
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence2H-Thiazolo[4,5-d][1,2,3]triazole: synthesis, functionalization, and application in scaffold-hopping†
Ryuya
Miyazaki
 a,
Fumito
Takada‡
a,
Takunari
Kikuchi‡
a,
Yuya
Oguro
b,
Makoto
Kamata
b,
Takafumi
Yukawa
b,
Kenta
Kato
a,
Kei
Muto
*c and
Junichiro
Yamaguchi
*a
a,
Fumito
Takada‡
a,
Takunari
Kikuchi‡
a,
Yuya
Oguro
b,
Makoto
Kamata
b,
Takafumi
Yukawa
b,
Kenta
Kato
a,
Kei
Muto
*c and
Junichiro
Yamaguchi
*a
aDepartment of Applied Chemistry, Waseda University, 513 Wasedatsurumakicho, Shinjuku, Tokyo 162-0041, Japan. E-mail: junyamaguchi@waseda.jp
bTakeda Pharmaceutical Company Limited, 2-26-1 Muraoka-Higashi, Fujisawa, Kanagawa 251-8555, Japan
cInstitute of Transformative Bio-molecules, Nagoya University, Furo-cho, Chikusa, Nagoya 464-8601, Japan. E-mail: muto.kei.v4@f.mail.nagoya-u.ac.jp
First published on 3rd September 2024
Abstract
This manuscript unveils the synthesis of 2H-thiazolo[4,5-d][1,2,3]triazole (ThTz), an unprecedented [5-5]-fused heteroaromatic system, and established a scalable synthetic procedure for producing large quantities of the ThTz ring bearing a sulfone group on the thiazole ring. The sulfone moiety proves to be a versatile reactive tag, facilitating diverse transformations such as SNAr reactions, metal-catalyzed couplings, and radical-based alkylations. Furthermore, functionalization of the triazole ring highlights the potential of this newly developed heteroaromatic compound as a valuable heteroaryl building block, promoting scaffold hopping strategies in medicinal chemistry.
Introduction
Heteroarenes have long been celebrated for their pivotal role in synthetic chemistry, serving as the backbone for groundbreaking developments in medicinal, agrochemical, and materials chemistry. Their allure in medicinal chemistry stems from the inclusion of heteroatoms within rigid aromatic rings, which imbue compounds with desirable properties like enhanced hydrophilicity, hydrogen bonding potential, and improved pharmacokinetics.1 These attributes are crucial for medicinal chemists striving to enhance protein binding, boost target selectivity, and innovate in fragment-based drug design. Additionally, the structural diversity and modifiability of heteroarenes make them invaluable in agrochemical design for developing potent and selective pesticides and fungicides with favorable environmental properties.2Among the diverse landscape of aromatic configurations, [5-5]-bicyclic heteroarenes stand out and have garnered significant attention within the pharmaceutical and agrochemical industries (Fig. 1A). Notable pharmaceutical examples include AstraZeneca's GPi688, which utilizes the 6H-thieno[2,3-b]pyrrole scaffold to inhibit glycogen phosphorylase,3 and Merck's samatasvir, which incorporates thieno[3,2-b]thiophene in its quest to inhibit NS5A.4 Imidazo[2,1-b]thiazole has also been embedded in bioactive molecules like PF-5190457 5 and SRT2104,6 showcasing its utility in drug development. In agrochemistry, these scaffolds are engineered to target specific enzymes or pathways in pests, enhancing the efficacy and prolonging the efficacy offered by agrochemical products.
 | ||
| Fig. 1 (A) [5-5]-Bicyclic heteroarenes in bioactive compounds. (B) 2H-Thiazolo[4,5-d][1,2,3]triazole (ThTz): an innovative and previously unexplored aromatic system. (C) Sulfonylated ThTz (1): synthesis, functionalization, and application in scaffold-hopping. | ||
In this study, we delve into the realm of [5-5]-bicyclic heteroarenes, particularly focusing on thiazole-fused systems, given thiazole's prominence as a privileged structure in medicinal chemistry. Thiazoles are celebrated not only for their pharmacological benefits but also for their robustness and functional versatility, which make them ideal candidates for agrochemical applications where environmental stability and broad-spectrum activity are required.7
Our attention is captivated by 2H-thiazolo[4,5-d][1,2,3]triazole (ThTz), an innovative and previously unexplored aromatic system that brings together the thiazole and triazole rings—both of which are known for their remarkable chemical stability and biological relevance (Fig. 1B). Triazoles are particularly noted for their bioisosteric replacement of amide functionality and their capacity to alter compound architecture through substitution at various nitrogen positions, offering a vast landscape for chemical exploration and optimization. This attribute enables the design of molecules with enhanced metabolic stability and improved binding characteristics, crucial for developing more effective pharmaceuticals and agrochemicals.
