyridylamides of zirconium and hafnium with [C,N,N]-ligands reversibly insert nitriles into M–CAr bonds leading to an observable equilibrium between the starting [C,N,N]-complexes and newly formed [N,N,N]-complexes with a ketimide moiety in a 7-membered metallacycle. The discovered reversible insertion of nitriles into M–CAr bonds represents an unprecedented example of β-aryl elimination from a ketimide ligand in early transition metal complexes. Experimental and computational studies suggest thermodynamic and electronic reasons for this reactivity. Weak orbital overlap between the ketimide nitrogen and the metal, and an unfavorable 7-membered metallacycle destabilize the product of insertion into the M–CAr bond, while the pyridylamide moiety acts as a directing group making the reverse process viable. The influence of non-chelate spectator ligands on the metal center and substituents in nitrile on the thermodynamic stability of the [N,N,N]-complexes was also studied. Exploiting β-carbon elimination in complexes of early transition metals may extend the range of catalysts that are accessible for C–C activation processes in the future.
Introduction
Over the recent decades, the cleavage of C–C bonds has garnered significant attention due to its wide-ranging applications in catalytic transformations,1–6 including highly sought after chemical methods for polyolefin degradation.7 Since the first seminal reports8–11 this area of research has witnessed substantial advancements and continues to be a subject of immense interest in the scientific community. One of the main strategies for transition metal catalysed C–C bond activation is β-carbon elimination,4,12,13 the microscopic reverse of migratory insertion (Fig. 1A; X = C, heteroatom). In terms of mechanisms, the process of β-carbon elimination is akin to that of β-hydride elimination, though it is less explored due to the challenges in designing systems for its study.12 Generally, migratory insertion is thermodynamically favoured; reversing this transformation requires an additional driving force: (a) formation of stronger Csp–M, Csp2–M14,15 or C X bonds,16 (b) release of ring strain,17 and (c) relief of steric strain.2,3 β-Carbon elimination processes do not require a change in metal oxidation states and are known for early transition metals.18
X bonds,16 (b) release of ring strain,17 and (c) relief of steric strain.2,3 β-Carbon elimination processes do not require a change in metal oxidation states and are known for early transition metals.18
 | ||
| Fig. 1 β-Carbon elimination and migratory insertion in complexes of zirconium and hafnium. | ||
Since the first reports of β-aryl elimination employing palladium and rhodium alkoxy and ketimide complexes by Uemura,19,20 Miura21 and Hartwig16,22,23 (Fig. 1B) in the 2000s, this reaction has gained importance for catalytic C–C bond activation.4,24 Presently, this type of reactivity has been demonstrated for alcoholates of Mn, Pd, Rh, Co, Ni, Cu,4 Ru25 and Re15 and for ketimides of Pd20,24 and Rh.16,22 The driving force of β-carbon elimination in complexes of these metals is the irreversible release of π-bond containing molecules, while examples of reversible abstraction of ketones or nitriles via these mechanisms are still very rare.16,26 Meanwhile, β-alkyl elimination is an important (endergonic) chain release mechanism in olefin polymerization catalysed by cationic group 4 metal complexes (Fig. 1C);27 however, β-carbon elimination from alcoholate or ketimide complexes of these metals has not been reported yet. The strength of M–X (X = O or N) bonds in these cases28 presents a significant hurdle in designing systems that can provide an adequate driving force.
One of the approaches to facilitate C–C bond activation is the promotion of metal–carbon interactions via directing groups which dates back to the 1980s with the use of nitrogen heterocycles.8 The choice of directing group not only influences the selectivity for a specific C–C bond activation but also reduces activation barriers by participating in the formation of stable metallacyclic intermediates. Pyridine is one of the most popular directing groups in C–H activation;29 unsurprisingly, it found application in C–C activation as well. Thus, while complexes of Pd19–21 and Rh16 do not require directing groups in the substrate to extrude a nitrile or a ketone, the corresponding reactions of Co30 and Mn31 complexes necessitate the presence of a coordinating nitrogen heterocycle.
Recently, some of us explored the migratory insertion of small polar molecules such as ketones, nitriles, isocyanides, isocyanates, azides, and imines into the M–CAr bond of pyridylamide32–34 (Fig. 1D, I and II) and heteroarylamide35,36 (Fig. 1D, III) complexes of hafnium and zirconium with [C,N,N]-ligands.36–38 These complexes are renown olefin polymerization precatalysts which upon activation via cationization with methylaluminoxane (MAO) or boron-based cocatalysts are modified in situ by initial monomer insertion into the M–CAr bond.39–41 Similarly, dimethylated complexes of type III, even in the presence of Hf–CH3 moieties, insert nitriles exclusively into the Hf–CAr bond forming an 8-membered metallacycle with an N(ketimide)–Hf bond.36
Here, we report an intriguing case of reversible carbon–carbon bond formation via nitrile insertion and extrusion in complexes of types I and II (Fig. 1E). While migratory insertion of nitriles36 and isonitriles42,43 in group 4 metal–carbon bonds is a well-known transformation, β-carbon elimination with the release of nitrile has never been reported for group 4 metal ketimides. Nitrile release via [4 + 2]-retrocycloaddition has been reported by Frye et al.44 while benzonitrile extrusion has been observed for a hafnium complex by Ghana et al.45 however, in both examples the reactions are irreversible. The reversibility of migratory insertion in our case allowed us to study the β-carbon elimination process in detail using NMR spectroscopy and DFT calculations establishing the reasons for such unique reactivity.
Results and discussion
Reactions of 1-HfCl2 and 1-HfMe2 with iPrCN: experimental study
Late-stage functionalization of hafnium [C,N,N]-ligated heteroarylamide complexes via insertion of small polar molecules into Hf–CAr bonds36 was demonstrated to be a convenient way to obtain a series of variously substituted precatalysts for olefin polymerization from a common precursor in one step. We decided to apply this method for functionalization of recently studied [C,N,N]-complexes of type II (Fig. 1D).34Reaction of dichloride complex 1-HfCl2 with isobutyronitrile smoothly gives [N,N,N]-ligated product 1iPrCN-HfCl2 in 80% isolated yield (Scheme 1A). In order to obtain dimethyl complex 1iPrCN-HfMe2, which could potentially be activated with borate cocatalysts (such as B(C6F5)3, [Ph3C][B(C6F5)4], and [Me2HNPh][B(C6F5)4]), dichloride 1iPrCN-HfCl2 was treated with MeMgBr in diethyl ether at room temperature. Surprisingly, after extraction of the product from the crude mixture with hot hexane, we isolated dimethyl complex 1-HfMe2 with the original [C,N,N]-ligand as in 1-HfCl2 in 60% yield instead of the expected 1iPrCN-HfMe2 (Scheme 1B). This suggests that formation of 1-HfMe2 must have occurred via extrusion of the nitrile during the methylation of 1iPrCN-HfCl2.
