
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTrimodal operation of a robust smart organic crystal†
Wenbo
Wu‡
a,
Kui
Chen‡
a,
Hui
Yu
a,
Jiaxuan
Zhu
a,
Yaoguang
Feng
a,
Jingkang
Wang
ab,
Xin
Huang
 ab,
Liang
Li
cd,
Hongxun
Hao
ab,
Ting
Wang
*ab,
Na
Wang
*ab and
Panče
Naumov
*cefg
ab,
Liang
Li
cd,
Hongxun
Hao
ab,
Ting
Wang
*ab,
Na
Wang
*ab and
Panče
Naumov
*cefg
aNational Engineering Research Center of Industrial Crystallization Technology, School of Chemical Engineering and Technology, Tianjin University, Tianjin 300072, China. E-mail: wang_ting@tju.edu.cn; wangna224@tju.edu.cn
bChina State Key Laboratory of Chemical Engineering, Tianjin University, 300072, China
cSmart Materials Lab, New York University Abu Dhabi, PO Box 129188, Abu Dhabi, UAE. E-mail: pance.naumov@nyu.edu
dDepartment of Sciences and Engineering, Sorbonne University Abu Dhabi, PO Box 38044, Abu Dhabi, UAE
eCenter for Smart Engineering Materials, New York University Abu Dhabi, PO Box 129188, Abu Dhabi, UAE
fResearch Center for Environment and Materials, Macedonian Academy of Sciences and Arts, Bul. Krste Misirkov 2, MK-1000 Skopje, Macedonia
gDepartment of Chemistry, Molecular Design Institute, New York University, 100 Washington Square East, New York, NY 10003, USA
First published on 13th May 2024
Abstract
We describe a dynamic crystalline material that integrates mechanical, thermal, and light modes of operation, with unusual robustness and resilience and a variety of both slow and fast kinematic effects that occur on very different time scales. In the mechanical mode of operation, crystals of this material are amenable to elastic deformation, and they can be reversibly morphed and even closed into a loop, sustaining strains of up to about 2.6%. Upon release of the external force, the crystals resume their original shape without any sign of damage, demonstrating outstanding elasticity. Application of torque results in plastic twisting for several rotations without damage, and the twisted crystal can still be bent elastically. The thermal mode of operation relies on switching the lattice at least several dozen times. The migration of the phase boundaries depends on the crystal habit. It can be precisely controlled by temperature, and it is accompanied by both slow and fast motions, including shear deformation and leaping. Parallel boundaries result in a thermomechanical effect, while non-parallel boundaries result in a thermosalient effect. Finally, the photochemical mode of operation is driven by isomerization and can be thermally reverted. The structure of the crystal can also be switched photochemically, and the generation of a bilayer induces rapid bending upon exposure to ultraviolet light, an effect that further diversifies the mechanical response of the material. The small structural changes, low-energy and weak intramolecular hydrogen bonds, and shear deformation, which could dissipate part of the elastic energy, are considered to be the decisive factors for the conservation of the long-range order and the extraordinary diversity in the response of this, and potentially many other dynamic crystalline materials.
1. Introduction
Being equivalent to human sensing organs for visual, olfactory, audial, and gustative senses, artificial sensing materials and devices play a critical role in all intelligent systems as the primary point and the main method in the process of data collection. An optimal smart sensing device is precise, small, well-integrated, and can be networked with other components.1,2 However, multifunctionality increases the number of internal components required to sense different stimuli and, therefore, requires a larger volume, which is disparate from the intended miniaturization of sensors for implementation in mobile smart devices, which requires reduced overall volume.3 Multisensing devices incorporate multiple sensing units having different sensing functionalities, require complex, costly fabrication protocols, and suffer from difficulties with the physical or operational integration of the individual components.4 Moreover, multifunctionality of the sensors is only advantageous if it is coupled with reduced size and volume for portable applications, high accuracy for reliable detection, ease of integration with other components, and low power consumption and fabrication cost.5 The methods available for the preparation of multicomponent sensors are currently only available for demonstration at a research scale and are not available at an industrial scale.6 Traditional methods cannot be used, while the multiple sensing abilities require multiple sensing components that are prepared separately and sometimes difficult to integrate into an integrated, durable device.The current approach to respond to these challenges is to turn to the use of multifunctional materials where a single sensing material is capable of detecting and responding linearly to multiple stimuli. In the age of molecular engineering materials, adaptive molecular crystals7 are emerging as such materials that could mechanistically replicate some kinematic traits of biological systems.8 From the approaches that are currently available to change the morphology of an object, it has been demonstrated that molecular crystals or their composites can respond to practically all external effectors, including light,9–12 heat,13–19 force,20–24 solvent,25 chemical agents,26 and humidity.27 Having nearly perfectly ordered and anisotropic supramolecular structures, the well-defined molecular arrangement in dynamic molecular crystals facilitates direct, atomic-scale observation of the dynamic structure at work. In crystals, the concerted motion of molecules in an ordered structural setting offers the opportunity to amplify the changes that occur at a molecular level to tangible kinematic effects that are expressed on a macroscopic scale, oftentimes causing readily observable changes of morphology or deformation of crystals, that could be accompanied by kinematic effects akin to some biological gaits of locomotion, such as jumping, crawling, and walking.28–31 Moreover, the transfer of momentum across the directional intermolecular interactions and molecular migration available via the weak interactions could elicit reversibility of the dynamic processes, and even recuperation abilities such as self-healing.32–36 Outside of the biomimetic context, some of these dynamic crystals could be considered single-material replicas of energy-harvesting devices, soft robots, or artificial muscles that come with high energy conversion ratio, work output, and fast response, while they are also very light in weight.37,38
Among the prerequisites towards the design of actual devices that could range in complexity from simple actuators to complex autonomous soft robots, controllability, reversibility and fatigueless operation during the motion and deformation are not only required, but also are essential assets for any practical application.39 Of these, the question of control over the kinematics is crucial, as it could enable reliable operation of the device where the response is related to the stimulus in a reproducible, predictable and reliable manner, much alike muscular motoric functions. However, the quest for a robust molecular single-crystal machine with controllable motions is a visionary, yet—at least at the present stage of maturity of this field—an arduous pursuit. Regardless of the stimulus used, the mechanical effects that are related to sufficiently large deformations, such as bending or twisting, are usually slower than those that result in small strokes. Faster actuation that occurs on a sub-millisecond scale, on the other hand, similar to that available with martensitic transitions, is prohibitive for efficient elastic energy dissipation, resulting in disintegration that is only suitable for single-event (single stroke) operations. There have been attempts to integrate orthogonal functionalities into one single material by applying crystal engineering approaches, such as cocrystallization, coordination polymers, and hybridization,40–42 however at present, the success of such approaches is unpredictable,43 and the actual design aspect can be hardly reliably extended to the mechanical properties. This is partly due to fact that the structure–activity relationships between molecular arrangement, the strength and directionality of intermolecular interactions, and the mechanical properties remain unclear and hard to design.44
This conundrum could possibly be resolved by resorting to adaptive crystals capable of sensing multiple stimuli, whereby the effects of one stimulus could be accelerated, augmented or suppressed by the other to instigate gated operation, complex kinematics, or synergism. More importantly, the combined effects could compensate for the aforementioned challenges with complete control over the operation. Here, we report an exceedingly robust dynamic crystalline material that overcomes some of these challenges. The material, (E)-4-chloro-N′-(4-(trifluoromethoxy)benzylidene)benzohydrazide (CTBB), integrates mechanical response to heat, light, and mechanical force with unusual robustness in operation; for instance, in thermal mode of operation, crystals of this material retain their quality even after the structure has been switched several dozen times. The thermomechanical response relies on a phase transition where the migration of the phase boundaries can be precisely controlled by temperature, and it is accompanied by both slow and fast motions, including shear deformation and leaping. Most importantly, these effects that occur on very different time scales can be induced selectively by manipulating the migration of the phase boundary. Since the chemical structure includes a chromophore, the structure of the crystal can also be switched photochemically, and the generation of a bilayer induces rapid bending upon exposure to ultraviolet light, an effect that further diversifies the mechanical response of the material. Finally, the crystals are also mechanically flexible upon deformation, and they can be morphed into elastically or plastically bent or twisted morphologies, with notable retention of the elasticity over multiple cycles of deformation.