The design of ThTz was guided by its potential as a versatile building block, facilitating assembly with various molecular fragments. We introduced a sulfone group on the ThTz scaffold, enhancing its utility as a leaving group and thus increasing its versatility in synthetic applications (Fig. 1C).8 Herein, we report the synthesis of C5-sulfonylated ThTz (1) and its successful deployment as a robust building block. With a high scalability in synthesis and high applicability as a heteroarene building block, 1 has been commercialized through Tokyo Chemical Industry Ltd (TCI: catalogue number M3881).
Results and discussion
Retro-synthetically, we envisioned that heteroarene 1 could be synthesized through S-alkylation and oxidation of thione 2 (Scheme 1A). The synthesis of 2, in turn, was envisaged via the annulation of bromoaminotriazole 3 with a CS2 equivalent. In line with this retro-synthetic analysis, we first embarked on the preparation of bromoaminotriazole 3 (Scheme 1B). The N-alkylation of readily prepared dibromotriazole 49 with p-methoxybenzyl (PMB) chloride afforded PMB-triazole 5 in 70% yield along with its positional isomer in 21% yield, both of which could be separated by silica-gel purification. The reason for the preference for alkylation at the N2 position is the steric hindrance caused by the bromo group.10 Subsequently, an attempt to directly install an NH2 group from the obtained 5 using NH3 under copper-catalyzed conditions led to diamination, proving unsuccessful for our targeted monoamination (Scheme 1C, see the ESI†).11 We envisaged that the use of a bulky NH2-surrogate could suppress the undesired diamination. Pursuing this approach, we employed palladium-catalyzed conditions with benzophenoneimine,12 followed by an acidic workup to achieve the desired monoamination, delivering bromoaminotriazole 3 in 60% yield with a tiny amount of diamine