 | ||
| Scheme 1 Synthesis of 1iPrCN-HfCl2 and methylation with nitrile extrusion. | ||
Next, we attempted to substitute the two chloride ligands sequentially to elucidate at which step the nitrile extrusion takes place. The first methylation of 1iPrCN-HfCl2 with 1 equiv. of MeMgBr (Scheme 2A) resulted in selective substitution of the first chloride, and partial substitution of the second one with bromide from the Grignard reagent, giving a mixture of monomethylated complexes 1iPrCN-HfMe(Cl/Br). The X-ray structure (Fig. S130†) confirmed the presence of the inserted nitrile forming a 7-membered metallacycle. The second methylation of 1iPrCN-HfMe(Cl/Br) with 1 equiv. of MeMgBr gave dimethyl complex 1iPrCN-HfMe2 still containing the nitrile. As a result, 1iPrCN-HfMe2 was obtained from 1iPrCN-HfCl2 in 57% yield over two steps (Scheme 2B).
 | ||
| Scheme 2 Stepwise methylation of 1iPrCN-HfCl2. | ||
An alternative attempt to synthesize 1iPrCN-HfMe2 from dimethyl complex 1-HfMe2 through the addition of 1 equiv. of isobutyronitrile to a solution of 1-HfMe2 gave a mixture of the following complexes: 1iPrCN-HfMe2 as a major product, 1iPrCN-HfMe(N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CMeiPr) – a product of double insertion of the nitrile into the Hf–CAr and Hf–CH3 bonds – as a minor product, and unreacted 1-HfMe2 in molar ratio = 7/2/1 (Scheme 3A). This experiment demonstrates that the insertion of the nitrile into the Hf–CAr bond is faster in comparison with insertion into Hf–CH3, analogous to what we found earlier for complexes of type III (Fig. 1D).36
CMeiPr) – a product of double insertion of the nitrile into the Hf–CAr and Hf–CH3 bonds – as a minor product, and unreacted 1-HfMe2 in molar ratio = 7/2/1 (Scheme 3A). This experiment demonstrates that the insertion of the nitrile into the Hf–CAr bond is faster in comparison with insertion into Hf–CH3, analogous to what we found earlier for complexes of type III (Fig. 1D).36
 | ||
| Scheme 3 Reactions of 1-HfMe2 with isobutyronitrile. a Molar ratio of the products. | ||
Addition of two equivalents of nitrile to 1-HfMe2 (Scheme 3B) led to quantitative formation of 1iPrCN-HfMe(NCMeiPr) (isolated yield 58%), whose structure was further confirmed by 2D NMR (Fig. S23–S33†) and X-ray diffraction crystallography (vide infra). Addition of three equivalents of the nitrile to 1-HfMe2 yields exclusively 1iPrCN-HfMe(NCMeiPr); no product of triple insertion of isobutyronitrile was observed.
Heating a solution of 1iPrCN-HfMe2 in toluene-d8 in an NMR tube for 3 h led to formation of 1iPrCN-HfMe(NCMeiPr) along with 1-HfMe2 and 1-HfMe(NCMeiPr), a product of isobutyronitrile insertion into the Hf–CH3 bond only (Fig. S35†). Overnight heating of the reaction mixture resulted in exclusive formation of 1-HfMe(NCMeiPr) (Scheme 4A), whose structure was confirmed by 2D NMR (Fig. S18–S20†). This experiment convincingly demonstrates that nitrile insertion into the Hf–CAr bond of 1-HfMe2 is indeed reversible. Thus, complex 1-HfMe(NCMeiPr) is the thermodynamic product of the reaction of 1-HfMe2 with isobutyronitrile, whereas 1iPrCN-HfMe2 is the kinetic product. Additionally, 1iPrCN-HfMe2 was dissolved in toluene-d8 at three concentrations, and the solutions were kept at room temperature and analysed by 1H NMR at four time points from 20 min to 2 days. The identical character of the kinetic curves (Fig. S139†) regardless of the concentration, allows us to conclude that the rate-limiting step is unimolecular, and that the migration of the nitrile from CAr to methyl occurs via the nitrile release and reinsertion rather than via a bimolecular reaction between two molecules of complexes exchanging the nitrile fragment.
 | ||
| Scheme 4 β-Carbon elimination from 1iPrCN-HfMe2 and 1iPrCN-HfMe(NCMeiPr). Quant. conv. = quantitative conversion. a Molar ratio. | ||
Heating the mixture of 1-HfMe2, 1iPrCN-HfMe2, and 1iPrCN-HfMe(NCMeiPr), prepared from 1-HfMe2 and iPrCN in toluene (Scheme 3A), resulted in exclusive formation of 1-HfMe(NCMeiPr) as well (Scheme 4B) which was isolated in 60% yield. These observations indicate that nitrile insertion into the Hf–CAr bond of 1-HfMe(NCMeiPr) giving 1iPrCN-HfMe(NCMeiPr) is reversible, too. Indeed, the 1H NMR spectrum of complex 1iPrCN-HfMe(NCMeiPr) in toluene-d8 at 87 °C evidences the formation of complex 1-HfMe(NCMeiPr) (Scheme 4C and Fig. 2) which disappears again upon cooling to room temperature.
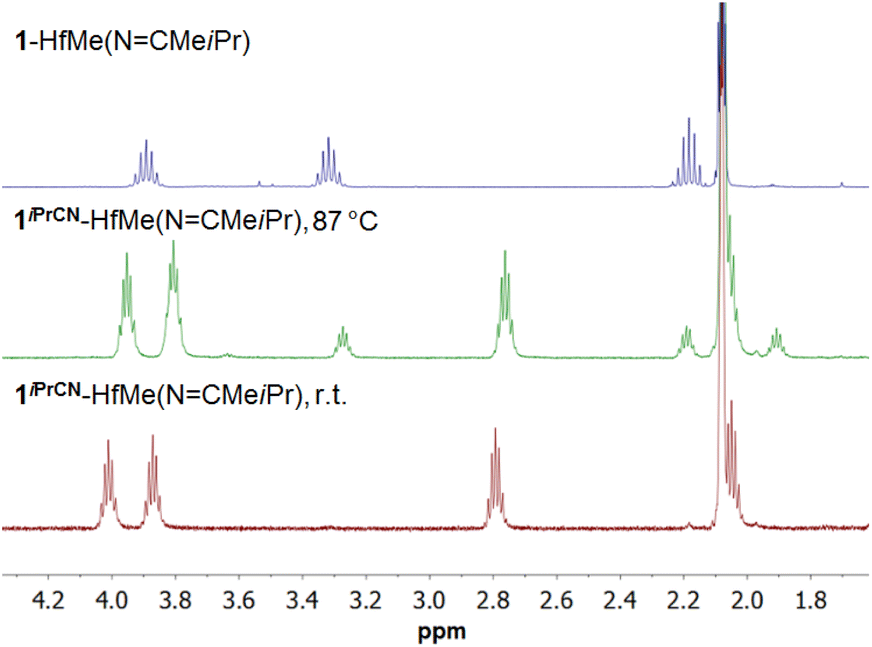 | ||
| Fig. 2 Fragments of 1H NMR spectra of complexes 1iPrCN-HfMe(NCMeiPr) and 1-HfMe(NCMeiPr) in toluene-d8 at room temperature (r.t.) and complex 1iPrCN-HfMe(NCMeiPr) in toluene-d8 at 87 °C. | ||
The observed nitrile release can be classified as an example of a β-aryl elimination reaction. While well-known for complexes of Pd4,14,21,24 and Rh,4,16,22,23 the process has not been reported for group 4 metal complexes before.