2. Results and discussion
2.1. Slow thermomechanical response
The molecule of CTBB (Fig. 1a) contains two aromatic rings that are connected by a flexible bridge that provides rotational flexibility. Differential scanning calorimetry (DSC) revealed that it has three polymorphs, I, II and III. The three forms of CTBB crystallize in the monoclinic P21/c space group, with two of them having one molecule in their asymmetric units, while form III has two molecules in the asymmetric unit. However, their lattice parameters are different (form I: a = 18.847(6) Å, b = 10.089(3) Å, c = 8.313(2) Å, and β = 90.9(8)°; form II: a = 19.031(16) Å, b = 9.539(8) Å, c = 8.813(7) Å, β = 93.7(3)°, and form III: a = 19.074(6) Å, b = 9.480(3) Å, c = 8.983(3) Å, and β = 94.741(10)°) (Table S1†). Form I can undergo a phase transition to form II at 374.3–385.8 K, and form II transforms to form III at 394.7–422.5 K (Fig. S3†). The transition between forms I and II is reversible, and form II reverts to form I upon cooling to 377.3–369.7 K (Fig. 1d and S4†). The reversible phase transition was further confirmed by the variable temperature powder X-ray diffraction. As shown in Fig. S5,† in the diffraction pattern of form II at 378.15 K, the characteristic peaks of (211), (120) and (400) of form I were obviously shifted, and the new peak of (202) of form II appeared. Moreover, the diffraction pattern was recovered after cooling down to room temperature. Additionally, if we take the DSC data (Fig. 1d) into consideration, together, these experiments indicate the reversible nature of the phase transition between forms I and II. Unlike most of the other thermomechanical crystals, which are related to wide thermal hysteresis (>10 K, Table S2†)13–15,18,45 the transition I ↔ II is related to a very narrow hysteresis of about 2–3 K (Fig. 1f and Movies 1–3†), which could be related to small changes in the molecular conformation and crystal lattices during the phase transition (vide infra). On the contrary, the transition of form II to form III, II → III, is irreversible; heating of the former to 396 K leads to form III that does not revert to form II by cooling (Fig. S6 and S7†). Upon heating from 374 to 378 K, the crystal of form I exerts shear deformation and a linear stroke of about 6% along its longest axis (Fig. S8†). The crystals are mechanically robust, and they retain their macroscopic structural integrity without any visible deterioration even after more than twenty heating/cooling cycles (Fig. 1e). As shown in Fig. 1f and h, the migration of the phase boundary during the transition I → II is clearly visible with hot-stage microscopy. The direction of progression of the phase boundary can be reversed by cooling, whereupon the crystal recovers its original shape (Fig. S9a and Movies 1–3†). | ||
| Fig. 1 Structure and thermal switching of CTBB between forms I and II. (a) Chemical structure of CTBB. (b and c) Optical microscopic images of parallelepiped (b) and trapezoid (c) crystals. (d) Two cycles in the DSC curves of CTBB crystals recorded at heating/cooling rate 10 K min−1 showing the reversibility of the process with small change in the peak temperature. (e) Cyclability of a good-quality single crystal over the phase transition monitored by the change of the crystal length under heating/cooling. The expansion and shrinkage of the crystal were recorded over 20 thermal cycles. (f) Progression of the phase transition observed by the migration of parallel phase boundaries induced by a temperature variation at a rate of 5 K min−1. (g) Schematic showing simplified mechanism of phase transition in a parallelepiped crystal. (h) Thermosalient effects of a crystal during a phase transition that occurs by migration of non-parallel phase boundaries induced by heating at a rate of 5 K min−1. (i) Schematic showing simplified mechanism of the phase transition in a trapezoid crystal. | ||
Unlike some fast phase transitions,46 we observed that the transition between forms I and II is slow, and the thermomechanical response of the transitioning crystal can be thermally controlled (Fig. S9†). Upon heating, two phase boundaries develop on the opposite termini of the crystal and progress towards each other, resulting in an expansion of the crystal (Fig. S8b and c†). At low heating rate (1 or 2 K min−1) the progression of the parallel phase boundaries can be precisely controlled by controlling the temperature. Their migration towards each other can be paused by stopping the heating or cooling (Movie 4†); if the temperature is kept constant (e.g., 375.40 K), the boundaries can be kept static over a prolonged period of time (Movie 4†). The motion of the two phase fronts can be restored and adjusted by additional heating or cooling (Fig. S9a, S9b, Movies 5 and 6†). The switching of the unit cell that accompanies the transition I → II is related to shortening of the crystallographic b axis by 5.5% and expansion of the c axis by 6.0% (Table S1†). The related changes in the crystal lattice generate a shear force along the edge of the phase boundaries, which inhibits the progression of the transition. However, additional heating or cooling can overcome the energy barrier generated by the lattice mismatch and ultimately drives the advancement of the phase boundary.47,48 In turn, the mode of temperature variation can be preset to control the expansion or the contraction of the crystal. Since the two polymorphs are expected to have different properties,49–53 we suggest that this thermal control over the phase transition could be applied for continuous, bidirectional actuation of other objects.
