
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceKinetic resolution of 1-(1-alkynyl)cyclopropyl ketones via gold-catalyzed divergent (4 + 4) cycloadditions: stereoselective access to furan fused eight-membered heterocycles†
Xunhua
Wang
a,
Ruifeng
Lv
a and
Xiaoxun
Li
 *ab
*ab
aDepartment of Medicinal Chemistry, Key Laboratory of Chemical Biology (Ministry of Education), NMPA Key Laboratory for Technology Research and Evaluation of Drug Products, School of Pharmaceutical Sciences, Cheeloo College of Medicine, Shandong University, Jinan, Shandong 250012, China. E-mail: xli@sdu.edu.cn
bSuzhou Research Institute of Shandong University, NO. 388 Ruoshui Road, SIP, Suzhou, Jiangsu 215123, China
First published on 22nd May 2024
Abstract
Chiral eight-membered heterocycles comprise a diverse array of natural products and bioactive compounds, yet accessing them poses significant challenges. Here we report a gold-catalyzed stereoselective (4 + 4) cycloaddition as a reliable and divergent strategy, enabling readily accessible precursors (anthranils and ortho-quinone methides) to be intercepted by in situ generated gold-furyl 1,4-dipoles, delivering previously inaccessible chiral furan/pyrrole-containing eight-membered heterocycles with good results (56 examples, all >20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr, up to 99% ee). Moreover, we achieve a remarkably efficient kinetic resolution (KR) process (s factor up to 747). The scale-up synthesis and diversified transformations of cycloadducts highlight the synthetic potential of this protocol. Computational calculations provide an in-depth understanding of the stereoselective cycloaddition process.
:1 dr, up to 99% ee). Moreover, we achieve a remarkably efficient kinetic resolution (KR) process (s factor up to 747). The scale-up synthesis and diversified transformations of cycloadducts highlight the synthetic potential of this protocol. Computational calculations provide an in-depth understanding of the stereoselective cycloaddition process.
Introduction
Eight-membered rings, fused polycyclic ones in particular, represent a structurally diverse group of heterocycles distributed in a wide range of natural products and pharmaceuticals, such as hippobrine, FR-900482, heliannuol A, protosapannin, and PDK inhibitors (Scheme 1a).1 They possess various interesting biological activities such as anticancer, antiviral, and anti-inflammatory effects. Despite the steady progress in synthetic methods achieved over past decades, the construction of these complex structures is still quite challenging due to the unfavorable enthalpy and entropy factors, as well as the transannular interactions.2 Thus, it is highly desirable to develop concise and robust strategies to access these prestigious motifs with a high level of stereo-control.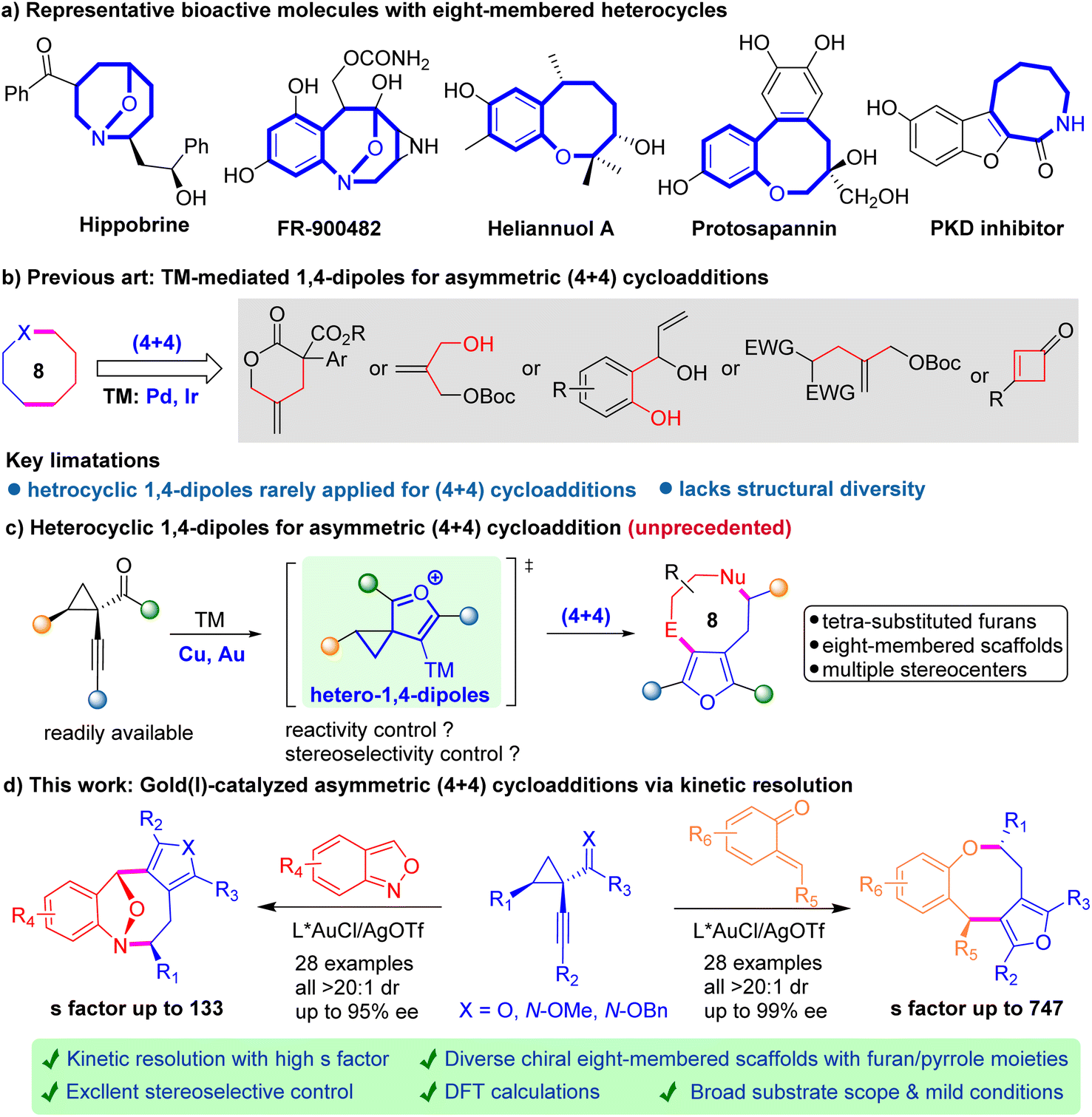 | ||
| Scheme 1 Asymmetric (4 + 4) cycloadditions for constructing eight-membered scaffolds. | ||
Among recent methods developed for the construction of eight-membered rings, the intermolecular (4 + 4) cycloaddition has evolved as a prevailing strategy.3 In this scenario, transition-metal (TM) mediated zwitterionic 1,4-dipolar species have been recognized as privileged building blocks for divergent preparation of chiral eight-membered scaffolds, especially with regard to high diastereo- and enantio-control (Scheme 1b).4 However, these dipolar species often lack structural diversity and heterocyclic 1,4-dipoles are seldom studied, hampering the exploration of the (4 + 4) strategy in a broader manner.5 To address this long-standing challenge, we wondered whether TM-based furyl species could serve as heterocyclic 1,4-dipoles, which would furnish challenging eight-membered cycloadducts with tetrasubstituted furan scaffolds (Scheme 1c). In 2008, Zhang and co-workers presented one remarkable study of Au-catalyzed racemic (4 + 2) cycloaddition of 1-(1-alkynyl)-cyclopropyl ketones with indoles, where the alkynyl cyclopropyl ketones serve as the Au-furyl 1,4-dipole precursors.6 Later, several related gold-catalyzed (4 + 2) and (4 + 3) cycloadditions involving 1-(1-alkynyl)-cyclopropyl ketones have been disclosed, yet mainly in the racemic version.7 Despite the considerable successes, catalytic asymmetric (4 + 4) cycloaddition of Au-furyl 1,4-dipoles to afford chiral eight-membered heterocycles remains elusive in the literature.
With the growing demand for eight-membered motifs and the desirable bioactivities associated with furan-containing molecules in the pharmaceutical area, we envisioned that integrating Au-furyl 1,4-dipoles with various four-atom synthons could chart a novel path for the rapid assembly of furan-fused eight-membered skeletons, further expanding their chemical space for drug discovery. Even strategically ideal, this proposal still faces several challenges, including the particular issue of controlling diastereo- and enantioselectivity during the cycloaddition process due to the unfavorable entropic factors and transannular interactions. Another issue is that it is uncertain whether the Au-furyl 1,4-dipoles are compatible with readily available four-atom acceptors to generate unprecedented eight-membered heterocycles instead of the competitive six-membered rings. In an outgrowth of our ongoing interest,4b herein we report a highly enantio- and diastereoselective gold-catalyzed (4 + 4) cycloaddition for the facile access to valuable chiral eight-membered rings bearing tetrasubstituted furan moieties with diverse structural features (Scheme 1d). This asymmetric approach enables the construction of two types of optically active furan/pyrrole-fused ring systems through the efficient kinetic resolution of 1-(1-alkynyl)-cyclopropyl ketones.8 Of note, density functional theory (DFT) investigations shed light on the mechanism of the chiral gold-catalyzed stereoselective cycloaddition. Furthermore, the versatility of this strategy is demonstrated through a series of synthetic transformations of the resulting enantioenriched eight-membered products.
Results and discussion
To validate our working hypothesis, we began the investigation by evaluating the (4 + 4) cycloaddition reaction involving racemic cyclopropyl ketone 1a and anthranil 2a as the model substrates (Table 1). Various metal catalysts were initially screened, and the superior result was obtained with the Au(I) catalyst and AgOTf.9 We next optimized the asymmetric cycloaddition by using various chiral gold complexes. To our delight, the desired eight-membered cycloadduct 3a was formed in reasonable enantiomeric excess (ee) with commercially available bidentate phosphine ligands via the KR process (see the ESI† for details). The reaction proceeded smoothly with (S)-SEGPHOS, affording the desired 3a in 55% ee. Encouraged by this result, a variety of sterically more demanding SEGPHOS derivatives were investigated (entries 1–3), and it was found that the enantioselectivity was improved to 61% ee with (S)-DM-SEGPHOS L1 (entry 1) and 65% ee with (S)-DTBM-SEGPHOS L2 (entry 2), respectively. Surprisingly, the enantioselectivity was not further improved by subsequent modification of the ligand (entry 3). Then, different solvents were examined with (S)-DTBM-SEGPHOS as the chiral ligand, showing that the cycloaddition was more efficient in halogenated solvents (entries 4–6). Finally, the reaction temperature was optimized to furnish the product 3a, the best result, in 45% yield with 87% ee at −20 °C (s = 45, entry 9). Notably, excellent diastereoselectivity (all >20:1 dr) was obtained for this transformation.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Ligand | Solvent | 3a yieldb | eec | s |
| a All reactions were performed with 1a (0.1 mmol) and 2a (0.07 mmol) in the presence of Me2SAuCl (4.0 mol%), ligand (2.0 mol%), and AgOTf (6.0 mol%) in solvent (1.0 mL) at room temperature or indicated temperature under a nitrogen atmosphere. b Isolated yields. c Determined by HPLC analysis. d Selectivity (s) values were calculated through the equation s = ln[(1 – C)(1 – ee1a)]/ln[(1 – C)(1 + ee1a)], where C is the conversion; C = ee1a/(ee1a + ee3a). e The reaction temperature was 0 °C. f The reaction temperature was −10 °C. g The reaction temperature was −20 °C. | |||||
| 1 | L1 | DCM | 21% | 61% | 6 |
| 2 | L2 | DCM | 43% | 65% | 14 |
| 3 | L3 | DCM | 35% | 55% | 7 |
| 4 | L2 | Toluene | 15% | 60% | 10 |
| 5 | L2 | DCE | 36% | 60% | 12 |
| 6 | L2 | CHCl3 | 30% | 61% | 12 |
| 7e | L2 | DCM | 44% | 70% | 19 |
| 8f | L2 | DCM | 46% | 77% | 25 |
| 9g | L2 | DCM | 45% | 87% | 45 |
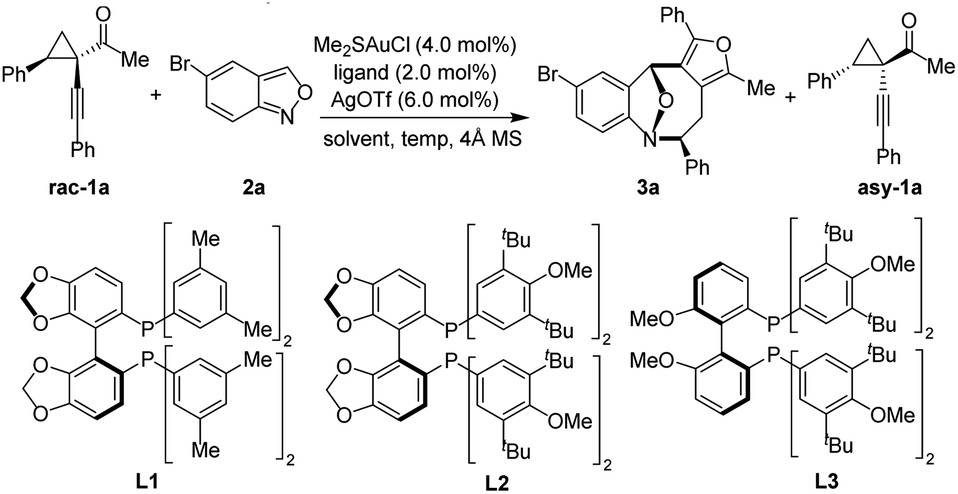
Under the optimal reaction conditions, we investigated the scope of anthranils and cyclopropyl ketones/oximes in this asymmetric (4 + 4) cycloaddition. As demonstrated in Table 2, this method tolerates a broad range of electronically distinct groups at different positions on aryl moieties, delivering the desired furan-fused eight-membered heterocyles 3a–3a′ in good yields with generally high levels of stereoselectivities (s factor up to 133, up to 45% yield, up to 95% ee, all >20:1 dr). Various functional groups, including fluoro, chloro, bromo, alkoxyl, methyl ester, benzoate and nitro, were all compatible (3a–3m). Note that the substrate bearing a strong electron-donating group (methoxyl) afforded the eight-membered adduct 3e in lower yield (33%) and chiral cyclopropyl ketone 1c in moderate enantioselectivity (76% ee). Subsequently, we explored the versatility of cyclopropyl ketones 1 with anthranil 2d as the model component (Table 2). Racemic cyclopropyl ketones bearing methyl or various halogens, regardless of the position on the aromatic ring, were well tolerated, generating the desired products 3n–3w′ in 31–45% yields with 80–95% ee and the corresponding chiral cyclopropyl ketones 1b–1j′′ in 38–45% yields with 82–99% ee. 3-Thiophenyl substrate 1j led to the cycloadduct 3u in 37% yield with 90% ee.
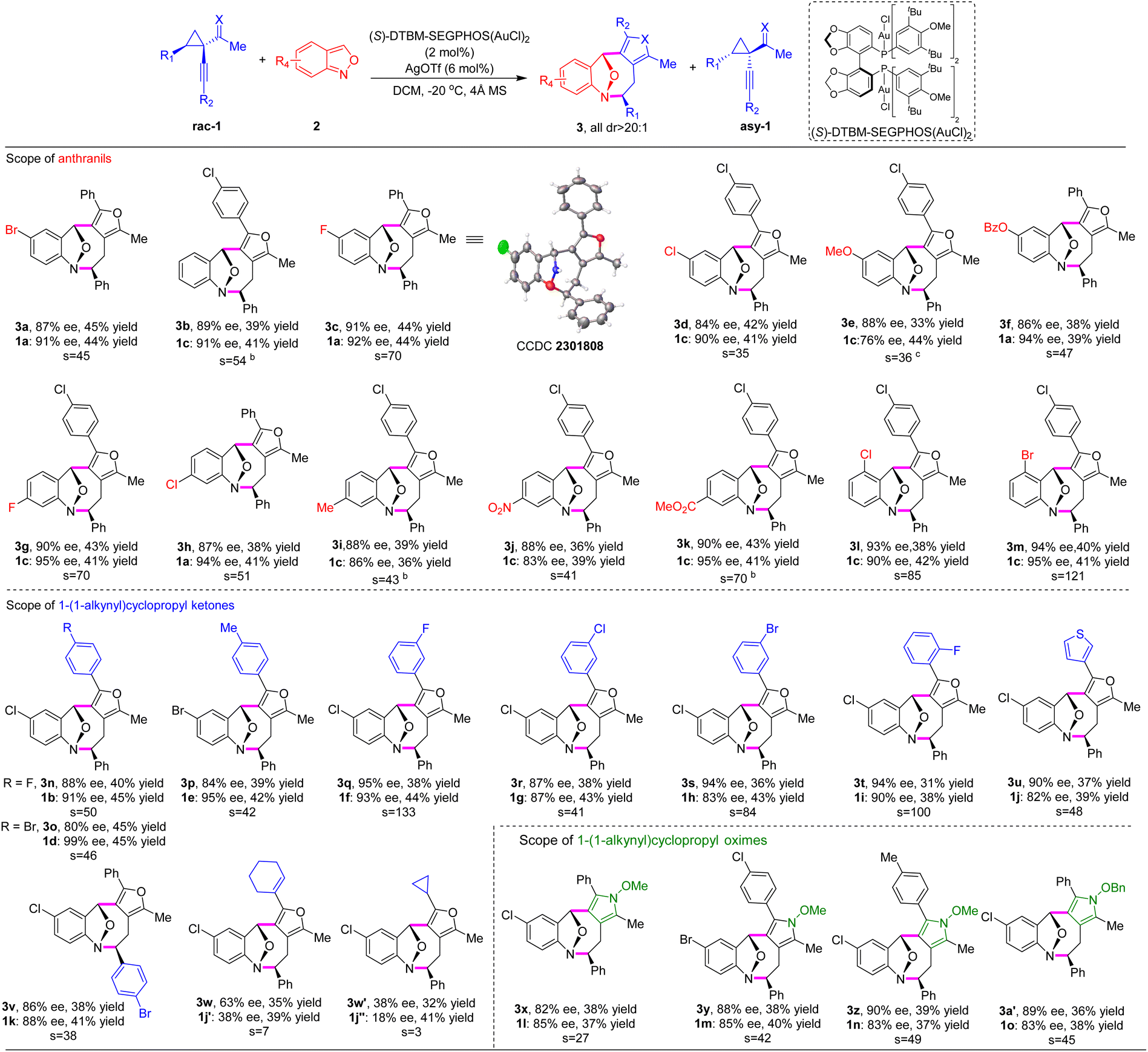
Furthermore, a similar level of efficiency was observed for cyclopropyl ketone 1k with 4-bromo phenyl substitution as R1 (3v, 38% yield, and 86% ee). Remarkably, substrate 1j′ bearing a cyclohexenyl group and 1j′′ featuring a cyclopropyl moiety performed well under optimized reaction conditions, affording the corresponding products 3w and 3w′ in 35% yield, >20:1 dr, 63% ee, and 32% yield, >20:1 dr, 38% ee, respectively. In addition, the absolute configuration of product 3c was confirmed as (5S, 6R, 11R) by single-crystal X-ray structure analysis.10
Pyrroles are ubiquitous in natural products and exhibit diverse biological activities.11 It is intriguing to construct complex pyrrole-containing molecules that are needed for biological investigation.12 We anticipated that the development of new asymmetric methods to build pyrrole-containing scaffolds bearing multistereogenic centers could be valuable yet underdeveloped. When alkynylcyclopropyl oximes were employed, this (4 + 4) cycloaddition strategy proceeded smoothly under the aforementioned reaction conditions, affording the densely substituted pyrrole-fused benzo[b]azocines 3x–3a′ with up to 39% yield and 90% ee (Table 2).
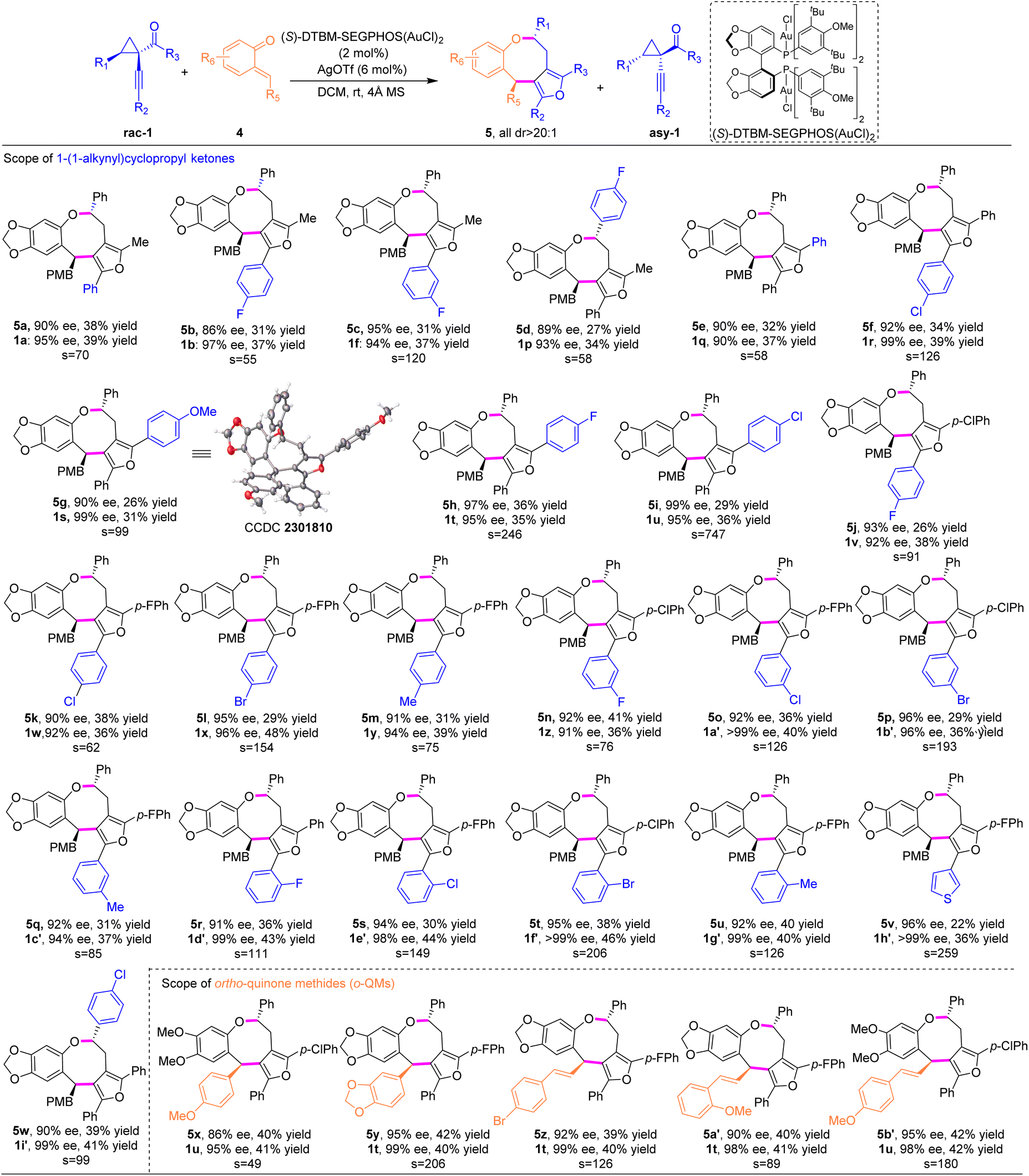
To demonstrate the synthetic diversity of this strategy, we continued to extend this method to the stereoselective (4 + 4) cycloaddition with another versatile reactive intermediate ortho-quinone methides (o-QMs),13 providing straightforward access to synthetically challenging furan-fused benzo[b]oxocines, which are important structural motifs found in biologically active compounds.14 Pleasingly, o-QMs were suitable cycloaddition partners for this asymmetric (4 + 4) reaction under similar catalytic conditions to those for the anthranil substrates, representing the first gold-catalyzed asymmetric higher-order cycloaddition to construct furan-containing eight-membered cyclic ethers. Initially, the substrate scope of the R3 substituent of cyclopropyl ketones was examined. As shown in Table 3, for R3, both alkyl and aryl groups participated well to deliver the cycloadducts 5a–5i in good yields and excellent enantioselectivities (26–38% yields, 86–99% ee), albeit substrates with aryl groups showed better resolution efficiency than ones with alkyl groups under the optimized conditions. Meanwhile, cycloadduct 5h with an electron-withdrawing group was obtained in 36% yield with 97% ee, while product 5g with an electron-donating group was isolated in lower yield with 90% ee. The s factor could be further improved to 747 when the p-chlorophenyl substituent was introduced (5i). With this information in hand, cyclopropyl ketones bearing R3 with an electron-deficient aryl substituent were selected to explore the scope of R2 substituents. Regardless of electronic and steric properties of the R2 substituents on the phenyl group, for example, F, Cl, Br, and Me, at ortho-, meta- and para-positions, cyclopropyl ketones 1 participated in the corresponding (4 + 4) cycloadditions smoothly, producing a series of furan-fused eight-membered heterocycles 5j–5v with satisfactory results (up to 41% yield, all >20:1 dr and 90–96% ee). The chiral cyclopropyl ketones 1v–1h′ were prepared in 91–99% ee. In addition, 3-thiophene-cycloadduct 5v was synthesized in 36% yield and >99% ee. Substitution at the R1 fragment was also compatible (5w). Unfortunately, substrates with R2 as aliphatic substituents, such as n-butyl and cyclopropyl groups, were sluggish in the current reaction system. Next, we turned our attention to explore the feasibility of o-QMs.
| a All reactions were performed with 1 (0.2 mmol) and 4 (0.13 mmol) in the presence of (S)-DTBM-SEGPHOS(AuCl)2 (2.0 mol%), and AgOTf (6.0 mol%) in DCM at room temperature. PMB: p-methoxybenzyl; p-FPh: p-fluorobenzyl; p-ClPh: p-chlorobenzyl. |
|---|
|
Notably, o-QMs bearing electron-rich phenyl substituents and various styryl-substituted groups were transformed into the densely substituted products (5x–5b′) in excellent yields and stereoselectivities. The absolute configuration of eight-membered cyclic ether 5g was determined by single-crystal X-ray analysis.10
To evaluate the synthetic utility of this strategy, gram-scale reactions and derivatization of the obtained eight-membered rings were conducted (Scheme 2). The catalytic asymmetric (4 + 4) cycloadditions proceeded well with 1 mol% chiral gold complex, while maintaining the high efficiency and excellent stereochemical outcome as the corresponding small-scale reactions. Subjecting cyclopropyl ketone 1a (5.0 mmol) and anthranil 2a (3.5 mmol) under the standard conditions led to chiral cycloadduct 3a in 38% yield with 90% ee. Another product 5h was prepared in 1.1 gram by a similar process on a 5.0 mmol scale (37% yield, 95% ee, >20:1 dr, Scheme 2A). Upon subjecting the optically active 1-(1-alkynyl)cyclopropyl ketones asy-1a to the (4 + 4) cycloaddition with racemic gold catalysis, the resulting chiral eight-membered heterocycles 3aa and 5aa were obtained in favorable yields with 83% ee and 86% ee, respectively. This outcome indicates that the cycloaddition predominantly proceeds via an SN2 process (Scheme 2B). Next, synthetic transformations of the product were explored. The furan moiety in products 3d and 5h underwent (4 + 2) cycloadditions with in situ generated benzyne to provide polycyclic scaffolds 6 and 7 in 62% yield and 72% yield, respectively (Scheme 2C). Cycloadduct 3a was functionalized via Buchwald–Hartwig amination with aniline to generate 8 in 67% yield (Scheme 2D). Moreover, compound 3a can be further elaborated with Zn powder under acidic conditions to deliver the tetrahydrobenzo[b]azocine derivative 9 in 93% yield via cleavage of the N–O bond. Methylation of 9 with MeI/K2CO3 gave N-methylated compound 10 in 85% yield. The complex bridged scaffold 11 could be accessed in 89% yield by treating 9 with formaldehyde. Treatment of 9 with triphosgene afforded another bridged carbamate 12.
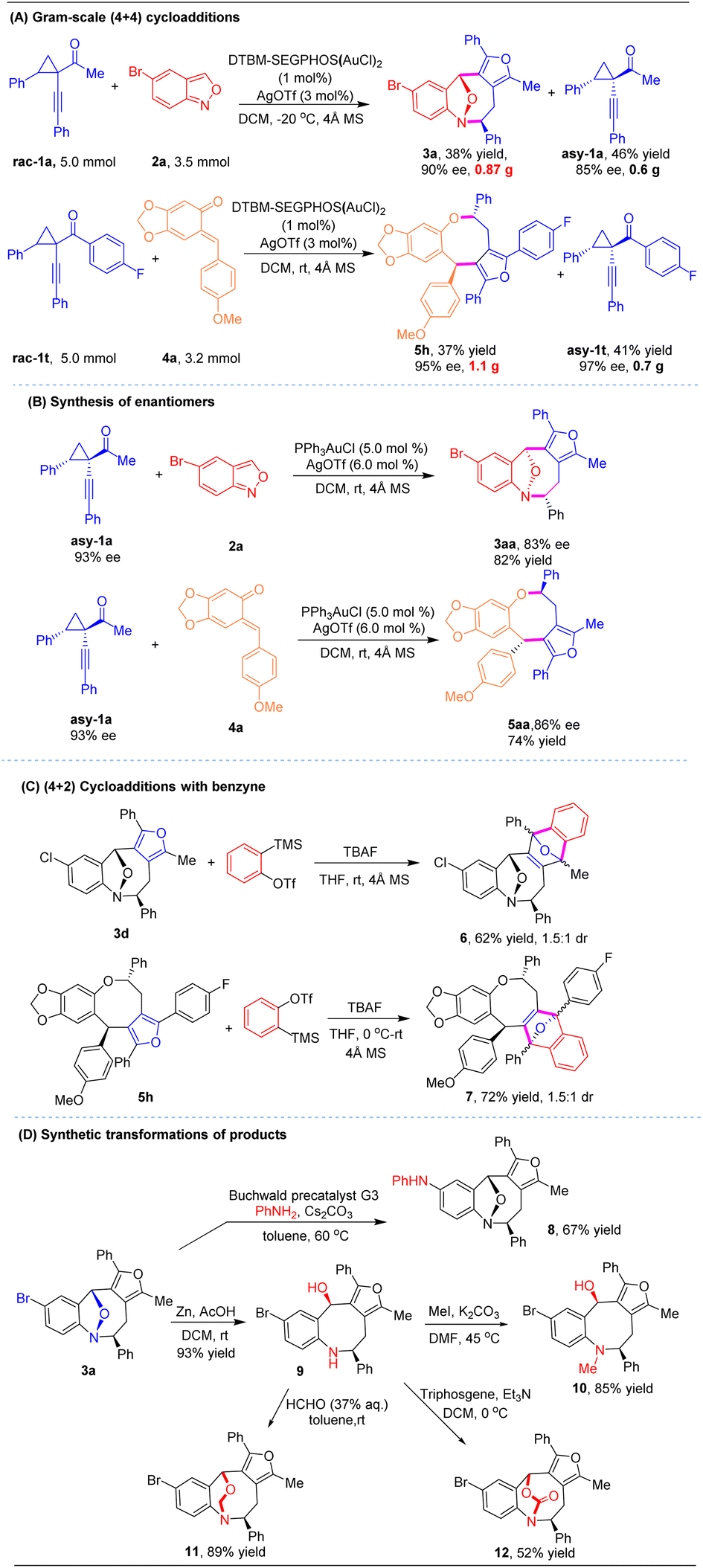 | ||
| Scheme 2 Synthetic transformations. | ||
We next carried out density functional theory (DFT) calculations to rationalize the mechanism of the asymmetric (4 + 4) cycloaddition method (see the ESI† for details). The energy profile is outlined in Scheme 3. The kinetic resolution process was realized by the activation of racemic 1-(1-alkynyl)cyclopropyl ketone 1 with the (S)-DTBM-SEGPHOS gold complex, generating two gold-π-alkyne intermediates INT1 and INT 2 with the energy barrier of 2.22 kcal mol−1. The formation of INT2 with cyclopropyl ketone 1 as (1R, 2S) configuration has lower energy compared to INT1. The following intramolecular ring closure to generate gold-mediated 1,4-dipole intermediate INT4 is more favored than the formation of INT3 by 5.26 kcal mol−1. For INT4, the subsequent stereoselective (4 + 4) cycloaddition is highly exothermic and energetically feasible with the energy barrier of 6.29 kcal mol−1. Comparing the energies of TS2 and TS2′ reveals that the observed cycloadduct (5S, 6R, 11R)-3 is 7.41 kcal mol−1 more favorable than the formation of cycloadduct (5R, 6S, 11S)-3. These results are consistent with the synthetic observations.
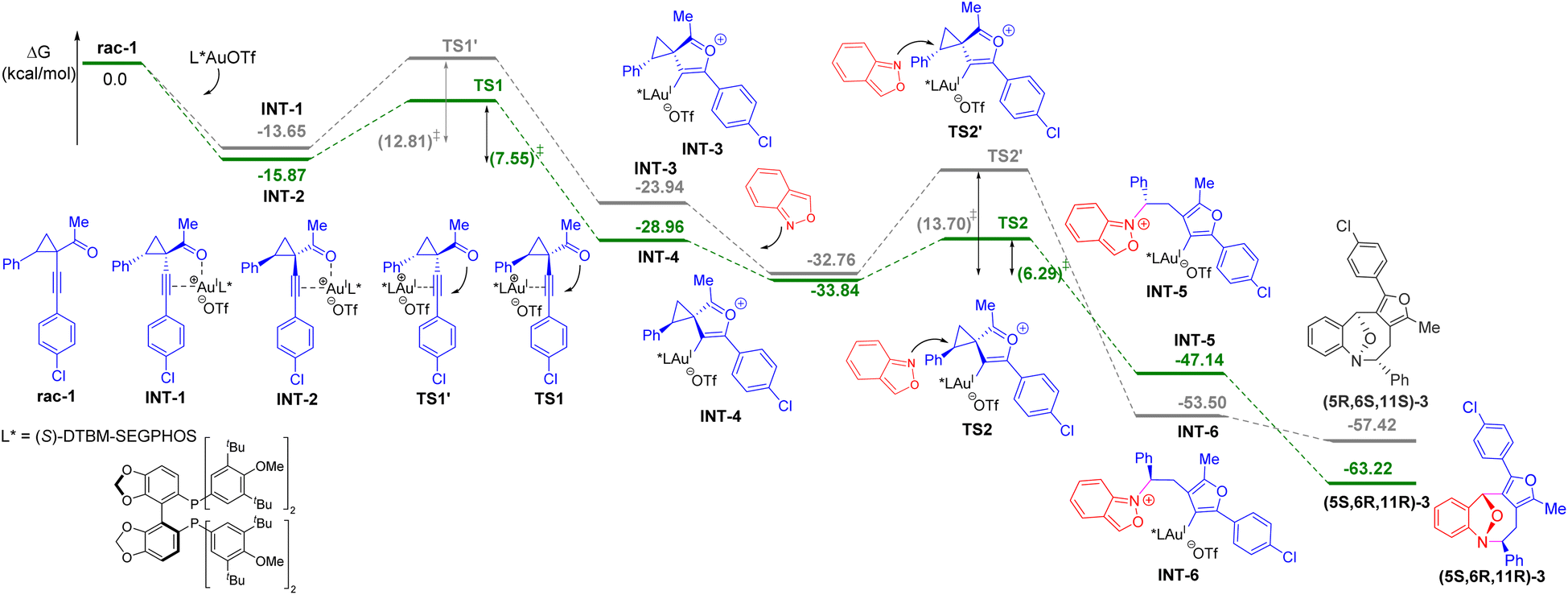 | ||
| Scheme 3 DFT calculations. | ||
Conclusions
We herein present two distinct chiral gold-catalyzed stereoselective (4 + 4) cycloadditions of cyclopropyl ketones with anthranils/o-QMs under mild reaction conditions, which offers a straightforward and promising approach for preparing densely substituted furan/pyrrole-fused eight-membered scaffolds featuring multiple stereocenters. High reaction efficiency (s factor up to 747) and excellent stereoselectivities were obtained, which are challenging within the asymmetric intermolecular higher-order cycloaddition arsenal. The broad substrate scope as well as good functional group tolerance makes this strategy appealing to the synthetic community. DFT calculations illustrated the kinetic resolution and (4 + 4) cycloaddition model. This strategy would not only expand the current library of furan/pyrrole-containing molecules with pharmaceutical importance, but also offer insights to explore unconventional chiral medium-sized structures.Data availability
The data (experimental procedures, characterization data and DFT calculations) that support this article is available within the article and its ESI.†Author contributions
X. Li conceived and supervised the project. X. Wang and R. Lv conducted the experiments and organized all data. X. Li wrote the manuscript with the consultation of all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the financial support for this work from the National Natural Science Foundation of China (21901142), the Natural Science Foundation of Jiangsu Province (BK20220097) and the Fundamental Research Funds of Shandong University (2020QNQT007 and 2020QNQT009).Notes and references
- (a) T.-Y. Ke, S.-W. Wang, S.-R. Chen, D.-M. Huang, Y.-S. Lin, T.-L. Hwang, W.-C. Sun and Y.-B. Cheng, J. Nat. Prod., 2023, 86, 719 CrossRef CAS PubMed; (b) T. Fukuyama, L. Xu and S. Goto, J. Am. Chem. Soc., 1992, 114, 383 CrossRef CAS; (c) B. Harrison and P. Crews, J. Org. Chem., 1997, 62, 2646 CrossRef CAS PubMed; (d) F. A. Macías, R. M. Varela, A. Torres, J. M. G. Molinillo and F. R. Fronczek, Tetrahedron Lett., 1993, 34, 1999 CrossRef; (e) F. A. Macías, J. M. G. Molinillo, R. M. Varela, A. Torres and F. R. Fronczek, J. Org. Chem., 1994, 59, 8261 CrossRef; (f) L.-C. Fu, X.-A. Huang, Z.-Y. Lai, Y.-J. Hu, H.-J. Liu and X.-L. Cai, Molecules, 2008, 13, 1923 CrossRef CAS PubMed; (g) K. M. George, M.-C. Frantz, K. Bravo-Altamirano, C. R. LaValle, M. Tandon, S. Leimgruber, E. R. Sharlow, J. S. Laz, Q. J. Wang and P. Wipf, Pharmaceutics, 2011, 3, 186 CrossRef CAS PubMed.
- (a) L. Yet, Chem. Rev., 2000, 100, 2963 CrossRef CAS PubMed; (b) I. Shiina, Chem. Rev., 2007, 107, 239 CrossRef CAS PubMed; (c) A. Hussain, S. K. Yousuf and D. Mukherjee, RSC Adv., 2014, 4, 43241 RSC; (d) J. R. Donald and W. P. Unsworth, Chem.–Eur. J., 2017, 23, 8780 CrossRef CAS PubMed; (e) D. McLeod, M. K. Thøgersen, N. I. Jessen, K. A. Jørgensen, C. S. Jamieson, X.-S. Xue, K. N. Houk, F. Liu and R. Hoffmann, Acc. Chem. Res., 2019, 52, 3488 CrossRef CAS PubMed; (f) Y.-J. Hu, L.-X. Li, J.-C. Han, L. Min and C.-C. Li, Chem. Rev., 2020, 120, 5910 CrossRef CAS PubMed; (g) R. L. Reyes, T. Iwai and M. Sawamura, Chem. Rev., 2021, 121, 8926 CrossRef CAS PubMed.
- (a) S. M. N. Sieburth and N. T. Canard, Tetrahedron, 1996, 52, 6251 CrossRef CAS; (b) Z.-X. Yu, Y. Wang and Y. Wang, Chem.–Asian J., 2010, 5, 1072 CrossRef CAS PubMed; (c) T. A. Palazzo, R. Mose and K. A. Jørgensen, Angew. Chem., Int. Ed., 2017, 56, 10033 CrossRef CAS PubMed; (d) N. De and E. J. Yoo, ACS Catal., 2018, 8, 48 CrossRef CAS; (e) B. Niu, Y. Wei and M. Shi, Org. Chem. Front., 2021, 8, 3475 RSC; (f) M.-M. Zhang, B.-L. Qu, B. Shi, W.-J. Xiao and L.-Q. Lu, Chem. Soc. Rev., 2022, 51, 4146 RSC; (g) J. Otevrel, M. Eugui, S. Ričko and K. A. Jørgensen, Nat. Synth., 2023, 2, 1142 CrossRef; (h) B. Xu, Q. Wang, C. Fang, Z.-M. Zhang and J. Zhang, Chem. Soc. Rev., 2024, 53, 883 RSC; (i) M. Tori and R. Mizutani, Molecules, 2010, 15, 4242 CrossRef CAS PubMed; (j) K. Clarke and W. P. Unsworth, Chem. Sci., 2020, 11, 2876 RSC; (k) S.-S. Qi, H. Yin, Y.-F. Wang, C.-J. Wang, H.-T. Han, T.-T. Man and D.-Q. Xu, Org. Lett., 2021, 23, 2471 CrossRef CAS PubMed; (l) T. Yao, J. Li, C. Jiang and C. Zhao, Chem Catal., 2022, 2, 2929 CrossRef CAS; (m) V. Corti, C. L. Barløse, N. L. Østergaard, A. Kristensen, N. I. Jessen and K. A. Jørgensen, J. Am. Chem. Soc., 2023, 145, 1448 CrossRef CAS PubMed.
- (a) M. Mathiew, J. K. Tan and P. W. H. Chan, Angew. Chem., Int. Ed., 2018, 57, 14235 CrossRef CAS PubMed; (b) C. Gao, X. Wang, J. Liu and X. Li, ACS Catal., 2021, 11, 2684 CrossRef CAS; (c) Q. Li, R. Pan, M. Wang, H. Yao and A. Lin, Org. Lett., 2021, 23, 2292 CrossRef CAS PubMed; (d) M.-M. Zhang, P. Chen, W. Xiong, X.-S. Hui, L.-Q. Lu and W.-J. Xiao, CCS Chem., 2021, 3, 3383 Search PubMed; (e) W.-L. Yang, Y.-L. Wang, W. Li, B.-M. Gu, S.-W. Wang, X.-Y. Luo, B.-X. Tian and W.-P. Deng, ACS Catal., 2021, 11, 12557 CrossRef CAS; (f) G.-H. Chen, Y. Ye, D.-X. Zhang, H.-J. Li, N. Zhang, G.-J. Liang, D. Zhang, J. Zhou and H. Zhou, Org. Chem. Front., 2023, 10, 4698 RSC; (g) J. Pan, T. O. Ho, Y.-C. Chen, B.-M. Yang and Y. Zhao, Angew. Chem., Int. Ed., 2023, 62, e202317703 Search PubMed.
- (a) B. M. Trost, Z. Jiao, Y. Liu, C. Min and C.-I. J. Hung, J. Am. Chem. Soc., 2020, 142, 18628 CrossRef CAS PubMed; (b) B. M. Trost and Z. Jiao, J. Am. Chem. Soc., 2020, 142, 21645 CrossRef CAS PubMed; (c) J. Du, Y.-D. Hua, Y.-J. Jiang, S. Huang, D. Chen, C.-H. Ding and X.-L. Hou, Org. Lett., 2020, 22, 5375 CrossRef CAS PubMed; (d) Y. Chen, M. Zang, W. Wang, Y.-Z. Liu, X. Luo and W.-P. Deng, Chin. J. Chem., 2023, 41, 2825 CrossRef CAS.
- (a) G. Zhang, X. Huang, G. Li and L. Zhang, J. Am. Chem. Soc., 2008, 130, 1814 CrossRef CAS PubMed . For selected reviews, see:; (b) R. Antonio and M. Echavarren, Chem. Rev., 2015, 115, 9028 CrossRef PubMed; (c) T. Wang and A. S. K. Hashmi, Chem. Rev., 2021, 121, 8948 CrossRef CAS PubMed; (d) L. De, W. Zang, M. J. Bird, C. J. T. Hyland and M. Shi, Chem. Rev., 2021, 121, 8685 CrossRef PubMed; (e) M. Mato, A. Franchino, C. García-Morales and A. M. Echavarren, Chem. Rev., 2021, 121, 8613 CrossRef CAS PubMed; (f) C. C. Chintawar, A. K. Yadav, A. Kumar, S. P. Sancheti and N. T. Patil, Chem. Rev., 2021, 121, 8478 CrossRef CAS PubMed; (g) D. Campeau, D. F. L. Rayo, A. Mansour, K. Muratov and F. Gagosz, Chem. Rev., 2021, 121, 8756 CrossRef CAS PubMed; (h) L.-W. Ye, X.-Q. Zhu, R. L. Sahani, Y. Xu, P.-C. Qian and R.-S. Liu, Chem. Rev., 2021, 121, 9039 CrossRef CAS PubMed; (i) L. Rocchigiani and M. Bochmann, Chem. Rev., 2021, 121, 8364 CrossRef CAS PubMed; (j) S. Witzel, A. S. K. Hashmi and J. Xie, Chem. Rev., 2021, 121, 8868 CrossRef CAS PubMed; (k) A. F. L. Rojas, S. H. Kyne and P. W. H. Chan, Acc. Chem. Res., 2023, 56, 1406 CrossRef PubMed.
- (a) J. F. Bai, J. Ren and Z. Wang, Chem.–Eur. J., 2009, 15, 8975 CrossRef PubMed; (b) Y. Bai, W. Tao, J. Ren and Z. Wang, Angew. Chem., Int. Ed., 2012, 51, 4112 CrossRef CAS PubMed; (c) Y. Zhang and J. Zhang, Adv. Synth. Catal., 2012, 354, 2556 CrossRef CAS; (d) Y. Zhang and J. Zhang, Chem. Commun., 2012, 48, 4710 RSC; (e) Z.-M. Zhang, P. Chen, W. Li, Y. Niu, X.-L. Zhao and J. Zhang, Angew. Chem., Int. Ed., 2014, 53, 4350 CrossRef CAS PubMed; (f) Y. Zhang and Y. X. J. Zhang, Synthesis, 2016, 48, 512 CrossRef CAS.
- T. Peng, S. Li, D. Yang and L. Wang, Org. Chem. Front., 2023, 10, 3401 RSC.
- See ESI† for details.
- Crystallographic data for the structure reported in this paper have been deposited with the Cambridge Crystallographic Data Centre as 1c (CCDC 2301807), 3c (CCDC 2301808) and 5g (CCDC 2301810).
- (a) V. Bhardwaj, D. Gumber, V. Abbot, S. Dhiman and P. Sharma, RSC Adv., 2015, 5, 15233 RSC; (b) G. Li Petri, V. Spanò, R. Spatola, R. Holl, M. V. Raimondi, P. Barraja and A. Montalbano, Eur. J. Med. Chem., 2020, 208, 112783 CrossRef CAS PubMed.
- (a) V. Estévez, M. Villacampa and J. C. Menéndez, Chem. Soc. Rev., 2014, 43, 4633 RSC; (b) N. Singh, S. Singh, S. Kohli, A. Singh, H. Asiki, G. Rathee, R. Chandra and E. A. Anderson, Org. Chem. Front., 2021, 8, 5550 RSC; (c) T. Shi, G. Yin, X. Wang, Y. Xiong, Y. Peng, S. Li, Y. Zeng and Z. Wang, Green Synth. Catal., 2023, 4, 20 CrossRef CAS.
- (a) W.-J. Bai, J. G. David, Z.-G. Feng, M. G. Weaver, K.-L. Wu and T. R. R. Pettus, Acc. Chem. Res., 2014, 47, 3655 CrossRef CAS PubMed; (b) B. Yang and S. Gao, Chem. Soc. Rev., 2018, 47, 7926 RSC; (c) C. Dorsch and C. Schneider, Synthesis, 2022, 54, 3125 CrossRef CAS; (d) X. Li, Z. Li and J. Sun, Nat. Synth., 2022, 1, 426 CrossRef.
- (a) F. A. Macías, J. M. G. Molinillo, R. M. Varela and A. Torres, J. Org. Chem., 1994, 59, 8261 CrossRef; (b) C.-J. Wu, C.-W. Li and C.-B. Cui, Mar. Drugs, 2014, 12, 1815 CrossRef PubMed; (c) M. Mueller, D. Weinmann, S. Toegel, W. Holzer, F. M. Unger and H. Viernstein, Food Funct., 2016, 7, 1671 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2301807, 2301808 and 2301810. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc02763a |
| This journal is © The Royal Society of Chemistry 2024 |