The well-defined solution equilibrium of 1iPrCN-HfMe(NCMeiPr) (Table 1) at temperatures higher than 60 °C provided an opportunity to study the thermodynamics of the β-aryl elimination process. Dissolving pure 1iPrCN-HfMe(NCMeiPr) in toluene-d8 and heating the solution gives an equilibrium mixture of 1iPrCN-HfMe(NCMeiPr), 1-HfMe(NCMeiPr) and iPrCN. A Van't Hoff analysis over a 30 K range yielded thermal parameters shown in Table 1 (entry 1) with ΔG298 estimated to be 6.4 kcal mol−1. Repeating the experiment with dichloride complex 1iPrCN-HfCl2 in toluene-d8 yields an equilibrium mixture of 1iPrCN-HfCl2, 1-HfCl2 and iPrCN upon heating it at temperatures higher than 70 °C. A Van't Hoff analysis over a 20 K range yielded thermal parameters shown in Table 1 (entry 2) with ΔG298 estimated to be 9.2 kcal mol−1. Interestingly, switching the solvent to ortho-dichlorodeuterobenzene shifts the equilibrium slightly, ΔG298 = 7.8 kcal mol−1 (Table 1, entry 3) for 1iPrCN-HfCl2 ⇆ 1-HfCl2 + iPrCN, which can be observed already at 60 °C. These findings demonstrate that the spectator ligands on hafnium and the solvent affect the reaction energetics: (a) electron acceptors like chloride stabilize the product of nitrile insertion towards β-aryl elimination and (b) a solvent with a higher dielectric constant destabilizes the product of nitrile insertion towards β-aryl elimination.
CMeiPr) in toluene-d8 or ortho-dichlorodeuterobenzene (o-C6D4Cl2)

![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif)
The rate of interconversion of 1iPrCN-HfCl2 ⇆ 1-HfCl2 + iPrCN was measured through spin saturation transfer difference (SSTD) experiments in ortho-dichlorodeuterobenzene.46–48 On-resonance frequency was selected at 2.319 ppm (CH proton of iPrCN), which affected the resonance at 2.758 ppm (CH proton of the isopropyl group in 1iPrCN-HfCl2). SSTD data were collected at temperatures between 72.0 and 92.0 °C, and the rate constants were plotted according to the Eyring equation (see the ESI†). This method yields activation parameters for the transformation of ΔH‡ = 20 ± 2 kcal mol−1, and ΔG‡298 = 22.7 kcal mol−1 (Table 1, entry 5). The entropy of activation was found to be slightly negative ΔS‡ = −10 ± 5 cal mol−1, possibly due to the higher polarizing effect of o-C6D4Cl2 and/or coordination of the latter to the hafnium.
The activation energy for nitrile extrusion from 1iPrCN-HfCl2 implies that the equilibrium 1iPrCN-HfCl2 ⇆ 1-HfCl2 + iPrCN is already viable at room temperature. Indeed, after addition of PhCN to a solution of 1iPrCN-HfCl2 in o-C6D4Cl2, isobutyronitrile was almost completely substituted by benzonitrile in the chelate ligand after 94 h at room temperature (Fig. S120†).
Reaction of 1iPrCN-HfMe2 with iPrCN: DFT calculations
DFT calculations, according to established protocols,49,50 at the TPSSh-D0(SMD)/cc-pVTZ(-PP)//TPSSh/cc-pVTZ level of theory interrogate the reversible insertion of iPrCN into the Hf–CAr bond.51–62 Thermal and activation parameters for the 1iPrCN-HfCl2 ⇆ 1-HfCl2 + iPrCN equilibrium are well reproduced (see Table 1). The competition of insertion into the Hf–CAr and Hf–CH3 bonds of 1-HfMe2 is shown in Fig. 3. Coordination of iPrCN to 1-HfMe2 is endergonic by 5.2 kcal mol−1, in line with the observation of free nitrile. Insertion into the Hf–CAr bond viaTS-1iPrCN-HfMe2 has a barrier of 21.3 kcal mol−1 and is preferred by 5.2 kcal over insertion into the Hf–CH3 bond viaTS-1-HfMe(NCMeiPr). However, formation of 1iPrCN-HfMe2 is only exergonic by 5.1 kcal mol−1 and the reversible C–C elimination barrier (26.4 kcal mol−1) is accessible at elevated temperatures. Conversely, insertion into the Hf–CH3 bond and formation of 1-HfMe(NCMeiPr) are highly exergonic (−19.9 kcal mol−1) and irreversible even at 100 °C (C–C elimination barrier 46.4 kcal mol−1).
 | ||
| Fig. 3 Potential energy surface for insertion of iPrCN into Hf–C bonds of 1iPrCN-HfMe2. Gibbs free energies in kcal mol−1 at 298 K, 1 atm and 1 equiv. of iPrCN. | ||
Intrinsic bond orbital (IBO)63–65 and transition state analyses were employed to analyse the differences in insertion barriers (kinetics). The Hf–CAr insertion TS leading to 1iPrCN-HfMe2 (TS-1iPrCN-HfMe2) is characterized by smaller distortion energies than the Hf–CH3 insertion TS leading to 1-HfMe(NCMeiPr) (TS-1-HfMe(NCMeiPr), 27.9 vs. 32.0 kcal mol−1). Both TSs are energetically early66 with respect to the 1-HfMe2-iPrCN adduct but geometrically central with TS-1iPrCN-HfMe2 showing more short C–C contacts below the van-der-Waals limit than TS-1-HfMe(NCMeiPr), indicating higher steric strain. The electron flow along the reaction coordinate for the IBOs with the largest change for insertion into the Hf–CAr bond (TS-1iPrCN-HfMe2) is depicted in Fig. 4. The IBO associated with the Hf–CAr bond (C: 1.798 e−, Hf 0.128 e−) becomes the new C–C bond while the IBO associated with the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N π-bond transforms into a σ-bond that is largely centred on N (N: 1.856 e−, Hf 0.131 e−). Therefore, the transformation of the Hf–CAr bond into the C–C bond in the product can be identified as a nucleophilic attack by the aryl ligand on the electrophilic carbon of the nitrile ligand.