2.2. Fast thermomechanical response
Contrary to the slow migration of boundaries that results in crystal expansion, as shown in Fig. 1h–i, when the phase boundaries are not parallel to each other, the crystal undergoes a thermosalient effect,13 that is, it propels itself and leaps (Fig. S9c, Movies 7 and 8†). Our observations based on a large number of experiments showed that the orientation of phase boundaries is closely related to the facets that define the shape of the crystal. For parallelepiped crystals the transition occurs via migration of two phase boundaries that are parallel to each other (Fig. 1b, f and g), while in crystals with irregular or trapezoidal shape, the boundaries are not parallel (Fig. 1c, h and i and S9†). As shown in Fig. 1h–i, initially such non-parallel phase boundaries move forward rapidly at the heating rate of 5 K min−1. When they meet, they stop moving, and if the sample is heated further, above certain temperature the boundaries suddenly disappear and the crystal jumps off its original position. To examine the effect of heating/cooling rate on the thermomechanical response, a total of 30 crystals were heated or cooled at 1, 5, 10, 20 or 30 K min−1 (Fig. S10 and S11†). Most of the crystals having parallel phase boundaries underwent shear deformation instead of motion. Occasionally, at a heating rate of 5, 10 or 20 K min−1 some crystals showed splitting. At 30 K min−1, only 10% of the crystals jumped, and 24% split. When the phase boundaries were not parallel, almost all crystals jumped, except when the rate was 1 K min−1. These results are suggestive of two factors that contribute to the type of mechanical effect: the crystallographic planes along which the phase transition occurs and the heating rate.For crystals with parallel phase boundaries, the shear force caused by lattice distortion is parallel to the phase boundaries. As shown in Fig. S12a,† in this case, the parallel shear forces drive the advancement of the phase boundaries. The relationships between the two phases across the two boundaries are identical, and the two advancing phase fronts do not interfere with each other. Close to the point of contact, however, molecules at the phase boundaries cannot be transformed, since the two parallel, opposite shear forces offset each other, leaving a lattice gap in the crystal. Thus, the crystal responds to heating by slow shear deformation. Besides, the reverse phase transition (on cooling) starts in the middle of the crystal because of the incomplete phase transition at the lattice gap.47 On the contrary, when the non-parallel phase boundaries meet, the junction leaves a large area where crystal remains untransformed (Fig. S12b†). Although the molecular rearrangements at the crossed phase boundaries hinder each other, the crystal can still be transformed due to the synergistic effect of shear forces. This, however, requires additional energy, which results in temporary stagnancy of the phase boundary progression. Eventually, the energy barrier is exceeded, the remaining domain of form I is rapidly converted into form II, the stored elastic energy is released instantaneously, and this propels the crystal to jump.13
We noted that at lower or higher heating rates, such as 1 and 30 K min−1 the crystals usually exhibit only one type of thermomechanical behavior, that is, they undergo slow shearing or sudden jumping. At relatively higher heating rate, the rapid change in temperature facilitates the mechanical reconfiguration, causing more dramatic and violent mechanical response.19,54–56 Hence, at faster heating or cooling rate, the thermosalient effect occurs regardless of the mode of migration of the phase boundaries. The lattice parameters during the heating process were monitored, and the variation of lattice axis length as function of temperature in the range of 298–388 K (10 K increment) is plotted in Table S3† and Fig. 2a. Thermally induced expansion was observed up to the phase transition temperature. At 374.4 K, the length of the c axis increased by 6.07% and that of the b axis decreased by 5.20% (Fig. 2a and b). The abrupt anisotropic changes in the unit cell parameters are consistent with the occurence of the phase transition during heating, which provides energy for triggering of the rapid motion of the crystals. Based on these results, we propose that the difference in migration of the phase boundaries relative to each other is one of the reasons that accounts for the observation that crystals of the same compound are capable of different thermomechanical effects (for example, deformation, flipping, and jumping). In prospect, this variation in the mechanical response could be used as an approach to control the type of thermomechanical behaviors by selecting or controlling the crystal habit or by selecting crystals with favorable facets.
 | ||
| Fig. 2 Crystal structure and phase transition between forms I and II of CTBB. (a) Variation of the unit cell axes between 298 and 388 K. (b) Comparison of the unit cell parameters of forms I and II. (c) Crystal structure of form I (blue) and form II (red) viewed along the [001] direction. (d) Optical microscopic image of a partially transitioned single crystal with two phases. (e) Overlapped representation of the CTBB molecules of form I (blue) and II (red). The numbers are the dihedral angles between the aromatic rings. (f) Molecular packing motifs in the structures of form I (368 K, blue) and form II (380 K, red) in a partially transformed crystal. | ||
2.3. Thermomechanical switching of the crystals structure
The structures of forms I and II were determined by single crystal X-ray diffraction at 365 K and 380 K, respectively (Table S1†). The crystals of form I are monoclinic (P21/c, a = 18.847(6) Å, b = 10.089(3) Å, c = 8.313(2) Å, and β = 90.9(8)°). The CTBB molecules are stacked along the c axis, while the molecules are cross-stacked with each other when viewed from the (001) plane (Fig. 2c). The adjacent layers are stacked atop each other at a distance of 4.545 Å along the c axis via strong hydrogen bonds, N–H⋯O (D, d, θ = 2.868 Å, 2.025 Å, 166.2(9)°) and C–H⋯O (3.407 Å, 2.628 Å, 141.8(1)°) (Fig. 2f). The crystals of form II are also monoclinic (P21/c, a = 19.031(16) Å, b = 9.539(8) Å, c = 8.813(7) Å, β = 93.7(3)°), and the cross-stacked molecular arrangement from form I is preserved in its structure (Fig. 2c). From form I to from II, the distance between the adjacent sheets increases from 4.545 to 4.819 Å (Fig. 2f), and the dihedral angle between the aromatic rings decreases from 35.21° to 23.20° due to rotation of the aromatic rings around the C–C (Fig. 2e). This difference in crystal structures indicates the mechanism of the phase transition (Fig. S2a and S5†). Although the distance between the adjacent molecules in form II is relatively larger than that in form I, the hydrogen bond distances are shorter due to rotation around the central bond, with N–H⋯O (2.853 Å, 2.025 Å, 161.2(1)°) and C–H⋯O (3.334 Å, 2.555 Å, 141.5(3)°), indicating strengthening of the hydrogen bonds. In line with the above structural comparison, three factors might be taken to account for the reversibility of the phase transition I ↔ II: (1) the molecular packing patterns of the two polymorphs maintain a high degree of similarity. Small changes in the crystal structure during the phase transition could reduce the elastic strain so as to avoid the onset of disintegrative processes and destruction of the crystal, (2) the low-energy and weak intramolecular hydrogen bonds confer the soft properties of the crystals, which in turn render them adaptive to the changes in strain without damage, and (3) macroscopically, the phase transition occurs via motion of phase boundaries with small shear deformation, which could dissipate part of the elastic energy.2.4. Actuation performance
To assess the actuation performance of the CTBB crystals, the forces generated during the phase transition were measured and analyzed. The technical details of the force analysis setup are summarized in Fig. S13 and the Experimental section.† Briefly, a crystal was placed between two parallel glass plates on a flat substrate, and during the phase transition, the plate was displaced by the shear deformation (Fig. S10 and Movie 9†). The force exerted by the crystal was found to be about 1.1025 × 10−3 N, corresponding to about thousand times its gravitational force. The corresponding maximum force density (maximum force generated per unit volume) of the crystals was approximately 3.49 × 107 N m−3, a value that is comparable to forces obtained from electroactive polymers.16 In addition to the force, the work output is an important metric for actuation. It was found that during the phase transition, the sudden elongation of the fixed crystal can actuate an iron bead to roll, and the position of the bead was recorded by using a camera operating at a frame rate of 30 frames pers second (Fig. S14 and Movie 10†). Based on the average velocity and the mass of the iron bead, the work capacity (work output per unit volume) of the crystal was estimated to be >240 J m−3. Moreover, we note that the operating temperature of this simple actuating material is far below its melting point (484 K), and its thermal hysteresis is narrow (2–3 K), which ensures both favorable stability and high efficiency of its operation.2.5. Photomechanical effects
It is well known that acylhydrazone derivatives can undergo reversible isomerization under UV light57–59 whereby the trans-isomer (E) isomerizes to the cis-isomer (Z). The cis-isomer is stable at room temperature. As shown in Fig. 3a, b and Movie 11,† exposure of an acicular (needle-like) CTBB crystal (10.22 mm × 135 μm × 24 μm) to UV light (365 nm, 960 mW cm−2) results in reversible photomechanical deformation. Upon photoirradiation on the (010) face, the crystal visibly deforms within 2 s, and upon further irradiation it gradually bends away from the light source. After continuous irradiation for 15 s, the bending suddenly accelerates and the bending angle approaches 40°. Thinner crystals (9.74 mm × 26 μm × 11 μm) can even be bent into a circle by light (Fig. 3c and d). The photodeformed crystals do not recover their original shape at room temperature even after the irradiation has been terminated. | ||
| Fig. 3 Photomechanical deformation. (a–d) Acicular crystals of CTBB bend away from the light source upon irradiation on their (010) facet. (e–h) Recovery by heating of the shape of crystals that had been previously bent by exposure to light. (i) A schematic showing a simplified model of the photoinduced bending and thermally induced straightening of the crystal. | ||
Photoexcitation of microcrystalline CTBB induces blueshift of the absorption in the UV-vis spectrum at 216 nm, and enhances the absorption in the range 366–444 nm (Fig. 4a). This is paralleled by appearance in the infrared spectrum of bands at 1692 and 754 cm−1 that are mainly from the ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) and δ(C–H) modes, respectively (Fig. 4b, c and S15†). When the irradiated microcrystals were heated to 374 K, the UV-vis absorption and infrared spectra of form I were almost completely restored. The above spectral changes indicate that the crystals undergo reversible trans–cis isomerization (Fig. 4h), and the original phase and conformation can be recovered thermally. The thermal recovery was confirmed by powder X-ray diffraction analysis (Fig. 4d). To investigate the possible transformation(s) that occur during irradiation, the irradiated crystals were analyzed by DSC. As illustrated in Fig. 4e, upon heating, the endothermic phase transition peak at 374.5 K disappears, and a new exothermic peak appears at 359.0 K. However, when the crystal is cooled, the phase transition peak appears again, indicating that the material has been transformed back to form I. This was further confirmed by a second DSC measurement on the same sample (Fig. 4f). We therefore suggest that initially the trans-isomer (E) is transformed to the cis-isomer (Z) upon irradiation. The irreversibility of the process at room temperature is in accordance with the expected stability of the cis-isomer. However, the cis-isomer (Z) is transformed back to the trans-isomer (E) upon heating above 374 K, and the crystal reverts to form I again. Accordingly, a crystal that was previously bent by light and heated to 383 K at a rate of 10 K min−1 became straight again around 374 K (Fig. 3e–h, Movies 12 and 13†). This experiment shows the possibility of controlling the shape of the crystal by using both light and heat, where the effect of one stimulus is gated by that of the other.
N) and δ(C–H) modes, respectively (Fig. 4b, c and S15†). When the irradiated microcrystals were heated to 374 K, the UV-vis absorption and infrared spectra of form I were almost completely restored. The above spectral changes indicate that the crystals undergo reversible trans–cis isomerization (Fig. 4h), and the original phase and conformation can be recovered thermally. The thermal recovery was confirmed by powder X-ray diffraction analysis (Fig. 4d). To investigate the possible transformation(s) that occur during irradiation, the irradiated crystals were analyzed by DSC. As illustrated in Fig. 4e, upon heating, the endothermic phase transition peak at 374.5 K disappears, and a new exothermic peak appears at 359.0 K. However, when the crystal is cooled, the phase transition peak appears again, indicating that the material has been transformed back to form I. This was further confirmed by a second DSC measurement on the same sample (Fig. 4f). We therefore suggest that initially the trans-isomer (E) is transformed to the cis-isomer (Z) upon irradiation. The irreversibility of the process at room temperature is in accordance with the expected stability of the cis-isomer. However, the cis-isomer (Z) is transformed back to the trans-isomer (E) upon heating above 374 K, and the crystal reverts to form I again. Accordingly, a crystal that was previously bent by light and heated to 383 K at a rate of 10 K min−1 became straight again around 374 K (Fig. 3e–h, Movies 12 and 13†). This experiment shows the possibility of controlling the shape of the crystal by using both light and heat, where the effect of one stimulus is gated by that of the other.
