
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTransition metal-free decarboxylative olefination of carboxylic acid salts†
Ebbin
Joseph
,
Deshkanwar S.
Brar
,
Gaven
Stuhlsatz
and
Jon A.
Tunge
 *
*
Department of Chemistry, The University of Kansas, 1567 Irving Hill Road, Lawrence, Kansas, USA. E-mail: tunge@ku.edu
First published on 15th May 2024
Abstract
The cost-effective and efficient synthesis of alkenes is highly significant due to their extensive applications in both synthetic and polymer industries. A transition metal-free approach has been devised for the chemoselective olefination of carboxylic acid salts. This modular approach provides direct access to valuable electron-deficient styrenes in moderate to good yields. Detailed mechanistic studies suggest anionic decarboxylation is followed by halogen ion transfer. This halogen transfer leads to an umpolung of reactant electronics, allowing for a rate-limiting rebound elimination.
Introduction
Alkenes are appealing, low-cost synthetic precursors. While unfunctionalized olefins are easily obtained in bulk quantities from petrochemical feedstocks,1 the efficient and low-cost synthesis of electronically varied alkenes is of great interest because of their widespread application in synthesis and polymerization.2 The rising demand and escalating financial costs of petroleum products3 have emphasized the necessity of synthesizing functionalized alkenes from more oxidized, readily available intermediates, including those obtained from biomass. Carboxylic acids are particularly attractive oxidized feedstocks since a large number of carboxylic acids are prepared on an industrial scale and are commercially available.4Decarboxylation is a burgeoning strategy for forming reactive intermediates that avoids preformed organometallic reactants by leveraging the extrusion of carbon dioxide, an innocuous by-product, to form carbanions, carbocations, and radicals (Scheme 1).5 Since the pioneering work of Kolbe demonstrating the formation of carbon-centered radicals through decarboxylation from carboxylic acids,6 decarboxylative strategies have been shown to provide access to a large variety of functionally interesting products.7 Classically, the barton decarboxylation allows for the direct access of alkanes and arenes from carboxylic acids,8 and the Hunsdiecker–Borodin reaction allows for the direct conversion of silver carboxylates into halides.9 More recently, coupling reactions have utilized decarboxylative metalation to generate organometallics that couple to form new C–C, C–N, C–S, and C–O bonds, providing alternatives to traditional cross-coupling reactions.7a,10 The decarboxylative elimination of carboxylic acids to produce alkenes, on the other hand, still represents a relatively underexplored reaction class.
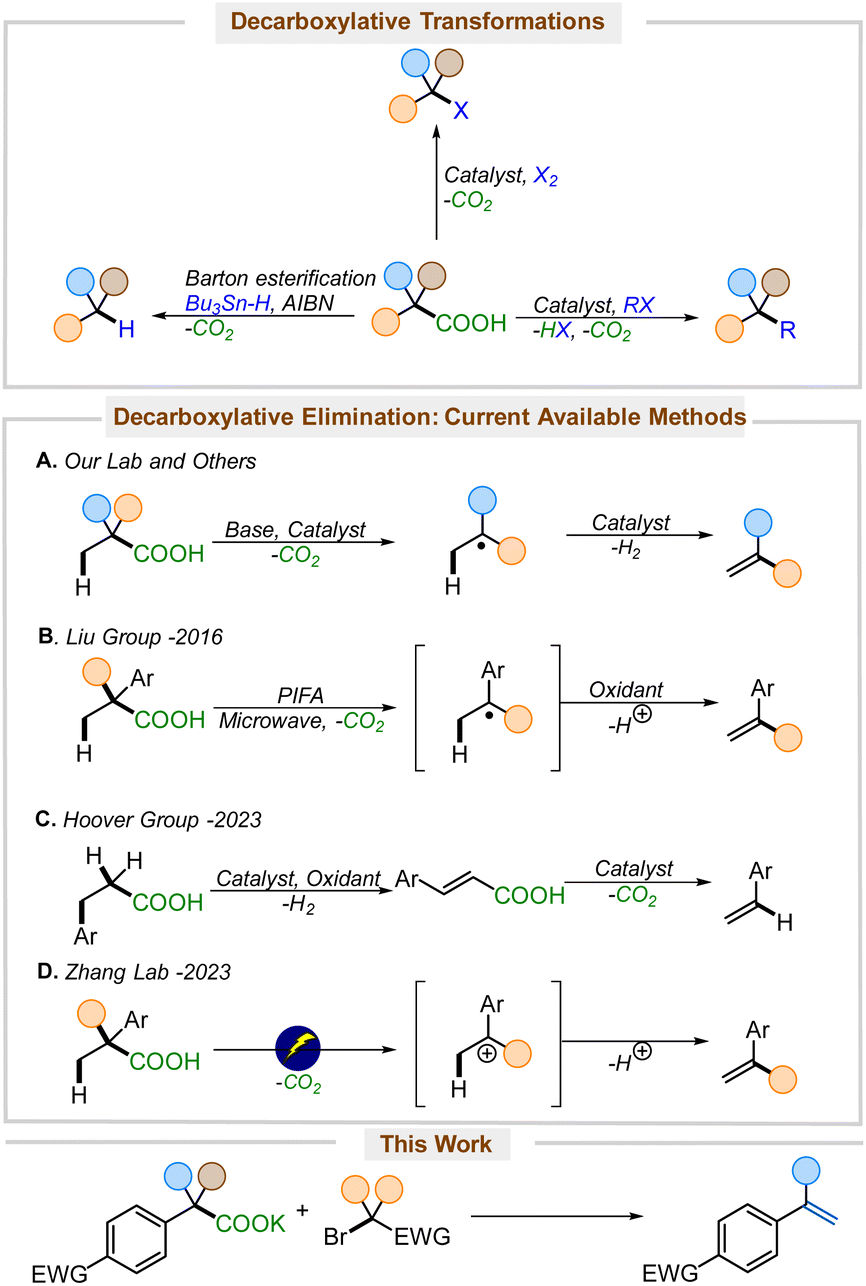 | ||
| Scheme 1 Decarboxylative transformations. | ||
Kochi pioneered the direct conversion of carboxylic acids into olefins, although the method was limited by the requirement of a stoichiometric lead oxidant and forcing reaction conditions.11 Building upon Kochi's work, more reports have emerged demonstrating more efficient and practical versions of decarboxylative elimination.12 Our lab and others have developed a decarboxylative-dehydrogenation strategy for the direct conversion of carboxylic acids to alkenes (Scheme 1A).12b,13 However, these methods rely on a single-electron-transfer pathway, which often requires expensive iridium-based14 or specialized photocatalysts,15 that also engage styrenes and thus are often incompatible with the formation of styrene products. In 2016, Liu and co-workers reported a metal-free microwave-assisted approach for the synthesis of styrenes from carboxylic acids (Scheme 1B).12a Later in 2023, the Hoover group developed a copper-catalyzed decarboxylative elimination of carboxylic acids to styrenes (Scheme 1C).16 These important contributions do, however, suffer from several drawbacks. PIFA is a relatively high molecular weight oxidant that promotes the formation of radicals and cations, and thus is not effective for the generation of electron-deficient styrenes;12a electrochemical decarboxylative elimination is similarly limited (Scheme 1D).12e,f In contrast, copper-catalyzed elimination works well for the synthesis of nitrostyrenes, but it also involves intermediate radicals and is mechanistically limited by the need for benzylic deprotonation with LiOAc.16
We set forth to develop a metal-free reagent for decarboxylative elimination that would be compatible with the formation of electron-deficient styrenes. Here, we envisioned an anionic intermediate, generated by thermal decarboxylation, could undergo oxidation via halogen ion transfer.17 The resulting umpoled intermediates would be poised for elimination to furnish styrenes (Scheme 2). The key to unlocking this methodology would lie in the selection of an appropriate reagent that would initially serve as a halogen ion source, generating a stabilized anion, which could rebound to effect an E2-elimination. While there are numerous reports of decarboxylative halogenation reactions in the literature,8a,18 none leverage this one-pot halogenation-rebound elimination approach starting directly from carboxylic acids.
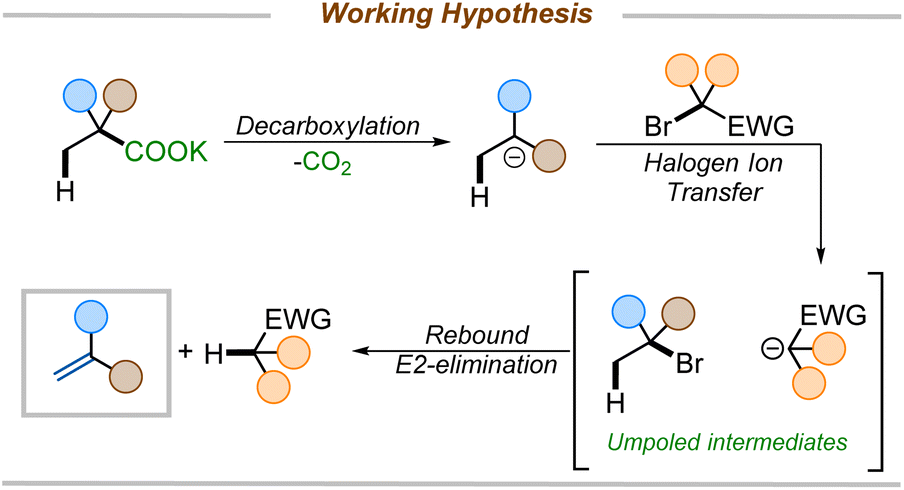 | ||
| Scheme 2 Our working hypothesis. | ||
Results and discussion
Initial optimizations
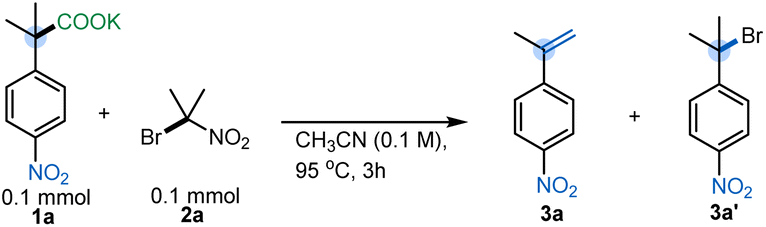
To evaluate the feasibility of umpolung elimination, we initiated our investigations by screening reagents that would serve as the halogen ion donor. We chose NBS as our initial reagent, and fortunately we were able to observe the corresponding styrene 3a in 39% yield (Table 1). We hypothesized that the low yield was due to the relatively low basicity of the succinimide anion (conj. acid pKa 14.7) formed after the initial halogen ion transfer. With this in mind, 2-bromo-2-nitropropane19 (nitronate pKaca. 17) was chosen as the halogen ion transferring reagent,17,20 and styrene 3a was furnished in 77% yield. Interestingly, a further increase in the conjugate basicity of the halogen source resulted in a preferential substitution rather than the expected elimination. Specifically, when ethyl 2-bromoisobutyrate (pKaca. 29) was utilized, hindered substitution product 4a was isolated in good yield (Table 1).| a Yields determined by quantitative 1H NMR analysis using anisole as a standard. Reaction conditions: 1a (1 equiv), reagent (1 equiv), CH3CN (0.1 M), 95 °C, 3 h. |
|---|
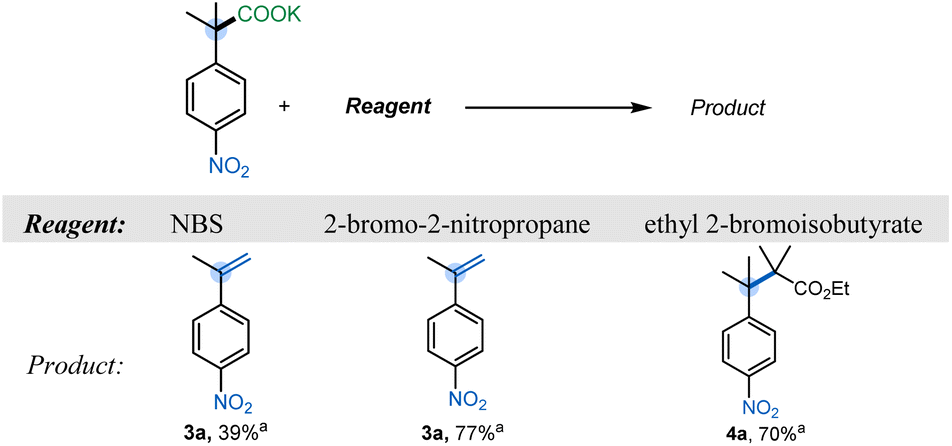
|
With elimination conditions in hand, control studies confirmed the necessity of bromo nitropropane 2a for effective reaction (Table 2A, entry 2). Acetonitrile was found to be the best solvent for the reaction, although dry MeCN was necessary for the efficient formation of 3a (Table 2A, entries 3 & 4). Furthermore, a more dilute reaction concentration led to drastic yield decrement (Table 2A, entry 5). Additionally, we found that the temperature and reaction time had a significant influence on the yield of the decarboxylative elimination reaction. For example, running the reaction for shorter periods or at lower temperatures resulted in a considerable amount of the brominated product 3a′ (Table 2A, entries 6 & 7). Furthermore, increasing the loading of 2a to 1.2 equiv with respect to the carboxylate salt gave the best results: 83% isolated yield of α-methyl-4-nitrostyrene (Table 2A, entry 8). Additionally, different counter cations of 1a were screened, although K+ was found to be better compared to Cs+, Na+ or Li+ (see Table S6, ESI†). Moreover, these conditions provide superior results compared to the state-of-the-art photochemical13c or microwave12a methods for synthesizing electron-deficient styrenes from carboxylic acids (Table 2B, entries 10 & 11) (see ESI† for more details). Only the copper-catalyzed elimination is comparable (Table 2B, entry 12), but that reaction requires isomeric β-nitroaryl acids and has lower atom economy.16
| A: Optimizations | |||
|---|---|---|---|
| a Yields determined by quantitative 1H NMR analysis using anisole as a standard. Numbers in parenthesis are isolated yields. b 0.12 mmol of 2a instead of 0.1 mmol. | |||
|
|||
| Entry | Variation in conditions | % Yielda | |
| 1 | — | 77:– | |
| 2 | No 2a | — | |
| 3 | DMF, DCM, THF instead of MeCN | <36:– | |
| 4 | Wet CH3CN | 60:– | |
| 5 | 0.05 M instead of 0.1 M | 39:– | |
| 6b | 90 min instead of 3 h | 73:10 | |
| 7 | 70 °C instead of 95 °C | 47:27 | |
| 8 | 0.12 mmol of 2a | 88(83):– | |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||
| B: State-of-the-art-conditions | |||
| Entry | Elimination | Reaction conditions | % Yield (3a)a |
| 9 | This work | 2a (1.2 equiv), CH3CN, 95 °C, 3 h | 88(83) |
| 10 | Co/Acr hν, (ref. 13c) | [Co] cat., [Acr] cat., STAB cat., Na2CO3 cat., H2O cat., MeOH, blue LED, rt, 18 h | 6 |
| 11 | PIFA μW, (ref. 12a) | PIFA (1.2 equiv), CH3CN, μW 120 °C, 20 min | — |
| 12 | Cu/oxidant, (ref. 16) | [Cu] cat., [bpy] cat., MnO2 (2 equiv), LiOAc (2 equiv), 120 °C, 24 h | 89(80) |
Scope of decarboxylative elimination
With the optimized conditions in hand, a series of substituted styrenes were made starting from different carboxylate salts (Scheme 3). Several different 4-nitrophenylacetic acid salts bearing alkyl or aryl substituents in the benzylic position were screened under our reaction conditions. Remarkably, in all cases the alkene formation was chemoselective. An electron-rich vinyl ether (3c), often prone to electrochemical or photochemical oxidation,21 was readily synthesized using the current method. Additionally, a wide range of substituted 1,1-diphenylethylenes was also rapidly accessed using the current protocol (3d–3h). The internal alkene, resulting from an α,α-diethyl carboxylate provided product in excellent yield, but resulted in a stereoisomeric mixture of alkene (3i). Carboxylate salts bearing other important functional groups such as nitrile (3j; 91%), keto (3k; 71%), and ester groups (3l; 76%) were all tolerated well under the reaction conditions, furnishing the internal alkenes selectively. Notably, as the steric bulk of the substituent increased, the Z alkene isomer diminished, providing the E alkene exclusively (3j–l). Interestingly, the ethyl-substituted salt 1m gave a regioisomeric mixture of the internal and external olefin (76:24) along with the E and Z isomers (89:11) for the internal alkene (see ESI† for details). However, as the chain length increased, the selectivity for the internal to the external alkene decreased (3n and 3o). While a steric influence in the elimination is clear, a slight electronic bias resulted in controlled formation of internal alkenes. Specifically, alkyl chains with more acidic hydrogens (3j–3l) provided the conjugated olefins, as did allyl (3p), benzyl (3q), naphthyl (3r), and propargyl (3s) substituted carboxylates. Moreover, further isomerization studies have revealed that the regioisomeric and geometric mixtures of olefins stem from E2 elimination, as very minimal isomerization was observed when the alkenes were subjected to basic conditions (see ESI† for more details).
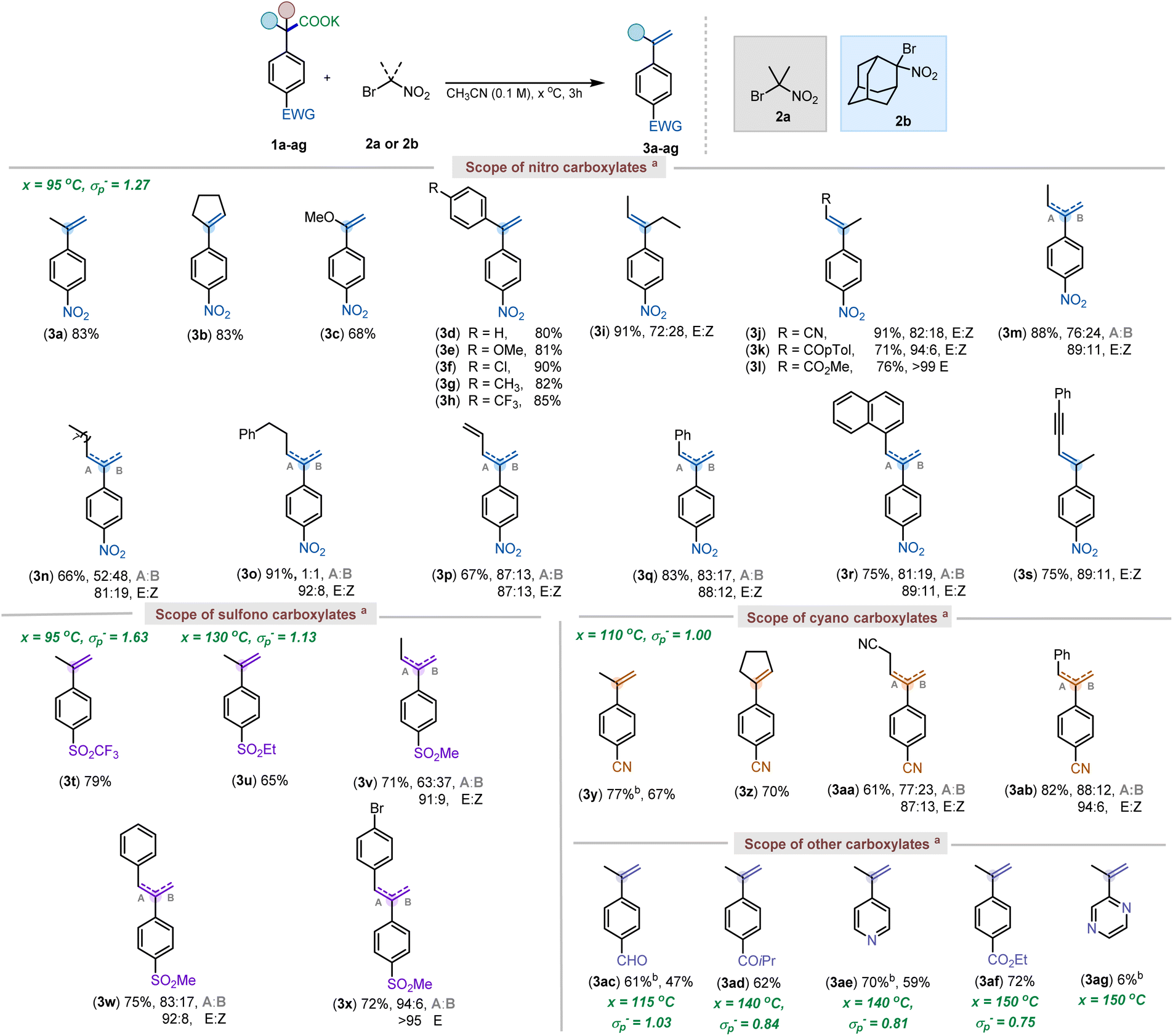 | ||
| Scheme 3 Scope of carboxylates. aReactions were run on a 0.1–0.2 mmol scale, and yields reported are isolated, unless otherwise denoted. Regioisomer ratio was determined using 1H NMR & COSY. bYields reported are quantitative 1H NMR yields with anisole as the internal standard. Reaction conditions: 1a–1ag (1 equiv), 2a or 2b (1.2 equiv), CH3CN (0.1 M), 3–6 hours, temperature as specified in ESI.† | ||
Having established the feasibility of our elimination conditions with respect to nitrophenyl acetates, the scope of other weakly activated aryl acetic acid salts was explored. Functional groups that provide adequate stabilization of an anionic intermediate formed after decarboxylation, such as SO2Me (σp− = 1.13), CN (σp− = 1.00), keto (σp− = 0.84), pyridyl (σp− = 0.81) and ester groups (σp− = 0.75) were expected to undergo the elimination between temperature range of 110 and 150 °C.10d,22 However, at the higher temperatures, required for decarboxylation of these substrates, it was observed that the parent carboxylic acids were recovered from the reaction mixture, with minimal to no elimination occurring (Scheme 4a). This was attributed to competitive E2-elimination to form 2-nitropropene with the carboxylates serving as the base (Scheme 4b). Thus, it was anticipated that choosing a gem-bromonitro alkane that could not undergo such an elimination should facilitate the elimination of 1y (Scheme 4c). In support of this hypothesis, utilization of an adamantane-derived gem-bromonitro alkane (2b) led to decarboxylative elimination of 1y in 77% yield (Scheme 4d).
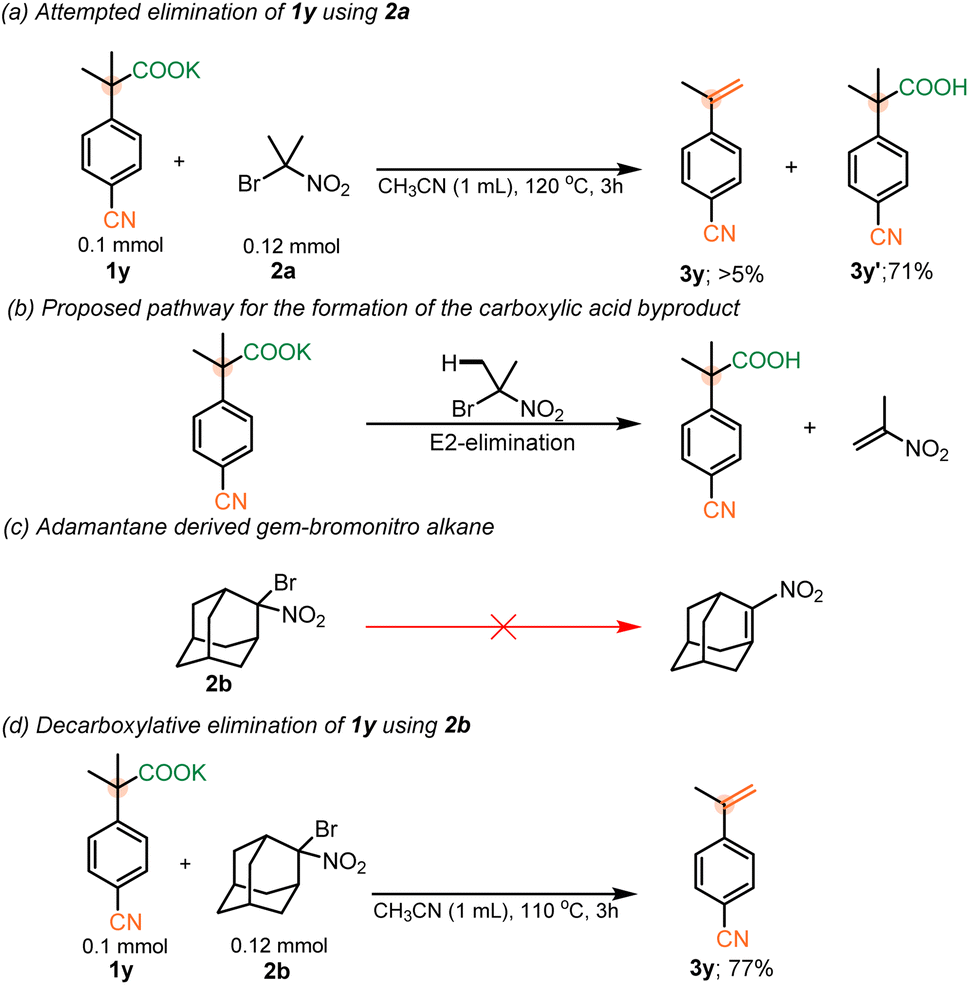 | ||
| Scheme 4 (a) Attempted elimination of 1y with 2a. (b) Mechanistic proposal for the formation of 3y′. (c) Anti-bredt olefin formation from 2b. (d) Elimination of 1y using 2b. | ||
With the newer reagent, the decarboxylative elimination of 4-SO2CF3-phenylacetic acid salt (1t) and other aryl acetic acids were evaluated (Scheme 3). The reaction with 1t delivered the corresponding styrene 3t in good yield (79%). Other activating groups such as –SO2Et (3u; 65%), –CN (3y; 77%), –CHO (3ac; 61%), –COR (3ad; 62%), –pyridine (3ae; 70%), and –CO2Me (3af; 72%) gave moderate to good yields for the corresponding styrenes. Overall, the observed functional group tolerance can, in part, be attributed to use of bromonitroalkanes as mild oxidants.
Importantly, the method was readily scaled to gram-scale, yielding 3a in 78% yield (Scheme 5a). To further illustrate the utility of this decarboxylative elimination approach, we subjected 3a to several different transformations (Scheme 5b). Specifically, 3a was subjected to radical cyclization under oxidative conditions to furnish the dihydrofuran derivative 4c in 61% yield.23 Additionally, dimerization was achieved under acidic conditions (4e, 50%),24 and allylic halogenation was performed using NBS (4b, 42%).25 Nucleophilic addition to styrene 3a was achieved under basic conditions to deliver 4d in 67% yield.26
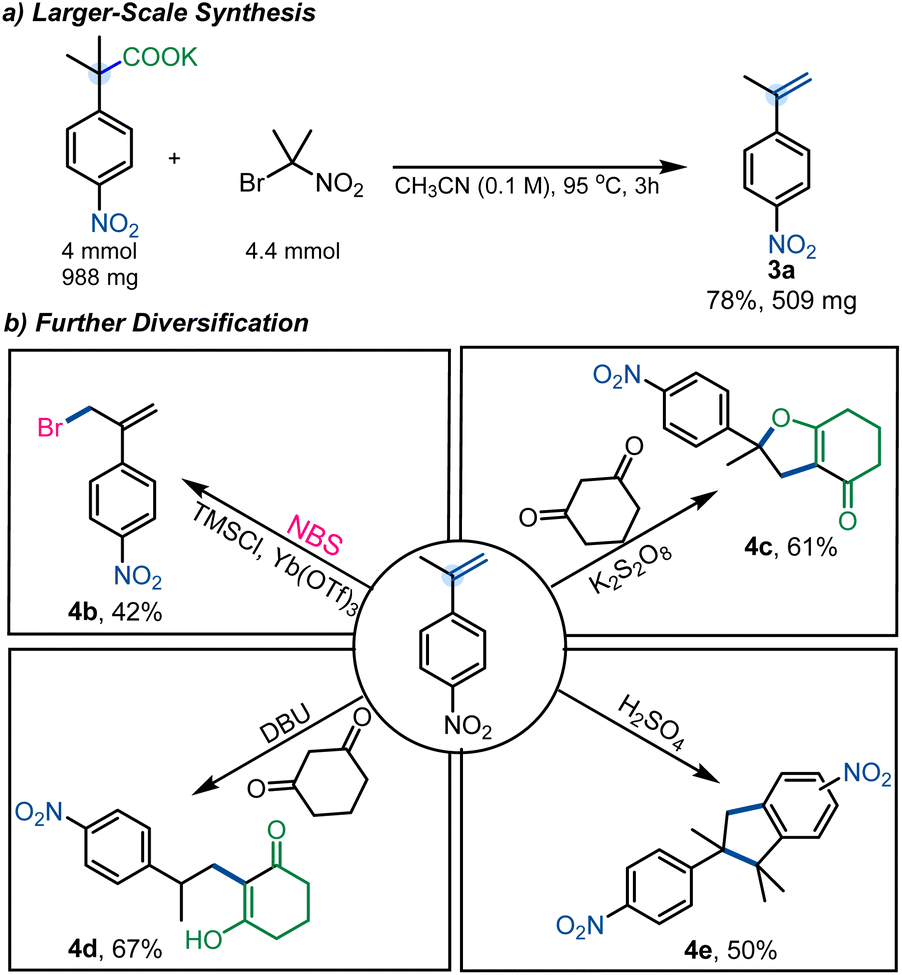 | ||
| Scheme 5 (a) Larger-scale synthesis. (b) Diversification. | ||
Mechanistic studies
Despite our reaction design, we considered three possible pathways for the formation of alkenes under the reaction conditions (Scheme 6). Pathway A involves a thermal decarboxylation followed by Halogen-ion-transfer from 2-bromo-2-nitropropane and a rebound E2-elimination to furnish the styrene. Pathway B would involve a thermal decarboxylation followed by a single-electron-transfer (SET) to 2-bromo-2-nitropropane followed by hydrogen-atom-transfer (HAT). SETs to gem-bromonitroalkanes are known and are a common pathway in radical nucleophilic substitution (SRN1) reactions.27 Another possible pathway, pathway C, would begin with a halogen ion exchange to form an acyl hypohalide, as in the Hunsdiecker–Borodin reaction.9c Subsequent bond homolysis, radical decarboxylation, and recombination would form a halide intermediate. This intermediate would then undergo elimination to generate the olefin.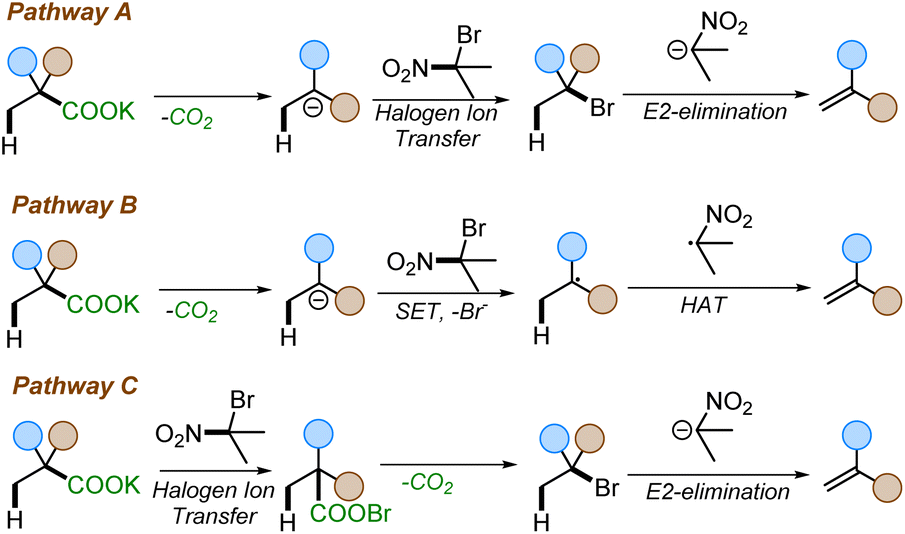 | ||
| Scheme 6 Possible mechanistic pathways. | ||
A series of mechanistic experiments were conducted to eliminate one or more of these potential mechanistic pathways (Scheme 7). To examine the possibility of radical intermediates, we employed TEMPO as radical trapping agent. Addition of TEMPO into the reaction mixture did not yield any TEMPO trapped product nor did TEMPO inhibit the reaction (Scheme 7a). Next, a radical clock experiment was performed to probe for the formation of nitro-alkyl radicals. However, no ring-opened products were observed when 2c was subjected to our reaction. Instead, 3a and 2c′ were isolated in 71% and 70% yield, respectively (Scheme 7b). This lies in contrast with existing decarboxylative eliminations, where radicals are involved;12,13 for example the Liu group observed the formation of cyclopropyl ring-opened products in their decarboxylation methodology.12a Moreover, if pathway B were operating, the competitive SRN1 reaction products were expected to be observed in addition to the olefins.27b However, no such products were observed under our reaction conditions (see ESI† for details). Additionally, we were able to isolate and characterize the intermediate halide 3a′ (Scheme 7B), which is inconsistent with reaction through pathway B. Finally, the reaction with 2-phenylisobutyric acid salt failed to give the corresponding styrene 4aj (Scheme 7c), suggesting the need for an electron withdrawing group to facilitate reaction. In contrast, the Hunsdiecker–Borodin reaction is known to proceed with unactivated phenylacetic acid salts.28 The observed requirement for higher temperatures to effect reaction of substrates with less electron-withdrawing activating groups further suggests that pathway C may not be operative.22 Thus, we favor pathway A as it is consistent with the observation of the intermediate halide 3a′ and the temperature dependence for attaining the decarboxylation with other weakly activating groups.
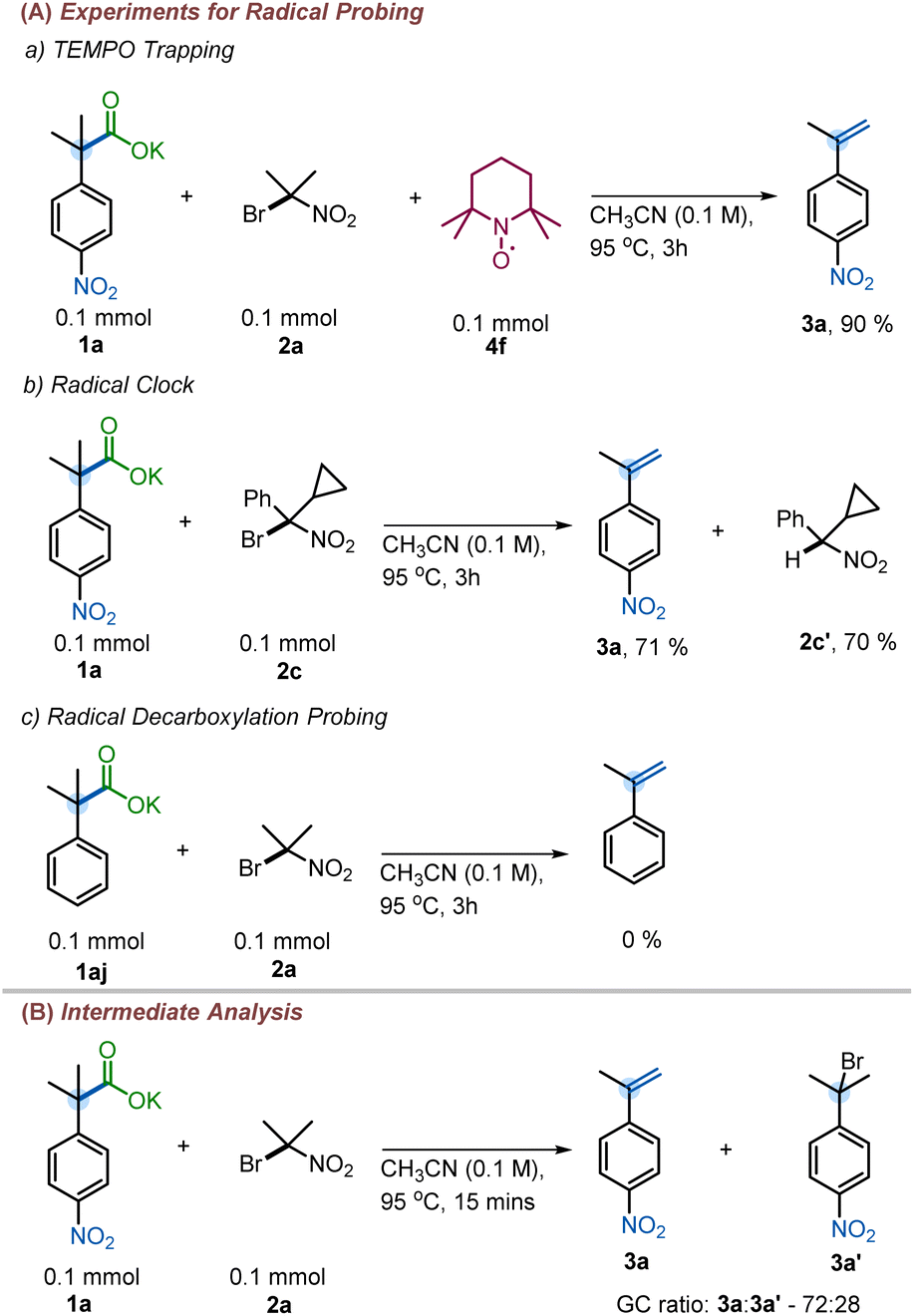 | ||
| Scheme 7 Mechanistic experiments. (A) Radical probing. (B) Intermediate analysis. | ||
With the goal of obtaining further insight into the mechanism of the reaction, KIE experiments were undertaken. Kinetic isotopic effects were obtained for a series of substrates (1a, 1d and 1h) and their deuterated derivates through the analysis of initial rates of independent reactions. For 1a and 1a-d6, independent rate studies derived a KIE of 2.8, and the intermolecular competition experiment resulted in a KIE of 2.0 (Scheme 8a and b). These values suggest the elimination to be the rate-determining step in pathway A. Furthermore, these KIE values are consistent with those reported in the literature for the elimination reaction of α-phenylethyl bromide systems.29 Independent rate studies of 1d and 1d-d3 revealed a KIE of 2.67, again suggesting a rate-determining elimination (Scheme 8c).
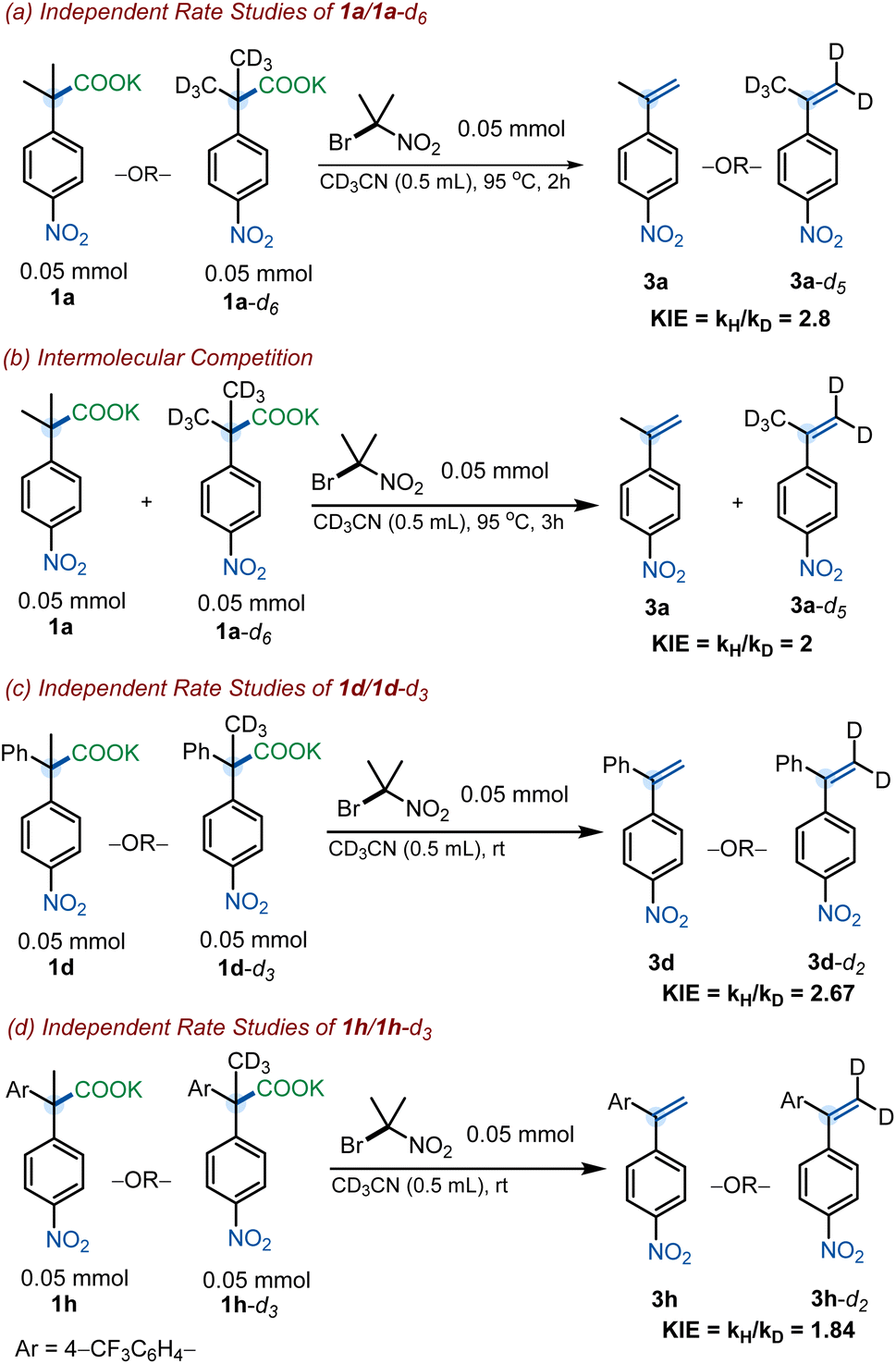 | ||
| Scheme 8 Kinetic isotopic studies. | ||
To further gain an understanding of the mechanism and the nature of the transition state of this elimination reaction, we conducted a Hammett competition study of various 4-substituted-2,2-diphenylpropanoate salts (Scheme 9). The competition studies were carried out under the standard conditions reported in Table 1, although an excess of the competing 4-substituted-2,2-diphenylpropanoate salts was added to the reaction mixture (5 equivalents each) to ensure that reactant concentration did not affect product selectivity (see ESI† for more details). The Hammett competition study suggests positive charge buildup in the product-determining transition state (ρ = −1.63). Such negative slopes have previously been observed in the gas phase elimination of substituted 1-phenylethyl chlorides30 and is indicative of a moderate degree of charge separation through an E2 transition state with significant E1 character.
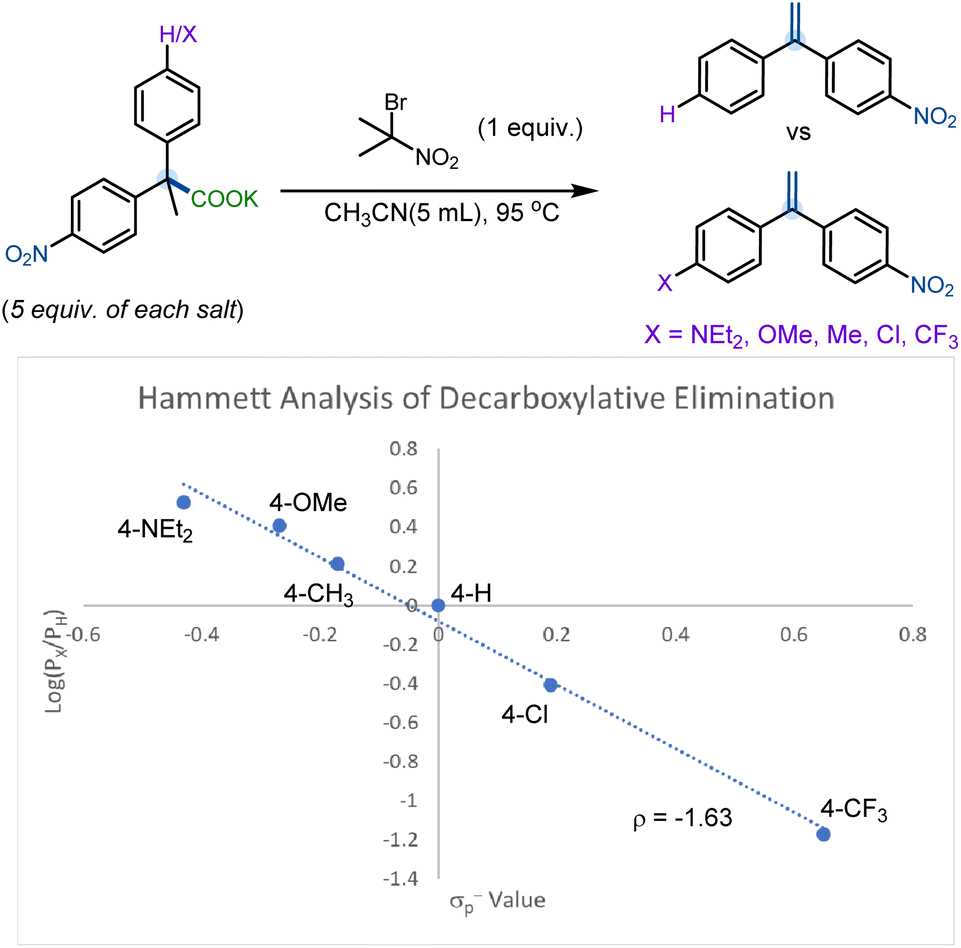 | ||
| Scheme 9 Hammett plot. | ||
Since Hammett competition experiments only reflect electronic differences in the product-determining step, additional kinetic investigations were conducted to verify that the rate-determining step (RDS) and the step governing product formation (PDS) are identical (see ESI† for additional details). Here, it was evident that the individual rate of reaction of 1g (4-CH3) was much faster rate than that of 1d (4-H) and, it was also evident, that the rate decreased drastically for 1f (4-Cl) and 1h (4-CF3) (see ESI† for details). These relative rates are consistent with a negative slope for the Hammett study. Thus, it was confirmed that the ‘E1-like’ E2-elimination was indeed the rate-determining step of this decarboxylative elimination reaction, as outlined in the Bunnett spectrum.31
Conclusions
In summary, we have developed a simple and efficient method for the olefination of potassium salts of carboxylic acids. This transition metal free approach allows for the facile construction of olefins in a chemoselective fashion in moderate to good yields. The green approach proceeds with good efficacy, good functional group tolerance, and broad substrate scope. Mechanistic studies demonstrated decarboxylation, followed by a halogen ion transfer and rebound E2 elimination as the likely pathway for this transformation. KIE and Hammett studies revealed a rate-determining ‘E1-like’ transition state for the E2-elimination.Data availability
The data underlying this study are available in the published article and its ESI.†Author contributions
J. T. and E. J. conceptualized and initiated the project. E. J., D. B. & G. S. performed the experiments and analyzed the data. J. T. and E. J. co-wrote the paper.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the National Science Foundation (CHE-2247708). Support for the NMR instrumentation was provided by NSF Academic Research Infrastructure Grant No. 9512331, NIH Shared Instrumentation Grant No. S10RR024664, and NSF Major Research Instrumentation Grant No. 0320648.Notes and references
- (a) I. Amghizar, L. A. Vandewalle, K. M. Van Geem and G. B. Marin, Engineering, 2017, 3, 171–178 CrossRef CAS; (b) W. Wang, S. Qu, X. Li, J. Chen, Z. Guo and W.-H. Sun, Coord. Chem. Rev., 2023, 494, 215351 CrossRef CAS; (c) R. Zhao, G. L. Haller and J. A. Lercher, Microporous Mesoporous Mater., 2023, 358, 112390 CrossRef CAS; (d) X.-Y. Wang, Y. Gao and Y. Tang, Prog. Polym. Sci., 2023, 143, 101713 CrossRef CAS.
- (a) G. Yin, X. Mu and G. Liu, Acc. Chem. Res., 2016, 49, 2413–2423 CrossRef CAS PubMed; (b) J. Lin, R.-J. Song, M. Hu and J.-H. Li, Chem. Rec., 2019, 19, 440–451 CrossRef CAS PubMed; (c) H. Jiang and A. Studer, Chem. Soc. Rev., 2020, 49, 1790–1811 RSC; (d) J.-H. Qin, N. Nan and J.-H. Li, Synthesis, 2023, 55, 2843–2859 CrossRef CAS; (e) H. Yang and Y. Ye, Top. Curr. Chem., 2023, 381, 23 CrossRef CAS PubMed; (f) M. Sun, Y. Xiao, K. Liu, X. Yang, P. Liu, S. Jie, J. Hu, S. Shi, Q. Wang, K. H. Lim, Z. Liu, B.-G. Li and W.-J. Wang, Can. J. Chem. Eng., 2023, 101, 4886–4906 CrossRef CAS; (g) F. Li and W. Liu, Can. J. Chem. Eng., 2023, 101, 4992–5019 CrossRef CAS; (h) Z. Dong, L. Song and L.-A. Chen, ChemCatChem, 2023, 15, e202300803 CrossRef CAS; (i) J. Han, R. He and C. Wang, Chem Catal., 2023, 3, 100690 CrossRef CAS; (j) C.-X. Liu, S.-Y. Yin, F. Zhao, H. Yang, Z. Feng, Q. Gu and S.-L. You, Chem. Rev., 2023, 123, 10079–10134 CrossRef CAS PubMed.
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044–4098 CrossRef CAS PubMed.
- (a) E. Scott, F. Peter and J. Sanders, Appl. Microbiol. Biotechnol., 2007, 75, 751–762 CrossRef CAS PubMed; (b) P. Gallezot, Chem. Soc. Rev., 2012, 41, 1538–1558 RSC; (c) Y. Zabolotna, D. M. Volochnyuk, S. V. Ryabukhin, D. Horvath, K. S. Gavrilenko, G. Marcou, Y. S. Moroz, O. Oksiuta and A. Varnek, J. Chem. Inf. Model., 2022, 62, 2171–2185 CrossRef CAS PubMed.
- (a) S. Senaweera, K. C. Cartwright and J. A. Tunge, J. Org. Chem., 2019, 84, 12553–12561 CrossRef CAS PubMed; (b) J. A. Tunge, ISR J. Chem., 2020, 60, 351–359 CrossRef CAS; (c) L. Li, Y. Yao and N. Fu, Eur. J. Org Chem., 2023, 26, e202300166 CrossRef CAS; (d) S. B. Beil, T. Q. Chen, N. E. Intermaggio and D. W. C. MacMillan, Acc. Chem. Res., 2022, 55, 3481–3494 CrossRef CAS PubMed.
- (a) H. Kolbe, Liebigs Ann. Chem., 1849, 69, 257–294 CrossRef; (b) H. Koble, Liebigs Ann. Chem., 1849, 64, 339–341 Search PubMed; (c) A. K. Vijh and B. E. Conway, Chem. Rev., 1967, 67, 623–664 CrossRef CAS.
- (a) N. Rodríguez and L. J. Goossen, Chem. Soc. Rev., 2011, 40, 5030–5048 RSC; (b) D. Kong, P. J. Moon, E. K. J. Lui, O. Bsharat and R. J. Lundgren, Science, 2020, 369, 557–561 CrossRef CAS PubMed; (c) W.-L. Xing, J.-X. Wang, M.-C. Fu and Y. Fu, Chin. J. Chem., 2022, 40, 323–328 CrossRef CAS; (d) W.-L. Xing, D.-G. Liu and M.-C. Fu, RSC Adv., 2021, 11, 4593–4597 RSC.
- (a) D. H. R. Barton, H. P. Faro, E. P. Serebryakov and N. F. Woolsey, J. Chem. Soc., 1965, 2438–2444 RSC; (b) D. H. R. Barton and S. W. McCombie, J. Chem. Soc., Perkin Trans. 1, 1975, 1574–1585 RSC; (c) D. H. R. Barton, D. Crich and W. B. Motherwell, J. Chem. Soc., Chem. Commun., 1983, 939–941 RSC.
- (a) A. Borodine, Liebigs Ann. Chem., 1861, 119, 121–123 CrossRef; (b) H. Hunsdiecker, C. Hunsdiecker and B. Dtsch, Chem. Ges., 1942, 75, 291–297 Search PubMed; (c) R. G. Johnson and R. K. Ingham, Chem. Rev., 1956, 56, 219–269 CrossRef CAS; (d) C. V. Wilson, in Organic Reactions, 2011, pp. 332–387 Search PubMed.
- (a) J. D. Weaver, A. Recio, A. J. Grenning and J. A. Tunge, Chem. Rev., 2011, 111, 1846–1913 CrossRef CAS PubMed; (b) T. Patra and D. Maiti, Chem.–Eur. J., 2017, 23, 7382–7401 CrossRef CAS PubMed; (c) X.-Q. Hu, Z.-K. Liu, Y.-X. Hou and Y. Gao, iScience, 2020, 23, 101266 CrossRef CAS PubMed; (d) P. J. Moon and R. J. Lundgren, ACS Cat., 2020, 10, 1742–1753 CrossRef CAS; (e) Z. Zeng, A. Feceu, N. Sivendran and L. J. Gooßen, Adv. Synth. Catal., 2021, 363, 2678–2722 CrossRef CAS; (f) E. Joseph, R. D. Hernandez and J. A. Tunge, Chem.–Eur. J., 2023, 29, e202302174 CrossRef CAS PubMed; (g) E. Joseph, I. Smith and J. A. Tunge, Chem. Sci., 2023, 14, 13902–13907 RSC.
- (a) J. D. Bacha and J. K. Kochi, J. Org. Chem., 1968, 33, 83–93 CrossRef CAS; (b) J. D. Bacha and J. K. Kochi, Tetrahedron, 1968, 24, 2215–2226 CrossRef CAS; (c) J. K. Kochi and J. D. Bacha, J. Org. Chem., 1968, 33, 2746–2754 CrossRef CAS.
- (a) S.-W. Wu, J.-L. Liu and F. Liu, Org. Lett., 2016, 18, 1–3 CrossRef CAS PubMed; (b) X. Sun, J. Chen and T. Ritter, Nat. Chem., 2018, 10, 1229–1233 CrossRef CAS PubMed; (c) A. Tlahuext-Aca, L. Candish, R. A. Garza-Sanchez and F. Glorius, ACS Cat., 2018, 8, 1715–1719 CrossRef CAS; (d) K. C. Cartwright, S. B. Lang and J. A. Tunge, J. Org. Chem., 2019, 84, 2933–2940 CrossRef CAS PubMed; (e) A. F. Garrido-Castro, Y. Hioki, Y. Kusumoto, K. Hayashi, J. Griffin, K. C. Harper, Y. Kawamata and P. S. Baran, Angew. Chem., Int. Ed., 2023, 62, e202309157 CrossRef CAS PubMed; (f) J. Yu, T. Liu, W. Sun and Y. Zhang, Org. Lett., 2023, 25, 7816–7821 CrossRef CAS PubMed.
- (a) K. C. Cartwright and J. A. Tunge, ACS Cat., 2018, 8, 11801–11806 CrossRef CAS; (b) V. T. Nguyen, V. D. Nguyen, G. C. Haug, H. T. Dang, S. Jin, Z. Li, C. Flores-Hansen, B. S. Benavides, H. D. Arman and O. V. Larionov, ACS Cat., 2019, 9, 9485–9498 CrossRef CAS PubMed; (c) K. C. Cartwright, E. Joseph, C. G. Comadoll and J. A. Tunge, Chem.–Eur. J., 2020, 26, 12454–12471 CrossRef CAS PubMed.
- Ir[dF(CF3)ppy]2(dtbbpy)PF6 can be purchased from Sigma for $1050 per 500 milligram..
- (a) A. R. White, L. Wang and D. A. Nicewicz, Synlett, 2019, 30, 827–832 CrossRef CAS PubMed; (b) B. Zilate, C. Fischer and C. Sparr, Chem. Commun., 2020, 56, 1767–1775 RSC.
- M. P. Stanton and J. M. Hoover, J. Org. Chem., 2023, 88, 1713–1719 CrossRef CAS PubMed.
- (a) P. K. Sazonov, G. A. Artamkina and I. P. Beletskaya, Russ. Chem. Rev., 2012, 81, 317 CrossRef CAS; (b) N. S. Zefirov and D. I. Makhonkov, Chem. Rev., 1982, 82, 615–624 CrossRef CAS.
- (a) S. Cristol and J. W. Firth, J. Org. Chem., 1961, 26, 280 CrossRef CAS; (b) J. K. Kochi, J. Am. Chem. Soc., 1965, 87, 2500–2502 CrossRef CAS; (c) J. I. Concepcion, C. G. Francisco, R. Freire, R. Hernandez, J. A. Salazar and E. Suarez, J. Org. Chem., 1986, 51, 402–404 CrossRef CAS; (d) A. Naruse, K. Kitahara, S. Iwasa and K. Shibatomi, Asian J. Org. Chem., 2019, 8, 691–693 CrossRef CAS; (e) A. Varenikov, E. Shapiro and M. Gandelman, Chem. Rev., 2021, 121, 412–484 CrossRef CAS PubMed.
- 2-Bromo-2-nitropropane can be purchased from Sigma for $89 per 25 grams..
- (a) M. J. Leonard, P. G. McKay and A. R. Lingham, RSC Adv., 2015, 5, 76401–76418 RSC; (b) X. Zhang, J. Ren, S. M. Tan, D. Tan, R. Lee and C.-H. Tan, Science, 2019, 363, 400–404 CrossRef CAS PubMed; (c) J. Ren, X. Ban, X. Zhang, S. M. Tan, R. Lee and C.-H. Tan, Angew. Chem., Int. Ed., 2020, 59, 9055–9058 CrossRef CAS PubMed; (d) X. Zhang and C.-H. Tan, Chem, 2021, 7, 1451–1486 CrossRef CAS.
- (a) Y. Tanaka, S. Kubosaki, K. Osaka, M. Yamawaki, T. Morita and Y. Yoshimi, J. Org. Chem., 2018, 83, 13625–13635 CrossRef CAS PubMed; (b) M.-J. Luo, Q. Xiao and J.-H. Li, Chem. Soc. Rev., 2022, 51, 7206–7237 RSC.
- D. Kong, P. J. Moon, W. Qian and R. J. Lundgren, Chem. Commun., 2018, 54, 6835–6838 RSC.
- S. Wang, L.-Y. He and L.-N. Guo, Synthesis, 2015, 47, 3191–3197 CrossRef CAS.
- I. V. Farr, D. Kratzner, T. E. Glass, D. Dunson, Q. Ji and J. E. McGrath, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 2840–2854 CrossRef CAS.
- J. Häfliger, K. Livingstone, C. G. Daniliuc and R. Gilmour, Chem. Sci., 2021, 12, 6148–6152 RSC.
- K. K. Gnanasekaran, J. Yoon and R. A. Bunce, Tetrahedron Lett., 2016, 57, 3190–3193 CrossRef CAS.
- (a) S. I. Al-Khalil, W. R. Bowman, K. Gaitonde, M. A. Marley and G. D. Richardson, J. Chem. Soc., Perkin Trans. 2, 2001, 1557–1565 RSC; (b) R. A. Rossi, A. B. Pierini and A. B. Peñéñory, Chem. Rev., 2003, 103, 71–168 CrossRef CAS PubMed.
- (a) Z. Wang, L. Zhu, F. Yin, Z. Su, Z. Li and C. Li, J. Am. Chem. Soc., 2012, 134, 4258–4263 CrossRef CAS PubMed; (b) X. Tan, T. Song, Z. Wang, H. Chen, L. Cui and C. Li, Org. Lett., 2017, 19, 1634–1637 CrossRef CAS PubMed.
- (a) T. Yoshida, Y. Yano and S. Oae, Tetrahedron, 1971, 27, 5343–5351 CrossRef CAS; (b) T. Hasan, L. B. Sims and A. Fry, J. Am. Chem. Soc., 1983, 105, 3967–3975 CrossRef CAS.
- M. R. Bridge, D. H. Davies, A. Maccoll, R. A. Ross, B. Stephenson and O. Banjoko, J. Chem. Soc. B, 1968, 805–808 RSC.
- J. F. Bunnett, Angew. Chem., Int. Ed. Engl., 1962, 1, 225–235 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc01905a |
| This journal is © The Royal Society of Chemistry 2024 |