N π-bond transforms into a σ-bond that is largely centred on N (N: 1.856 e−, Hf 0.131 e−). Therefore, the transformation of the Hf–CAr bond into the C–C bond in the product can be identified as a nucleophilic attack by the aryl ligand on the electrophilic carbon of the nitrile ligand.
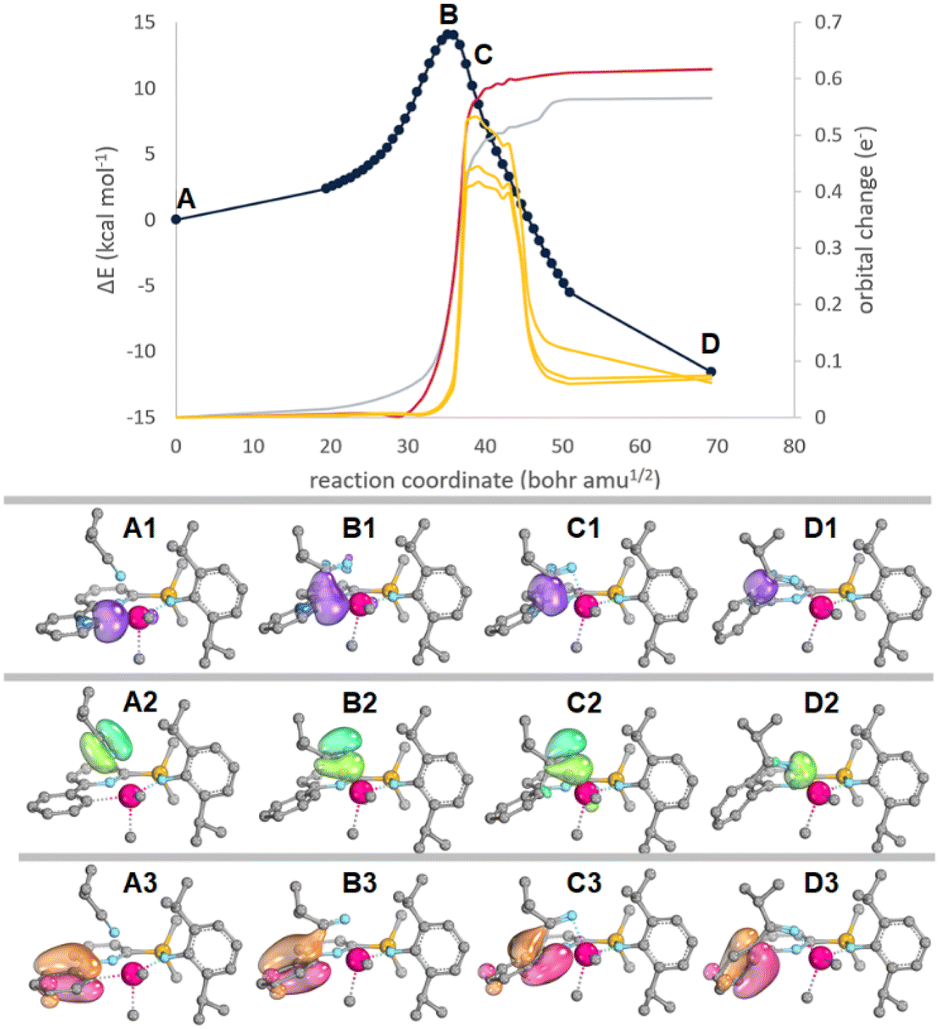 | ||
| Fig. 4 Top: Plot of the root of the sum of square deviations (RSSD) of the partial charge distribution changes along the IRC for iPrCN insertion into the Hf–CAr bond leading to 1-HfMe(NCMeiPr) viaTS-1-HfMe(NCMeiPr). Hf–CAr (red), the CN π (grey) and the three aryl based π IBOs. Bottom: Depiction of the Hf–CAr (purple, top), CN π (green, middle), and one selected aryl π IBO (orange-red) along the IRC. H-atoms omitted for clarity. Movies showing IBOs along the reaction coordinate for iPrCN insertion into 1-HfCl2 and 1-HfMe2 can be found as gif files in ESI (see also page S115).† | ||
These findings are mirrored for insertion into the Hf–CH3 bond (TS-1-HfMe(NCMeiPr)), with one notable exception: the aryl π-orbitals also significantly change temporarily along the reaction coordinate indicating stabilizing π-donation into metal-based orbitals (Fig. 4, C3). In our opinion, this stabilizing IBO overlap of the aromatic pi-system with the Hf-centre compensates for electronic changes occurring in the Hf–CAr and N based orbitals along the reaction coordinate. Subsequently, TS-1iPrCN-HfMe2 is shifted and occurs earlier on the reaction coordinate than would be expected without these stabilizing interactions. In fact, the sum of the charges of Hf–CAr/CH3 and the N-based orbital is much smaller at the TS for TS-1iPrCN-HfMe2 (0.36 e−) than for TS-1-HfMe(NCMeiPr) (0.46 e−). It appears likely that the lower insertion barrier leading to product 1iPrCN-HfMe2 is due to underlying electronic factors, rather than steric differences. Meanwhile, the much lower exergonicity of the insertion into the Hf–CAr bond forming 1iPrCN-HfMe2 compared to insertion into the Hf–CH3 bond forming 1-HfMe(NCMeiPr) (thermodynamics) results from steric strain and a worse orbital overlap in the former. Formation of 1-HfMe(NCMeiPr) results in a strain free system with a Hf–N–CiPr angle of 176° while this angle reaches 144° in 1iPrCN-HfMe2. Wiberg bond indices for the largely ionic bonds are lower for 1iPrCN-HfMe2 than 1-HfMe(NCMeiPr) (IBO: 0.334 vs. 0.362; NBO 0.701 vs. 0.856).