byproduct (<5% yield). On the other hand, the amination did not proceed at all for the isomer of 5. It is clear that the reactivity of the triazole is significantly influenced by the position of the substituent. With the desired 3 in hand, we next aimed to construct the thiazole ring. Although the utilization of CS2 as well as isothiourea resulted in failure, potassium ethylxanthate under the influence of a copper catalyst gave thiocarbonyl 2 through a carbonodithioate intermediate 6, which spontaneously cyclized.13 The final steps involved S-methylation of 2, followed by oxidation with mCPBA, which furnished the desired novel heteroaromatic compound 1 as a stable solid. This synthetic route proved amenable to scale-up, allowing for the production of up to 5.7 g of 1 in a single batch.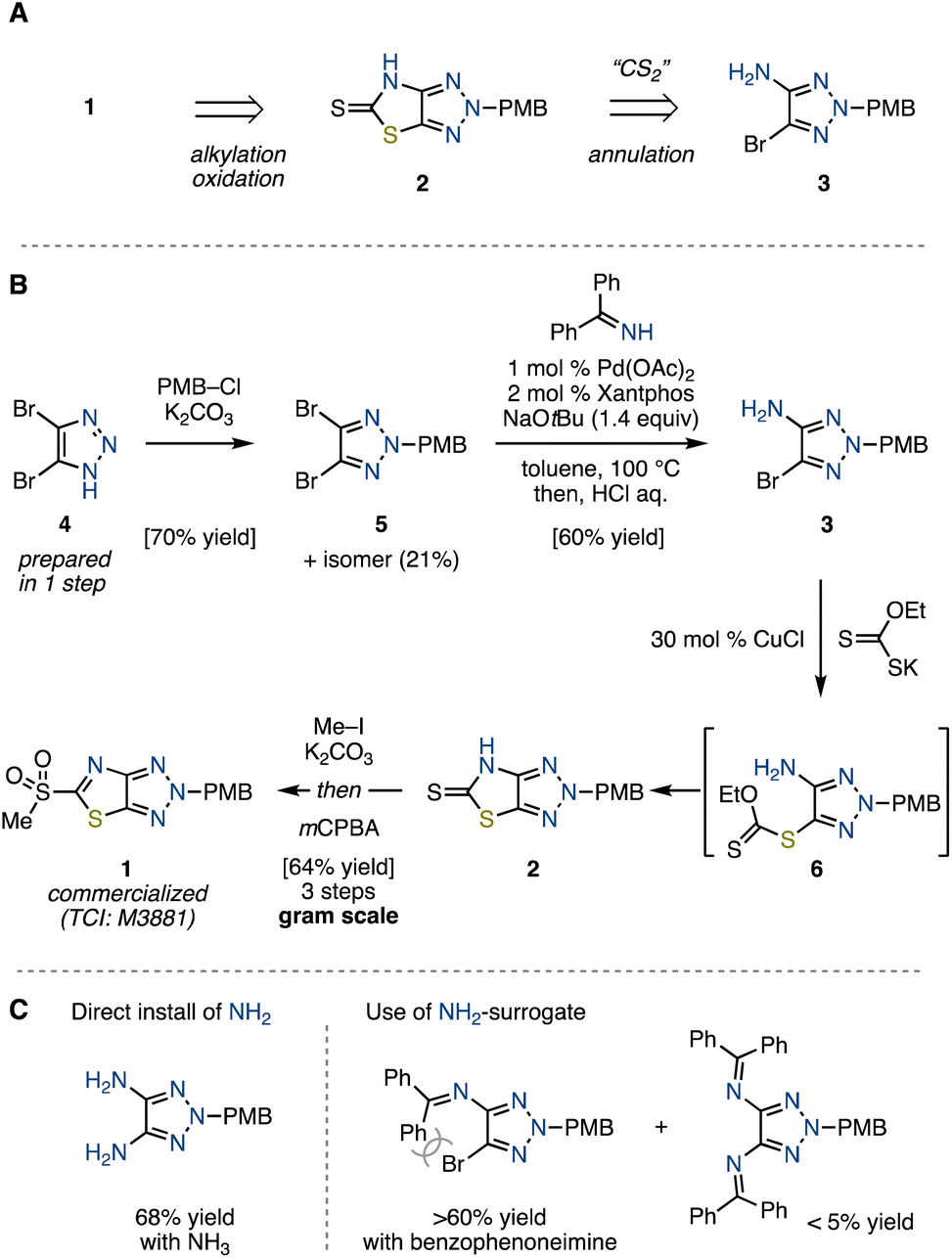 | ||
| Scheme 1 (A) Retrosynthetic analysis of sulfonylated ThTz (1). (B) Synthesis of 1. (C) Amination of PMB-triazole 5. | ||
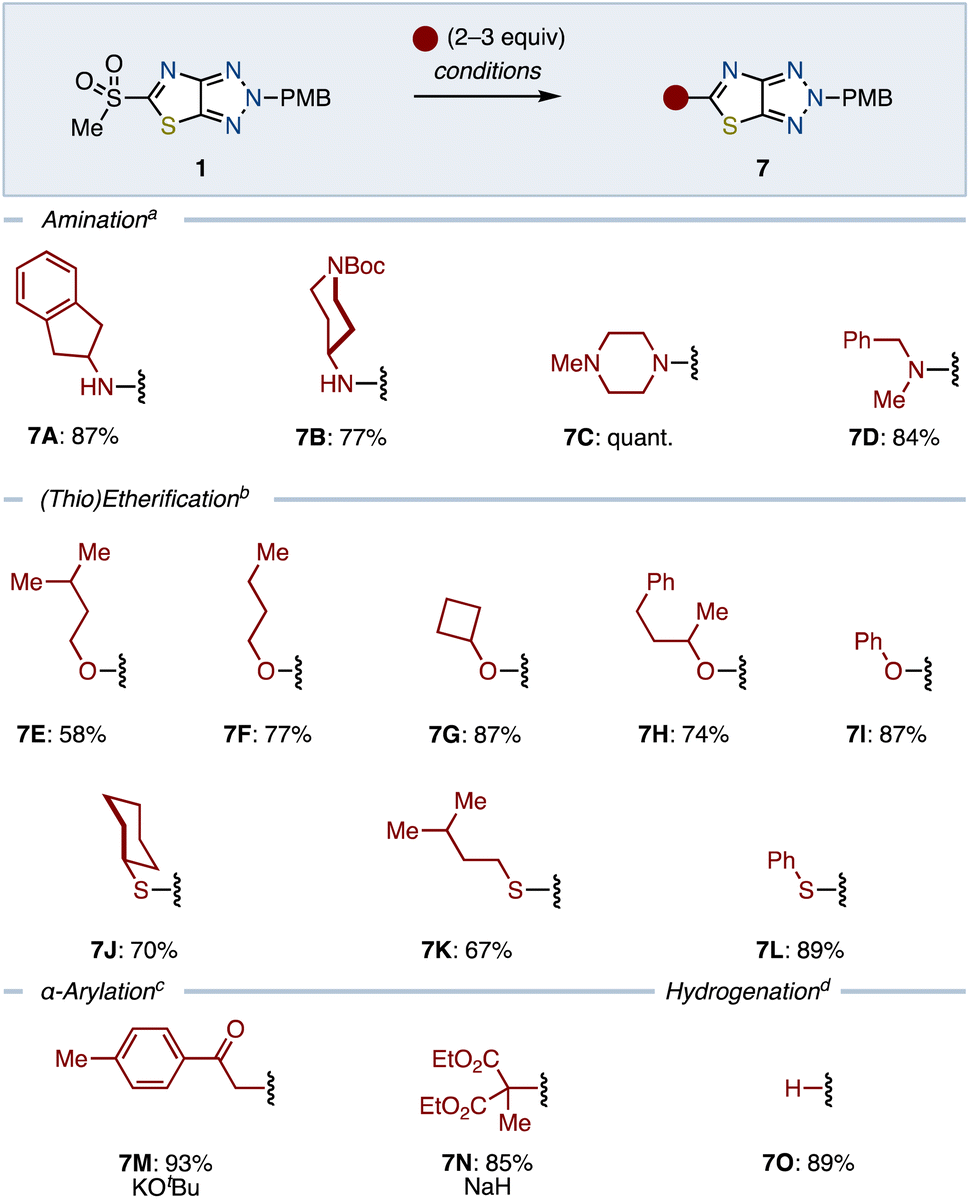
To readily introduce a variety of substituents, we next embarked on functionalizing the sulfone moiety of 1 (Scheme 2). We initiated this process by exploring the SNAr reaction capabilities with various amines in THF at 60 °C. Amination using both primary and secondary amines proceeded smoothly to give aminated ThTz 7A–7D generally in good yields. Etherification using alcohol in the presence of NaH in THF was also found to be applicable. The use of primary and secondary alcohols as well as phenol efficiently provided the corresponding heteroaryl ethers (7E–7I). However, less nucleophilic tertiary alcohol furnished the product in a low yield (<20%, see the ESI†).
 | ||
| Scheme 2 Functionalization of 1 by the SNAr reaction. aConditions: amine (2.0 equiv.), THF, 60 °C. bConditions: alcohol or thiol (2.0 equiv.), NaH (2.0 equiv.), THF, 0 °C. cConditions: carbonyl compound, base, RT. dConditions: NaBH4 (2.0 equiv.), MeOH/THF, 0 °C. | ||