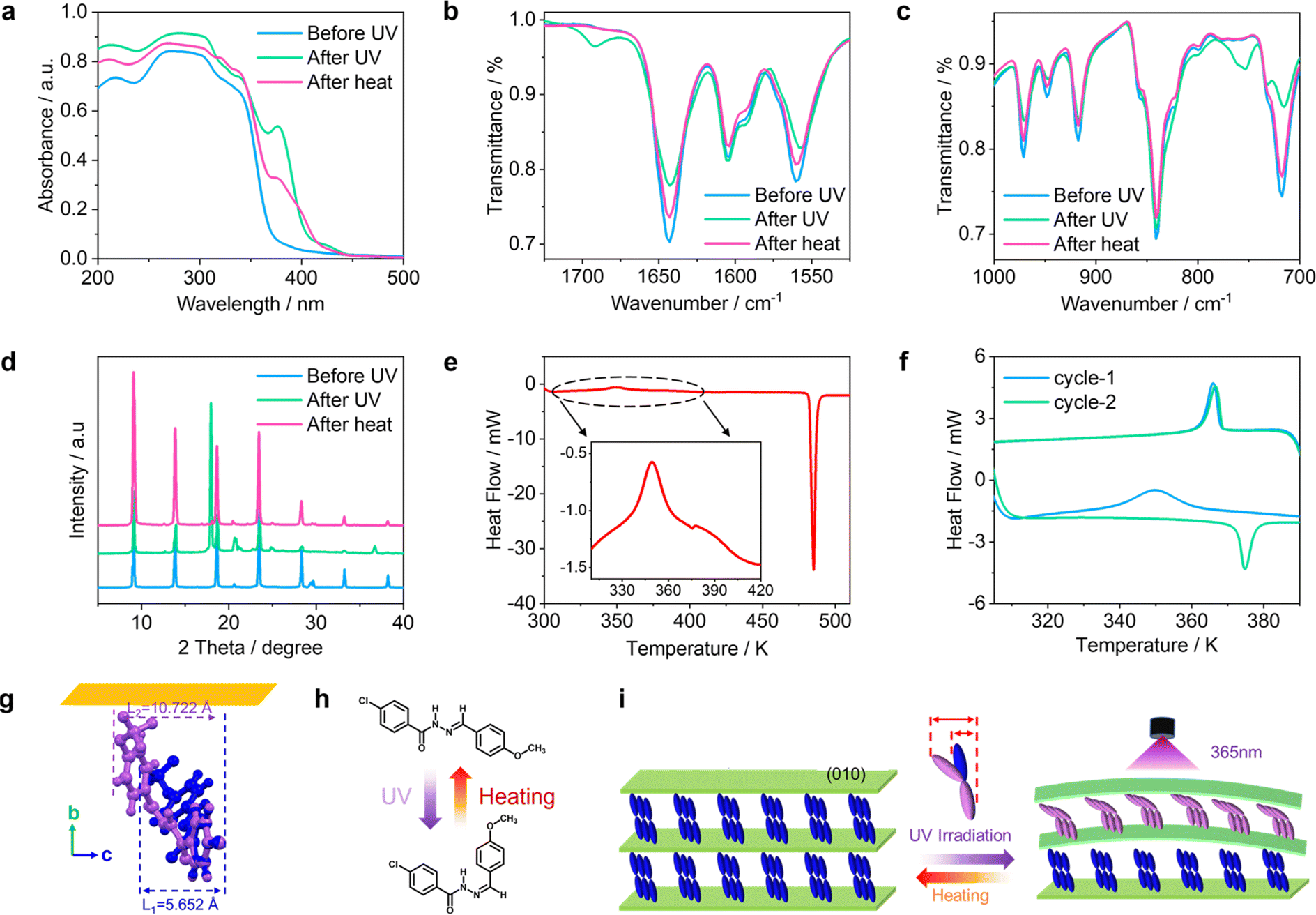 | ||
| Fig. 4 Photochemical and thermal transformations. (a–c) UV-vis absorption spectra (a) and infrared spectra (b and c) of CTBB microcrystals in form I before and after irradiated by 365 nm light for 5 minutes, and after heating for 5 min at 374 K. (d) Powder X-ray diffraction (PXRD) patterns of form I microcrystals before and after irradiation by 365 nm light for 5 min, and after heating for 5 minutes at 374 K. (e) Differential Scanning Calorimetry (DSC) curves of irradiated crystals of form I. (f) DSC evidence of the recovery to the irradiated crystals to form I after heating to 373 K. The heating and cooling rates in all experiments were 10 K min−1. (g) Molecular orientation in the CTBB crystals. (h) Photothermal isomerization of CTBB. (i) Schematic illustration of the reactions in the crystal upon irradiation with 365 nm light, followed by heating above 374 K. | ||
Photoinduced bending of molecular crystals has been well studied because it could be used to fabricate actuators and energy-conversion devices, whose mechanism of operation is oftentimes simplified as the “bimetal mechanism”.37 Generally, the bending direction depends on the nonuniform change in the unit volume that is caused by change of molecules on the irradiated surface during photoirradiation. For CTBB, the molecular volume variation caused by UV irradiation was calculated based on Bondi's atomic van der Waals radii,60 and it was found that the width of the molecule increased by 89.70% (+5.070 Å) after E → Z isomerization (Fig. 4g and S16†). This would further lead to the elongation of the b axis (Fig. 4i). Therefore, we hypothesize that when UV light is shone on the (010) facet, the crystal expands in its longest direction that results in bending away from the light source.
2.6. Mechanical properties
With regards to the excellent reproducibility of CTBB during the thermomechanical and photomechanical experiments (Fig. S17 and Movie 13†), we expected that it was the low-barrier, weak intermolecular interactions in the crystal that were dominant in the enhancement in the mechanically robustness of the crystal under external force, which would be able to absorb and dissipate the energy caused by the changes in shape. Indeed, as shown in Fig. 5a and b, when external force is applied on the well-developed facet (010), crystals of form I can be bent and even closed into a loop. Upon the release of the external force, the crystal resumes its original shape without any sign of damage, demonstrating its outstanding elasticity. What is more, this process can be performed many times (Movie 14†). The maximum elastic strain of form I crystal was calculated to be approximately 2.6% (Fig. S18†).61 Moreover, when subject to a torque, form I can be twisted for several rotations without visible damage (Fig. 5e–g). However, upon retraction of the torque, the crystal of form I cannot recover its original shape, and therefore becomes plastically twisted. What is even more impressive is that the twisted crystal can be bent elastically, demonstrating an extraordinary mechanical compliance (Fig. 5h–j and Movie 15†). In addition, since form I is transformed to II at high temperature, the flexibility of form II was also tested, and it was found to be elastic (Fig. S19 and Movie 16†). We envisage that these and similar crystals, being capable of multiple modes of deformation (Fig. 5m) could expand the prospective applications of flexible crystalline materials. | ||
| Fig. 5 Mechanical responses of CTBB crystals under external forces. (a and b) Elastic bending of a CTBB crystal. (c and d) Scanning electron micrographs images of a bent crystal. (e–g) Plastic twisting of a CTBB crystal. (h–j) Elastic bending of a twisted CTBB crystal. (k and l) Scanning electron micrographs of a twisted crystal. (m) Schematic showing the elastic bending and plastic twisting of a single crystal. | ||
To rationalize the mechanism of the deformations, the crystal structure of form I was analyzed (Table S1†). Although the flexible chain between the two aromatic rings possesses a significant degree of geometric freedom, the CTBB molecules maintain planar conformation due to the intramolecular hydrogen bonds C–H⋯N (2.866 Å, 2.590 Å, 97.6(8)°), C–H⋯N (2.233 Å, 2.342 Å, 71.7(3)°), and C–H⋯O (2.868 Å, 2.615 Å, 96.1(3)°). The dihedral angles between the aromatic rings in form I is 35.21°. As shown in Fig. 6a, the nearly planar molecules form columns by π⋯π interactions along the crystallographic c axis, and the columnar structure is strengthened by intermolecular hydrogen bonds N–H⋯O (2.854 Å, 2.011 Å, 166.5(4)°) and C–H⋯O (3.259 Å, 2.760 Å, 114.6(1)°). The adjacent columns are linked by intermolecular hydrogen bonds C–H⋯N (3.513 Å, 2.969 Å, 118.8(2)°; 3.721 Å, 2.937 Å, 142.9(2)°) along the crystallographic b axis, thereby generating a bendable (010) plane (Fig. S20†). These columns could serve as a spring to absorb the strain that has been generated by the compression of the inner arc and expansion of the outer arc of the crystal during bending.
 | ||
| Fig. 6 Structural basis of the mechanical deformation of CTBB crystals. (a) Molecular packing of form I viewed perpendicular to the (001), (010), and (100) planes. (b) Schematic of the mechanism for the elastic bending of CTBB crystals. (c) Schematic showing the mechanism of plastic twisting of form I. (d and e) Energy frameworks of form I shown together with the π⋯π (d) and hydrogen bonding (e) interactions. The pink and green envelopes represent intermolecular interactions related to the π-stacked columns along different directions. | ||
The intermolecular interaction energy within the π-stacked columns is Etotal = −130.0 kJ mol−1. However, the intermolecular interaction energies in the other directions are Etotal = −123.5 kJ mol−1 and −39.2 kJ mol−1 along the crystallographic b and a axes, respectively (Fig. S21 and S22†), which are lower than that of the π-stacked columns. As shown in Fig. 6b, the molecules can undergo slight rotations and translations when the crystal is bent; at the outer arc, the molecules are expected to move away from each other, causing expansion along the extended axis, whereas in the inner arc, the molecules would shift closer to each other, causing contraction along the extended axis. Since the crystal packing and intermolecular interactions in the two polymorphs are not very different (Fig. S23–S25;†Etotal = −31.9 kJ mol−1, −124.1 kJ mol−1 and −115.4 kJ mol−1 along the crystallographic a, b and c axes, respectively), it is not surprising that form II can also be bent elastically (Fig. S19†).62–64 Nanoindentation on the (010) plane showed that the elastic modulus (E) and hardness (H) of crystals of form I are E = 6.18 ± 0.01 GPa and H = 0.37 ± 0.02 GPa (Fig. S26†), which places the CTBB crystals in the category of soft materials.65
During the twisting, the torque is tangential to the cross-section of the (001) plane, so that the shearing momenta applied on the π-stacked columns along the c axis are quite small. Therefore, the two adjacent molecules in one π-stacked columns could be rotated relative to each other during the twisting (Fig. 6c).23,66 However, since the intermolecular interactions along the crystallographic c axis are sufficiently strong to resist the force (Etotal = −130.0 kJ mol−1, Fig. 6d), the macroscopic integrity of the crystal remains intact. The C–H⋯N hydrogen bonds that connect the π-stacked columns are almost coplanar with the twisting forces, and thus they are expected to play an important role in the twisting. Moreover, the hydrogen bonds parallel to the cross-section are weak (Etotal = −123.5 kJ mol−1 and −39.2 kJ mol−1, Fig. 6e), so that they might be destroyed under torque, resulting in plastic twisting.67 The above mechanism is in line with the observation that a twisted crystal can still be bent elastically, because the π⋯π interactions, being the key component of elastic bending, are well preserved after the plastic twisting.
3. Conclusions
In summary, we report robust dynamic crystalline material that can respond to multiple stimuli (heat, light, and mechanical force). When heated, crystals of this material undergo single-crystal-to-single-crystal phase transition by migration of phase boundaries, and this process is accompanied by several thermomechanical effects, including shear deformation, flipping, and jumping. The reversible migration of the phase boundaries can be precisely controlled by changing the temperature, and this also varies across different crystal habits. When the phase boundaries are parallel, the transition is accompanied by slow thermomechanical behavior. On the contrary, when the phase boundaries are not parallel, a pronounced thermosalience was observed. Moreover, the crystal can undergo reversible bending by combined exposure to UV radiation and heat, which is attributed to cis–trans isomerization around the CN double bonds. Apart from the remarkable response to heat and UV stimuli, these crystals also exhibit excellent mechanical flexibility under external forces. The crystals are capable of both elastic bending and plastic twisting, and they maintain their elasticity even in the twisted state. Altogether, the results show that even a simple organic crystalline material can exhibit a multitude of dynamic mechanical responses.
Data availability
The data supporting the findings of this study are available within the article and in the ESI.†Author contributions
Wenbo Wu and Kui Chen contributed equally to this work. Wenbo Wu: conceptualization (equal); data curation (lead); formal analysis (lead); methodology (lead); validation (lead); visualization (lead) writing – original draft (lead). Kui Chen: conceptualization (lead); data curation (equal); formal analysis (equal); visualization (equal); writing – review and editing (lead). Hui Yu: data curation (equal); formal analysis (equal); investigation (equal). Jiaxuan Zhu: data curation (equal); formal analysis (equal); investigation (equal). Jiaxuan Zhu: data curation (equal); formal analysis (equal); investigation (equal). Yaoguang Feng: data curation (equal); formal analysis (equal); investigation (equal). Jingkang Wang: project administration (equal); resources (equal); supervision (equal). Xin Huang: formal analysis (equal); investigation (equal); project administration (equal); resources (equal). Liang Li: data curation (equal); resources (equal); supervision (equal); validation (equal). Hongxun Hao: funding acquisition (lead); project administration (lead); resources (equal); writing – review and editing (equal). Ting Wang: conceptualization (equal); formal analysis (equal); funding acquisition (lead); supervision (lead); writing – original draft (equal); writing – review and editing (equal). Na Wang: conceptualization (equal); data curation (equal); funding acquisition (lead); supervision (equal); writing – review and editing (equal). Panče Naumov: project administration (lead); formal analysis (equal); writing – original draft (equal); writing – review and editing (equal).Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are very grateful for the financial support by the National Natural Science Foundation of China (Grant number 22108196) and the National College Student Innovation and Entrepreneurship Training Program (Grant number 202110056036). This material is based upon works supported by Tamkeen under NYUAD RRC Grant No. CG011. The authors thank the School of Pharmaceutical Science and Technology, Faculty of Medicine, Tianjin University, for the support with the single crystal X-ray diffraction measurements.Notes and references
- M. W. M. Tan, H. Wang, D. Gao, P. Huang and P. S. Lee, Chem. Soc. Rev., 2024, 53, 3485–3535 RSC.
- Y. Huang, L. Ning, X. Zhang, Q. Zhou, Q. Gong and Q. Zhang, Chem. Soc. Rev., 2024, 53, 1090–1166 RSC.
- S. Shrivastava, T. Q. Trung and N.-E. Lee, Chem. Soc. Rev., 2020, 49, 1812–1866 RSC.
- P. Tonkaev, I. S. Sinev, M. V. Rybin, S. V. Makarov and Y. Kivshar, Chem. Rev., 2022, 122, 15414–15449 CrossRef CAS PubMed.
- H. Chen, Y. Jing, J.-H. Lee, D. Liu, J. Kim, S. Chen, K. Huang, X. Shen, Q. Zheng, J. Yang, S. Jeon and J.-K. Kim, Mater. Horiz., 2020, 7, 2378–2389 RSC.
- E. Benvenuti, A. Lanfranchi, S. Moschetto, M. Natali, M. Angelini, P. Lova, F. Prescimone, V. Ragona, D. Comoretto, M. Prosa, M. Bolognesi and S. Toffanin, J. Mater. Chem. C, 2024, 12, 4243–4252 RSC.
- E. Ahmed, D. P. Karothu and P. Naumov, Angew. Chem., Int. Ed., 2018, 57, 8837–8846 CrossRef CAS PubMed.
- P. Naumov, D. P. Karothu, E. Ahmed, L. Catalano, P. Commins, J. Mahmoud Halabi, M. B. Al-Handawi and L. Li, J. Am. Chem. Soc., 2020, 142, 13256–13272 CrossRef CAS PubMed.
- S. Kobatake, S. Takami, H. Muto, T. Ishikawa and M. Irie, Nature, 2007, 446, 778–781 CrossRef CAS PubMed.
- A. K. Bartholomew, I. B. Stone, M. L. Steigerwald, T. H. Lambert and X. Roy, J. Am. Chem. Soc., 2022, 144, 16773–16777 CrossRef CAS PubMed.
- Y.-S. Chen, C.-H. Wang, Y.-H. Hu, C.-Y. D. Lu and J.-S. Yang, J. Am. Chem. Soc., 2023, 145, 6024–6028 CrossRef CAS PubMed.
- X. Yang, L. Lan, L. Li, J. Yu, X. Liu, Y. Tao, Q.-H. Yang, P. Naumov and H. Zhang, Nat. Commun., 2023, 14, 3627 CrossRef CAS PubMed.
- Ž. Skoko, S. Zamir, P. Naumov and J. Bernstein, J. Am. Chem. Soc., 2010, 132, 14191–14202 CrossRef PubMed.
- S. C. Sahoo, M. K. Panda, N. K. Nath and P. Naumov, J. Am. Chem. Soc., 2013, 135, 12241–12251 CrossRef CAS PubMed.
- S. C. Sahoo, S. B. Sinha, M. S. R. N. Kiran, U. Ramamurty, A. F. Dericioglu, C. M. Reddy and P. Naumov, J. Am. Chem. Soc., 2013, 135, 13843–13850 CrossRef CAS PubMed.
- D. P. Karothu, R. Ferreira, G. Dushaq, E. Ahmed, L. Catalano, J. Mahmoud Halabi, Z. Alhaddad, I. Tahir, L. Li, S. Mohamed, M. Rasras and P. Naumov, Nat. Commun., 2022, 13, 2823 CrossRef CAS PubMed.
- L. Lan, L. Li, Q. Di, X. Yang, X. Liu, P. Naumov and H. Zhang, Adv. Mater., 2022, 34, 2200471 CrossRef CAS PubMed.
- M. K. Panda, T. Runčevski, S. Chandra Sahoo, A. A. Belik, N. K. Nath, R. E. Dinnebier and P. Naumov, Nat. Commun., 2014, 5, 4811 CrossRef CAS PubMed.
- D. P. Karothu, J. Weston, I. T. Desta and P. Naumov, J. Am. Chem. Soc., 2016, 138, 13298–13306 CrossRef CAS PubMed.
- S. Ghosh and C. M. Reddy, Angew. Chem., Int. Ed., 2012, 51, 10319–10323 CrossRef CAS PubMed.
- S. Ghosh, M. K. Mishra, S. B. Kadambi, U. Ramamurty and G. R. Desiraju, Angew. Chem., Int. Ed., 2015, 54, 2674–2678 CrossRef CAS PubMed.
- G. R. Krishna, R. Devarapalli, G. Lal and C. M. Reddy, J. Am. Chem. Soc., 2016, 138, 13561–13567 CrossRef CAS PubMed.
- H. Liu, Z. Lu, B. Tang, C. Qu, Z. Zhang and H. Zhang, Angew. Chem., Int. Ed., 2020, 59, 12944–12950 CrossRef CAS PubMed.
- B. Tang, X. Yu, K. Ye and H. Zhang, Adv. Opt. Mater., 2022, 10, 2101335 CrossRef CAS.
- Y. Sun, Y. Lei, H. Dong, Y. Zhen and W. Hu, J. Am. Chem. Soc., 2018, 140, 6186–6189 CrossRef CAS PubMed.
- X. Zheng, X. Liu, L. Liu, X. Li, S. Jiang, C. Niu, P. Xie, G. Liu, Z. Cao, Y. Ren, Y. Qin and J. Wang, Angew. Chem., Int. Ed., 2022, 61, e202113073 CrossRef CAS PubMed.
- L. Lan, X. Yang, B. Tang, X. Yu, X. Liu, L. Li, P. Naumov and H. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202200196 CrossRef CAS PubMed.
- D. Kitagawa, H. Nishi and S. Kobatake, Angew. Chem., Int. Ed., 2013, 52, 9320–9322 CrossRef CAS PubMed.
- S. Bhandary, M. Beliš, A. M. Kaczmarek and K. Van Hecke, J. Am. Chem. Soc., 2022, 144, 22051–22058 CrossRef CAS PubMed.
- B. B. Rath, G. Gallo, R. E. Dinnebier and J. J. Vittal, J. Am. Chem. Soc., 2021, 143, 2088–2096 CrossRef CAS PubMed.
- T. Taniguchi, H. Sugiyama, H. Uekusa, M. Shiro, T. Asahi and H. Koshima, Nat. Commun., 2018, 9, 538 CrossRef PubMed.
- P. Commins, H. Hara and P. Naumov, Angew. Chem., Int. Ed., 2016, 55, 13028–13032 CrossRef CAS PubMed.
- G. Liu, J. Liu, X. Ye, L. Nie, P. Gu, X. Tao and Q. Zhang, Angew. Chem., Int. Ed., 2017, 56, 198–202 CrossRef CAS PubMed.
- P. Commins, M. B. Al-Handawi, D. P. Karothu, G. Raj and P. Naumov, Chem. Sci., 2020, 11, 2606–2613 RSC.
- S. Bhunia, S. Chandel, S. K. Karan, S. Dey, A. Tiwari, S. Das, N. Kumar, R. Chowdhury, S. Mondal, I. Ghosh, A. Mondal, B. B. Khatua, N. Ghosh and C. M. Reddy, Science, 2021, 373, 321–327 CrossRef CAS PubMed.
- S. Mondal, P. Tanari, S. Roy, S. Bhunia, R. Chowdhury, A. K. Pal, A. Datta, B. Pal and C. M. Reddy, Nat. Commun., 2023, 14, 6589 CrossRef CAS PubMed.