Scope of the reaction of RCN with Zr and Hf pyridylamides
Next, we investigated how changes in the pyridylamide ligand, the metal, and the nitrile influence the reactivity pathways. Thus, we chose MeCN, tBuCN, and PhCN to study their reactions with 1-HfCl2/1-HfMe2. Additionally, we studied the reactivity of pyridylamide complexes 2-HfMe2, 2-ZrMe2, and 3-HfMe2 (Scheme 5–7) with iPrCN. | ||
| Scheme 5 Reactions of L-HfMe2 complexes with 1 equiv. of nitriles. a Quantitative conversion by NMR. b Unresolved mixture of products. | ||
 | ||
| Scheme 6 Reactions of L-HfMe2 complexes with 2 equiv. of nitriles. a NMR yield. | ||
 | ||
| Scheme 7 Nitrile extrusion and reinsertion in complex 2iPrCN-Zr(NCMeiPr). | ||
Reactions of dimethyl complexes L-HfMe2 with 1 equiv. of RCN at r.t. gave mixtures of products of insertions LRCN-HfMe2 and LRCN-HfMe(NCMeR) with the starting complexes (Scheme 5). Upon stirring at 100 °C, most of these mixtures were converted into the thermodynamic products L-HfMe(NCMeR), suggesting that the β-carbon elimination from both LRCN-HfMe2 and LRCN-HfMe(NCMeR) took place.
Contrary to expectations, in the case of reaction between 1-HfMe2 and PhCN, heating the mixture did not lead to the formation of pure 1-HfMe(NCMePh), and after stirring at 100 °C for two days, we found that the major component of the mixture was 1PhCN-HfMe(NCMePh) (Fig. S73†). Accumulation of this double insertion product indicates that the extrusion of PhCN from 1PhCN-HfMe(NCMePh) did not occur at 100 °C, whereas it did from 1PhCN-HfMe2, which was initially observed in the mixture. Indeed, NMR experiments demonstrated that 1PhCN-HfMe(NCMePh) prepared separately from 1-HfMe2 and 2 equiv. of PhCN (Scheme 6) did not release nitrile even at elevated temperatures (Fig. S76†).
One more common transformation was the reaction of complexes L-MMe2 with 2 equiv. of nitrile (Scheme 6) giving products of double insertion LRCN-MMe(NCMeR) after few hours at r.t. except for the reaction with bulky tBuCN, which required additional stirring at 100 °C for 1.5 days to complete. Complexes 1tBuCN-HfMe(N=CMetBu) and 2iPrCN-HfMe(NCMeiPr) extruded the nitriles reversibly and cleanly upon heating. Meanwhile, complexes 1MeCN-HfMe(NCMe2) and 3iPrCN-HfMe(NCMeiPr) gave unresolved mixtures after heating their solutions in toluene-d8 at 100 °C.
Another notable exception was 2iPrCN-ZrMe(NCMeiPr), which, upon heating, was found to transform into 2-Zr(NCMeiPr)2, a product of double insertion of iPrCN into both Zr–Me bonds, which was obviously formed via nitrile extrusion from the former complex and isolated in 71% preparative yield (Scheme 7).
Molecular structures
In total, we have been able to obtain single crystals for seven complexes, representative of all variations of the ligand framework studied herein (Table 2). Structurally, the complexes can be divided into two groups: with chelate ligands of [C,N,N]- (1-HfMe2, 1-HfMe(NCMe2), and 2-Zr(NCMeiPr)2) and [N,N,N]-types (1iPrCN-HfMe(Cl/Br), 1iPrCN-HfMe(NCMeiPr), 1tBuCN-HfMe2, and 1PhCN-HfMe(NCMePh).
CMe2), 1iPrCN-HfMe(NCMeiPr), 1PhCN-HfMe(NCMePh) and 2-Zr(NCMeiPr)2
| Metric | 1-HfMe2 | 1iPrCN-HfMe(Cl/Br) | 1tBuCN-HfMe2 |
1-HfMe(NCMe2) |
1iPrCN-HfMe(NCMeiPr) |
1PhCN-HfMe(NCMePh) |
2-Zr(NCMeiPr)2 |
|---|---|---|---|---|---|---|---|
|
a M = Hf for 1-HfMe2, 1iPrCN-HfMe(Cl/Br), 1tBuCN-HfMe2, 1-HfMe(NCMe2), 1iPrCN-HfMe(NCMeiPr), and 1PhCN-HfMe(NCMePh) and M = Zr for 2-Zr(NCMeiPr)2.
|
|||||||
| d[Hf–C1] | 2.204(5) | 2.320(11) | 2.221(10) | 2.208(4) | 2.249(5) | 2.248(4) | — |
| d[Hf–C2] | 2.242(5) | — | 2.286(9) | — | — | — | — |
| d[Ma–C3] | 2.271(5) | — | — | 2.285(3) | — | — | 2.316(4) |
| d[Ma–N1] | 2.328(4) | 2.335(8) | 2.434(7) | 2.362(3) | 2.404(4) | 2.421(3) | 2.341(3) |
| d[Ma–N2] | 2.105(4) | 2.054(9) | 2.090(9) | 2.114(2) | 2.097(3) | 2.083(3) | 2.114(3) |
| d[Hf–N3] | — | 2.034(8) | 2.034(9) | — | 2.053(4) | 2.068(3) | — |
| d[Ma–N4]/d [Ma–N5] | — | — | — | 1.979(3) | 1.997(4) | 1.998(3) | 2.023(4)/1.999(3) |
| d[C3–C4] | — | 1.496(15) | 1.526(14) | — | 1.510(6) | 1.508(5) | — |
| d[N3–C4] | — | 1.273(13) | 1.250(12) | — | 1.267(6) | 1.267(5) | — |
| d[N4–C5]/d [N5–C6] | — | — | — | 1.259(4) | 1.257(6) | 1.272(5) | 1.262(5)/1.247(10) |
| ∠[Hf–N3–C4] | — | 134.5(8) | 142.0(8) | — | 136.3(3) | 138.9(3) | — |
| ∠[Ma–N4(N5)–C5(C6)] | — | — | — | 169.8(3) | 174.8(4) | 168.6(3) | 173.3(3)/168.4(6) |
All seven complexes share the pyridine-Y-N-diisopropylphenyl moiety (where Y = SiMe2 or CMe2) whose geometrical parameters fall in the range known for Zr and Hf complexes containing the same fragments (ca. 2.25–2.45 Å for M–N1 and 2.05–2.15 Å for M–N2, Fig. 5)33,34,67,68 and differ insignificantly between the two groups of the chelate ligands. Although NMR spectra of the symmetrically substituted [C,N,N]-complexes 1-HfMe2 and 2-Zr(NCMeiPr)2 exhibit Cs-symmetry in solution, the geometry of the coordination surrounding the metal in the solid-state is best described as a distorted square pyramid. Insertion of nitrile results in expansion of the 5-membered metallacycle containing the CAr–M bond (C3–M in 1-HfMe2, 1-HfMe(NCMe2) and 2-Zr(NCMeiPr)2), Fig. 5) to the 7-membered metallacycle, and change in the coordination polyhedron to a distorted trigonal bipyramid.