The versatility of the sulfone functionality of 1 was further demonstrated through its reactivity with thiols under the same conditions, leading to a range of thioethers 7J–7L in moderate to good yields. Enolizable nucleophiles were also accommodated, synthesizing 7M and 7N by using the corresponding ketone and ester in the presence of a base. Moreover, removal of the sulfonyl group with NaBH4 smoothly afforded 7O in a good yield.
Further explorations into C–C bond formations such as arylation and alkylation using 1 were conducted. However, attempts using organometallic reagents such as organolithiums and organocuprates unfortunately resulted in failure (see the ESI†). Expecting high reactivity of the sulfone moiety toward transition metal catalysts,8 we decided to use metal-catalyzed cross-couplings (Scheme 3A). As a result, we found that conditions with the Pd(PPh3)4 catalyst and copper thiophene-2-carboxylate (CuTC)14 realized cross-coupling between 1 and p-tolylboronic acid, furnishing 7P in 51% yield. Likewise, we uncovered that Sc(OTf)315 can catalyze coupling of 1 with N-methylindole, which gave 7Q in 92% yield. However, these two metal-catalyzed protocols given above were not applicable for alkylations. Focusing on the reactivity of sulfones toward radical ipso-substitution, we next explored radical-based reactions (Scheme 3B).16 Guided by Baran's protocol,17 we identified that Fe-catalyzed coupling of 1 with alkenes in the presence of silane can provide alkylating products 7R and 7S. This protocol allowed for constructing a quaternary carbon center, albeit in a low yield of 25% (7S). Furthermore, it was found that photoredox-catalyzed aminoalkylation was possible. Under the photo-irradiated conditions with a photosensitizer,181 and N,N-dimethylcyclohexylamine reacted to provide 7T in 79% yield.
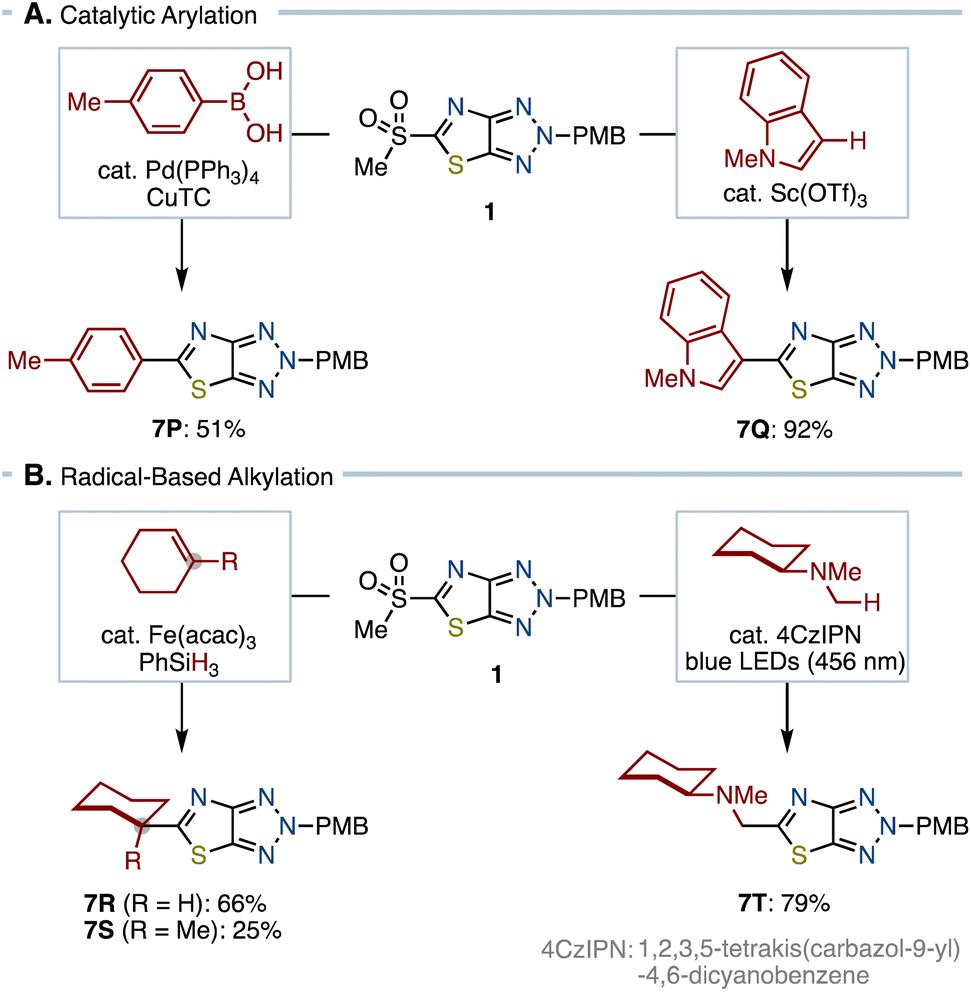 | ||
| Scheme 3 Arylation and alkylation of 1. | ||
Overall, the incorporation of a sulfone group on the thiazole moiety of 1 enabled a uniquely diverse range of molecular assemblies in generally good yields. This distinct reactivity, compared to that of other functional groups, highlights the potential of sulfone-functionalized thiazoles as versatile building blocks in synthetic chemistry, offering substantial flexibility for complex molecule construction.
Upon the success in installing a wide range of functional groups on the thiazole moiety, our next interest was aimed at the functionalization of the triazole ring (Scheme 4). Using 7D as a representative substrate, we first removed the PMB group using TFA to synthesize unprotected ThTz 8. Subsequently, the SNAr reaction of 8 with an electron-deficient dinitrofluorobenzene delivered heterobiaryl 9A in 80% yield with a remarkable regioselectivity. X-ray crystallographic analysis of 9A proved that this SNAr reaction proceeded at the N2 position selectively. Similarly, 2-chloropyridine was able to react with 8 to furnish 9B selectively in an excellent yield. Alkylation of the triazole moiety was also possible by conducting 1,4-addition of cyclohexenone, providing 9C. In this case, the N2-alkylated product was obtained as a major product, but the other positional isomers were also generated (N2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :N1:N3 = 82:10:8). The coupling reaction with aryl halides using a Pd catalyst did not proceed;19,20 however, the reaction with aryl iodonium salts was successful, yielding 9D in a moderate yield.21 Furthermore, the Chan–Lam coupling, although providing 9E in a low yield and with low regioselectivity, demonstrated that the reaction could indeed proceed under these conditions.22 A simple SN2 reaction using alkyl electrophiles such as butyl tosylate and benzyl chloride delivered the corresponding alkylated products 9F and 9G, respectively. We currently assume that this N2-selectivity would be rationalized by the steric factor. Functionalization at the N1 and N3 positions was not preferred due to the steric repulsion between an electrophile and the thiazole ring (nitrogen and sulfur atoms, respectively). Thus, in certain arylation and alkylation reactions of 8, perfect regioselectivity was not attained. However, from a positive perspective, the generation of these positional isomers is advantageous for the synthesis of diverse derivatives and for scaffold hopping applications. Notably, our preliminary investigations indicate that alkylation under acidic conditions, albeit with low yields, is selective for the N1 position (see the ESI† for details).
:N1:N3 = 82:10:8). The coupling reaction with aryl halides using a Pd catalyst did not proceed;19,20 however, the reaction with aryl iodonium salts was successful, yielding 9D in a moderate yield.21 Furthermore, the Chan–Lam coupling, although providing 9E in a low yield and with low regioselectivity, demonstrated that the reaction could indeed proceed under these conditions.22 A simple SN2 reaction using alkyl electrophiles such as butyl tosylate and benzyl chloride delivered the corresponding alkylated products 9F and 9G, respectively. We currently assume that this N2-selectivity would be rationalized by the steric factor. Functionalization at the N1 and N3 positions was not preferred due to the steric repulsion between an electrophile and the thiazole ring (nitrogen and sulfur atoms, respectively). Thus, in certain arylation and alkylation reactions of 8, perfect regioselectivity was not attained. However, from a positive perspective, the generation of these positional isomers is advantageous for the synthesis of diverse derivatives and for scaffold hopping applications. Notably, our preliminary investigations indicate that alkylation under acidic conditions, albeit with low yields, is selective for the N1 position (see the ESI† for details).
 | ||
| Scheme 4 Functionalization on the triazole ring. | ||
These products 9 pose challenges in structural determination due to the lack of protons on the present heterocycle. In this regard, results from X-ray crystallographic analysis as well as two-dimensional NMR analyses such as HMBC indicated that the 13C NMR peaks at the C7 and C8 positions of this heterocycle could provide valuable information. As shown in Scheme 4, when the N1 position is functionalized, the 13C NMR peaks of C7 and C8 appear at 145–153 ppm and 134–143 ppm, respectively. For N2-substituted derivatives, these peaks are observed at 158–167 ppm and 136–149 ppm, while for N3-substituted derivatives, they are found at 158–168 ppm and 120–131 ppm, respectively. It is evident that structural determination via13C NMR obviates the need for X-ray crystallographic analysis (see the ESI† for details).
Finally, aiming to harness the practical utility of our heteroaryl building block in medicinal chemistry, we explored the replacement of the heteroaromatic ring in known bioactive compounds (Scheme 5). This approach, commonly referred to as scaffold-hopping, is a strategic method used in medicinal chemistry to alter bioactivity, pharmacokinetics, and stability.23 First, we focused on the triazolopyridine compound 10, which is known as a positive allosteric modulator (PAM) of the metabotropic glutamate 2 receptor (mGlu2).24 Replacement of the aromatic core of 10 was initiated by photo-irradiated radical alkylation of 1 with N-methyl-4-phenylpiperidine, synthesizing aminomethylated product 7U. Upon removal of the PMB group, we performed N-alkylation using cyclopropylmethyl chloride, giving N2-alkylated 11 along with its positional isomers. These were successfully separated by silica-gel chromatography. In another example, we pursued the heteroarene replacement of benoxaprofen (13) with anti-inflammatory activity.25 First, Suzuki–Miyaura-type arylation (7V), followed by removal of the PMB group provided heterobiaryl 15. Subsequent alkylation using 2-bromopropionic acid resulted in the desired benoxaprofen mimic compound 14. Although for scaffold hopping, the isomer substituted at the N1 position may also be more suitable in these examples, they illustrate the flexibility and potential of our heteroaryl building block for modifying and enhancing the properties of existing pharmacologically active compounds through scaffold-hopping, underscoring its value in the discovery and optimization of new therapeutic agents.
 | ||
| Scheme 5 Heteroarene replacement of bioactive molecules. | ||
Conclusions
In conclusion, we have developed an unprecedented heteroaromatic system 2H-thiazolo[4,5-d][1,2,3]triazole (ThTz). Our developed synthesis of sulfonylated ThTz 1 proved to be scalable, enabling extensive functionalization of both the thiazole and triazole moieties. This demonstrates that 1 can serve as a highly versatile building block. The commercialization of our synthesized sulfonylated ThTz 1 paves the way for its use in scaffold-hopping strategies, with practical applications anticipated in medicinal chemistry. We believe that this work will provide a valuable tool for researchers seeking to explore new realms of chemical space in drug development.Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for 9A and 11-N1 have been deposited at the CCDC under CCDC 2360991 and CCDC 2360992. Data sharing is not applicable to this article as no datasets were generated or analysed during the current study.Author contributions
Y. O., M. K., and T. Y. conceived the thiazolotriazole ring. K. M. and J. Y. designed the sulfonylated heterocycle and directed this project. R. M., F. T., and T. K. performed all experiments. K. K. performed X-ray crystallographic analysis. K. M. and J. Y. wrote the manuscript with feedback from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JSPS KAKENHI Grant Number JP21H05213 (Digi-TOS) (to J. Y.), JP21K05079 (to K. M.), and JP24K17670 (to K. K.). This work was partly supported by JST ERATO Grant Number JPMJER1901 (to J. Y.) The Materials Characterization Central Laboratory in Waseda University is acknowledged for the support of HRMS measurement. We thank Dr Bunnai Saito for fruitful discussions and flexible management to facilitate the collaborative research between Waseda University and Takeda Pharmaceutical Company Limited and Dr Yasushi Miyazaki for contribution to the alliance management between Waseda University and Takeda Pharmaceutical Company Limited.Notes and references
- W. R. Pitt, D. M. Parry, B. G. Perry and C. R. Groom, J. Med. Chem., 2009, 52, 2952–2963 CrossRef CAS PubMed.
- (a) P. J. Hajduk and J. A. Greer, Nat. Rev. Drug Discovery, 2007, 6, 211–219 CrossRef CAS PubMed; (b) C. W. Murray and D. C. Rees, Nat. Chem., 2009, 1, 187–192 CrossRef CAS PubMed.
- (a) P. R. O. Whittamore, M. S. Addie, S. N. L. Bennett, A. M. Birch, M. Butters, L. Godfrey, P. W. Kenny, A. D. Morley, P. M. Murray, N. G. Oikonomakos, L. R. Otterbein, A. D. Pannifer, J. S. Parker, K. Readman, P. S. Siedlecki, P. Schofield, A. Stocker, M. J. Taylor, L. A. Townsend, D. P. Whalley and J. Whitehouse, Bioorg. Med. Chem. Lett., 2006, 16, 5567–5571 CrossRef CAS PubMed; (b) S. Freeman, J. B. Bartlett, G. Convey, I. Hardern, J. L. Teague, S. J. G. Loxham, J. M. Allen, S. M. Poucher and A. D. Charles, Br. J. Pharmacol., 2006, 149, 775–785 CrossRef CAS PubMed.
- B. Vince, J. M. Hill, E. J. Lawitz, W. O'Riordan, L. R. Webster, D. M. Gruener, R. S. Mofsen, A. Murillo, E. Donovan, J. Chen, J. F. McCarville, J. Z. Sullivan-Bólyai, D. Mayers and X.-J. Zhou, J. Hepatol., 2014, 60, 920–927 CrossRef CAS PubMed.
- S. K. Bhattacharya, K. Andrews, R. Beveridge, K. O. Cameron, C. Chen, M. Dunn, D. Fernando, H. Gao, D. Hepworth, V. M. Jackson, V. Khot, J. Kong, R. E. Kosa, K. Lapham, P. M. Loria, A. T. Londregan, K. F. McClure, S. T. M. Orr, J. Patel, C. Rose, J. Saenz, I. A. Stock, G. Storer, M. VanVolkenburg, D. Vrieze, G. Wang, J. Xiao and Y. Zhang, ACS Med. Chem. Lett., 2014, 5, 474–479 CrossRef CAS PubMed.
- J. C. Milne, P. D. Lambert, S. Schenk, D. P. Carney, J. J. Smith, D. J. Gagne, L. Jin, O. Boss, R. B. Perni, C. B. Vu, J. E. Bemis, R. Xie, J. S. Disch, P. Y. Ng, J. J. Nunes, A. V. Lynch, H. Yang, H. Galonek, K. Israelian, W. Choy, A. Iffland, S. Lavu, O. Medvedik, D. A. Sinclair, J. M. Olefsky, M. R. Jirousek, P. J. Elliott and C. H. Westphal, Nature, 2007, 450, 712–716 CrossRef CAS PubMed.
- S. Cascioferro, B. Parrino, D. Carbone, D. Schillaci, E. Giovannetti, G. Cirrincione and P. Diana, J. Med. Chem., 2020, 63, 7923–7956 CrossRef CAS PubMed.
