DOI:
10.1039/D4SC01180E
(Edge Article)
Chem. Sci., 2024,
15, 9298-9317
Sulphur-atom positional engineering in perylenimide: structure–property relationships and H-aggregation directed type-I photodynamic therapy†
Received
19th February 2024
, Accepted 10th May 2024
First published on 10th May 2024
Abstract
An innovative design strategy of placing sulfur (S)-atoms within the pendant functional groups and at carbonyl positions in conventional perylenimide (PNI-O) has been demonstrated to investigate the condensed state structure–property relationship and potential photodynamic therapy (PDT) application. Incorporation of simply S-atoms at the peri-functionalized perylenimide (RPNI-O) leads to an aggregation-induced enhanced emission luminogen (AIEEgen), 2-hexyl-8-(thianthren-1-yl)-1H-benzo[5,10]anthra[2,1,9-def]isoquinoline-1,3(2H)-dione (API), which achieves a remarkable photoluminescence quantum yield (ΦPL) of 0.85 in aqueous environments and established novel AIE mechanisms. Additionally, substitution of the S-atom at the carbonyl position in RPNI-O leads to thioperylenimides (RPNI-S): 2-hexyl-8-phenyl-1H-benzo[5,10]anthra[2,1,9-def]isoquinoline-1,3(2H)-dithione (PPIS), 8-([2,2′-bithiophen]-5-yl)-2-hexyl-1H-benzo[5,10]anthra[2,1,9-def]isoquinoline-1,3(2H)-dithione (THPIS), and 2-hexyl-8-(thianthren-1-yl)-1H-benzo[5,10]anthra[2,1,9-def]isoquinoline-1,3(2H)-dithion (APIS), with distinct photophysical properties (enlarged spin–orbit coupling (SOC) and ΦPL ≈ 0.00), and developed diverse potent photosensitizers (PSs). The present work provides a novel SOC enhancement mechanism via pronounced H-aggregation. Surprisingly, the lowest singlet oxygen quantum yield (ΦΔ) and theoretical calculation suggest the specific type-I PDT for RPNI-S. Interestingly, RPNI-S efficiently produces superoxide (O2˙−) due to its remarkably lower Gibbs free energy (ΔG) values (THPIS: −40.83 kcal mol−1). The non-toxic and heavy-atom free very specific thio-based PPIS and THPIS PSs showed selective and efficient PDT under normoxia, as a rare example.
Introduction
In recent years, PDT, a photochemical reaction-based treatment that utilizes light with appropriate PSs, has attracted significant attention due to its distinct advantages over most conventional therapies in cancer treatments.1,2 Yet, classical PSs have low excited state triplet formation, and the introduction of heavy-atoms to enlarge SOC and subsequently enhance intersystem crossing (ISC) into their structures often raises significant concerns about cost and high dark toxicity.3,4 In PDT, two distinct mechanisms, type-I and type-II, govern the reactions of PSs with ground-state oxygen (3O2). Type-I PDT involves the intermolecular electron transfer (IET) process, which transfers an electron from the triplet excitons of the PS to 3O2, leading to the formation of O2˙−. This process can occur independently of any reducing intermediary, generating hydrogen peroxide (H2O2) and hydroxyl radicals (HO˙) through superoxide disproportionation and the Franck–Condon transition.5 Crucially, this reaction requires an allowed electron transfer, indicated by a negative Gibbs free energy change (ΔG < 0).6 Conversely, type-II PDT relies on the energy transfer from PS triplet excitons to 3O2, forming 1O2. Comparatively, the type-I process in PDT mechanisms is more advantageous due to its lower dependence on oxygen content, and it remains relatively rare to date.7,8 The S-substituted nucleobase anticancer drugs have received increasing interest in the past few decades due to their potential chemotherapeutic applications. They are still being explored for their potential new therapeutic abilities.9–11 Additionally, the excellent excited triplet formation triggered by the thiocarbonyl group of thiobase PSs could result in more prospective PSs with promising PDT applications. However, numerous drawbacks of thiobase drugs, including long irradiation times, the need for higher concentrations, etc., have limited their obvious practical applications.12,13 Yet, numerous S-substituted derivatives in the carbonyl group have been comprehensively designed as heavy-atom-free materials.14,15 The development of such an S-substituted carbonyl group in tumor treatment has not been extensively studied. This motivated us to reflect upon the thionation approach for future exploration of PSs with clinical acceptance. While heavy-atom-free thio-based naphthalimide (NI) PSs were designed for hypoxic environments, they lack specificity, selectivity, and HO˙ generation ability. The study did not thoroughly explore type-I mechanisms and varied PS production. It was limited to one cell line, raising questions about broader applicability. Additional targeting agents were needed for prior heavy-atom-free PSs in biological environments. The absence of an S-atom at a functional position in their design is noteworthy, as it may significantly impact PDT-mediated cancer treatment and other unique properties, rather than solely relying on PDT applications (Table S1†).14
Considering these insights, the PNI-O core has been chosen as the potent core due to its exceptional ΦPL, chemical stability, and thermal resilience, as well as its promising potential applications in various fields, including sensors,16 therapeutics,17 optoelectronics,18 and bioimaging.19 Further, an innovative design strategy involving the incorporation of S-atoms into both pendant functional groups and carbonyl positions within a planar PNI-O core, distinct from the previous thio-based approach, has been demonstrated here. The aim is to explore the structure–property relationship in the condensed state, especially in the context of potential PDT applications for cancer treatment. This investigation aligns with a well-established and ongoing effort to understand condensed state structure–property relationships, driven by their significance across various fields. Incorporating S-atoms at both the functional and carbonyl positions results in distinct properties within PNI-O. The introduction of simple S-atoms at functional positions triggers a unique transformation from aggregation-caused quenching (ACQ)-to-AIEE behavior within the extremely planar PNI-O core, which results in ΦPL of 0.85 (with unique microrose supramolecular-assembly) and a significant Stokes shift of 146 nm, which are rarely observed. This modification also establishes a novel AIE mechanism. While S-atoms at the carbonyl position led to a complete drop in the fluorescence intensity (ΦPL ≈ 0.00) and enlarged SOC, it promoted type-I PDT reactions with efficient production of O2˙−. This strategy demonstrates broad PDT applicability, targeting cancer cells (HeLa and MCF7) and normal cells without additional targeting agents, emphasizing its newer therapeutic ability and cost-effectiveness. Notably, the biocompatible heavy-atom-free thio-based PSs exhibit selective and efficient PDT efficacy, marking the first reported instance. This work provides SOC enhancement mechanisms through prominent H-aggregation (Table S2†). While previous research showed improved SOC and ISC, this research unveils the critical role of pronounced H-aggregation in this enhancement, opening up new possibilities for future PS design.14,15 This approach allows for precise control over the generation of distinct variations of type-I and type-II ROS, expanding the potential applications of these PSs.
Hence, this research introduces a novel concept of condensed state emitters and highly specific and selective heavy-atom-free thio-based PSs, exhibiting a simple and precise platform to manipulate condensed state emissions, supramolecular assembly, and unique PS development, marking significant progress in the field.
Results and discussion
Design, synthesis, and characterization of RPNI-O and RPNI-S derivatives
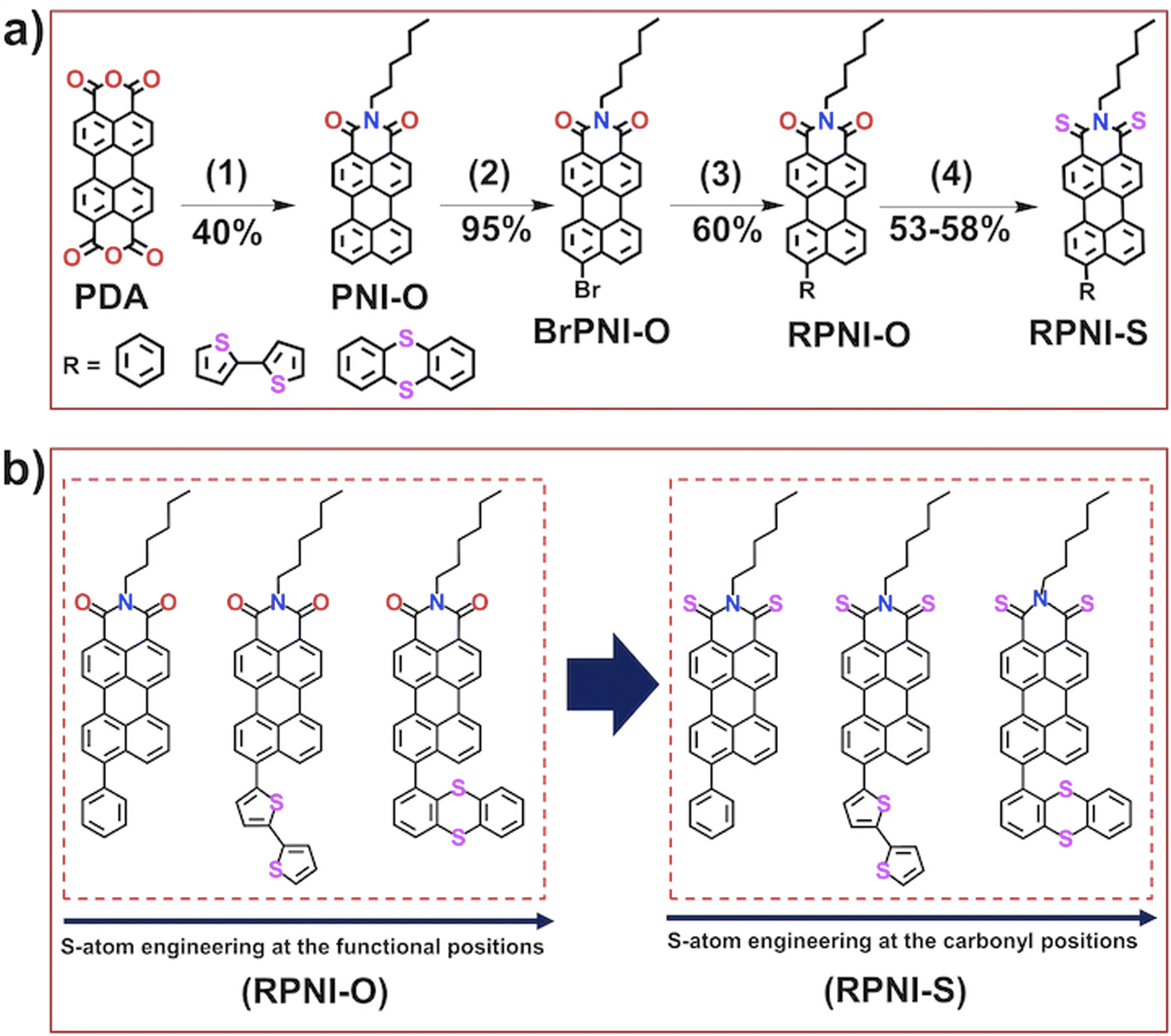
The primary objective of this research was to explore the unique condensed state photophysical behavior and the photosensitizing capabilities of newly designed heavy-atom-free RPNI-O and RPNI-S. To achieve this, RPNI-O (PPI, THPI, and API) and RPNI-S (PPIS, THPIS, and APIS) have been strategically developed by introducing S-atom positional engineering at the pendant functional group and carbonyl position in the PNI-O core (Schemes 1 and S1–S4†). For the synthesis, very economical perylene-3,4,9,10-tetracarboxylic acid anhydride (PDA) was selected as a versatile precursor material, which underwent a condensation reaction with hexylamine in good yields (Scheme S1†). Subsequently, BrPNI-O has been synthesized via bromination of the PNI-O core (yield-95%) (Scheme S2†), which underwent Suzuki coupling with various boronic acid derivatives, including phenyl, bithiophene, and thianthranyl, leading to the successful synthesis of RPNI-O derivatives (Scheme S3†). Additionally, thioperylenimide (RPNI-S) was prepared using a one-pot synthesis approach (Scheme S4†) involving the reaction between the parent RPNI-O and a commercial Lawesson’s reagent, resulting in the formation of RPNI-S compounds. The detailed synthetic procedure and reaction scheme can be found in the corresponding Scheme 1 and Fig. S1–S4 in the ESI.† The synthesized materials were extensively characterized using multinuclear NMR spectroscopy (1H and 13C) and matrix-assisted laser desorption/ionization (MALDI) techniques. Further details of the comprehensive characterization data can be found in the ESI.†
 |
| | Scheme 1 (a) Synthetic route for the preparation of RPNI-O (PPI, THPI, and API) and thioperylenimides (RPNI-S), viz.PPIS, THPIS, and APIS lack an S-atom; instead, the S-atom is positioned within the pendant rotor groups at the peri-position of the PNI-O core. (b) Chemical structure of the synthesized RPNI-O and RPNI-S [(1) hexyl amine, H2O, 20 h, (2) chlorobenzene, Br2, 4.5 h, (3) Pd(0), THF, H2O, (4) Lawesson's reagent, toluene, 20 h, and the values below the arrow represents the yield of the corresponding product]. | |
Photophysical properties
UV-vis absorption and fluorescence spectroscopy were employed to investigate the optical properties of RPNI-O and RPNI-S (Fig. 1, S1–S3† and Table 1). RPNI-S exhibited a pronounced 130 nm red shift in absorption maxima (λabs,max) from 506 to 636 nm, accompanied by a corresponding 125 nm red shift in emission maxima (λem,max) in solution (Fig. 1a and b). This shift is attributed to the stabilization of the lowest unoccupied molecular orbital (LUMO) and destabilization of the highest occupied molecular orbital (HOMO) in RPNI-S, indicating decreased LUMO energies (increased electron affinity) and increased HOMO energies, resulting in the observed red shift in optical absorption.14,15,20,21 In contrast, alteration of the S-atom within the pendant functional group (–R) in RPNI-O resulted in blue-shifted λabs,max in API compared to other RPNI-O derivatives, namely PPI and THPI. This blue shift can be attributed to reduced electronic communication in API in its solution state. Crucially, the International Commission on Illumination (CIE) chromaticity diagram vividly displayed a broad spectrum of emission colors in the solution state, ranging from yellow (580 nm) to near-infrared (NIR) at 705 nm (Fig. 1b′, Tables 1 and S3†). RPNI-O derivatives, such as PPI, showed yellow emission, while THPI and API displayed bright red emissive colors. In contrast, RPNI-S derivatives exhibited emission colors that remained entirely undetectable under 365 nm UV-light illumination, even though λem,max peaks were observed at 655 nm for PPIS, 647 nm for THPIS, and 705 nm for APIS, respectively (Fig. 1b′, Tables 1 and S3†). API exhibited notable condensed state emissive properties in its aggregated and solid states, owing to two S-atoms in one fused aromatic anthracene ring (rotor). In contrast, PPI and THPI, lacking an S-atom at the pendant R rotor and having two S-atoms in two different pendant aromatic rings (rotor), exhibited suppressed emission in their aggregated state. This finding contrasts what is expected based on the RIM mechanism, which typically leads to the generation of condensed state emitters. A detailed explanation of this mechanism is provided through single crystal X-ray diffraction (SCXRD) analysis to shed more light on this discrepancy and provide a comprehensive understanding. Furthermore, API exhibits a remarkably high ΦPL of 0.85 in its aggregated state, likely attributed to the unique intermolecular packing arrangement influenced by self-assembly behavior in its condensed state. Additionally, API showed ΦPL of 0.89 in its highest λem,max. In contrast, in their aggregated state, other RPNI-O derivatives, namely PPI and THPI, display significantly lower ΦPL of 0.10 and 0.04, respectively. Conversely, the RPNI-S compounds exhibited nearly non-fluorescent behavior (ΦPL ≈ 0.00) (Table 1, Fig. S2, S3 and eqn (S1)†). The fluorescence suppression indicated that thionation enhanced the ISC process, causing a more efficient transition from the singlet to the triplet excited state.14,15 The CIE chromaticity diagram highlights the distinct emissive colors of RPNI-O in their aggregated state. Specifically, PPI and THPI exhibited quenched emission despite having distinct λem,max peaks, whereas API displayed deep red emission. In contrast, PPIS, THPIS, and APIS showed an undetectable λem,max peak and non-emissive features under 365 nm UV irradiation (Fig. 1c′, Tables 1 and S3†). API showed a slightly blue-shifted λem,max in the solid state compared to other RPNI-O derivatives, with a similar trend observed overall (Fig. 1d and d′). However, when comparing the aggregated states, API exhibited a significant 50 nm blue shift in its λem,max compared to PPI and THPI (Fig. 1c). The validity of this observation was reinforced and elaborated upon using excitation–emission matrix (EEM) spectra. Moreover, API displayed a noteworthy 31 nm red shift in its λem,max (646 nm) when in the solid state, compared to the aggregated state (614 nm) (Fig. 1e). This difference can be attributed to the unique intermolecular packing arrangement influenced by the steric constraints of the pendant S-substituted thianthranyl rotor in RPNI-O. In contrast, the other derivatives exhibited minimal shifts between their aggregated state and solid state λem,max. Further, distinct variations in the powder colors were observed under white light and 365 nm UV irradiation, and this difference correlated well with the CIE chromaticity diagram. API showed strong and bright red emission, while PPI and THPI showed faint emission. RPNI-S showed complete emission quenching in its solid state under 365 nm UV light illumination (Fig. 1d′, f, g and Table S3†).
 |
| | Fig. 1 Photophysical characteristics of all the RPNI-O (PPI, THPI, and API) and RPNI-S (PPIS, THPIS, and APIS) derivatives. (a) Normalized absorbance spectra in their solution state (in DMSO, 100 μM). (b) The normalized PL spectra in their solution (λex = 500 nm). (b′) The CIE diagram at their solution state. (c) Normalized PL spectra at 99% fw in DMSO (λex = 500 nm). (c′) The CIE plot represents the aggregated-state emission color. (d) Normalized solid state PL spectra (λex = 500 nm). (d′) CIE plot represents the solid state emission color. (e) Comparison of aggregated-state and solid-state fluorescence spectra of the RPNI-O and RPNI-S derivatives (λex = 500 nm). Capturing of digital photographs (f) under daylight and (g) under UV irradiation (λex = 365 nm) of solid powder of RPNI-O derivatives (PPI, THPI, and API) and RPNI-S (PPIS, THPIS, and APIS), respectively. [a and s represent the aggregated and solid state, respectively, inset in CIE: 1, 2, 3, 4, 5, 6 = PPI, THPI, API, PPIS, THPIS, and APIS, respectively. In the (c′ and d′) CIE plot, points 4, 5, and 6 representing the PPIS, THPIS, and APIS were absent due to their non-emissive characteristics]. | |
Table 1 Photophysical and photosensitizing characteristics of RPNI-O and RPNI-S derivatives
| Materials |
λ
abs (nm) |
λ
ex (nm) |
λ
em (nm) |
λ
em (nm) |
λ
em (nm) |
Stokes shift (nm) |
Φ
PL
|
E
g (eV) |
Φ
Δ
|
ΔEST (eV) |
SOC (cm−1) |
|
λ
abs,max.
λ
ex.
λ
em,max represents absorption, excitation, and emission wavelength maxima in the solution state.
Aggregated and solid state λem.max.
Stokes-shift calculated between solid λem.max and λex.max.
Φ
PL quantum yield in the aggregated state.
E
g gaseous-state band-energies.
Φ
Δ singlet-oxygen quantum yield.
ΔEST is the first singlet and triplet state energy gap.
Spin–orbit coupling between singlet and triplet-states [concentration: 100 μM of RPNI-O and RPNI-S derivatives in DMSO solution (solution-state), 99% fw in DMSO (aggregated state), G = not observed].
|
|
|
|
|
|
|
|
|
|
|
|
|
PPI
|
506 |
500 |
580 |
646 |
646 |
146 |
0.10 |
2.60 |
0.015 |
1.19 |
3.81 |
|
THPI
|
510 |
500 |
655 |
664 |
670 |
170 |
0.04 |
2.51 |
0.019 |
0.65 |
3.50 |
|
API
|
517 |
500 |
559 |
614 |
646 |
146 |
0.85, 0.89 |
2.70 |
0.017 |
1.92 |
3.13 |
|
PPIS
|
590 |
550 |
655 |
G |
G |
G |
G |
2.19 |
0.04 |
0.40 |
10.88 |
|
THPIS
|
590 |
550 |
647 |
G |
G |
G |
G |
2.09 |
0.34 |
0.42 |
9.20 |
|
APIS
|
636 |
630 |
705 |
G |
G |
G |
G |
2.22 |
0.26 |
0.39 |
5.73 |
The obtained results provide strong evidence that the presence and positional manipulation of the S-atom within the pendant functional unit played a crucial role in controlling its unique AIE properties.22–25 Conversely, substituting the S-atom at the carbonyl position leads to distinct characteristics in the condensed state luminescence. Furthermore, all the RPNI-O compounds exhibited huge Stokes shifts, ranging from 146 to 170 nm (Table 1). This is noteworthy as it represents the rarest instance of such significant Stokes shifts reported in the literature. These shifts are attributed to an excited-state intramolecular charge transfer (ICT) between the electron donor and acceptor within the dye molecule.25 The substantial Stokes shifts make these materials highly suitable for bio-imaging applications, as they minimize interference between excitation and emission signals.26,27
ACQ-to-AIEE transformation
To explore the influence of aggregation on the fluorescence emission of RPNI-O and RPNI-S derivatives, UV-vis and photoluminescence (PL) spectra were measured at different water fractions (fw) in DMSO (Fig. 2, S1, S4, S5, Tables 1 and S4–S6†). RPNI-O derivatives (PPI, THPI, and API) displayed distinct absorption peaks at 506 nm, 517 nm, and 490 nm, respectively, characteristic of the π–π* transition associated with the substituted PNI-O core. API showed an additional long-wavelength absorption peak at 510 nm, attributed to the ICT process from the AIE donor to the acceptor PNI-O core. The consistent red-shifted UV and PL spectra in more polar solvents with increasing donor functional groups across RPNI-O derivatives suggest a more substantial ICT effect (Fig. S5, Tables S5 and S6†).28 However, API exhibited lesser red-shifted UV and PL spectra than other RPNI-O derivatives, indicating reduced D–A interaction and decreased electronic communication in its diluted state. This results in a blue-shifted λabs,max in API, distinguishing it from other derivatives. Significant alterations in absorption spectra of PPI and THPI occurred at 99% fw, with substantial blue shifts of 60 nm and 46 nm, respectively, indicating H-type aggregation and a strong π–π stacking planar core (Fig. S4a, b and Table S4†). On the other hand, as shown in Fig. S4c and c′† at 0% fw, API displayed characteristic peaks at 490 nm and 510 nm, attributed to the π–π* transition and the ICT peak from the thianthranyl segment to the acceptor PNI-O core, respectively.29–31 As the fw increased to 99%, the λabs,max at 490 nm gradually red-shifted by 5 nm, accompanied by a red shift of the ICT peak from 510 nm to 536 nm, intensifying more. This red-shifted λabs,max indicates the emergence of J-type aggregation, likely facilitated by the unique intermolecular arrangement of the thianthranyl unit in API (Fig. S4c, c′ and Table S4†).32–36 In contrast to the RPNI-O derivatives, the RPNI-S derivatives exhibited red-shifted λabs,max, with wavelengths of 590 nm, 590 nm, and 636 nm for PPIS, THPIS, and APIS, respectively, at 0% fw. However, with an increase in water content to 99%, these derivatives displayed remarkable blue shifts in their λabs,max, leading to 89 nm, 76 nm, and 23 nm blue shifts, all attributed to the H-type aggregation phenomenon (Fig. S4d–f and Table S4†). This marks the highest reported blue-shifted λabs,max in the aggregated state (Table S2†). The literature has reported the significance of H-aggregation in effectively capturing and stabilizing triplet excited states.37–39 This highlights the significant impact of the S-atom positional strategy at the functional unit and carbonyl position, as they drastically alter the absorption properties. It is intriguing to note that even though API and APIS share the same electronic structure, a simple modification involving the introduction of the S-atom at the carbonyl position led to a profound change in their aggregation behavior. In Fig. 2, at 0% fw, RPNI-O (PPI, THPI, and API) showed distinct λem,max peaks at 580 nm, 655 nm, and 559 nm, respectively (Fig. 2a–c, a′–c′, a′′–c′′ and S1†). With increasing fw, the emission intensity of PPI and THPI decreased, becoming completely quenched at 99% fw, accompanied by a progressive red shift in their λem,max to 646 nm and 664 nm, respectively, known as the ACQ phenomenon. Conversely, API exhibited different behavior, with λem,max (located at 559 nm) initially decreasing from 0% to 20% fw due to twisted intramolecular charge transfer (TICT) characteristics.28 At 40% fw, API reached maximum emission intensity, with red-shifted λem,max centered at 646 nm, signifying typical AIEE features. However, with more water added, emission intensity decreased due to agglomeration, as illustrated in the supramolecular self-assembly section. The abnormal ACQ behavior of PPI and THPI in their aggregated state highlights the significant role of the S-atom at the functional unit in RPNI-O, energized by intermolecular orientation and packing arrangement.22,23 In contrast, the RPNI-S derivatives exhibited non-emissive characteristics in their solution and aggregated state (Fig. 2d–f, d′–f′, d′′–f′′ and S1†). When diluted, RPNI-S derivatives such as PPIS, THPIS, and APIS exhibited λem,max peaks at 655 nm, 647 nm, and NIR (705) nm, respectively. However, the emission colors were markedly distinct from those of RPNI-O derivatives, rendering them undetectable. Furthermore, RPNI-S displayed an entirely undetectable λem,max in the aggregated state. This lack of emission can be attributed to the increased SOC and a higher population of excited state triplets.14,15
 |
| | Fig. 2 PL spectra of (a) PPI, (b) THPI, (c) API, (d) PPIS, (e) THPIS, and (f) APIS at various fw in DMSO (100 μM, λex = 500 nm, for PPI, THPI and API, respectively, λex = 550 nm for PPIS and THPIS, and λex = 630 nm for APIS, respectively). (a′–f′) plots of λem,max along with insets: (a′′–f′′) digital photographs under 365 nm UV illumination of the corresponding luminogens at 0%, 40%, and 99% fw in DMSO and chemical structure of the respective RPNI-O and RPNI-S luminogens, daylight photographs of all the derivatives have been placed in Fig. S6 of the ESI.† | |
Excitation–emission matrix spectra
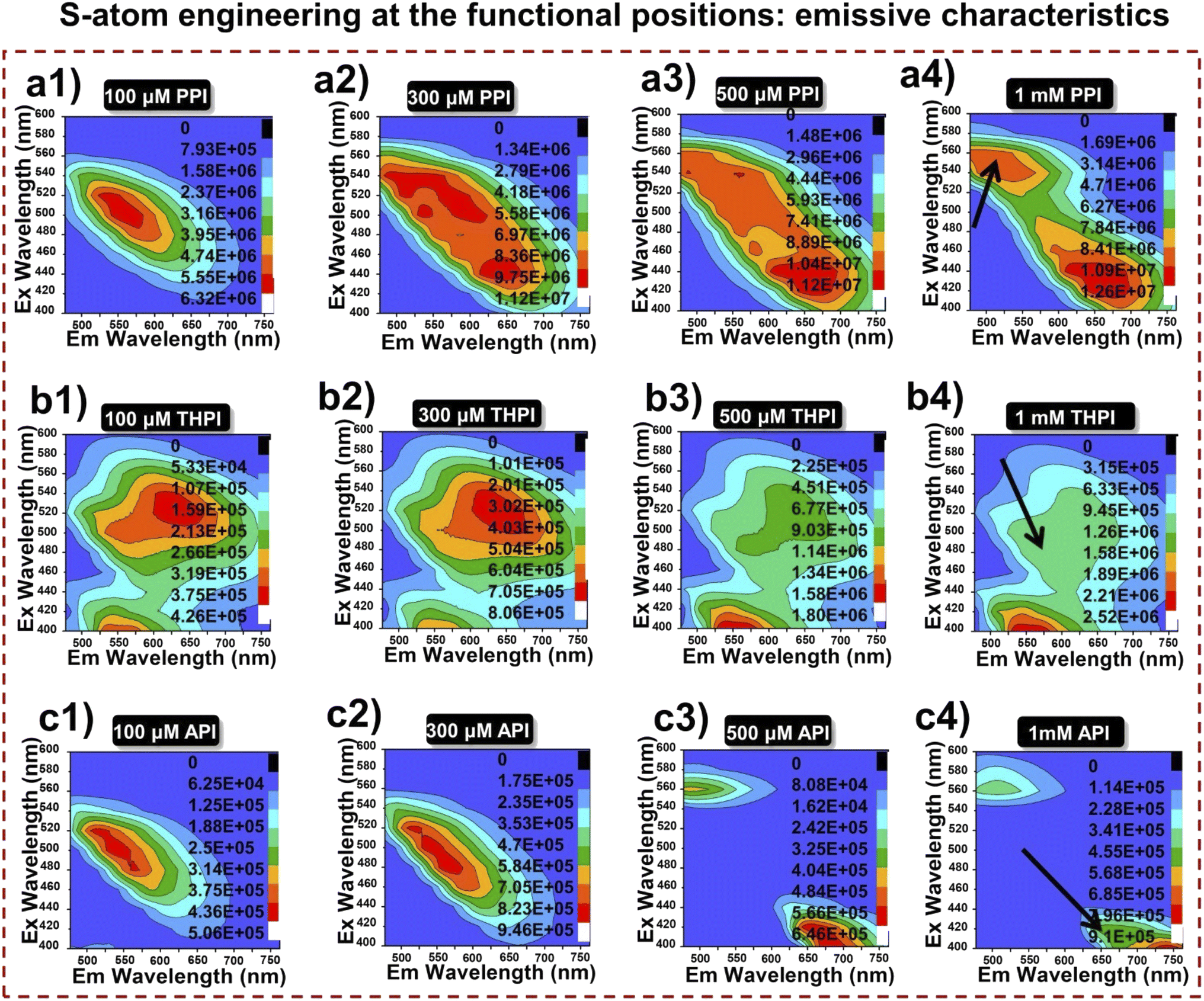
A comparative investigation of the photophysical characteristics of RPNI-O and RPNI-S derivatives in their condensed state was conducted using EEM spectra (Fig. 3 and S7†). This analysis involved solutions with varying concentrations, providing insights into their optical properties. EEM spectroscopy is recognized for characterizing systems with multiple fluorophores, with each spectrum offering valuable spectral information about chemical components in a mixture.40 Two-dimensional (2D) EEM spectra were collected for each compound at concentrations of 100 μM (diluted), 300 μM, 500 μM, and 1 mM (concentrated) to examine the effect of concentration on aggregation. For RPNI-O derivatives, excitation wavelengths were scanned from 400 to 600 nm in 10 nm increments, with emission values recorded from 500 to 750 nm. Conversely, RPNI-S experienced scanning from 500 to 700 nm for excitation, and emission values were measured within 600 to 850 nm. At 100 μM concentration, RPNI-O derivatives (PPI and API) exhibited distinct asymmetric peaks in their EEM spectra, while THPI showed partially separated peaks. With increasing concentrations (300 μM and 500 μM), PPI and THPI displayed broadening in their contour profiles along the Y-axis (representing λex), indicating the presence of multiple fluorophore components. Conversely, API showed two fluorophore systems responding differently to different λex, one at higher and the other at lower λex. PPI and THPI exhibited a red-shifted λex fluorophore system at higher concentrations, suggesting increased conjugation and enhanced electronic communication between pendant functional units and the PNI-O core.40 In a concentrated solution (1 mM), PPI and THPI showed weak emission with a red shift in λex. At the same time, API exhibited a highly emissive system with a blue-shifted λex and a non-fluorescent species at higher λex. API displayed a unique blue-shifted λex fluorophore system in its condensed state, attributed to reduced molecular conjugation influenced by the twisted structure of the thianthranyl group within the PNI-O core, verified further by SCXRD analysis. RPNI-S derivatives displayed weak fluorescence emission at lower concentrations, which diminished completely in a concentrated solution of 1 mM. THPIS exhibited a non-emissive fluorophore system with a blue-shifted λex compared to PPIS and APIS. The emission properties were influenced by intermolecular packing interactions and steric constraints imposed by S-atom substitution and positional manipulation at the pendant functional unit in RPNI-O. The introduction of the S-atom at the carbonyl position in RPNI-O played a critical role in determining non-fluorescent behavior at higher concentrations, differentiating emission characteristics of RPNI-S from those of RPNI-O derivatives.
 |
| | Fig. 3 (a1–c4) 2D EEM contour projections of RPNI-O luminogens at different concentrations (left: 100 μM; middle: 300 μM and 500 μM; right: 1 mM) in DMSO [inset: arrow represents the fluorescence intensity]. | |
Supramolecular self-assembly
The luminescent properties of RPNI-O and RPNI-S derivatives were investigated using field emission scanning electron microscopy (FESEM) (Fig. 4 and S8†). The FESEM images of the spontaneously formed self-assembled RPNI-O and RPNI-S derivatives were obtained using a simple, reliable, cost-effective drop-casting method, followed by air-drying at room temperature. Morphological analyses were carried out on the supramolecular assemblies formed at 99% fw to understand better the fluorescence behavior resulting from aggregation. The remarkable variation in morphological transformation emphasized the exceptional tunability of these RPNI-O and RPNI-S derivatives, allowing for a wide range of structural possibilities. RPNI-O derivatives like PPI and THPI formed spherical nanoparticles with sizes of 105 nm and 100 nm, respectively, as confirmed by dynamic light scattering (DLS) measurements (Fig. 4a, b, a′ and b′). API exhibited unique microrose self-assembly structures, leading to the highest ΦPL generation of 0.85, confirming the discovery of the highest reported ΦPL to date. It is worth noting that the existing literature reports the highest ΦPL value of 0.99 and 0.92, respectively (measurement was conducted in the solution state using an organic solvent such as toluene and dichloromethane as the solvent, respectively), unlike the present work performed in an aqueous environment (aggregated state).14,15 The unique intermolecular packing arrangement in API was determined to be the primary factor contributing to these distinct characteristics (Fig. 4c). RPNI-S derivatives, including PPIS and THPIS, exhibited self-assembly in the form of microcrystalline rods. At the same time, APIS displayed a different pattern characterized by micro-agglomeration (Fig. 4d–f). The degree of order in supramolecular self-assembly was directly correlated with the intensity of their emission in the aggregated state.41–43 In previous literature, it was hypothesized that the decrease in fluorescence emission of AIEgenic materials at higher fw was due to the formation of agglomerates resulting from stronger stacking interactions.44 Furthermore, existing literature extensively supports the essential role of energy-dependent endocytosis in the uptake of micro-sized particles. Additionally, numerous studies have highlighted the significant influence of particle morphology on their selective uptake within the intracellular environment.45,46
 |
| | Fig. 4 (a–f) The FESEM images of the micro and nano-assembly of RPNI-O and RPNI-S derivatives spontaneously formed at 99.9% fw in DMSO (100 μM). Insets: the chemical structure of RPNI-O and RPNI-S (left) and the magnified image at the selected area (right) and left for PPI, and (a′ and b′) hydrodynamic radius vs. intensity plot of PPI and THPI at 99.9% fw in DMSO (100 μM). | |
SCXRD analysis
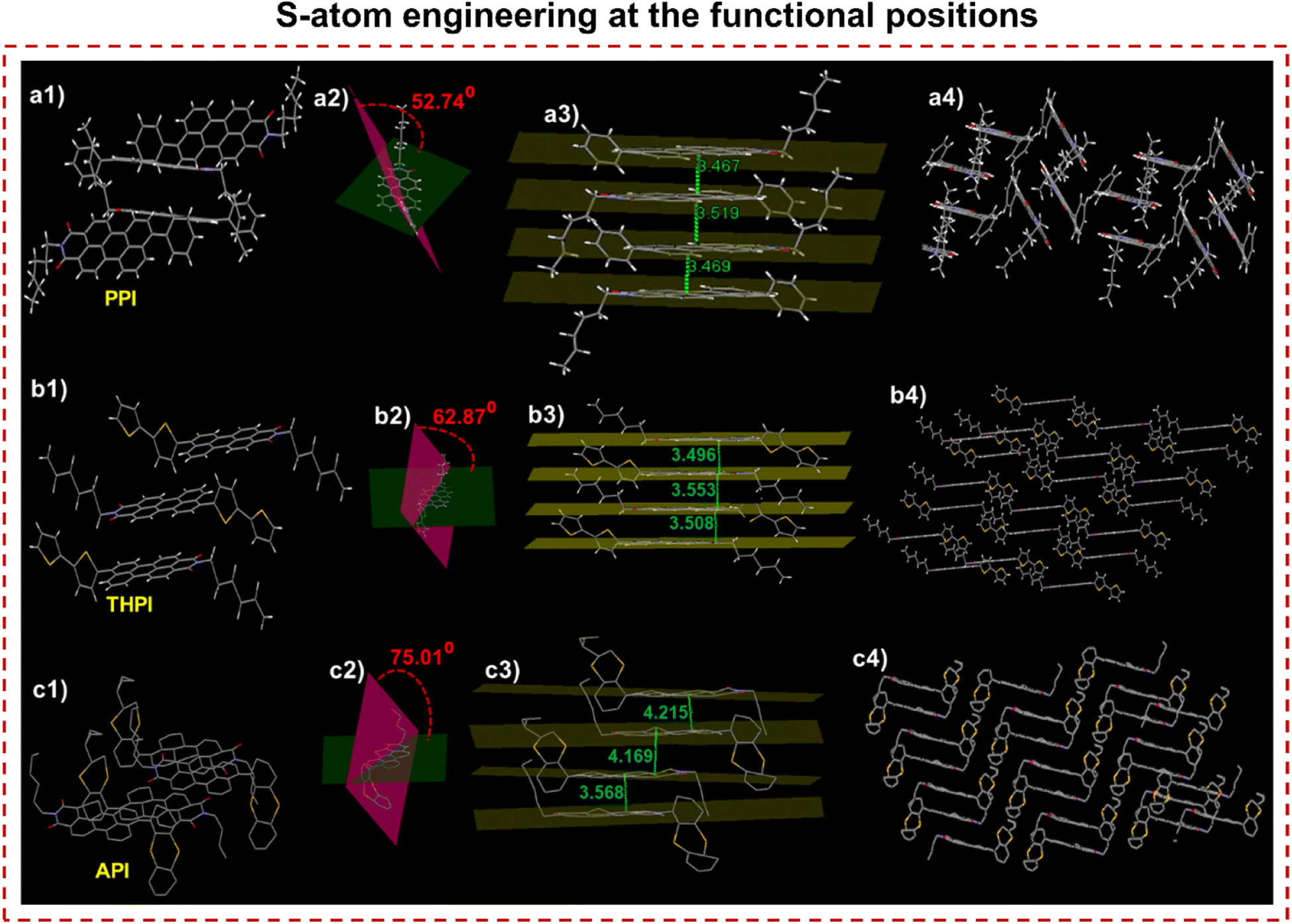
The effects of positional S-substitution within the functional unit were investigated to understand the tuning and triggering characteristics of RPNI-O derivatives in the condensed state. This exploration involved a comprehensive analysis of intermolecular interactions and packing arrangements by examining a single crystal (Fig. 5, S9 and Table S7†). PPI, THPI, and API were successfully crystallized from DMF, providing valuable single crystals for analysis. However, attempts to obtain single crystals of other RPNI-S derivatives have been unsuccessful. The S-substituted API, containing two S atoms within the one fused aromatic ring, exhibits a notably increased intermolecular π–π stacking distance of 4.169 Å compared to other RPNI-O derivatives. This phenomenon can be attributed to the highly twisted structure of the pendant thianthranyl unit, as reflected by the larger dihedral angle of 75.01° between the thianthranyl segment and the planar PNI-O core. Consequently, this twisted conformation of the thianthranyl segment enlarges the intermolecular distance by increasing the prominent steric hindrance of the planar PNI-O core. As a result, the emission quenching in the aggregated state can be prevented due to the remarkably reduced intermolecular π–π stacking interaction.25,47–50 In contrast, the remaining RPNI-O derivatives exhibited robust π–π interactions, likely attributed to their lower dihedral angles between the functional groups and the planar perylene core.25 Previous research has extensively documented that electron-donating substituents weaken these detrimental π–π interactions by increasing the π-electron density of the aromatic core.51,52 In contrast, THPI, which includes a bithiophene electron-donor functional group with two S-atoms present in two distinct aromatic rings, exhibited a contrary trend. Surprisingly, despite the electron-donating nature of the functional group, THPI demonstrated stronger π–π interactions. Moreover, the angle (θ) between the transition dipoles and the interconnected axis was 75.51° for PPI and 55.12° for THPI. These values exceed the critical threshold of 54.7°, indicating the presence of H-aggregates and further supporting the observed blue-shifted λabs,max. Conversely, for API, the θ value of 42.08° aligns with the findings of a red-shifted λabs,max, further supporting J-type aggregation (Fig. S9†).38
 |
| | Fig. 5 Single crystal structure of (a1) PPI, (b1) THPI, and (c1) API confirmed via SCXRD. (a1–c1) Insight of molecular packing arrangement. (a2–c2) Dihedral angle between two planes of the rotor and acceptor plane. Green plane: perylene core plane, pink plane: the plane of the rotor (phenyl, bithiophene, and thianthranyl in crystals of PPI, THPI, and API). (a3–c3) Intermolecular plane distances and (a4–c4) different molecular packing organization exhibiting J- and H-aggregation patterns, respectively. | |
This confirms that the strategic manipulation of the S-atom position at the functional unit plays a crucial role in generating distinct intermolecular packing orientations, forming unique and well-defined supramolecular assemblies. Consequently, this manipulation of intermolecular packing orientations leads to the corresponding modulation of their condensed state properties.53
Electronic properties driving the photophysical behavior
To validate the hypothesis concerning the distinct electronic properties of RPNI-O and RPNI-S derivatives and their impact on photophysical behavior, a computational analysis using density functional theory (DFT) using the B3LYP/6-31G(d,p) method was conducted (Fig. 6a) in a gaseous state. This analysis aimed to gain deeper insights into these derivatives’ electronic structure and characteristics. The investigation demonstrated that RPNI-S derivatives exhibited a significant stabilization of the LUMO and, simultaneously, a destabilization of the HOMO compared to RPNI-O. Consequently, the decrease in the energy gap (Eg) between the HOMO and LUMO from 2.70 to 2.09 eV supports the observed more pronounced red-shifted λabs,max.14,15 The presence of its electron-donating bithiophene group primarily contributed to THPIS exhibiting the smallest Eg among the derivatives. Conversely, API exhibited higher Eg compared to other RPNI-O derivatives, attributed to its more stabilized HOMO and less stabilized LUMO. This confirmation accounts for the blue-shifted λabs,max observed in API compared to PPI and THPI in a diluted state. The blue shift indicates reduced electronic communication, resulting from API’s more twisted thianthranyl moiety acting as a weaker donor.25 These results suggest that the S-atom positional engineering prominently influences the electronic properties and modulates its distinct photophysical behavior.
 |
| | Fig. 6 (a) Frontier molecular orbitals (HOMO and LUMO) with energies in eV calculated from the DFT/B3LYP method using the 6-31G(d,p) basis set by Gaussian 16 software in their ground state. Below is the chemical structure of the respective luminogens. (b and c) SOCME was calculated by the SOC-TDDFT method using ORCA 5.0 software for all the derivatives (RPNI-O: PPI, THPI, and API, respectively, and RPNI-S: PPIS, THPIS, and APIS, respectively). [PPI: ΔES1–T1 = 1.19 eV, ΔES2–T4 = 0.19 eV, ξ (S1, T1) = 0.3 cm−1, and ξ (S2, T4) = 3.53 cm−1; THPI: ΔES1–T1 = 0.98 eV and ξ (S1, T1) = 0.65 cm−1; API: ΔES1–T1 = 1.01 eV, ΔES2–T2 = 0.23 eV ξ (S1, T1) = 1.92 cm−1, and ξ (S2, T2) = 2.36 cm−1; PPIS: ΔES1–T1 = 0.40 eV, ΔES2–T4 = 0.096 eV, ξ (S1, T1) = 8.33 cm−1, and ξ (S2, T4) = 10.88 cm−1; THPIS: ΔES1–T1 = 0.42 eV, ΔES2–T4 = 0.094 eV, ξ (S1, T1) = 6.90 cm−1, and ξ (S2, T4) = 9.20 cm−1; APIS: ΔES1–T1 = 0.39 eV, ΔES2–T4 = 0.098 eV, ξ (S1, T1) = 4.44 cm−1, and ξ (S2, T4) = 5.73 cm−1]. | |
Theoretical calculation
The conversion of RPNI-O to RPNI-S led to remarkable fluorescence quenching, motivating further theoretical calculations on the optimized structures of RPNI-O and RPNI-S to understand the triplet state formation mechanism (Fig. 6b, c, Tables 1 and S8–S14†). The spin–orbit coupling matrix elements (SOCMEs) were calculated using ORCA 5.0 at the B3LYP/DEF2-SVP level, ensuring a reasonable TD-DFT error. Replacing oxygen with sulfur significantly increased SOC constants and decreased the singlet–triplet energy gap (ΔEST) for RPNI-S. The SOC from the S2 state to the T4 state was identified as the dominant contributor to the formation of the excited triplet population. Notably, SOC increased as the electron-donating ability of the –R groups increased. The ΔEST between the S2 and the nearest T4 states reduced gradually from APIS (0.098 eV) to PPIS (0.096 eV) and further to THPIS (0.094 eV), facilitating the efficient ISC process with SOC values of 10.88 cm−1, 9.20 cm−1, and 5.73 cm−1 for PPIS, THPIS, and APIS, respectively. Large SOC values were also observed between the lowest state transition of S1 to T1 for RPNI-S derivatives, with values of 8.33 cm−1 for PPIS, 6.90 cm−1 for THPIS, and 4.44 cm−1 for APIS with the lowest ΔEST values of 0.4 eV. Additionally, large SOC values were also found for PPIS between the transition of S7 to T7 (9.16 cm−1) and THPIS of S7 to T7 (6.19 cm−1), with ΔEST values of 0.23 eV and 0.25 eV, respectively (Tables S8 and S9†).39,54,55 It has been documented that H-aggregates efficiently trap and stabilize the excited state triplet through strong π–π coupling.37–39 Extensive research has focused on enhancing triplet excitons through photoinduced electron transfer or intramolecular charge transfer.56,57 Enhancing SOC through prominent H-aggregation represents a unique and innovative approach within this domain. Extensive research has consistently affirmed that H-aggregation significantly extends the excited electron's lifetime by suppressing the fluorescence, likely attributed to singlet wavefunction delocalization, which is subsequently energized by the strong π–π coupling. This extension affords sufficient time for the electrons to effectively engage in substantial SOC, which crucially enables the efficient ISC process.37–39 Interestingly, despite the presence of a less electron-donating phenyl group in PPIS compared to the bithiophene in THPIS, the SOC value of PPIS was found to be greater. The significant blue-shifted λabs,max corroborates the presence of H-aggregation. This difference can be attributed to the more prominent H-aggregation in PPIS compared to THPIS (Fig. S4, S9, Tables S2 and S4†). Unfortunately, single crystals for the RPNI-S derivatives could not be obtained. Nonetheless, a similar type of H-aggregation is anticipated to be observed as with PPI and THPI, with RPNI-S having a larger θ value, making the H-aggregation even more remarkable. RPNI-S is derived from RPNI-O through a minor modification involving substituting oxygen (O) atoms with S-atoms at the carbonyl position of the RPNI-O (Fig. S9†). Notably, Table S2† demonstrates that RPNI-S derivatives exhibited significant blue-shifted λabs,max in the aggregated state compared to the previously reported materials.38,39,55 Herein, the traditional triazine derivative (DPhCzT) and organoboronium (OB4)4 material demonstrate a distinct stabilization of the triplet excited state through H-aggregation, leading to the generation of ultralong phosphorescence.38,39 This research represents the crucial findings regarding the efficient generation of SOC through pronounced H-aggregation. It is widely explored that SOC plays a pivotal role across various fields, including achieving persistent PL,58 developing distinct light-emitting materials and devices,59,60 realizing ultralong room temperature phosphorescence,61 and development of efficient PSs.14,15 Extensive research has been devoted to enhancing SOC values in organic compounds, often achieved through strategies such as introducing heavy-atoms3 or by enhancing the D–A effect.62–64 In contrast, this study represents a cost-effective and rare design strategy to enhance SOC by incorporating S-atoms at the carbonyl position, avoiding costly heavy-atom incorporation. Importantly, this marks a significant instance of an elevated SOC value compared to previously explored PSs and other conventional organic materials, presented in Table S14a† (it is worth noting that distinct computational methods were employed for each study). In comparison, the reported dibenzofuran compound (DPBF) achieved a SOC value of 20.33 cm−1, but it is vital to note that this was based on theoretical studies conducted on aggregated molecules.61 On the other hand, the heavy-atom-free NI and PDI-4S PSs exhibited notably high SOC values of 27.8 and 85 cm−1, respectively. Importantly, these values were obtained using a distinct computational methodology, considering significantly more complex molecular systems.14,15 In contrast, this study unveils a precise and simple design strategy to enhance SOC through S-atom introduction at the carbonyl position, emphasizing the rare mechanism of SOC enhancement via promising H-aggregation, which could be energized by the distinct magnitude of the excited triplet state stabilization and prolonging the excited state electron's lifetimes.
Furthermore, the excited state singlet and triplet energies and the corresponding ΔES1–T1 values were lower in RPNI-S than RPNI-O (Fig. 6b, c, Tables 1, S8–S14 and S14b†). In contrast, RPNI-O demonstrated the highest state transition comprising a lower SOC value, which is considered less favorable from a quantum mechanical perspective under identical conditions.
Singlet oxygen (type 2 ROS) detection
Inspired by the remarkable SOC and abundant excited state triplet population in RPNI-S, an investigation into their ΦΔ was conducted using ABDA as a commercially available probe for quantifying 1O2 (Fig. 7a, S10, S11, S12a–c, S13b–d and eqn (S2)†). Table 1 presents the findings, demonstrating that RPNI-S displayed moderate ΦΔ for PPIS (0.04), THPIS (0.34), and APIS (0.26). In contrast, the signal for 1O2 was virtually undetectable for all RPNI-O derivatives tested under the same conditions. Notably, the ΦΔ showed a slight increase with an increase in the electron-donating ability of the –R groups. Thus, the lower ΦΔ indicates reduced type-II processes for THPIS and APIS and a complete absence of 1O2 generation for PPIS.
 |
| | Fig. 7 (a) Plots of ABDA (100 μM) degradation rates at λabs,max of 378 nm by the various PSs (100 μM). (b) Plot of fluorescence intensities of the DCFDA indicator in the presence of RPNI-O and RPNI-S derivatives. A0 and A are ABDA absorbance at λabs,max of 378 nm, where I0 and I are the PL intensities of the indicator at λem,max of 521 nm before and after irradiation of white light, respectively. (c and d) ESR signals of RPNI-O and RPNI-S derivatives in their solid state without using any trapping agent and TEMP for the 1O2 characterization in the presence of RPNI-O and RPNI-S derivatives in their aggregated state. (e) PL spectra of TA in the presence of RPNI-S derivatives at 528 nm. (f) Zeta potential versus pH values of RPNI-S derivatives. (g) EC and EV of RPNI-S derivatives at pH 5.6. The energy scale is expressed concerning NHE. EH values for H2O/HO˙, 1O2/O2, and O2/O2˙− are 2.2, 1.88, and −0.2 eV, respectively. (h) Gibbs free energy changes of (a′) PPIS, (b′) THPIS, and (c′) APIS, respectively, via the ORCA 5.0 SOCME module at the B3LYP DEF2-SVP level [aggregated-state: 99% PBS fraction in DMSO, fPBS, and [RPNI-S] or [RPNI-O] = 100 μM]. | |
Total ROS generation evaluation
In response to the lower ΦΔ, an evaluation of the overall ROS generation capabilities of RPNI-S derivatives was conducted (Fig. 7b, S12d–f, S13a and e–g†). For this purpose, a fluorescent indicator, 2,7-dichlorodihydrofluorescein (DCFDA), known for detecting various types of ROS, was utilized. Under white light irradiation for different durations, DCFDA, in the presence of RPNI-S materials, exhibited a gradual increase in PL intensity in their aggregated state (99% PBS fraction in DMSO, fPBS). Interestingly, as the donor functionality increased, the efficiency of ROS generation also increased. After 30 min of light irradiation, DCFDA showed approximately 39-fold, 40-fold, and 29-fold increase in PL intensity in the presence of PPIS, THPIS, and APIS, respectively (Fig. 7b). These results indicate the involvement of both type-I and type-II processes in the ROS generation of RPNI-S derivatives. Particularly noteworthy is the remarkable enhancement in PL intensity after just 5 min of white light irradiation, using just 10 μM of DCFDA: PPIS, THPIS, and APIS exhibited enhancements of 2.5 × 105, 1 × 106, and 2 × 105, respectively, while RPNI-O derivatives showed lower enhancements of PL intensity (Fig. S13a and e–g†). These findings highlight the rapid and highly efficient production of ROS observed in the THPIS PS. This efficiency is attributed to the promising donor bithiophene unit in THPIS, setting it apart from other RPNI-O and RPNI-S derivatives. Consequently, THPIS displays the smallest ΔEST value, facilitating a highly efficient triplet population in the aggregated state.
Type 1 ROS evaluation
To assess the ability of RPNI-S to generate free radicals, an electron spin resonance (ESR) experiment was conducted using 2,2,6,6-tetramethyl-4-piperidinol (TEMP) which is widely used as the 1O2 trapper (Fig. 7c and d).54,65,66 The experiments were performed on RPNI-S and RPNI-O derivatives in their condensed state (99% fPBS) under white light irradiation. No apparent ESR signal was observed upon white light irradiation, which confirmed the absence of the singlet oxygen generation ability (Fig. 7d). However, the observation of the ESR signal for RPNI-S even without the presence of any trapping agent in the solid state indicates the free radical generation ability and further confirms the effective stabilization of the triplet state (Fig. 7c). This stabilization is attributed to the notable H-aggregation phenomenon exhibited by these derivatives, which leads to the efficient energization of the triplet state, subsequently enhancing its stability.37–39 In contrast, no ESR signals were observed for RPNI-O derivatives under the same conditions. Building upon the type-I process observations, terephthalic acid (TA) was chosen as a fluorescence probe to detect the HO˙ radical (Fig. 7e and S14a†).5,55 The fluorescence intensity of RPNI-S probes showed no significant change when TA solutions were subjected to white light irradiation, indicating that RPNI-S did not produce the HO˙ radical. These findings align with the RPNI-S derivatives' electrochemical properties and support the type-I PDT mechanism, wherein O2˙− is generated through electron transfer from the triplet excited states to 3O2. Upon evaluating the ROS generation capabilities of these RPNI-S derivatives under white light irradiation, the band edge energy levels were estimated using EHOMO and ELUMO (Fig. 7f, g, S14b, c, S15, S16, Tables S15–S17 and eqn (S3)–(S6)†).55 The points of zero zeta potential for PPIS, THPIS, and APIS were determined to be 2.68, 2.18, and 4.35, respectively (Fig. 7f and Table S15†). The valence band energy (EV) was determined from X-ray photoelectron spectroscopy (XPS) data (Fig. S15 and Table S17†). The conduction band energy (EC) was estimated from the Eg obtained from the onset absorption data from UV-visible spectroscopy. It was referenced to the normal hydrogen electrode (NHE) (Fig. S16, Tables S16 and S17†).55 As a result, the EC values of PPIS, THPIS, and APIS were found to be −0.74228, −0.83178, and −0.96375 eV at a pH of 5.6, respectively. These values were lower than the redox potential (EH) of O2/O2˙− (−0.2 eV) (Fig. 7g and Table S17†). Consequently, all three RPNI-S derivatives could transfer electrons to oxygen and generate O2˙−, making them suitable PSs for type-I PDT. However, the EV values of the RPNI-S derivatives (0.92772 for PPIS, 0.777822 for THPIS, and 0.656225 eV for APIS, respectively) are smaller than the EH values of H2O/HO˙ (2.20 eV) and 1O2/O2 (1.88 eV) at a pH of 5.6, suggesting that HO˙ and 1O2 cannot be generated. Consequently, the type-II PDT process is not feasible for RPNI-S derivatives (Fig. 7g and Table S17†). On the other hand, RPNI-O does produce slight ROS, following the type-I process in PDT, as validated by the experiments involving TA and band energy calculations for NHE (Fig. S14a, c and Table S17†). This result indicates that the driving force for generating O2˙− continuously improves with larger atom sizes of sulfur compared to oxygen.55
Furthermore, the feasibility of type-I PDT, specifically in generating O2˙− through the IET mechanism, was investigated by calculating the ΔG values using the ORCA 5.0 quantum mechanics package at the B3LYP/DEF2-SVP level (Fig. 7h and Tables S18–S25†).54,55Fig. 7h illustrates that the ΔG value between THPIS and O2 is −40.83 kcal mol−1, which is smaller than the ΔG values of PPIS and APIS (−0.548 and −10.26 kcal mol−1, respectively). This indicates a higher probability of the IET reaction for THPIS. This signifies the exceptionally lower recorded instance of a ΔG value of THPIS compared to previously reported PSs. For example, tetraphenylethene (TPE) based PS (TPE-PTB) showed a ΔG value of −14.3839 kcal mol−1. At the same time, the tellurium (Te) comprising PS (PTTe) exhibited a ΔG value of −32.16 kcal mol−1.54,55 Based on the theoretical calculations, THPIS is theoretically predicted to be the most exceptional type 1 PDT material among the three RPNI-S derivatives, efficiently generating O2˙− and slight 1O2. On the other hand, PPIS is the second most promising type-I PDT material among the RPNI-S derivatives, efficiently generating only O2˙−.5,54,55
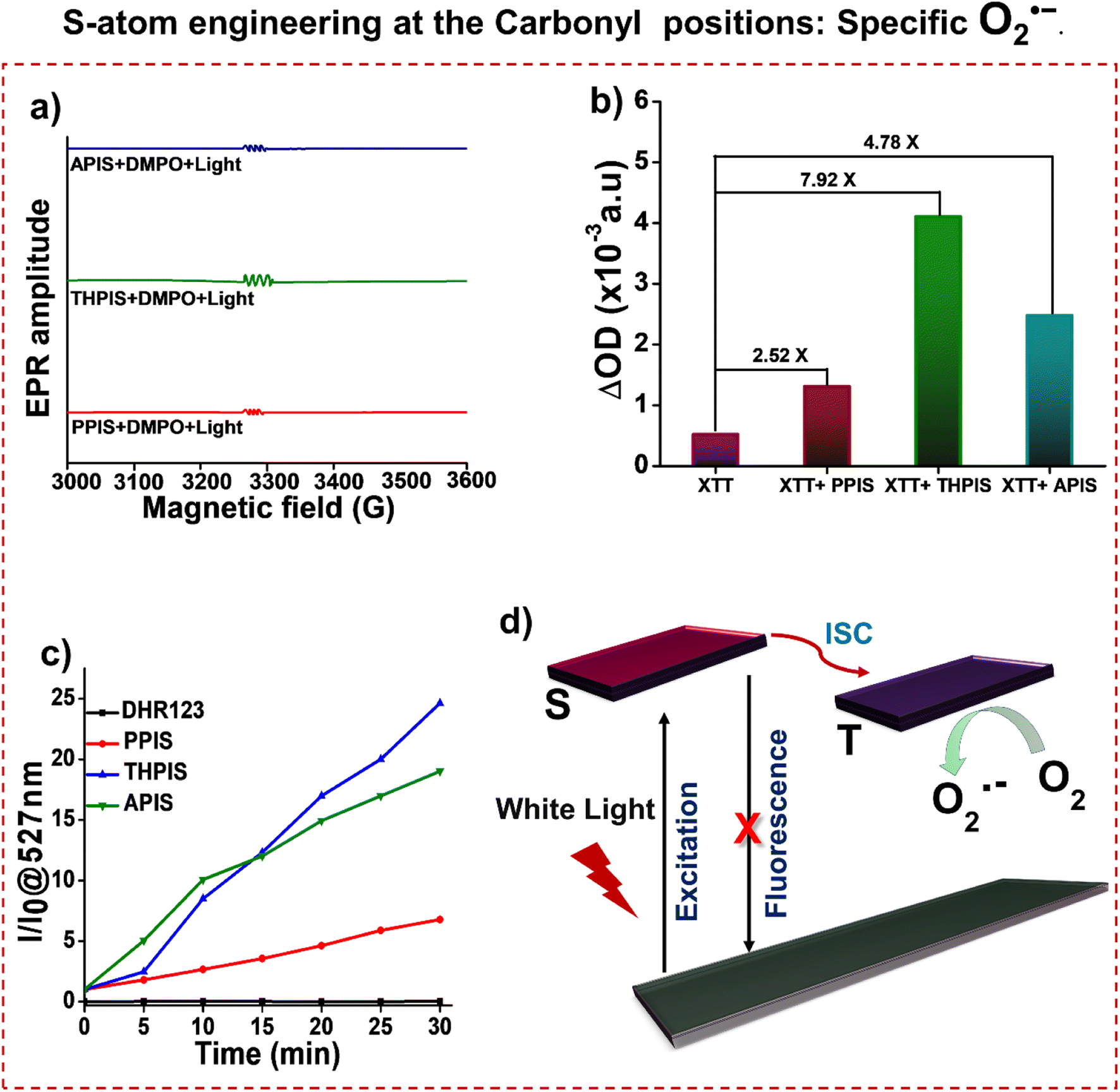
The O2˙− production was further confirmed through electron spin resonance (ESR) spectroscopy, employing 5,5-dimethyl-1-pyrroline-N-oxide (DMPO) as the spin-trapping agent. Upon exposure to white light irradiation in the presence of DMPO and RPNI-S PSs, the resulting ESR spectrum exhibited an obvious six-line ESR signal (Fig. 8a). These signals originate from the DMPO/O2˙− adduct.54,67,68 To further validate the O2˙− production ability of the three RPNI-S, 2,3-bis(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide (XTT) was utilized as an indicator for O2˙− detection. Note that the enhancement in absorption due to XTT formazan occurs upon the interaction of XTT with O2˙−. Fig. 8b and S17† showed that continuous enhancement in the absorption intensities of XTT at 470 nm was observed when PPIS, THPIS, or APIS PS were mixed with XTT and exposed to white light irradiation.69 The changes in probe XTT absorbance were modest but exceeded those documented in the relevant literature.55,70 Thus, it can be inferred that O2˙− are generated. Notably, the absorption intensity of pure XTT at 470 nm slightly increased. Further O2˙− production was confirmed using dihydrorhodamine 123 (DHR123), which, although non-fluorescent, reacts with O2˙− to emit intense green fluorescence centered at 527 nm.5,71 As illustrated in Fig. 8c and S18,† the PL intensity of DHR 123 in the presence of PPIS, THPIS, or APIS following 30 min of white light irradiation increased over 5-fold, 25-fold, and 20-fold, respectively, than before irradiation. This suggests a significant O2˙− generation efficiency for PPIS, THPIS, and APIS, respectively (Fig. 8d).
 |
| | Fig. 8 (a) ESR signals of DMPO for type-I ROS O2˙− characterization in the presence of PPIS, THPIS, or APIS after 10 min white light irradiation in the acetonitrile solution. (b) The UV-vis absorption spectra changes of XTT at 470 nm after being irradiated with white light for 30 min in the presence and absence of PPIS, THPIS, or APIS in the aggregated state. (c) DHR 123 for O2˙− detection in the presence of PPIS, THPIS, or APIS after white light irradiation in the aggregated state. (d) Schematic diagram of white light excitation of RPNI-S to produce O2˙− [aggregated state: 99% PBS fraction in DMSO, fPBS, and [RPNI-S] = 100 μM]. | |
Photodynamic therapy
Efficient ROS generation by RPNI-S derivatives prompted in vitro PDT studies on HeLa and MCF7 cancer cells, as well as normal cells (Fig. 9). Cellular uptake experiments revealed maximum enrichment of PPIS, THPIS, and APIS in HeLa cells after 4 hours of post-treatment (Fig. S19†).55RPNI-S derivatives exhibited superior cellular uptake towards HeLa cancer cells at 20 μM concentration while remaining non-toxic to normal cells even at concentrations up to 100 μM. Conversely, RPNI-O showed low cellular uptake at 20 μM concentration but was internalized by normal and HeLa cancer cells at 100 μM concentrations (Fig. S20†). Anticancer effects of RPNI-S (PPIS, THPIS, and APIS) were investigated in HeLa and MCF7 cancer cells, as well as normal cells, using the MTT assay under dark and light conditions (Fig. 9).14THPIS and PPIS significantly inhibited cell proliferation in HeLa cells with an IC50 of approximately 4.5 μM and 15.5 μM, respectively, under 20 minutes of white light irradiation (Fig. 9d–f). A PDT effect was observed in MCF-7 cells at higher concentrations (PPIS: ∼200 μM and THPIS: ∼75 μM) (Fig. 9a–c). Notably, THPIS and PPIS induced approximately 80% cell inhibition at 20 μM and 50 μM concentrations upon light exposure. At 100 μM concentration, THPIS exhibited complete cell inhibition, although it showed slight dark toxicity (Fig. 9d–f). APIS exhibited negligible cytotoxicity under both dark and light treatments due to its lower cellular uptake, regulated by its anomalous micro-aggregate morphology (Fig. 4f and S19†). RPNI-S derivatives showed minimal cytotoxicity towards normal cells in the absence and presence of light, attributed to their optimized cellular uptake mechanism (Fig. 9g–i), which has been strengthened by the precisely controlled morphology in aqueous media (Fig. 4d–f and S19†).45,46RPNI-O derivatives demonstrated minimal changes upon dark and light treatment towards normal and HeLa cancer cells (Fig. S21†). They predominantly exhibited dark toxicity towards HeLa cells with an IC50 of approximately 100 μM, suggesting reduced effectiveness in PDT due to decreased ROS production (Fig. S11, S13† and Table 1). Overall, RPNI-S derivatives show promise for broader biological applications due to their superior efficacy and minimal toxicity, supported by optimized cellular uptake mechanisms. Fluorescence microscopy imaging further confirmed the excellent photocytotoxicity of THPIS towards HeLa cells (Fig. 10 and 11), highlighting its potential as an exceptional PS. Ongoing research and optimizations hold the potential to enhance its efficacy further and extend its utility in cancer therapy and related fields. However, while PPIS has demonstrated the second most promising potential as a PDT material after THPIS, ongoing research in our laboratory is currently exploring this approach with APIS, utilizing liposomes to enhance its cellular uptake in a biological environment. This enhancement is expected to increase the efficacy of APIS PDT in cancer cells correspondingly.
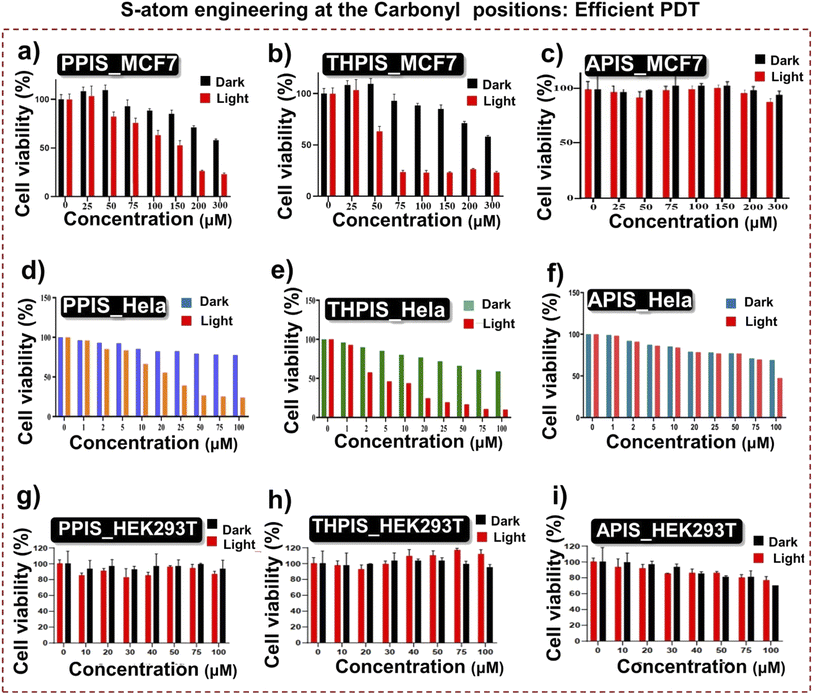 |
| | Fig. 9 (a–c) Cell viability of MCF 7 cells after treatment with different concentrations of (a) PPIS, (b) THPIS, and (c) APIS, respectively, along with white light irradiation. (d–f) Cell cytotoxicity of HeLa cells after treatment with different concentrations of (d) PPIS, (e) THPIS, and (f) APIS, respectively, along with white light irradiation. (g–i) Cell viability of HEK293Tcells after treatment with different concentrations of (g) PPIS, (h) THPIS, and (i) APIS, respectively, along with white light irradiation. | |
 |
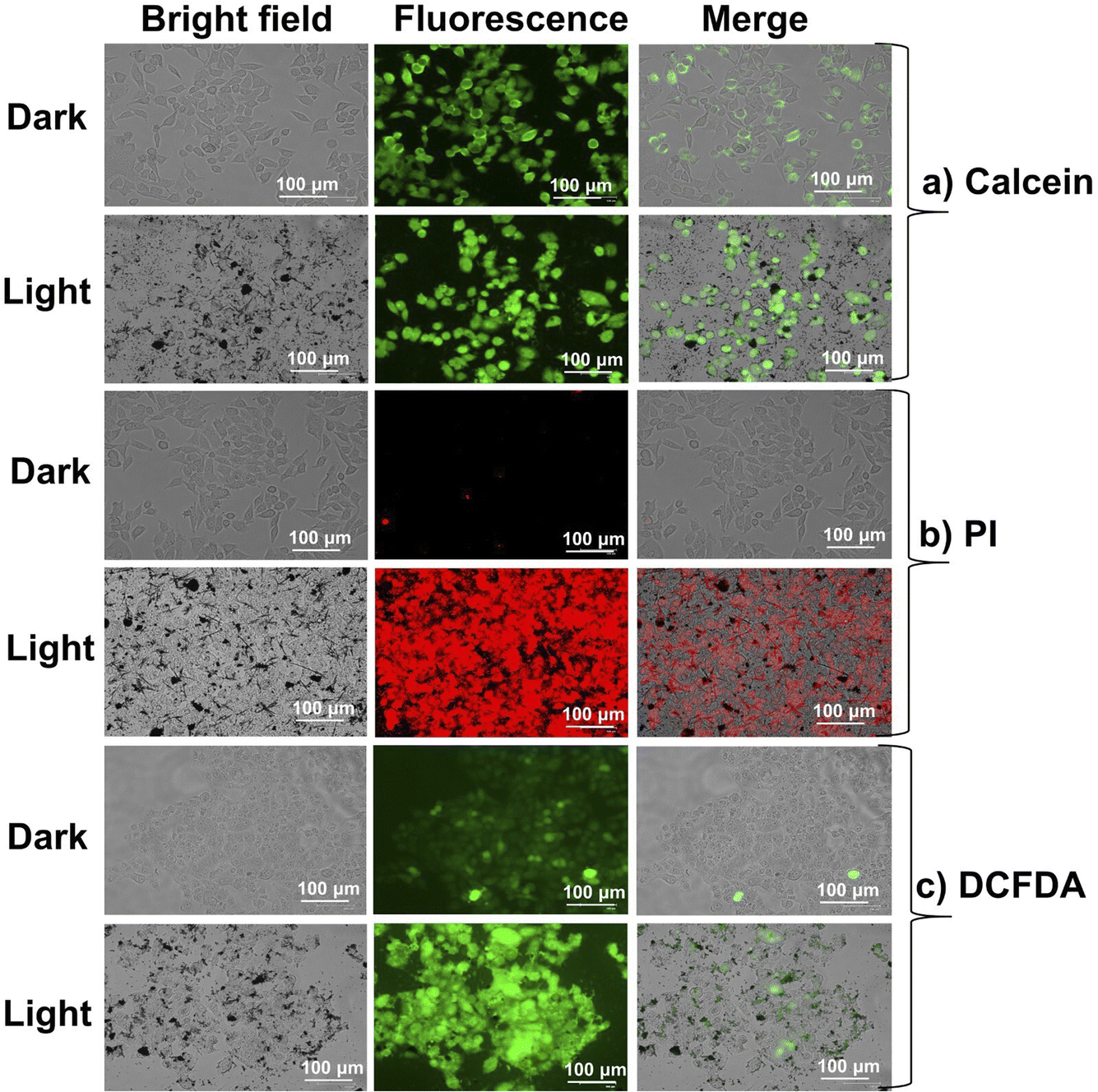
| | Fig. 10 Fluorescence microscope images of HeLa cells after treatment with PPIS under normoxia and loaded with (a) calcein-AM (2 μM, live cell marker), (b) PI (4 μM, dead cell marker), (c) DCFDA (10 μM, ROS detection probe) [concentration of PPIS: (50 μM), scale bar: 100 μm]. | |
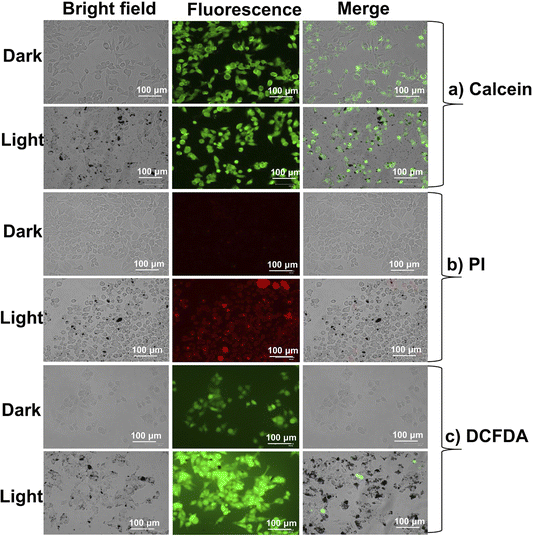
 |
| | Fig. 11 Fluorescence microscope images of HeLa cells after treatment with THPIS under normoxia and loaded with (a) calcein-AM (2 μM, live cell marker), (b) PI (4 μM, dead cell marker), and (c) DCFDA (10 μM, ROS detection probe) [concentration of THPIS: (50 μM), scale bar: 100 μm]. | |
Live cell/dead cell dual staining and intracellular ROS generation
The viability assay, visualized using calcein-AM/propidium iodide, clearly showed the impact of THPIS, PPIS, and APIS on HeLa cells upon white light irradiation (Fig. 10, 11 and S22–S24†).14 Additionally, intracellular ROS production was confirmed using DCFDA, where green fluorescence was a marker for generating total ROS (Fig. 10c and 11c). These results were consistent with the earlier findings (Fig. 7 and 8). Furthermore, the treated cells exhibited noticeable morphological changes, including reduced size, shrinkage, and cellular collapse under white light irradiation (Fig. 10a–c and 11a–c). These observations further substantiate that THPIS-induced photodynamic action enhances outcomes in photodynamic therapy for HeLa cells by inducing organelle destruction. This effect is likely amplified by the efficient accumulation of THPIS within HeLa cancer cells, which possess higher cell permeability than normal cells and MCF-7 cancer cells (Fig. S19†). This selective accumulation is facilitated by the precisely controlled morphology of THPIS in aqueous media (Fig. 4e and S19†).45,46 Thus, based on the theoretical and experimental studies regarding ROS generation efficacy, it is plausible to assert that the exceptional PDT efficacy observed in PPIS and THPIS can be attributed to their pronounced H-aggregation and effective accumulation towards cancer cells. This promising H-aggregation significantly amplifies the SOC and enhances the efficiency of the ISC process during PDT treatment (Fig. 6–8, S4, S19 and Table S2†).37–39 However, H-aggregation may limit the potential for condensed state applications, and this strategy showed how it could enhance the promising PDT application as well.22–24,72
Conclusion
This work suggests that meticulous modulation of the S-atom conformation can lead to enhanced SOC (10.88 cm−1), ΦPL (0.85), and the advancement of potent PSs. Precise incorporation of S-atoms at the pendant functional group in RPNI-O showed drastic photophysical properties: Stokes shift of 170 nm, a red-emitting AIEEgen, and established novel AIE mechanisms. Surprisingly, introducing S-atoms at the carbonyl position in RPNI-O results in the RPNI-S molecule, which exhibits unprecedented structure–property relationships (130 nm red-shifted λabs,max, a subsequent extension of λem,max into NIR, and ΦPL ≈ 0.00 in the aggregated state). Furthermore, RPNI-S established a novel SOC enhancement mechanism via prominent H-aggregation. Because the strong π–π coupling is intrinsic in H-aggregation, it efficiently traps and stabilizes the excited state triplet. RPNI-S exhibited an efficient IET reaction for type-I PDT, evidenced by an exceptionally lower ΔG value (THPIS: −40.83 kcal mol−1). Interestingly, the lower ΦΔ, ΔG, and energy levels suggest the efficient production of O2˙− and minimal type-II ROS (1O2) for THPIS. Herein, PPIS is very specific and mainly generates O2˙−. The biocompatible and very specific heavy-atom-free PPIS and THPIS PSs have been shown to have selective and efficient PDT efficacy under normoxia, which is exceptionally rare. Thus, this S-atom positional engineering provides a conceptually important perspective for the potential development of efficient PSs via pronounced H-aggregation and distinct condensed state emitters for unique applications.
Data availability
All the data has been included in the ESI file.†
Author contributions
MNK and PKI designed the experiments. MNK synthesized the materials and conducted all the experiments. MNK and PKI wrote and thoroughly revised the manuscript. SN and HR carried out the in vitro cellular studies. SN, HR, SK and SSG analysed the data. All authors discussed the results and contributed to the manuscript.
Conflicts of interest
The authors declare no conflicts of interest.
Acknowledgements
The authors extend their gratitude for financial support from the Department of Electronics & Information Technology, specifically DeitY project no. 5(9)/2012-NANO (Vol. II), the Department of Science and Technology (DST) with grant no. DST/SERB/EMR/2014/000034, and the DST-Max Planck Society, Germany, under grant no. IGSTC/MPG/PG(PKI)/2011A/48. Additionally, the authors express appreciation to the Centre for Nano-Technology and the Central Instruments Facility at IIT Guwahati for providing essential instrumental resources. The authors sincerely thank Prof. Aditya Narayan Panda (for his invaluable assistance with the intricate theoretical calculations) and Prof. Mohd Qureshi (for his significant help in conducting preliminary synthetic studies), whose expertise and guidance have been instrumental in shaping the direction and depth of this work.
References
- B. M. Luby, C. D. Walsh and G. Zheng, Angew. Chem., Int. Ed., 2019, 58, 2558–2569 CrossRef CAS PubMed
 .
.
- A. P. Castano, P. Mroz and M. R. Hamblin, Nat. Rev. Cancer, 2006, 6, 535–545 CrossRef CAS PubMed .
- S. Kolemen, M. Işık, G. M. Kim, D. Kim, H. Geng, M. Buyuktemiz, T. Karatas, X.-F. Zhang, Y. Dede, J. Yoon and E. U. Akkaya, Angew. Chem., Int. Ed., 2015, 54, 5340–5344 CrossRef CAS PubMed .
- Y. Cakmak, S. Kolemen, S. Duman, Y. Dede, Y. Dolen, B. Kilic, Z. Kostereli, L. T. Yildirim, A. L. Dogan, D. Guc and E. U. Akkaya, Angew. Chem., Int. Ed., 2011, 50, 11937–11941 CrossRef CAS PubMed .
- M. Li, J. Xia, R. Tian, J. Wang, J. Fan, J. Du, S. Long, X. Song, J. W. Foley and X. Peng, J. Am. Chem. Soc., 2018, 140, 14851–14859 CrossRef CAS PubMed .
- G. J. Kavarnos and N. J. Turro, Chem. Rev., 1986, 86, 401–449 CrossRef CAS .
- L. Shi, F. Hu, Y. Duan, W. Wu, J. Dong, X. Meng, X. Zhu and B. Liu, ACS Nano, 2020, 14, 2183–2190 CrossRef CAS PubMed .
- J. M. Brown and W. R. Wilson, Nat. Rev. Cancer, 2004, 4, 437–447 CrossRef CAS PubMed .
- M. Pollum, S. Jockusch and C. E. Crespo-Hernández, J. Am. Chem. Soc., 2014, 136, 17930–17933 CrossRef CAS PubMed .
- O. Reelfs, Y.-Z. Xu, A. Massey, P. Karran and A. Storey, Mol. Cancer Ther., 2007, 6, 2487–2495 CrossRef CAS PubMed .
- L. Martínez-Fernández, I. Corral, G. Granucci and M. Persico, Chem. Sci., 2014, 5, 1336–1347 RSC .
- K. M. Farrell, M. M. Brister, M. Pittelkow, T. I. Sølling and C. E. Crespo-Hernández, J. Am. Chem. Soc., 2018, 140, 11214–11218 CrossRef CAS PubMed .
- S. Mai, M. Pollum, L. Martínez-Fernández, N. Dunn, P. Marquetand, I. Corral, C. E. Crespo-Hernández and L. González, Nat. Commun., 2016, 7, 13077 CrossRef CAS PubMed .
- V.-N. Nguyen, S. Qi, S. Kim, N. Kwon, G. Kim, Y. Yim, S. Park and J. Yoon, J. Am. Chem. Soc., 2019, 141, 16243–16248 CrossRef CAS PubMed .
- C. Liu, C. Ji, Z. Fan, R. Ma and M. Yin, Chem. Commun., 2021, 57, 13126–13129 RSC .
- K. Pal, V. Sharma, D. Sahoo, N. Kapuria and A. L. Koner, Chem. Commun., 2018, 54, 523–526 RSC .
- N. Yang, S. Song, J. Ren, C. Liu, Z. Li, H. Qi and C. Yu, ACS Appl. Bio Mater., 2021, 4, 5008–5015 CrossRef CAS PubMed .
- C. Li and H. Wonneberger, Adv. Mater., 2012, 24, 613–636 CrossRef CAS PubMed .
- A. Jana, L. Bai, X. Li, H. Ågren and Y. Zhao, ACS Appl. Mater. Interfaces, 2016, 8, 2336–2347 CrossRef CAS PubMed .
- M. Hussain, J. Zhao, W. Yang, F. Zhong, A. Karatay, H. G. Yaglioglu, E. A. Yildiz and M. Hayvali, J. Lumin., 2017, 192, 211–217 CrossRef CAS .
- A. J. Tilley, R. D. Pensack, T. S. Lee, B. Djukic, G. D. Scholes and D. S. Seferos, J. Phys. Chem. C, 2014, 118, 9996–10004 CrossRef CAS .
- J. Mei, Y. Hong, J. W. Y. Lam, A. Qin, Y. Tang and B. Z. Tang, Adv. Mater., 2014, 26, 5429–5479 CrossRef CAS PubMed .
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed .
- N. Meher, S. Panda, S. Kumar and P. K. Iyer, Chem. Sci., 2018, 9, 3978–3985 RSC .
- K. Chen, R. Zhang, Z. Wang, W. Zhang and B. Z. Tang, Adv. Opt. Mater., 2020, 8, 1901433 CrossRef CAS .
- J. Luo, Z. Xie, J. W. Y. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhu and B. Z. Tang, Chem. Commun., 2001, 18, 1740–1741 RSC .
- B.-K. An, S.-K. Kwon, S.-D. Jung and S. Y. Park, J. Am. Chem. Soc., 2002, 124, 14410–14415 CrossRef CAS PubMed .
- S. Sasaki, G. P. C. Drummen and G.-I. Konishi, J. Mater. Chem. C, 2016, 4, 2731–2743 RSC .
- Y. Li, Z. Zhao, J. Zhang, R. T. K. Kwok, S. Xie, R. Tang, Y. Jia, J. Yang, L. Wang, J. W. Y. Lam, W. Zheng, X. Jiang and B. Z. Tang, Adv. Funct. Mater., 2018, 28, 1804632 CrossRef .
- S. Thazhathethil, T. Muramatsu, N. Tamaoki, C. Weder and Y. Sagara, Angew. Chem., Int. Ed., 2022, 134, e202209225 CrossRef .
- V. Sathish, A. Ramdass, Z.-Z. Lu, M. Velayudham, P. Thanasekaran, K.-L. Lu and S. Rajagopal, J. Phys. Chem. B, 2013, 117, 14358–14366 CrossRef CAS PubMed .
- D. G. Whitten, L. Chen, H. C. Geiger, J. Perlstein and X. Song, J. Phys. Chem. B, 1998, 102, 10098–10111 CrossRef CAS .
- S. J. Ananthakrishnan, E. Varathan, V. Subramanian, N. Somanathan and A. B. Mandal, J. Phys. Chem. C, 2014, 118, 28084–28094 CrossRef CAS .
- F. Tang, C. Wang, J. Wang, X. Wang and L. Li, ACS Appl. Mater. Interfaces, 2014, 6, 18337–18343 CrossRef CAS PubMed .
- B.-K. An, D.-S. Lee, J.-S. Lee, Y.-S. Park, H.-S. Song and S. Y. Park, J. Am. Chem. Soc., 2004, 126, 10232–10233 CrossRef CAS PubMed .
- S. Das, Y. Li, K. Junge and M. Beller, Chem. Commun., 2012, 48, 10742–10744 RSC .
- J. Yuan, R. Chen, X. Tang, Y. Tao, S. Xu, L. Jin, C. Chen, X. Zhou, C. Zheng and W. Huang, Chem. Sci., 2019, 10, 5031–5038 RSC .
- Z. An, C. Zheng, Y. Tao, R. Chen, H. Shi, T. Chen, Z. Wang, H. Li, R. Deng, X. Liu and W. Huang, Nat. Mater., 2015, 14, 685–690 CrossRef CAS PubMed .
- L. Yang, X. Wang, G. Zhang, X. Chen, G. Zhang and J. Jiang, Nanoscale, 2016, 8, 17422–17426 RSC .
- J. Dong, K. M. Solntsev and L. M. Tolbert, J. Am. Chem. Soc., 2009, 131, 662–670 CrossRef CAS PubMed .
- Y. Deng, M. Wang, Y. Zhuang, S. Liu, W. Huang and Q. Zhao, Light: Sci. Appl., 2021, 10, 76 CrossRef CAS PubMed .
- P. S. Marqués, M. Krajewska, B. D. Frank, K. Prochaska and L. Zeininger, Chem.–Eur. J., 2023, 29, e202203790 CrossRef PubMed .
- J. Ye, X. Huang, Y. Gao, X. Wang, T. Zheng, Y. Lin, X. Liu and G. Ning, CrystEngComm, 2015, 17, 9311–9317 RSC .
- M. Soni, S. K. Das, P. K. Sahu, U. P. Kar, A. Rahaman and M. Sarkar, J. Phys. Chem. C, 2013, 117, 14338–14347 CrossRef CAS .
- J. J. Rennick, A. P. R. Johnston and R. G. Parton, Nat. Nanotechnol., 2021, 16, 266–276 CrossRef CAS PubMed .
- D. Zhang, L. Wei, M. Zhong, L. Xiao, H.-W. Li and J. Wang, Chem. Sci., 2018, 9, 5260–5269 RSC .
- Q. Li, Y. Li, T. Min, J. Gong, L. Du, D. L. Phillips, J. Liu, J. W. Y. Lam, H. H. Y. Sung, I. D. Williams, R. T. K. Kwok, C. L. Ho, K. Li, J. Wang and B. Z. Tang, Angew. Chem., Int. Ed., 2020, 59, 9470–9477 CrossRef CAS PubMed .
- T. Zhang, J. Zhang, F.-B. Wang, H. Cao, D. Zhu, X. Chen, C. Xu, X. Yang, W. Huang, Z. Wang, J. Wang, Z. He, Z. Zheng, J. W. Y. Lam and B. Z. Tang, Adv. Funct. Mater., 2022, 32, 2110526 CrossRef CAS .
- D. Wang, M. M. S. Lee, G. Shan, R. T. K. Kwok, J. W. Y. Lam, H. Su, Y. Cai and B. Z. Tang, Adv. Mater., 2018, 30, 1802105 CrossRef PubMed .
- Y. Li, J. Zhuang, Y. Lu, N. Li, M. Gu, J. Xia, N. Zhao and B. Z. Tang, ACS Nano, 2021, 15, 20453–20465 CrossRef CAS PubMed .
- X. Zhang, L. Du, W. Zhao, Z. Zhao, Y. Xiong, X. He, P. F. Gao, P. Alam, C. Wang, Z. Li, J. Leng, J. Liu, C. Zhou, J. W. Y. Lam, D. L. Phillips, G. Zhang and B. Z. Tang, Nat. Commun., 2019, 10, 5161 CrossRef PubMed .
- F. Cozzi, M. Cinquini, R. Annuziata and J. S. Siegel, J. Am. Chem. Soc., 1993, 115, 5330–5331 CrossRef CAS .
-
A. S. Davydov, in Theory of Molecular Excitons, ed. A. S. Davydov, Springer US, Boston, MA, 1971, pp. 153–243 Search PubMed .
- Y. Li, R. Tang, X. Liu, J. Gong, Z. Zhao, Z. Sheng, J. Zhang, X. Li, G. Niu, R. T. K. Kwok, W. Zheng, X. Jiang and B. Z. Tang, ACS Nano, 2020, 14, 16840–16853 CrossRef CAS PubMed .
- K. Wen, H. Tan, Q. Peng, H. Chen, H. Ma, L. Wang, A. Peng, Q. Shi, X. Cai and H. Huang, Adv. Mater., 2022, 34, 2108146 CrossRef CAS PubMed .
- M. A. Filatov, S. Karuthedath, P. M. Polestshuk, H. Savoie, K. J. Flanagan, C. Sy, E. Sitte, M. Telitchko, F. Laquai, R. W. Boyle and M. O. Senge, J. Am. Chem. Soc., 2017, 139, 6282–6285 CrossRef CAS PubMed .
- Z. E. X. Dance, S. M. Mickley, T. M. Wilson, A. B. Ricks, A. M. Scott, M. A. Ratner and M. R. Wasielewski, J. Phys. Chem. A, 2008, 112, 4194–4201 CrossRef CAS PubMed .
- Y. Dou, C. Demangeat, M. Wang, H. Xu, B. Dryzhakov, E. Kim, T. Le Bahers, K.-S. Lee, A.-J. Attias and B. Hu, Nat. Commun., 2021, 12, 3485 CrossRef CAS PubMed .
- Z. He, W. Zhao, J. W. Y. Lam, Q. Peng, H. Ma, G. Liang, Z. Shuai and B. Z. Tang, Nat. Commun., 2017, 8, 416 CrossRef PubMed .
- J. Li, Q. Yao, L. Wu, Z. Hu, B. Gao, X. Wan and Q. Liu, Nat. Commun., 2022, 13, 919 CrossRef CAS PubMed .
- H. Ma, Q. Peng, Z. An, W. Huang and Z. Shuai, J. Am. Chem. Soc., 2019, 141, 1010–1015 CrossRef CAS PubMed .
- M. Wang, T. Chatterjee, C. J. Foster, T. Wu, C.-L. Yi, H. Yu, K.-T. Wong and B. Hu, J. Mater. Chem. C, 2020, 8, 3395–3401 RSC .
- Z. Yang, Z. Zhang, Y. Sun, Z. Lei, D. Wang, H. Ma and B. Z. Tang, Biomaterials, 2021, 275, 120934 CrossRef CAS PubMed .
- R. Singh, D.-G. Chen, C.-H. Wang, C.-C. Wu, C.-H. Hsu, C.-H. Wu, T.-Y. Lai, P.-T. Chou and C.-T. Chen, J. Mater. Chem. B, 2022, 10, 6228–6236 RSC .
- Y. Wan, G. Lu, W.-C. Wei, Y.-H. Huang, S. Li, J.-X. Chen, X. Cui, Y.-F. Xiao, X. Li, Y. Liu, X.-M. Meng, P. Wang, H.-Y. Xie, J. Zhang, K.-T. Wong and C.-S. Lee, ACS Nano, 2020, 14, 9917–9928 CrossRef CAS PubMed .
- X. Shi, S. H. P. Sung, J. H. C. Chau, Y. Li, Z. Liu, R. T. K. Kwok, J. Liu, P. Xiao, J. Zhang, B. Liu, J. W. Y. Lam and B. Z. Tang, Small Methods, 2020, 4, 2000046 CrossRef CAS .
- J. Wang, Y. Hou, W. Lei, Q. Zhou, C. Li, B. Zhang and X. Wang, ChemPhysChem, 2012, 13, 2739–2747 CrossRef CAS PubMed .
- P. Xiao, Z. Shen, D. Wang, Y. Pan, Y. Li, J. Gong, L. Wang, D. Wang and B. Z. Tang, Adv. Sci., 2022, 9, 2104079 CrossRef CAS PubMed .
- Y. Li, W. Zhang, J. Niu and Y. Chen, ACS Nano, 2012, 6, 5164–5173 CrossRef CAS PubMed .
- L. Brunet, D. Y. Lyon, E. M. Hotze, P. J. J. Alvarez and M. R. Wiesner, Environ. Sci. Technol., 2009, 43, 4355–4360 CrossRef CAS PubMed .
- J. S. Nam, M.-G. Kang, J. Kang, S.-Y. Park, S. J. C. Lee, H.-T. Kim, J. K. Seo, O.-H. Kwon, M. H. Lim, H.-W. Rhee and T.-H. Kwon, J. Am. Chem. Soc., 2016, 138, 10968–10977 CrossRef CAS PubMed .
- Z. Yan, H. Xu, S. Guang, X. Zhao, W. Fan and X. Y. Liu, Adv. Funct. Mater., 2012, 22, 345–352 CrossRef CAS .
Footnote |
| † Electronic supplementary information (ESI) available: Materials, instrumentation, methods, synthetic procedures, and characterization data (including multinuclear NMR and MALDI-TOF). Further experimental and computational data, including Schemes S1–S4, Fig. S1–S24, supporting Fig. S25–S48 and Tables S1–S25. CCDC 2325822, 2325824 and 2325826. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc01180e |
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Satyendu
Nandy
a,
Satyendu
Nandy