 | ||
| Fig. 5 Solid state molecular structure of 1tBuCN-HfMe2, 1-HfMe(NCMe2), 1iPrCN-HfMe(NCMeiPr), and 2-Zr(NCMeiPr)2 with ellipsoids drawn at the 50% probability level. The hydrogen atoms are removed for clarity. | ||
There are four types of metal–nitrogen bonds in the studied complexes (Table 2). The coordination bonds M–N1(pyridine) of 2.328(4)–2.434(7) Å are the longest. The shortest bonds are M–N4 (“linear” ketimides) of 1.979(3)–1.923(4) Å which are considerably shorter than M–N2(amide) bonds of 2.054(9)–2.114(3) Å. The bond shortening can be traced to a bond order higher than one. Indeed, sp-hybridization of the nitrogen atom in the ketimide is additionally witnessed by the large M–N4(N5)–C5(C6) angle of 168.4(6)–174.8(4)°. A similar geometry was observed previously for several other “linear” ketimides of group 4 metals.45,69–71 At the same time, Hf–N3 (“bent” ketimide) bonds of 2.034(9)–2.068(3) Å are notably longer than Hf–N4. In fact, Hf–N3 bond lengths have values that are in between those of Hf–N2 and Hf–N4 bonds. This may be related to the smaller Hf–N3–C4 angle of 134.5(8)–142.0(8)° which hinders donation of the N3-nitrogen lone pair into hafnium based orbitals. The M–N bond elongation on decreasing the M–N–C angle is typical for ketimides of group 4 metals and was reported for tethered45,72,73 and sterically hindered complexes74,75 previously. Lengths of double CN bonds (N3–C4, N4–C5 and N5–C6 in Table 2) of 1.250(12)–1.273(13) Å are virtually the same for “linear” and “bent” ketimides of Hf and Zr which is in line with previous observations for “linear” non-tethered ketimides (≈1.259 Å)69,71 and “bent” tethered ligands (≈1.261–1.269 Å).72,73
Conclusions
Reaction of pyridylamido hafnium complexes 1-HfCl2, 1-HfMe2, 2-HfMe2, 3-HfMe2 and 2-ZrMe2 with isobutyronitrile predominantly gives the products of the nitrile insertion into the Hf–CAr bond 1iPrCN-HfCl2, 1iPrCN-HfMe2, 2iPrCN-HfMe2, 3iPrCN-HfMe2 and 2iPrCN-ZrMe2. Surprisingly, these products were found to be unstable with respect to nitrile extrusion in solution already at room temperature. The observed reversible nitrile insertion represents the first example of β-aryl elimination in ketimides of early transition metals, previously reported only for complexes of Pd20 and Rh.22 Moreover, complexes 1iPrCN-HfCl2 and 1iPrCN-HfMe(NCMeiPr) are able to reversibly release isobutyronitrile in solution forming the well-defined equilibria (1iPrCN-HfCl2 ⇆ 1-HfCl2 + iPrCN and 1iPrCN-HfMe(NCMeiPr) ⇆ 1-HfMe(NCMeiPr) + iPrCN) which allowed us to study them in detail with NMR spectroscopy.
Analysis of the nitrile insertion and extrusion processes by DFT computations allowed us to conclude that the “bent” structure of ketimide plays a significant role in promoting the β-carbon elimination event in the described complexes but this is not the only prerequisite for this reactivity. While weak orbital overlap between N(ketimide) and metal and an unfavourable 7-membered metallacycle destabilize the product of insertion into the M–CAr bond, it is the backbone of the pyridylamide ligand that makes the reverse process viable. The pyridyl group linked with an amide fragment serves as a directing group to maintain proximity between the metal centre and the phenylene fragment required for Hf–CAr bond formation and, furthermore, assures the formation of the stable 5-membered metallacycle. The complexes described in the literature, which contain “bent” ketimide ligands,45,72,73 lack such a directing group, and as a result, they are unable to undergo β-carbon elimination.
Electronic properties of the spectator ligands markedly influence thermodynamic stability of the complexes as it was demonstrated for complexes 1iPrCN-HfCl2, 1iPrCN-HfMe2 and 1iPrCN-HfMe(NCMeiPr) in Table 1. Likely, electron withdrawing substituents on the metal increase the N(“bent” ketimide)–M bond energy and stabilize the complex. At the same time, substituents on the inserted nitrile were also found to be important. Thus, while complex 1tBuCN-HfMe2 with inserted tBuCN was isolated in good yield, complex 1MeCN-HfMe2 with inserted acetonitrile could only be traced in the NMR spectrum. Moreover, even though complexes 1iPrCN-HfMe(NCMeiPr) and 1PhCN-HfMe(NCMePh) have almost identical structural parameters (Table 2), 1iPrCN-HfMe(NCMeiPr) is in equilibrium with 1-HfMe(NCMeiPr) and iPrCN at 60 °C, whereas 1PhCN-HfMe(NCMePh) does not eliminate PhCN even at 100 °C, implying that the conjugation of the CN bond and phenyl group provides an additional stabilization of the inserted product.
The transformations reported in this work demonstrate for the first time that ketimides of Zr and Hf are able to undergo β-carbon elimination analogously to late transition metal complexes. Given the growing importance of such reactions in the field of catalysis for carbon–carbon bond activation, our study paves the path towards the application of cheap and earth abundant group 4 metals in these transformations in the future.
Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for 1iPrCN-HfMe(Cl/Br), 1-HfMe2, 1tBuCN-HfMe2, 1-HfMe(NCMe2), 1iPrCN-HfMe(NCMeiPr), 1PhCN-HfMe(NCMePh) and 2-Zr(NCMeiPr)2 have been deposited and are available free of charge from the Cambridge Crystallographic Data Center (CCDC No. 2301574–2301580) and can be obtained from https://www.ccdc.cam.ac.uk/.
Author contributions
PSK: investigation, writing original draft (organometallic part); GPG, ANY, and DYM: investigation (organometallic part); DVU: supervision (MSU team), writing – review & editing; CE: investigation and writing original draft (computational part); JAMC and JRH: conceptualization; AZV: project administration, funding acquisition, resources.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Partial financial support from ExxonMobil Technology and Engineering Company is gratefully acknowledged. The authors also thank the Ministry of Science and Higher Education for support of their work (project #121021000105-7). The authors are grateful to Dr K. A. Lyssenko for his assistance with the X-ray structure determination analysis. MS spectra were obtained using the Agilent Technologies 8890 GC/5977C MSD system of the Lomonosov Moscow State University Shared Research Equipment Centre “Technologies for obtaining new nanostructured materials and their complex study” and purchased by Lomonosov Moscow State University within Equipment Renovation Program (National Project “Science”). The authors express their gratitude towards Prof. Vincenzo Busico and Prof. Peter Budzelaar (University of Naples Federico II) for the gracious donation of computational time.Notes and references
- L. Souillart and N. Cramer, Chem. Rev., 2015, 115, 9410–9464 CrossRef CAS PubMed.
- Y. Xia and G. Dong, Nat. Rev. Chem., 2020, 4, 600–614 CrossRef CAS PubMed.
- G. Dong, Topics in Current Chemistry: C-C Bond Activation, 2014, vol. 346 Search PubMed.
- M. D. R. Lutz and B. Morandi, Chem. Rev., 2021, 121, 300–326 CrossRef CAS PubMed.
- F. Song, T. Gou, B. Q. Wang and Z. J. Shi, Chem. Soc. Rev., 2018, 47, 7078–7115 RSC.
- F. Chen, T. Wang and N. Jiao, Chem. Rev., 2014, 114, 8613–8661 CrossRef CAS PubMed.
- F. D. Cannavacciuolo, R. Yadav, A. Esper, A. Vittoria, G. Antinucci, F. Zaccaria, R. Cipullo, P. H. M. Budzelaar, V. Busico, G. P. Goryunov, D. V. Uborsky, A. Z. Voskoboynikov, K. Searles, C. Ehm and A. S. Veige, Angew. Chem., Int. Ed., 2022, 61, e202202258 CrossRef CAS PubMed.
- J. William Suggs and S. D. Cox, J. Organomet. Chem., 1981, 221, 199–201 CrossRef.
- R. H. Crabtree and R. P. Dion, J. Chem. Soc. Chem. Commun., 1984, 90, 1260–1261 RSC.
- R. A. Periana and R. G. Bergman, J. Am. Chem. Soc., 1986, 108, 7346–7355 CrossRef CAS.
- M. Gozin, A. Welsman, Y. Ben-David and D. Milstein, Nature, 1993, 364, 699–701 CrossRef CAS.
- C. Jun, Chem. Soc. Rev., 2004, 33, 610–618 RSC.
- M. Murakami and N. Ishida, Cleavage of Carbon-Carbon Single Bonds by Transition Metals, 2016, pp. 1–34 Search PubMed.
- A. D. Marchese, B. Mirabi, C. E. Johnson and M. Lautens, Nat. Chem., 2022, 14, 398–406 CrossRef CAS PubMed.
- K. F. Lee, W. Bai, H. H. Y. Sung, I. D. Williams, Z. Lin and G. Jia, Chem.–Eur. J., 2018, 24, 9760–9764 CrossRef CAS PubMed.
- P. Zhao and J. F. Hartwig, Organometallics, 2008, 27, 4749–4757 CrossRef CAS.
- G. Fumagalli, S. Stanton and J. F. Bower, Chem. Rev., 2017, 117, 9404–9432 CrossRef CAS PubMed.
- K. Ruhland, Eur. J. Org. Chem., 2012, 2683–2706 CrossRef CAS.
- T. Nishimura and S. Uemura, J. Am. Chem. Soc., 1999, 121, 11010–11011 CrossRef CAS.
- T. Nishimura, Y. Nishiguchi, Y. Maeda and S. Uemura, J. Org. Chem., 2004, 69, 5342–5347 CrossRef CAS PubMed.
- Y. Terao, H. Wakui, T. Satoh, M. Miura and M. Nomura, J. Am. Chem. Soc., 2001, 123, 10407–10408 CrossRef CAS PubMed.
- P. Zhao and J. F. Hartwig, J. Am. Chem. Soc., 2005, 127, 11618–11619 CrossRef CAS PubMed.
- P. Zhao, C. D. Incarvito and J. F. Hartwig, J. Am. Chem. Soc., 2006, 128, 3124–3125 CrossRef CAS PubMed.
- H. Li, B. Ma, Q. S. Liu, M. L. Wang, Z. Y. Wang, H. Xu, L. J. Li, X. Wang and H. X. Dai, Angew. Chem., Int. Ed., 2020, 59, 14388–14393 CrossRef CAS PubMed.
- J. F. Hartwig, R. G. Bergman and R. A. Andersen, Organometallics, 1991, 10, 3344–3362 CrossRef CAS.
- M. D. R. Lutz, V. C. M. Gasser and B. Morandi, Chem, 2021, 7, 1108–1119 CAS.
- M. E. O'Reilly, S. Dutta and A. S. Veige, Chem. Rev., 2016, 116, 8105–8145 CrossRef PubMed.
- J. Uddin, C. M. Morales, J. H. Maynard and C. R. Landis, Organometallics, 2006, 25, 5566–5581 CrossRef CAS.
- C. Sambiagio, D. Schönbauer, R. Blieck, T. Dao-Huy, G. Pototschnig, P. Schaaf, T. Wiesinger, M. F. Zia, J. Wencel-Delord, T. Besset, B. U. W. Maes and M. Schnürch, Chem. Soc. Rev., 2018, 47, 6603–6743 RSC.
- E. Ozkal, B. Cacherat and B. Morandi, ACS Catal., 2015, 5, 6458–6462 CrossRef CAS.
- H. Wang, I. Choi, T. Rogge, N. Kaplaneris and L. Ackermann, Nat. Catal., 2018, 1, 993–1001 CrossRef CAS.
- T. R. Boussie, G. M. Diamond, C. Goh, K. A. Hall, A. M. LaPointe, M. K. Leclerc, V. Murphy, J. A. W. W. Shoemaker, H. Turner, R. K. Rosen, J. C. Stevens, F. Alfano, V. Busico, R. Cipullo and G. Talarico, Angew. Chem., Int. Ed., 2006, 45, 3278–3283 CrossRef CAS PubMed.
- G. J. Domski, J. M. Eagan, C. De Rosa, R. Di Girolamo, A. M. LaPointe, E. B. Lobkovsky, G. Talarico and G. W. Coates, ACS Catal., 2017, 7, 6930–6937 CrossRef CAS.
- P. S. Kulyabin, D. V. Uborsky, A. Z. Voskoboynikov, J. A. M. Canich and J. R. Hagadorn, Dalton Trans., 2020, 49, 6693–6702 RSC.
- G. M. Diamond, K. A. Hall, A. M. Lapointe, M. K. Leclerc, J. Longmire, J. A. W. Shoemaker and P. Sun, ACS Catal., 2011, 1, 887–900 CrossRef CAS.
- P. S. Kulyabin, G. P. Goryunov, D. Y. Mladentsev, D. V. Uborsky, A. Z. Voskoboynikov, J. A. M. Canich and J. R. Hagadorn, Chem.–Eur. J., 2019, 25, 10478–10489 CrossRef CAS PubMed.
- J. R. Hagadorn, C. A. Faler and T. M. Boller, WO 2010/011435 A1, 2010.
- J. R. Hagadorn, R. N. Ganesh, D. V. Uborsky, I. S. Borisov, I. V. Pruss and A. Z. Voskoboynikov, WO 2012/134613 A2, 2012.
- R. D. J. Froese, P. D. Hustad, R. L. Kuhlman and T. T. Wenzel, J. Am. Chem. Soc., 2007, 129, 7831–7840 CrossRef CAS PubMed.
- C. Zuccaccia, A. Macchioni, V. Busico, R. Cipullo, G. Talarico, F. Alfano, H. W. Boone, K. A. Frazier, P. D. Hustad, J. C. Stevens, P. C. Vosejpka and K. A. Abboud, J. Am. Chem. Soc., 2008, 130, 10354–10368 CrossRef CAS PubMed.
- C. Zuccaccia, V. Busico, R. Cipullo, G. Talarico, R. D. J. Froese, P. C. Vosejpka, P. D. Hustad and A. Macchioni, Organometallics, 2009, 28, 5445–5458 CrossRef CAS.
- T. N. Valadez, J. R. Norton and M. C. Neary, J. Am. Chem. Soc., 2015, 137, 10152–10155 CrossRef CAS PubMed.
- J. Chen, N. Yassin, T. Gunasekara, J. R. Norton and M. Rauch, J. Am. Chem. Soc., 2018, 140, 8980–8989 CrossRef CAS PubMed.
- C. W. Frye, D. T. Egger, E. Kounalis, A. J. Pearce, Y. Cheng and I. A. Tonks, Chem. Sci., 2022, 13, 1469–1477 RSC.
- P. Ghana, S. Schrader, T. Rajeshkumar, T. P. Spaniol, U. Englert, L. Maron and J. Okuda, Angew. Chem., Int. Ed., 2021, anie.202103755 Search PubMed.
- M. T. Quirós, J. Angulo and M. P. Muñoz, Chem. Commun., 2015, 51, 10222–10225 RSC.
- J. Légaré Lavergne, A. Jayaraman, L. C. Misal Castro, É. Rochette and F. G. Fontaine, J. Am. Chem. Soc., 2017, 139, 14714–14723 CrossRef PubMed.
- S. Liu, M. P. Conley and R. R. Schrock, Organometallics, 2023, 42, 1087–1093 CrossRef CAS.
- C. Ehm, P. H. M. Budzelaar and V. Busico, J. Organomet. Chem., 2015, 775, 39–49 CrossRef CAS.
- E. N. T. Cuthbert, A. Vittoria, R. Cipullo, V. Busico and P. H. M. Budzelaar, Eur. J. Inorg. Chem., 2020, 2020, 541–550 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman, and D. J. Fox, Gaussian 16, revision A.03, Gaussian, Inc., Wallingford, CT, 2016 Search PubMed.
- P. H. M. Budzelaar, J. Comput. Chem., 2007, 28, 2226–2236 CrossRef CAS PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- E. D. Glendening, A. E. Reed, J. E. Carpenter and F. Weinhold, NBO Version 3.1, University of Wisconsin, Madison, WI, USA, 1998 Search PubMed.
- J. Tao, J. P. Perdew, V. N. Staroverov and G. E. Scuseria, Phys. Rev. Lett., 2003, 91, 146401 CrossRef PubMed.
- D. Figgen, K. A. Peterson, M. Dolg and H. Stoll, J. Chem. Phys., 2009, 130, 164108 CrossRef PubMed.
- B. P. Pritchard, D. Altarawy, B. Didier, T. D. Gibson and T. L. Windus, J. Chem. Inf. Model., 2019, 59, 4814–4820 CrossRef CAS PubMed.
- P. Schwerdtfeger, ChemPhysChem, 2011, 12, 3143–3155 CrossRef CAS PubMed.
- J. L. Whitten, J. Chem. Phys., 1973, 58, 4496–4501 CrossRef CAS.
- E. J. Baerends, D. E. Ellis and P. Ros, Chem. Phys., 1973, 2, 41–51 CrossRef CAS.
- M. Feyereisen, G. Fitzgerald and A. Komornicki, Chem. Phys. Lett., 1993, 208, 359–363 CrossRef CAS.
- O. Vahtras, J. Almlöf and M. W. Feyereisen, Chem. Phys. Lett., 1993, 213, 514–518 CrossRef CAS.
- G. Knizia, J. Chem. Theory Comput., 2013, 9, 4834–4843 CrossRef CAS PubMed.
- G. Knizia and J. E. M. N. Klein, Angew. Chem., Int. Ed., 2015, 54, 5518–5522 CrossRef CAS PubMed.
- J. S. Steen, G. Knizia and J. E. M. N. Klein, Angew. Chem., Int. Ed., 2019, 58, 13133–13139 CrossRef CAS PubMed.
- G. S. Hammond, J. Am. Chem. Soc., 1955, 77, 334–338 CrossRef CAS.
- K. A. Frazier, R. D. Froese, Y. He, J. Klosin, C. N. Theriault, P. C. Vosejpka, Z. Zhou and K. A. Abboud, Organometallics, 2011, 30, 3318–3329 CrossRef CAS.
- B. Han, Y. Liu, C. Feng, S. Liu and Z. Li, Organometallics, 2021, 40, 242–252 CrossRef CAS.
- G. Erker, W. Frömberg, J. L. Atwood and W. E. Hunter, Angew. Chem., Int. Ed. Engl., 1984, 23, 68–69 CrossRef.
- G. Erker, W. Frömberg, C. Krüger and E. Raabe, J. Am. Chem. Soc., 1988, 110, 2400–2405 CrossRef CAS.
- D. R. Armstrong, Organometallics, 2000, 19, 4369–4375 CrossRef CAS.
- M. Večeřa, V. Varga, I. Císařová, J. Pinkas, P. Kucharczyk, V. Sedlařík and M. Lamač, Organometallics, 2016, 35, 785–798 CrossRef.
- H. Ebert, V. Timmermann, T. Oswald, W. Saak, M. Schmidtmann, M. Friedemann, D. Haase and R. Beckhaus, Organometallics, 2014, 33, 1440–1452 CrossRef CAS.
- T. Zippel, P. Arndt, A. Ohff, A. Spannenberg, R. Kempe and U. Rosenthal, Organometallics, 1998, 17, 4429–4437 CrossRef CAS.
- T. Janssen, R. Severin, M. Diekmann, M. Friedemann, D. Haase, W. Saak, S. Doye and R. Beckhaus, Organometallics, 2010, 29, 1806–1817 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2301574–2301580. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc02173h |
| ‡ Present address: EaStCHEM, School of Chemistry, University of St Andrews, North Haugh, St Andrews, KY16 9ST, UK. |
| This journal is © The Royal Society of Chemistry 2024 |