- P. Naumov, S. Chizhik, M. K. Panda, N. K. Nath and E. Boldyreva, Chem. Rev., 2015, 115, 12440–12490 CrossRef CAS PubMed.
- W. M. Awad, D. W. Davies, D. Kitagawa, J. Mahmoud Halabi, M. B. Al-Handawi, I. Tahir, F. Tong, G. Campillo-Alvarado, A. G. Shtukenberg, T. Alkhidir, Y. Hagiwara, M. Almehairbi, L. Lan, S. Hasebe, D. P. Karothu, S. Mohamed, H. Koshima, S. Kobatake, Y. Diao, R. Chandrasekar, H. Zhang, C. C. Sun, C. Bardeen, R. O. Al-Kaysi, B. Kahr and P. Naumov, Chem. Soc. Rev., 2023, 52, 3098–3169 RSC.
- D. Rus and M. T. Tolley, Nature, 2015, 521, 467–475 CrossRef CAS PubMed.
- P. Gupta, D. P. Karothu, E. Ahmed, P. Naumov and N. K. Nath, Angew. Chem., Int. Ed., 2018, 57, 8498–8502 CrossRef CAS PubMed.
- B. B. Rath, M. Gupta and J. J. Vittal, Chem. Mater., 2022, 34, 178–185 CrossRef CAS.
- X. Yang, L. Lan, X. Pan, Q. Di, X. Liu, L. Li, P. Naumov and H. Zhang, Nat. Commun., 2023, 14, 2287 CrossRef CAS PubMed.
- W. Wu, K. Chen, X. Zhang, T. Wang, S. Li, H. Zhao, L. Zhou, X. Huang and H. Hao, Chem. - Eur. J., 2023, 29, e202202598 CrossRef CAS PubMed.
- W. Wu, K. Chen, T. Wang, N. Wang, X. Huang, L. Zhou, Z. Wang and H. Hao, J. Mater. Chem. C, 2023, 11, 2026–2052 RSC.
- Y. Duan, S. Semin, P. Tinnemans, H. Cuppen, J. Xu and T. Rasing, Nat. Commun., 2019, 10, 4573 CrossRef PubMed.
- E. Ahmed, D. P. Karothu, A. Slimani, J. Mahmoud Halabi, I. Tahir, K. Q. Canales and P. Naumov, Adv. Funct. Mater., 2022, 32, 2112117 CrossRef CAS.
- Y. Duan, S. Semin, P. Tinnemans, J. Xu and T. Rasing, Small, 2021, 17, 2006757 CrossRef CAS PubMed.
- J. Lin, Z. Guo, K. Zhang, P. Zhao, S. Wu, J. Xu, J. Gong and Y. Bao, Adv. Funct. Mater., 2022, 32, 2203004 CrossRef CAS.
- D. P. Karothu, G. Dushaq, E. Ahmed, L. Catalano, M. Rasras and P. Naumov, Angew. Chem., Int. Ed., 2021, 60, 26151–26157 CrossRef CAS PubMed.
- S. Chen, S. Zhu, Z. Lin and J. Peng, ACS Nano, 2022, 16, 11194–11203 CrossRef CAS PubMed.
- J.-J. Wu, S. Yu, Y. Liu, C.-C. Yan, W.-Y. Yang, W. Xie, X.-D. Wang, C. Sun and L.-S. Liao, Adv. Funct. Mater., 2023, 33, 2214308 CrossRef CAS.
- H. Chung, D. Dudenko, F. Zhang, G. D'Avino, C. Ruzié, A. Richard, G. Schweicher, J. Cornil, D. Beljonne, Y. Geerts and Y. Diao, Nat. Commun., 2018, 9, 278 CrossRef PubMed.
- D. W. Davies, B. Seo, S. K. Park, S. B. Shiring, H. Chung, P. Kafle, D. Yuan, J. W. Strzalka, R. Weber, X. Zhu, B. M. Savoie and Y. Diao, Nat. Commun., 2023, 14, 1304 CrossRef CAS PubMed.
- S. K. Park and Y. Diao, Chem. Soc. Rev., 2020, 49, 8287–8314 RSC.
- M. K. Panda, R. Centore, M. Causà, A. Tuzi, F. Borbone and P. Naumov, Sci. Rep., 2016, 6, 29610 CrossRef CAS PubMed.
- Y. Miura, T. Takeda, N. Yoshioka and T. Akutagawa, Cryst. Growth Des., 2022, 22, 5904–5911 CrossRef CAS.
- P. Gupta, T. Panda, S. Allu, S. Borah, A. Baishya, A. Gunnam, A. Nangia, P. Naumov and N. K. Nath, Cryst. Growth Des., 2019, 19, 3039–3044 CrossRef CAS.
- P. Gupta, S. Allu, P. J. Hazarika, N. R. Ray, A. K. Nangia and N. K. Nath, CrystEngComm, 2022, 24, 7261–7265 RSC.
- J. Peng, J. Xing, J. Bai, Y. Ren, T. Wang and J. Jia, Dyes Pigm., 2021, 194, 109529 CrossRef CAS.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- A. J. Thompson, A. I. Chamorro Orué, A. J. Nair, J. R. Price, J. McMurtrie and J. K. Clegg, Chem. Soc. Rev., 2021, 50, 11725–11740 RSC.
- B. Bhattacharya, A. A. L. Michalchuk, D. Silbernagl, N. Yasuda, T. Feiler, H. Sturm and F. Emmerling, Chem. Sci., 2023, 14, 3441–3450 RSC.
- R. Samanta, S. Das, S. Mondal, T. Alkhidir, S. Mohamed, S. P. Senanayak and C. M. Reddy, Chem. Sci., 2023, 14, 1363–1371 RSC.
- H. Zhao, X. Zhang, K. Chen, W. Wu, S. Li, T. Wang, X. Huang, N. Wang, L. Zhou and H. Hao, CrystEngComm, 2023, 25, 2600–2606 RSC.
- D. P. Karothu, J. Mahmoud Halabi, E. Ahmed, R. Ferreira, P. R. Spackman, M. A. Spackman and P. Naumov, Angew. Chem., Int. Ed., 2022, 61, e202113988 CrossRef CAS PubMed.
- K. Chen, J. Wang, Y. Feng, H. Liu, X. Zhang, Y. Hao, T. Wang, X. Huang and H. Hao, J. Mater. Chem. C, 2021, 9, 16762–16770 RSC.
- K. Chen, J. Wang, W. Wu, H. Shan, H. Zhao, N. Wang, T. Wang, X. Huang and H. Hao, Dyes Pigm., 2023, 219, 111536 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental section, supplementary figures and movies. CCDC 2286243, 2286247, 2286249, 2286528, 2286530 and 2341752. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc02152e |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |