
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOrganocatalytic diastereo- and atropo-selective construction of eight-membered bridged (hetero)biaryls via asymmetric intramolecular [3 + 2] cycloaddition†
Yue
Wang
a,
Yue
Huang
a,
Xiaoze
Bao
 b,
Xingfu
Wei
a,
Shiqiang
Wei
a,
Jingping
Qu
a and
Baomin
Wang
*a
b,
Xingfu
Wei
a,
Shiqiang
Wei
a,
Jingping
Qu
a and
Baomin
Wang
*a
aDepartment of Pharmaceutical Engineering, State Key Laboratory of Fine Chemicals, School of Chemical Engineering, Dalian University of Technology, Dalian 116024, P. R. China. E-mail: bmwang@dlut.edu.cn
bCollege of Pharmaceutical Science & Collaborative Innovation Center of Yangtze River Delta Region Green Pharmaceuticals, Zhejiang University of Technology, Hangzhou 310014, P. R. China
First published on 7th May 2024
Abstract
An unprecedented and straightforward route for the asymmetric construction of privileged atroposelective bridged (hetero)biaryl eight-membered scaffolds has been accomplished through chiral phosphoric acid catalyzed asymmetric intramolecular [3 + 2] cycloaddition of innovative (hetero)biaryl aldehydes with 3-aminooxindole hydrochlorides. A class of eight-membered bridged (hetero)biaryl lactones fused to spiro[pyrrolidine-oxindole] derivatives, possessing both chiral C–C/C–N axes and multiple contiguous stereocenters, were obtained in good yields with excellent enantioselectivities and diastereoselectivities in one step through this direct strategy. In addition, the good scalability and derivatization of the title compounds demonstrated their synthetic utility.
Introduction
Atropisomeric biaryl skeletons featuring restricted rotation around a stereogenic axis constitute an important class of structural motifs found in a large variety of natural products, pharmaceutical agents, bioactive molecules and chiral ligands (Fig. 1a).1 Over the past two decades, considerable research efforts have been devoted to the asymmetric construction of diverse biaryl atropisomers.2 Axially chiral medium-sized bridged (hetero)biaryls, wherein the biaryl moiety is embedded in a medium-sized ring, are a unique subclass possessing more rigid structures (Fig. 1a). Notably, medium-sized bridged (hetero)biaryls are well featured in bioactive natural products (Fig. 1b). For example, spirombandakamine A1 and A2 exhibit strong antiprotozoal properties,3 (–)-steganacin is a lignan compound known for its anti-leukemic activity,4 and alkaloid (–)-rhazinilam, first isolated in 1965 from Melodinus Australia, has significant potential as a tubulin inhibitor.5 However, in sharp contrast to the creation of common axially chiral biaryls, the enantioselective synthesis of atropisomeric medium-sized bridged (hetero)biaryls is still underdeveloped,6 reflecting the difficulties and challenges in the enantiocontrol of the biaryls within the entropically and enthalpically unfavorable formation of medium-sized rings.7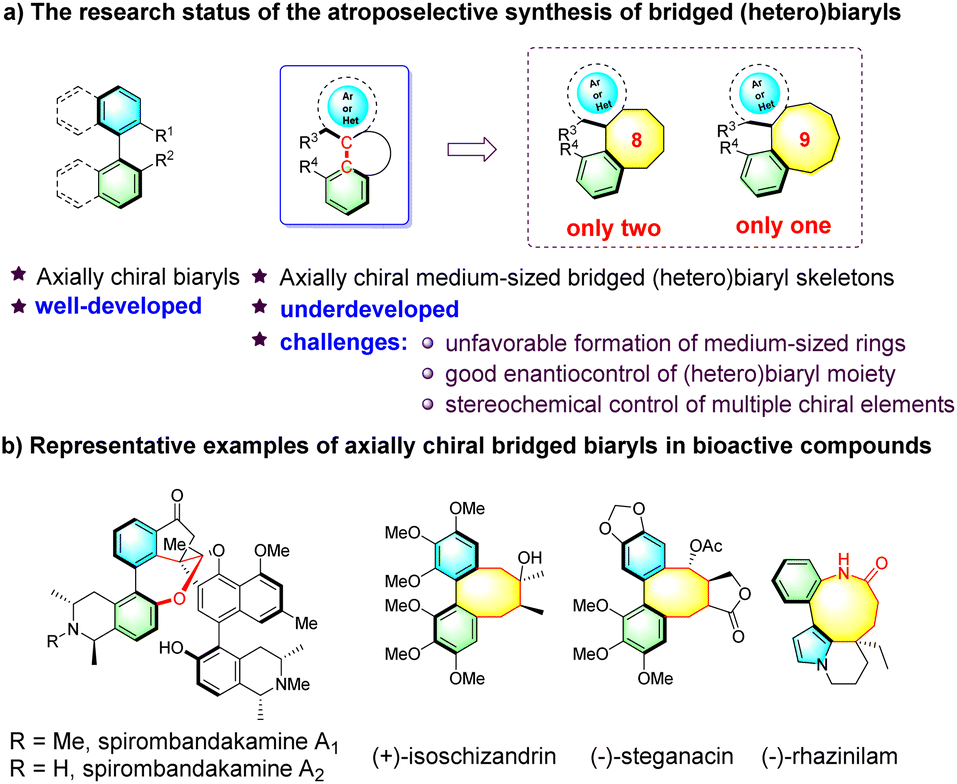 | ||
| Fig. 1 Catalytic asymmetric construction of axially chiral bridged (hetero)biaryls. | ||
To date, only two reports for the asymmetric construction of eight-membered and one for nine-membered bridged biaryls have been documented. In seminal work, Zhao's group reported the atroposelective synthesis of eight-membered bridged heterobiaryl lactones through an NHC-catalyzed cascade reaction (Scheme 1a).8 Very recently, Smith's group disclosed a process to construct eight-membered bridged biaryl lactams via an asymmetric intramolecular counterion-directed C-alkylation reaction (Scheme 1b).9 Meanwhile, Yan's group reported a quinine-thiourea-catalyzed nucleophilic cyclization to access axially chiral bridged nine-membered heterobiaryl carbonates through vinylidene ortho-quinone methide (VQM) intermediates (Scheme 1c).10 It should be noted that both the C–C chiral axis and one single chiral center were created in the former two reports, while catalytic methods of the enantioselective synthesis of medium-sized atropisomeric bridged (hetero)biaryls containing a C–C or C–N stereogenic axis and multiple contiguous stereocenters in one step have not been realized to date. As is known, significant challenges exist in the preparation of this kind of skeleton, such as the development of new strategies, the design of reasonable substrates and good enantio- and diastereocontrol of multiple chiral elements, which hindered the development of this topic.11
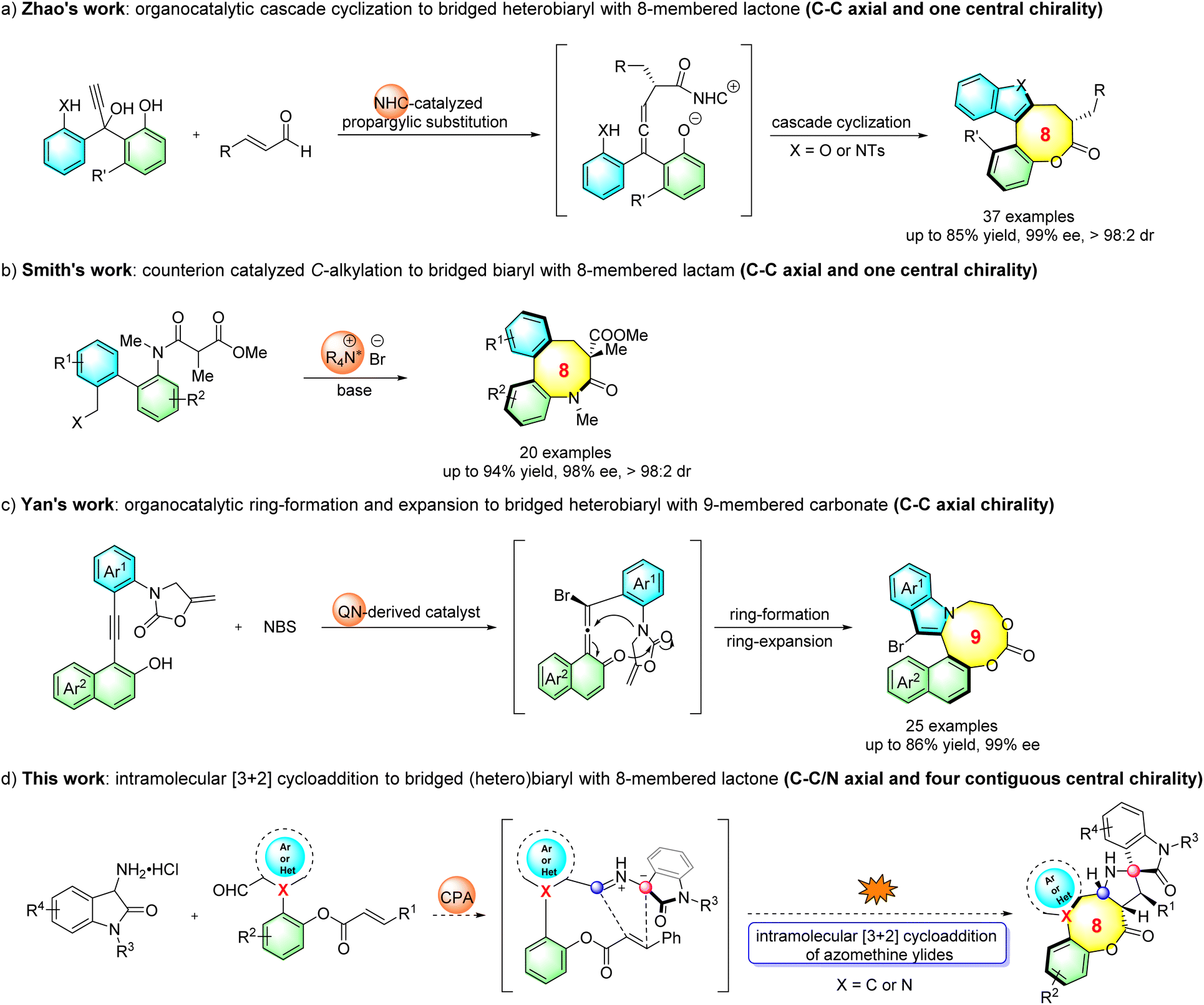 | ||
| Scheme 1 Current research on axially chiral medium-sized bridged (hetero)biaryls. | ||
Organocatalytic 1,3-dipolar cycloaddition involving azomethine ylides and electron-deficient unsaturated bonds is capable of furnishing biologically relevant nitrogenous five-membered heterocycles in asymmetric syntheses.12 Despite great advances achieved in organocatalytic asymmetric intermolecular [3 + 2] cycloaddition reactions, the corresponding intramolecular variants have barely been developed since the pioneering work by Gong in 2010.13 Inspired by the above-mentioned study and our previous work,14 herein, we report an organocatalytic intramolecular [3 + 2] cycloaddition process with in situ generation of azomethine ylides from 3-aminooxindole hydrochlorides and newly designed (hetero)biaryl aldehydes (Scheme 1d), which not only provides a straightforward asymmetric approach for the atroposelective synthesis of medium-membered bridged (hetero)biaryls, but also affords an array of structurally congested eight-membered bridged (hetero)biaryl lactones fused to spiro[pyrrolidine-oxindole] scaffolds bearing both C–C/C–N axial chirality and four contiguous chiral centers.
Results and discussion
As shown in Table 1, we commenced our study with 3-amino oxindole hydrochloride 2a and biaryl aldehyde 3a as model substrates in the presence of an appropriate inorganic base and 3 Å molecular sieves (MSs) to conduct the intramolecular [3 + 2] cycloaddition reaction in dichloromethane at room temperature with 10 mol% diphenyl phosphate (DPP). To our delight, the cycloaddition reaction proceeded smoothly to give the desired product 4aa in 46% yield, which is consistent with our design (entry 1). Encouraged by this preliminary result, we turned our attention to the asymmetric version by examining an array of BINOL-derived chiral phosphoric acids (CPAs) with different substituents and steric environments (entries 2–6). Among them, the catalyst 1e bearing a 9-phenanthryl group at 3,3′-positions was found to be the optimal choice to afford the target product 4aa with a stereogenic axis and multiple stereogenic centers in 64% yield and excellent diastereoselectivity and enantioselectivity (>20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr, 98% ee, entry 6). In addition, the amount of base was then evaluated, considering that an additional equivalent of inorganic base may have an effect on the formation of dipoles. The reactivity and efficiency of the reaction gradually decreased, when the amount of base was 1.2 equiv. or without the base, although excellent enantioselectivities and diastereoselectivities were maintained (entries 7 and 8). Subsequently, attempts to further improve the reactivity through screening of solvents (entries 9–13) proved to be effective, providing encouraging results and identifying Et2O as the best solvent for this reaction to furnish the product in 92% yield with excellent diastereo- and enantio-selectivity (>20:1 dr, 98% ee, entry 12).
:1 dr, 98% ee, entry 6). In addition, the amount of base was then evaluated, considering that an additional equivalent of inorganic base may have an effect on the formation of dipoles. The reactivity and efficiency of the reaction gradually decreased, when the amount of base was 1.2 equiv. or without the base, although excellent enantioselectivities and diastereoselectivities were maintained (entries 7 and 8). Subsequently, attempts to further improve the reactivity through screening of solvents (entries 9–13) proved to be effective, providing encouraging results and identifying Et2O as the best solvent for this reaction to furnish the product in 92% yield with excellent diastereo- and enantio-selectivity (>20:1 dr, 98% ee, entry 12).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Cat. | Solvent | T [h] | Yieldb [%] | drc | eed [%] | |
| a The reaction was carried out on a 0.1 mmol scale with Na2CO3 (1.5 equiv.), 3 Å MS (100 mg), cat. (10 mol%) in 1.0 mL solvent at 25 °C under nitrogen, and the ratio of 2a/3a was 1.5/1. b Isolated yield. c The dr was determined by 1H NMR of the crude reaction mixture. d The ee was determined by chiral HPLC. e In the presence of 1.2 equiv. Na2CO3. f No base. | |||||||
| 1 | DPP | DCM | 10 | 46 | >20:1 |
— | |
| 2 | 1a | DCM | 12 | 4 | >20:1 |
79 | |
| 3 | 1b | DCM | 48 | 31 | >20:1 |
95 | |
| 4 | 1c | DCM | 16 | 17 | >20:1 |
96 | |
| 5 | 1d | DCM | 16 | 47 | >20:1 |
96 | |
| 6 | 1e | DCM | 24 | 64 | >20:1 |
98 | |
| 7e | 1e | DCM | 28 | 44 | >20:1 |
98 | |
| 8f | 1e | DCM | 36 | 40 | >20:1 |
97 | |
| 9 | 1e | CHCl3 | 48 | 24 | >20:1 |
97 | |
| 10 | 1e | THF | 36 | 37 | >20:1 |
97 | |
| 11 | 1e | Tol. | 48 | 65 | >20:1 |
98 | |
| 12 | 1e | Et 2 O | 48 | 92 |
>20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1 :1
|
98 | |
| 13 | 1e | DCE | 40 | 40 | >20:1 |
98 | |
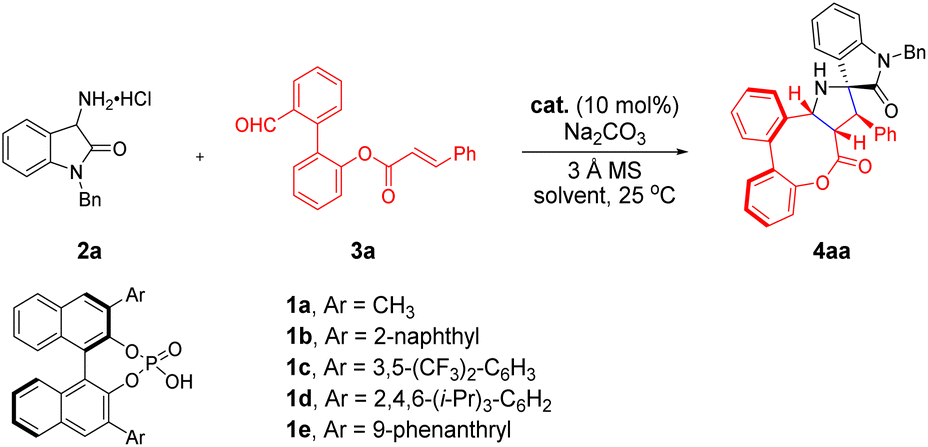
With the optimized reaction conditions established, we next set out to investigate the scope and generality of the intramolecular [3 + 2] cycloaddition strategy (Scheme 2). Initially, biaryl aldehydes 3 featuring diverse substituents, easily prepared via classic Pd-catalyzed Suzuki–Miyaura cross coupling reactions from readily available starting materials, were examined by reacting with substrate 2a under the standard conditions. In detail, substituents, including electron-donating (–Me and –OMe) and electron-withdrawing (–F) groups, were well tolerated at different positions (ortho, meta and para positions) of the aromatic R1 group, affording the corresponding products 4aa–4ag in good to excellent yields (62–92%) with consistently high diastereo- and enantio-selectivities (all >20:1 dr, 91–>99% ee). Meanwhile, the R1 group could be switched to heteroaryl or 1-naphthalene groups, which generally afforded products with good results (4ai and 4ah; 60% and 85% yields, 95% and >99% ee, respectively, all >20:1 dr). Notably, biaryl aldehyde 3j bearing a methyl functional group also performed well, delivering the expected product 4aj in 98% yield and 92% ee. In addition, we evaluated the scope with respect to the biaryl moiety under the optimized reaction conditions (4ak–4as). It is noteworthy that diverse substrates (3k and 3m–3p) proceeded smoothly with 2a to furnish desired atroposelective bridged biaryls, maintaining moderate to good yields and excellent ee values in this catalysis system. Among them, the absolute configuration of product 4am was determined by X-ray crystallographic analysis. However, when the C6 position of the biaryl moiety was the methoxy group, 4al could be applied in the current transformation, but in relatively lower yield and diminished enantioselectivity, perhaps due to the presence of bulky methoxyl. The result shows that the steric hindrance of substituents at the C6 position has a significant impact on both the efficiency and the stereoselectivity of the process. Moreover, biaryl aldehyde 3q containing the naphthalene group gives a racemic product under the standard conditions, and the reason might be that the larger naphthalene group adversely affects hydrogen-bonding interaction between the hydroxyl of catalyst 1e and the carbonyl group of the part of dipolarophile in organizing the transition state. Gratifyingly, biaryl aldehydes 3r and 3s bearing substitutes at the para- and meta-positions of the formyl were well accommodated, and desired products were successfully generated with high enantioselective excess.
 | ||
| Scheme 2 Substrate scope of 2 and 3. Reaction conditions: the reactions were conducted with 2 (0.3 mmol), Na2CO3 (0.3 mmol), 3 Å MS (200 mg), 1e (10 mol%) and 3 (0.2 mmol) in Et2O (2.0 mL) at 25 °C under nitrogen. Yields of the isolated products are given. The dr was determined by 1H NMR of the crude reaction mixture. The ee was determined by chiral HPLC. | ||
Next, the substrate scope for the organocatalytic asymmetric [3 + 2] cycloaddition of various 3-amino oxindole hydrochlorides 2 with biaryl aldehyde 3a was explored. Firstly, we probed the influence of the N-protecting group of the oxindole ring, and the result indicated that an alkyl group such as an N–Me-substituted reactant was fully compatible with the reaction, resulting in the anticipated product 4ba in good yield, albeit with lower enantioselectivity compared to model substrate 2a. It might be that N-Bn-substituted substrate 2a has π–π interaction with chiral phosphoric acid catalyst 1e, thereby achieving better stereochemical control. Ultimately, when incorporating electron-donating or electron-withdrawing substituents at the C5-position of the amino-oxindole benzene ring, the related products 4ca and 4da were also uneventfully obtained with excellent stereocontrol in good yields, respectively.
In addition, in parallel with atropisomers bearing a C–C axis, C–N axially chiral frameworks also frequently occur in natural products, pharmaceuticals and ligands with widespread applications.15 However, the construction of enantioenriched C–N atropisomers remains rare due to a lower rotation barrier and higher rotational degree of freedom around C–N bonds in comparison to the corresponding C–C bonds.16 Therefore, the enantioselective synthesis of C–N axially chiral skeletons containing additional stereogenic elements is more challenging.17 Inspired by the above results, we envisage that further expanding this methodology to the preparation of C–N axially chiral eight-membered bridged heterobiaryls with four stereogenic centers by fine-tuning of substrates may be possible. Hence, N-aryl-2-formylpyrroles 5 were successfully prepared through the key Clauson–Kaas reaction and subsequent transformations. As shown in ESI Table 1,† a preliminary investigation was performed by reacting newly designed heterobiaryl aldehyde 5a and 3-amino oxindole hydrochloride 2a in the presence of DPP, affording the desired product 6aa in 66% yield. Subsequently, the optimized reaction conditions were discovered via screening of diverse CPAs, solvents, reactant concentrations and bases. Under these conditions, the corresponding product 6aa was obtained in 84% yield and 93% ee.
Then, we investigated the substrate scope with regard to the substitution pattern of N-aryl-2-formylpyrroles 5. As highlighted in Scheme 3, newly designed and simply synthesized substrates bearing alkyl groups (5d and 5g), alkoxy groups (5b and 5h) and halogen groups (5c, 5e, 5f, 5i, 5j, and 5k) on the benzene ring of substituent R1 worked efficiently, generating the structurally varied products 6ab–6ak in 75–95% yields with 87–97% ee and >20:1 dr in all cases, which indicated that the substitution position and electronic feature affected neither the yield nor the diastereo- and enantio-selectivity. Among them, the absolute configuration of product 6ac was determined by X-ray crystallographic analysis. Particularly, both the introduced naphthyl and thienyl groups on the R1 moiety also participated well in this transformation, delivering the corresponding products 6al and 6am in 90% and 95% isolated yields with 97% ee and 95% ee, respectively. In addition to aryl and heteroaryl groups, the substrate with an aliphatic substituent (–Me) at this position was accommodated as well, furnishing the desired product 6an in good yield and excellent enantioselectivity. After that, pleasingly, different substituents at the C4 or C5 positions of the R2 group, such as methyl, chlorine and bromine, were all compatible with this reaction to give optically pure products 6ao–6as. In parallel, the influence of various substituents on 3-amino oxindole hydrochlorides 2 was then studied. Those substrates containing methyl and fluorine groups incorporated on the C5 position of the oxindole ring underwent the reaction smoothly under standard conditions, producing the products 6ba and 6ca in both 91% ee with 70% and 88% yields, respectively. Moreover, replacing the N–Bn of 2a with other electron-donating groups, such as N–Me, could also successfully afford product 6da with excellent yield, diastereo- and enantio-selectivity. In contrast, substrates 5 without a N-protecting group or with an electron-withdrawing group at the N1 position were not applicable at the current stage probably due to the poor nucleophilicity of the 3-amino group.
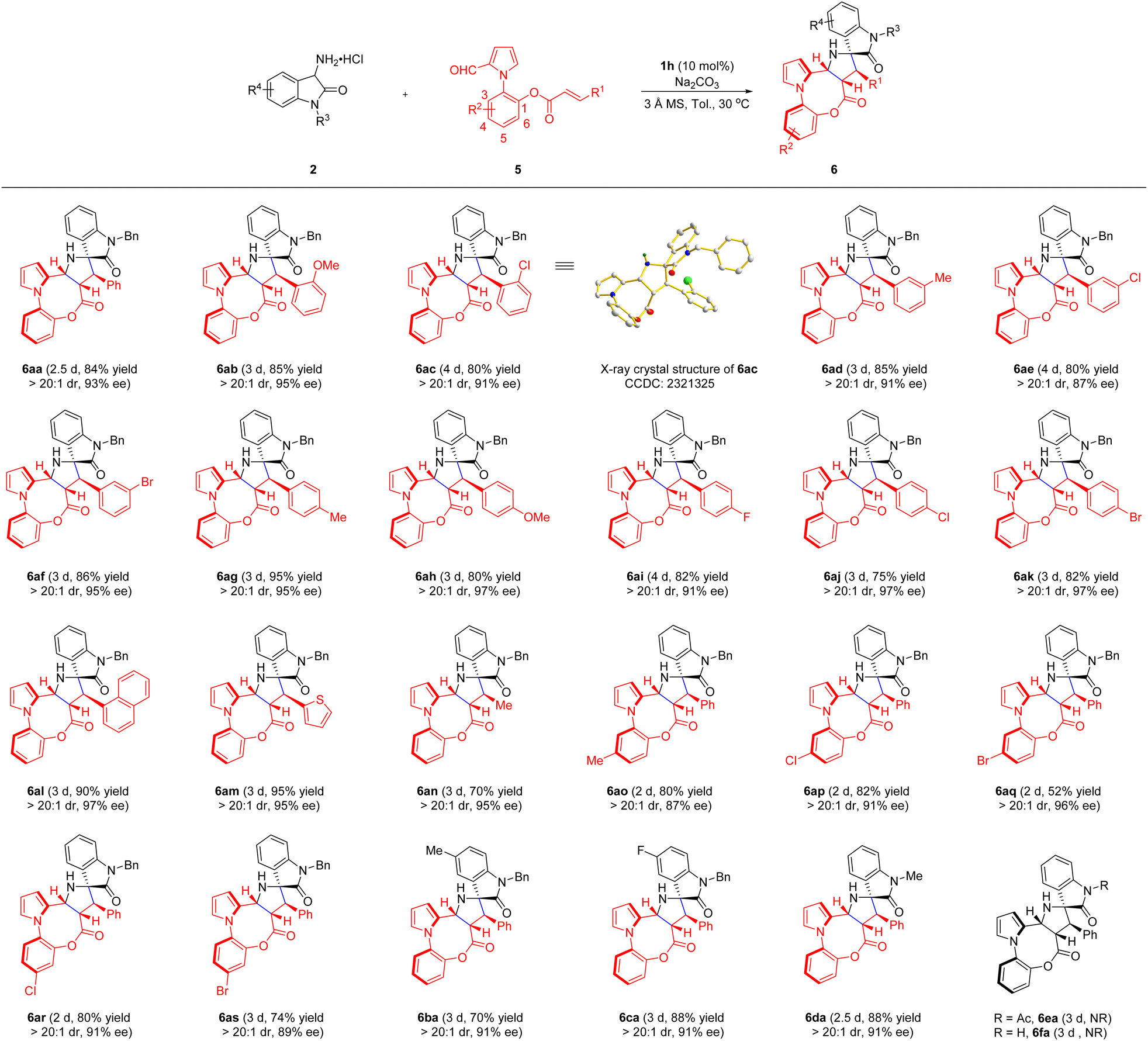 | ||
| Scheme 3 Substrate scope of 2 and 5. Reaction conditions: the reactions were conducted with 2 (0.3 mmol), Na2CO3 (0.3 mmol), 3 Å MS (200 mg), 1h (10 mol%) and 5 (0.2 mmol) in toluene (4.0 mL) at 30 °C under nitrogen. Yields of the isolated products are given. The dr was determined by 1H NMR of the crude reaction mixture. The ee was determined by chiral HPLC. | ||
To further demonstrate the synthetic practicality of this formal intramolecular [3 + 2] cycloaddition process (Scheme 4), the reaction of substrates 2a and 3d was conducted on a gram scale under the optimized conditions, producing the expected product 4ad in 75% yield with a similar level of enantioselectivity and diastereoselectivity observed for a lower-scale reaction. Subsequently, the 1.0 mmol scale reaction of 5m and 5s with 2a proceeded smoothly, achieving the atropisomers 6am and 6as in good yields with 96% ee and 87% ee, respectively.
 | ||
| Scheme 4 The scale-up experiments. | ||
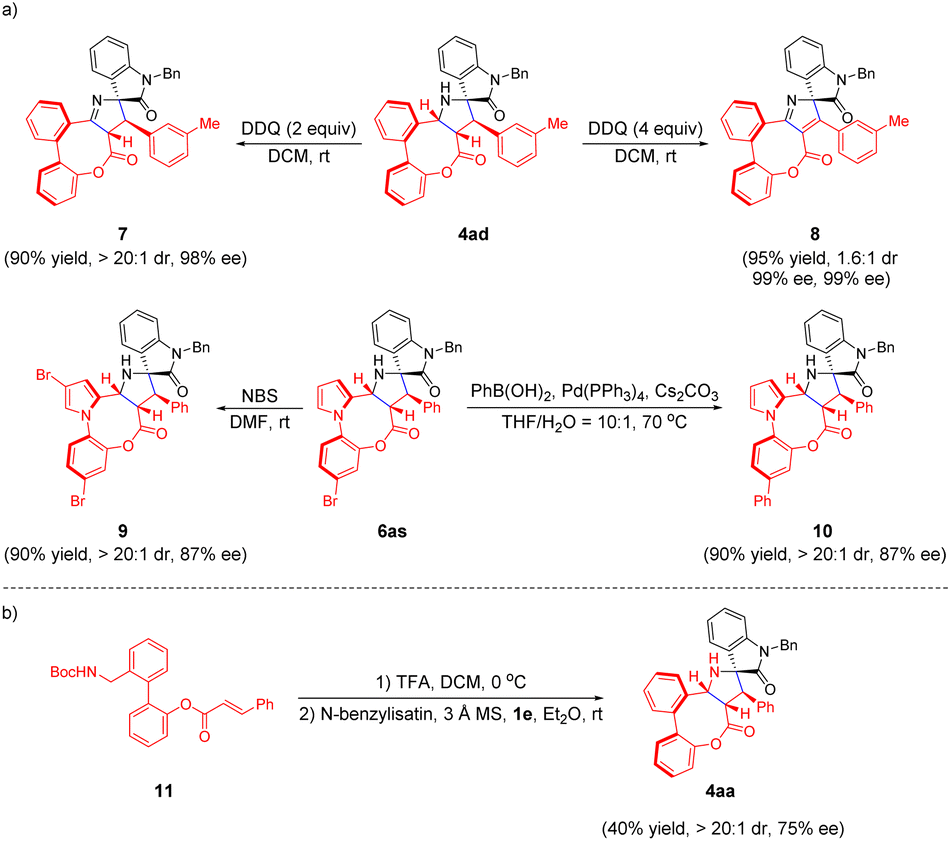
In addition, facile derivatization of the eight-membered bridged (hetero)biaryl lactones was carried out to showcase the potential application value of this protocol (Scheme 5a). The compound 4ad underwent dehydrogenative oxidation in the presence of 2.0 equiv. DDQ to provide 2H-pyrrole product 7 smoothly in 90% yield with high diastereoselectivity and enantioselectivity retained. Moreover, when 4ad was treated with 4.0 equiv. DDQ, the corresponding product 8 was achieved in 1.6:1 dr with 99% ee and 99% ee. The regioselective mono-bromination of 6as with NBS in DMF at room temperature afforded the compound 9 in 90% yield and 87% ee. Meanwhile, compound 10 could also be obtained in 90% yield, >20:1 dr and 87% ee, through a Pd-catalyzed Suzuki–Miyaura cross coupling reaction. Later, inspired by previous work,13b we explored the feasibility of the umpolung intramolecular 1,3-dipolar cycloaddition reaction of N-Boc biaryl benzylamine 11 and N-benzylisatin in the presence of catalyst 1e in Et2O at room temperature, affording compound 4aa in 40% yield with 75% ee (Scheme 5b).
 | ||
| Scheme 5 Synthetic transformations and umpolung intramolecular 1,3-dipolar cycloaddition reaction. | ||
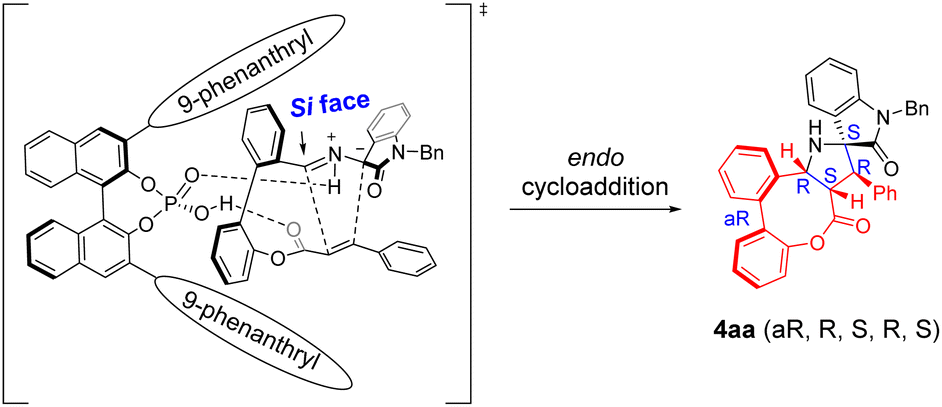
Combining the experimental results of our work with previous reports on chiral phosphoric acid catalysis, the plausible mode of dual activation was proposed and is depicted in Scheme 6 to interpret the observed outcome of stereocontrol. The high degree of diastereocontrol is attributed to the stereospecificity and stereoselectivity of [3 + 2] cycloaddition involving azomethine ylide18 together with the trend of eight-membered bridged biaryls to form a stable boat conformation.7f,19 On the other hand, chiral phosphoric acid (1e), a bifunctional catalyst, has hydrogen-bonding interaction with the reaction partner in this transformation. The hydroxyl of catalyst 1e is believed to act as a Brønsted acid to activate the carbonyl group of the part of dipolarophile, while the P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O moiety acts as a Brønsted base to activate the part of 1,3-dipole. In the meantime, the transition state undergoes intramolecular [3 + 2] cycloaddition from the Re face to afford the final product bearing multiple stereogenic elements with excellent enantioselectivity.
O moiety acts as a Brønsted base to activate the part of 1,3-dipole. In the meantime, the transition state undergoes intramolecular [3 + 2] cycloaddition from the Re face to afford the final product bearing multiple stereogenic elements with excellent enantioselectivity.
 | ||
| Scheme 6 Proposed transition state. | ||
Conclusions
In summary, we have successfully disclosed a chiral phosphoric acid catalyzed asymmetric intramolecular [3 + 2] cycloaddition reaction of (hetero)biaryl aldehydes bearing an unsaturated double bond with 3-amino oxindole hydrochlorides, which led to the efficient construction of highly enantiomerically enriched medium-membered bridged (hetero)biaryls. The reaction proceeded smoothly under mild conditions, which uneventfully delivered a wide range of atroposelective eight-membered bridged (hetero)biaryl lactones fused to spiro[pyrrolidine-oxindole] scaffolds containing both a C–C/C–N chiral axis and four contiguous stereocenters in good yields with high diastereoselectivities and enantioselectivities. In addition, further structural modifications demonstrated the promising utility of this methodology. Notably, only a single catalyst was used to realize stereochemical control of both central chirality and axial chirality. Therefore, this work provides a firm foundation for constructing other axially chiral bridged (hetero)biaryls bearing a larger-sized ring and further related studies are currently being conducted in our laboratory.Data availability
General information, detailed experimental procedures, characterization data for compounds, and NMR and HPLC spectra are available in the ESI.†Author contributions
Y. W. performed the experiments and analyzed the experimental data. B. W., Y. H., X. B., X. W., S. W. and J. Q. conceptualized and directed the project. B. W. and Y. W. wrote the manuscript with proofreading from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Fundamental Research Funds for the Central Universities (No. DUT21LAB134) for support of this work.Notes and references
-
(a) G. Bringmann, T. Gulder, T. A. M. Gulder and M. Breuning, Chem. Rev., 2011, 111, 563–639 CrossRef CAS PubMed
; (b) G. Bringmann and D. Menche, Acc. Chem. Res., 2001, 34, 615–624 CrossRef CAS PubMed
-
(a) G. Bringmann, A. J. Price Mortimer, P. A. Keller, M. J. Gresser, J. Garner and M. Breuning, Angew. Chem., Int. Ed., 2005, 44, 5384–5427 CrossRef CAS PubMed
- B. K. Lombe, T. Bruhn, D. Feineis, V. Mudogo, R. Brun and G. Bringmann, Org. Lett., 2017, 19, 6740–6743 CrossRef CAS PubMed
- S. M. Kupchan, R. W. Britton, M. F. Ziegler, C. J. Gilmore, R. J. Restivo and R. F. Bryan, J. Am. Chem. Soc., 1973, 95, 1335–1336 CrossRef CAS PubMed
-
(a) D. J. Abraham, R. D. Rosenstein, R. L. Lyon and H. H. S. Fong, Tetrahedron Lett., 1972, 13, 909–912 CrossRef
-
(a) S. L. Pira, T. W. Wallace and J. P. Graham, Org. Lett., 2009, 11, 1663–1666 CrossRef CAS PubMed
-
(a) B. Greve and P. Imming, J. Org. Chem., 1997, 62, 8058–8062 CrossRef CAS PubMed
- S. Lu, J. Y. Ong, H. Yang, S. B. Poh, X. Liew, C. S. D. Seow, M. W. Wong and Y. Zhao, J. Am. Chem. Soc., 2019, 141, 17062–17067 CrossRef CAS PubMed
- J. Y. Du, T. Balan, T. D. W. Claridge and M. D. Smith, J. Am. Chem. Soc., 2022, 144, 14790–14797 CrossRef CAS PubMed
- S. Jia, Y. Tian, X. Li, P. Wang, Y. Lan and H. Yan, Angew. Chem., Int. Ed., 2022, 61, e202206501 CrossRef CAS PubMed
-
(a) S. Krautwald and E. M. Carreira, J. Am. Chem. Soc., 2017, 139, 5627–5639 CrossRef CAS PubMed
-
(a) G. Pandey, P. Banerjee and S. R. Gadre, Chem. Rev., 2006, 106, 4484–4517 CrossRef CAS PubMed
-
(a) N. Li, J. Song, X. F. Tu, B. Liu, X. H. Chen and L. Z. Gong, Org. Biomol. Chem., 2010, 8, 2016–2019 RSC
-
(a) G. Zhu, B. Wang, X. Bao, H. Zhang, Q. Wei and J. Qu, Chem. Commun., 2015, 51, 15510–15513 RSC
-
(a) G. Bringmann, B. Hertlein-Amslinger, I. Kajahn, M. Dreyer, R. Brun, H. Moll, A. Stich, K. N. Ioset, W. Schmitz and L. H. Ngoc, Phytochemistry, 2011, 72, 89–93 CrossRef CAS PubMed
-
(a) D. Bonne and J. Rodriguez, Chem. Commun., 2017, 53, 12385–12393 RSC
-
(a) S. Huang, H. Wen, Y. Tian, P. Wang, W. Qin and H. Yan, Angew. Chem., Int. Ed., 2021, 60, 21486–21493 CrossRef CAS PubMed
-
(a) I. Coldham and R. Hufton, Chem. Rev., 2005, 105, 2765–2810 CrossRef CAS PubMed
- H. Tabata, H. Suzuki, K. Akiba, H. Takahashi and H. Natsugari, J. Org. Chem., 2010, 75, 5984–5993 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2234268 and 2321325. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc01892c |
| This journal is © The Royal Society of Chemistry 2024 |