- J. Corpas, S.-H. Kim-Lee, P. Mauleón, R. G. Arrayás and J. C. Carretero, Chem. Soc. Rev., 2022, 51, 6774–6823 RSC.
- Y. Wu, D. Feng, M. Gao, Z. Wang, P. Yan, Z. Gu, Q. Guan, D. Zuo, K. Bao, J. Sun, Y. Wu and W. Zhang, Sci. Rep., 2017, 7, 17120 CrossRef PubMed.
- X.-J. Wang, L. Zhang, D. Krishnamurthy, C. H. Senanayake and P. Wipf, Org. Lett., 2010, 12, 4632–4635 CrossRef CAS PubMed.
- M. K. Elmkaddem, C. Fischmeister, C. M. Thomas and J.-L. Renaud, Chem. Commun., 2010, 46, 925–927 RSC.
- (a) J. P. Wolfe, J. Åhman, J. P. Sadighi, R. A. Singer and S. L. Buchwald, Tetrahedron Lett., 1997, 38, 6367–6370 CrossRef CAS; (b) G. Mann and J. F. Hartwig, J. Am. Chem. Soc., 1998, 120, 827–828 CrossRef CAS.
- L. Liu, N. Zhu, M. Gao, X. Zhao, L. Han and H. Hong, Phosphorus, Sulfur Silicon Relat. Elem., 2016, 191, 699–701 CrossRef CAS.
- V. Koolma, R. Staiger, M. Schühle, A. Bixenmann, E. Bauschatz, M. Schmid, F. M. Miloserdov and B. Herlé, Org. Lett., 2024, 26, 2852–2856 CrossRef CAS PubMed.
- M. Nambo, Z. T. Ariki, D. Canseco-Gonzalez, D. D. Beattie and C. M. Crudden, Org. Lett., 2016, 18, 2339–2342 CrossRef CAS PubMed.
- X.-Q. Chu, D. Ge, Y.-Y. Cui, Z.-L. Shen and C.-J. Li, Chem. Rev., 2021, 121, 12548–12680 CrossRef CAS PubMed.
- J. C. Lo, Y. Yabe and P. S. Baran, J. Am. Chem. Soc., 2014, 136, 1304–1307 CrossRef CAS PubMed.
- T. Zhang and H. Huang, Angew. Chem., Int. Ed., 2023, 62, e202310114 CrossRef CAS PubMed.
- S. Ueda, M. Su and S. L. Buchwald, Angew. Chem., Int. Ed., 2011, 50, 8944–8947 CrossRef CAS PubMed.
- P. Beletskaya, D. V. Davydov and M. Moreno-Mañas, Tetrahedron Lett., 1998, 39, 5621–5622 CrossRef.
- S. Roshandel, M. J. Lunn, G. Rasul, D. S. M. Ravinson, S. C. Suri and G. K. S. Prakash, Org. Lett., 2019, 21, 6255–6258 CrossRef CAS PubMed.
- (a) M. T. Chan, K. L. Monaco, R.-P. Wang and M. P. Winters, Tetrahedron Lett., 1998, 39, 2933–2936 CrossRef; (b) Y. S. Lam, C. G. Clark, S. Saubern, J. Adams, M. P. Winters, D. M. T. Chan and A. Combs, Tetrahedron Lett., 1998, 39, 2941–2944 CrossRef.
- (a) Y. Hu, D. Stumpfe and J. Bajorath, J. Med. Chem., 2017, 60, 1238–1246 CrossRef CAS PubMed; (b) T. B. Callis, T. R. Garrett, A. P. Montgomery, J. J. Danon and M. Kassiou, J. Med. Chem., 2022, 65, 13483–13504 CrossRef CAS PubMed.
- J. M. Cid, G. Tresadern, J. A. Vega, A. I. de Lucas, E. Matesanz, L. Iturrino, M. L. Linares, A. Garcia, J. I. Andrés, G. J. Macdonald, D. Oehlrich, H. Lavreysen, A. Megens, A. Ahnaou, W. Drinkenburg, C. Mackie, S. Pype, D. Gallacher and A. A. Trabanco, J. Med. Chem., 2012, 55, 8770–8789 CrossRef CAS PubMed.
- D. W. Dunwell, D. Evans, T. A. Hicks, C. H. Cashin and A. Kitchen, J. Med. Chem., 1975, 18, 53–58 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures and spectroscopic data for compounds including 1H and 13C NMR spectra. CCDC 2360991 (for 9A) and 2360992 (for 11-N1). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc03874f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |