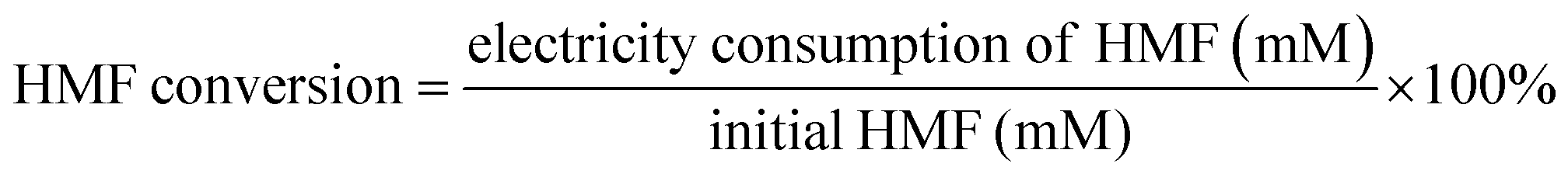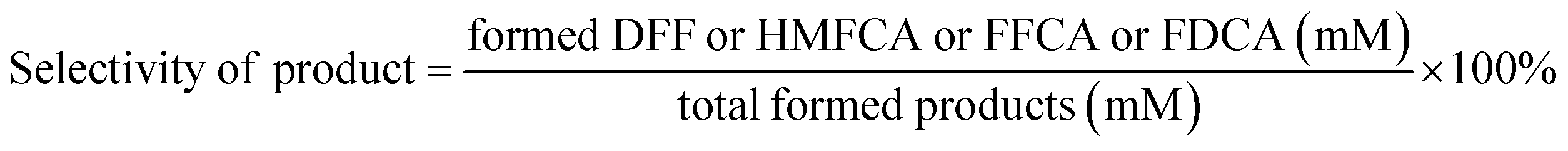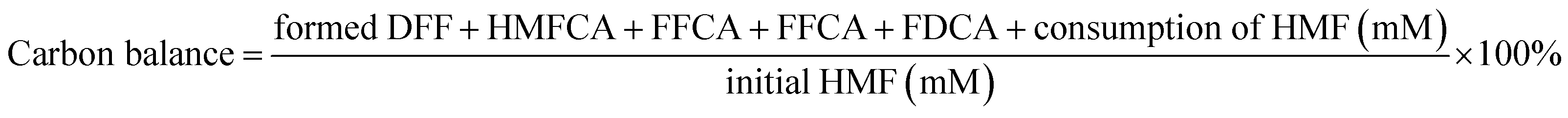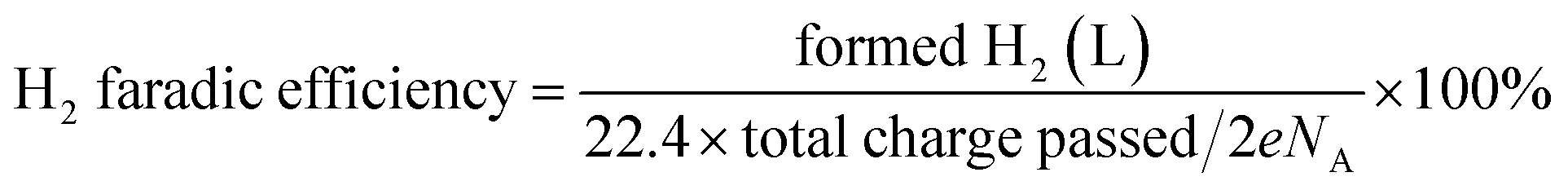DOI:
10.1039/D4SC01752H
(Edge Article)
Chem. Sci., 2024,
15, 11043-11052
Selective electrooxidation of 5-hydroxymethylfurfural to 5-formyl-furan-2-formic acid on non-metallic polyaniline catalysts: structure–function relationships†
Received
15th March 2024
, Accepted 12th June 2024
First published on 13th June 2024
Abstract
The biomass-derived HMF oxidation reaction (HMFOR) holds great promise for sustainable production of fine chemicals. However, selective electrooxidation of HMF to high value-added intermediate product 5-formyl-furan-2-formic acid (FFCA) is still challenging. Herein, we report the electrocatalytic HMFOR to selectively produce FFCA using carbon paper (CP) supported polyaniline (PANI) as a catalyst. The PANI/CP non-metallic hybrid catalyst with moderate oxidation capacity exhibitsoptimized FFCA selectivity up to 76% in alkaline media, which has reached the best performance in reported literature studies. Identification and quantification of active sites for the HMFOR are further realized via linking the activity to structural compositions of PANI; both polaronic-type nitrogen (N3) and positively charged nitrogen (N4) species are proved responsible for adsorption and activation of HMF, and the intrinsic activity of N4 is higher than that of N3. The present work provides new physical–chemical insights into the mechanism of the HMFOR on non-metallic catalysts, paving the way for the establishment of structure–function relations and further development of novel electrochemical synthesis systems.
1. Introduction
With the depletion of global fossil resources, the development and utilization of renewable resources have been focused intensively on the fields of chemical engineering and materials science.1 Biomass has become an ideal alternative to fossil resources due to its abundant reservation, renewability, and carbon neutrality.2 5-Hydroxymethylfurfural (HMF) is one of the main derivatives of biomass resources, which can be oxidized to produce a variety of high value-added chemicals.3 Among them, 5-formyl-furan-2-formic acid (FFCA) contains both aldehyde and carboxyl groups, which has a wide application prospect in the polyester industry and biomedicine field.4 Currently, the synthesis of FFCA from HMF mainly relies on liquid-phase catalytic reaction systems, the process of which normally requires noble metal catalysts (e.g., Au, Rh, and Pd) and toxic oxidants (e.g., MnO2 and KClO3) under harsh reaction conditions (>5 atm, 100–300 °C) leading to high cost and energy consumption.5 The key scientific challenge in this field is to develop an environmentally friendly and economical route to convert HMF to FFCA efficiently and economically.
The electrochemical reforming reaction process is an emerging technology for the efficient conversion of HMF to FFCA using H2O as an oxidant under mild conditions (room temperature and pressure),6 which has attracted increasing attention recently due to its obvious merits.7 On one hand, the electrooxidation reaction of HMF (HMFOR) can replace the oxygen evolution reaction (OER) with sluggish kinetics to reduce the onset potential of the H2O electrolysis reaction.8 On the other hand, H2 production through the hydrogen evolution reaction (HER) is typically considered an emission-free alternative to the conventional steam methane reforming process.9 The integration of the electrocatalytic HER and HMFOR is not only thermodynamically favorable but also produces valuable chemical feedstocks and clean H2 energy at the same time.10 However, the HMFOR still faces the critical issue that the target product (FFCA) selectivity is difficult to control.
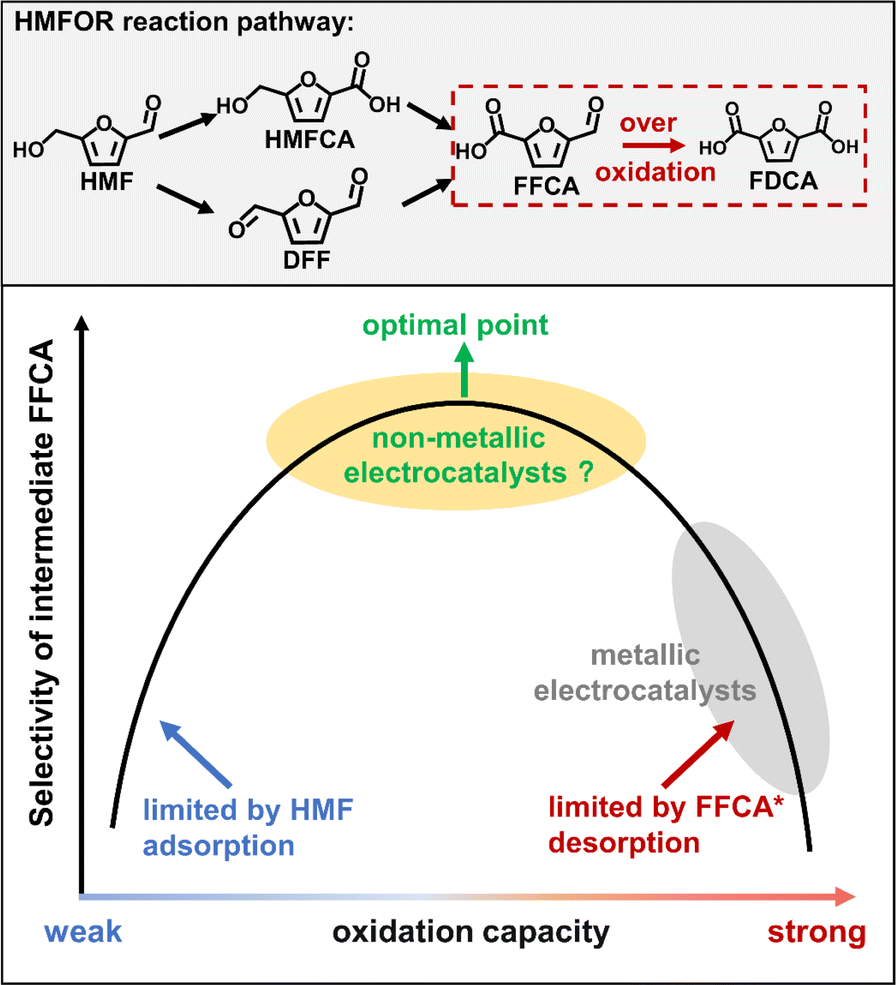
At present, most reported literature studies have focused on the direct oxidation of HMF to the final product 2,5-furan dicarboxylic acid (FDCA).11 However, it has the lowest market value among all the products shown in Scheme 1. As an intermediate product of the HMFOR, the market price of FFCA is 26 times higher than that of FDCA,12 so it is meaningful to selectively produce FFCA. It is very challenging to adopt a catalyst with moderate oxidation capacity (e.g., non-metallic materials) to achieve the selective formation of FFCA.13 There is one successful example reported that selectivity to FFCA could be enhanced in neutral electrolyte,14 but this system suffers from serious problems of poor reaction kinetics of the HMFOR compared to alkaline electrolyte due to the limited OH− concentration.15 To our best knowledge, there is barely any previous literature reporting the selective electrosynthesis of FFCA in an alkaline medium via the HMFOR route.
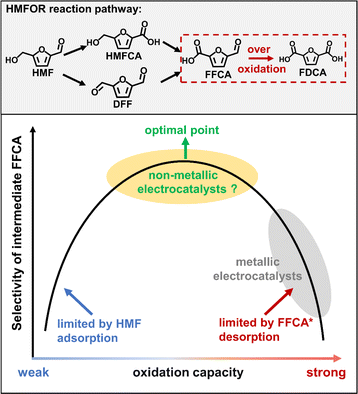 |
| | Scheme 1 Reaction pathway of the HMFOR process and the Sabatier principle for electrochemical synthesis of FFCA. | |
The key scientific challenge of this research topic is that the selective generation of intermediate FFCA requires the catalyst to exhibit a moderate oxidation capacity, which could perfectly balance the chemical adsorption of HMF and the timely desorption of FFCA. However, transition metal catalysts (e.g., Co3O4, FeOOH, NiPx, CuO etc.), which are normally used for the HMFOR process in most literature studies,11 have shown over-strong oxidation capacity for producing FFCA (Scheme 1). A transition metal catalyst normally exhibits high adsorption strength for FFCA* due to its strong electron withdrawing ability of the 3d orbital. In this case, the desorption of FFCA is seriously inhibited leading to the over-oxidation of HMF to fully oxidized product FDCA. It is crucial to design a series of catalysts with moderate and adjustable oxidation capacity. We believe that a non-metallic carbon catalyst is a suitable candidate for this reaction system.16 As a non-metallic catalyst, polyaniline has the advantages of low cost, easy structure regulation and mild oxidation capacity. However, the application of polyaniline in HMF electrooxidation has never been reported, and the exploration of its active site and structure–function relationship is also the key to achieve its high performance.
Herein, we selected polyaniline (PANI) with high conductivity and a tunable chemical structure as a novel non-metallic catalyst for realizing this reaction process,17,18 and the structure–function relationship of oxidation of HMF was revealed through combining active site regulation and quantitative calculations. In a typical procedure, PANI is in situ electro-polymerized and deposited on carbon paper (CP) with different deposition potentials yielding a series of PANI/CP hybrid electrode catalysts with various redox capacities, and the optimized PANI/CP catalyst could exhibit a relatively high FFCA selectivity of 76% at 1.96 VRHE in alkaline electrolyte. This is the first successful application of the non-metallic PANI catalyst in electrocatalytic selective oxidation of HMF. More importantly, the reaction pathway is well established, and the active sites for the HMFOR are identified and quantified via linking the catalytic activity to the chemical structure of the catalysts. The polaronic-type nitrogen (N3) and positively charged nitrogen (N4) species in PANI are proved responsible for the catalytic activation of HMF, and the intrinsic activity (rate constant) of N4 is higher than that of N3, which contributes over 80% overall activity. The establishment of structure–function relations provides the theoretical basis and guidance for designing novel non-metallic catalysts for the HMFOR process, shedding light on the potential practical applications of electro-chemical biomass refinery.
2. Experimental methods
2.1 Materials
Aniline (C6H7N, >99.0%) was purchased from Adamas. p-Toluenesulfonic acid (C7H8O3S, 90.0%) was purchased from Acros. Potassium hydroxide (KOH, >85.0%) and methanol (CH3OH, ≥99.9%) were all purchased from Sinopharm Chemical Reagent Co., Ltd. 5-Hydroxymethylfurfural (HMF, 97.0%), 2,5-furandicarboxylic acid (FDCA, 98.0%), 2,5-diformylfuran (DFF, >98.0%), 5-formyl-2-furan carboxylic acid (FFCA, 98.0%) and 5-hydroxymethyl-2-furancarboxylic acid (HMFCA, 98.0%) were all purchased from Alfa Aesar. Ammonium formate (CH5NO2, ≥99.0%) was purchased from Aladdin and the carbon paper was purchased from Shanghai Hesen Co., Ltd. Deionized water was dispensed from Hitech (Master-S). All the chemicals used in the experiments were at least of analytical grade and were used without further purification unless mentioned.
2.2 Synthesis of PANI/CP
The carbon paper (CP) substrate was first washed via sonication with deionized water and absolute ethanol for 10 min to remove residual impurities before experiments. Electro-polymerization deposition of polyaniline (PANI) on CP was conducted using a chronoamperometry method. Typically, the electro-polymerization experimental setup consisted of a standard three-electrode system in an undivided cell controlled by a CHI920D electrochemical workstation under ambient conditions. A piece of CP withdimensions of 1.5 cm × 2 cm was used as the working electrode, a Pt wire (PT0537, Gaoss Union) was used as the counter electrode and the Ag/AgCl containing saturated KCl solution was used as the reference electrode. The electrolyte was 50 mL H2O containing 0.3 g p-toluenesulfonic acid and 460 μL aniline solution. The deposition potential was maintained at 1.37, 1.57, 1.87, 2.17, 2.37 and 2.87 VRHE when the charge was transferred by 40 coulombs, which are named PANI1/CP, PANI2/CP, PANI3/CP, PANI4/CP, PANI5/CP and PANI6/CP, respectively. The potentials were converted to the RHE scale using ERHE = EAg/AgCl + 0.0591 × pH + 0.1976 V. These samples were rinsed with water and ethanol, and then the samples were vacuum dried before further characterization and electrochemical measurements.
2.3 Electrocatalytic measurements
All electrochemical measurements were performed in a divided cell with a CHI920D electrochemical workstation on a three-electrode setup under ambient conditions. The as-prepared PANI/CP samples with a platinum electrode clamp were directly used as the working electrode, coupled with Pt foil (3 cm × 4 cm, 0.1 mm thick, 99.8%, Gaoss Union) clamped with a glassy carbon electrode clamp as the counter electrode and the Hg/HgO containing 1 M KOH solution as the reference electrode. The Pt foil counter electrode area is four times the working electrode area, resulting in a small interfacial polarization, which ensures that the external polarization is mainly applied to the working electrode. All of the reported potentials were converted to the RHE scale using ERHE = EHg/HgO + 0.0591 × pH + 0.095 V.11 Linear sweep voltammetry (LSV) was conducted in a position scan at a rate of 10 mV s−1 at room temperature with stirring. Cyclic voltammetry (CV) for electrochemical surface area (ECSA) was conducted in a position scan at a rate of 100, 200, 300, 400, 500 and 600 mV s−1, respectively in the potential range of 0.65–1.45 VRHE at room temperature. Constant-potential electro-reforming of HMF was carried out at 1.96 VRHE using an H-cell reactor with anode (WE compartment) and cathode (CE compartment) chambers separated using a Nafion 117 proton exchange membrane. The anolyte (WE compartment) contained 15 mL of 0.1 M KOH solution (pH 13) with 10 mM HMF, while the catholyte (CE compartment) contained 15 mL of 0.1 M KOH solution. All electrochemical reactions were carried out at room temperature (25 ± 1 °C), and the electrolyte was stirred at 300 rpm with a magnetic stir bar at a constant potential after transferring 86 C of electrons. Then the HMF concentration was measured by HPLC and the loss of HMF was calculated. All electrochemical analyses reported in this study were performed without correcting for iR drop, providing a holistic evaluation without manipulation. The anode, cathode, and overall reactions could be summarized as follows:11
Anode reaction:
| HMF + 4OH−–4e− → FFCA + 3H2O |
Cathode reaction:
Overall reaction:
2.4 Characterization
The surface morphology and crystal structure of the electrodes were first characterized by scanning electron microscopy (SEM, LEO 1530 at an accelerating voltage of 5 kV). The transmission electron microscopy (TEM) images were recorded with an FEI Tecnai G2 T12 microscope. The thermal gravimetric analysis (TGA) was performed on a NETZSCH STA 449C. Fourier transform infrared spectroscopy (FT-IR) was performed on a SMART OMNI-TRANSMISSION (Thermo SCIENTIFIC). The surface chemistry of the electrodes was studied by X-ray photoelectron spectroscopy (XPS, ESCALAB250) with monochromatized Al Kα radiation. The XPS data were analyzed using CASA XPS software, and the binding energy was calibrated using the C 1s peak at 284.6 eV as a reference. The Raman spectra were measured on a LabRam HR 800 using a laser wavelength of 532 nm. The liquid chromatograph mass spectrometry spectrum (LC-MS) was measured on an Agilent Infinity II QTOF G6545.
2.5 Quantitative product analysis
High performance liquid chromatography (HPLC, Elite) with an online degassing machine (DG230-2) was used and two high pressure constant current pumps (P230) and an UV-Vis detector (UV 230+) were used for the quantitative analysis of the product and evaluation of the HMF oxidation efficiency. For every 20 C electrons transferred, 5 μL of the anode electrolyte containing the substrate and product was withdrawn and injected into high-performance liquid chromatography equipped with an UV-Vis detector set at λ = 265 nm and a waters organic acid column (SinoChrom ODS-BP 5 μm, 4.6 mm × 200 mm, E2715082) at room temperature. The analysis was performed under the following conditions: the mobile phase was a mixture of 5 mM ammonium formate (≥99.0%, Aladdin) aqueous solution and methanol (≥99.9%, Sinophare Chemical Reagent Co., Ltd). The separation was accomplished using an isocratic elution by using 50% ammonium formate aqueous solution and 50% methanol for 10 min with the flow rate set at 0.5 mL min−1. The identification and quantification of the oxidation products were performed based on a standard calibration curve of the pure component with the determined concentration. Hydrogen was quantified using a drainage-gas-collection method. The conversion of HMF (%), selectivity (%) of the oxidation products and carbon balance (%) were calculated using eqn (1)–(3). The reduction product H2 was measured by the drainage-gas collection method, and the faradaic efficiency of H2 was calculated using eqn (4).| |  | (1) |
| |  | (2) |
| |  | (3) |
| | 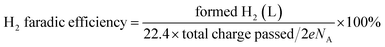 | (4) |
in which e is the electron charge (1.6 × 10−19 C) and NA is Avogadro's number (6.02 × 1023 mol−1).
3. Results & discussion
3.1 Morphology and chemical structure characterization
The proposed polyaniline/carbon paper (PANI/CP) hybrid electrodes were prepared by a simple electro-polymerization process with aniline as the precursor at a deposition potential of 1.37, 1.57, 1.87, 2.17, 2.37 and 2.87 VRHE, respectively and are named PANIx/CP (x = 1 to 6) in the sequence of their deposition potential. The synthesized PANI catalysts exhibit spherical particle-like morphology with a diameter of around 50 nm as shown in Fig. 1a and S1,† and they are firmly attached onto the carbon fiber (Fig. 1b and S2†). The C, O and N elements distribute uniformly on the surface of PANI/CP (Fig. 1c–f). The thermogravimetric analysis (TGA) results show that the PANIs begin to decompose at 350 °C and will be completely combusted at 650 °C in an air atmosphere (Fig. S3†).19 TGA analysis proves that these series of catalysts exhibit similar mass loss at 650 °C, indicating the similar loading content of PANI in these samples, which excludes the effect of catalyst dosage on catalytic performance.
 |
| | Fig. 1 Microscopic morphology characterization. (a) TEM (b) and (c) SEM images of PANI1/CP. SEM-EDAX images of (d) C element, (e) N element and (f) O element of the PANI1/CP surface. | |
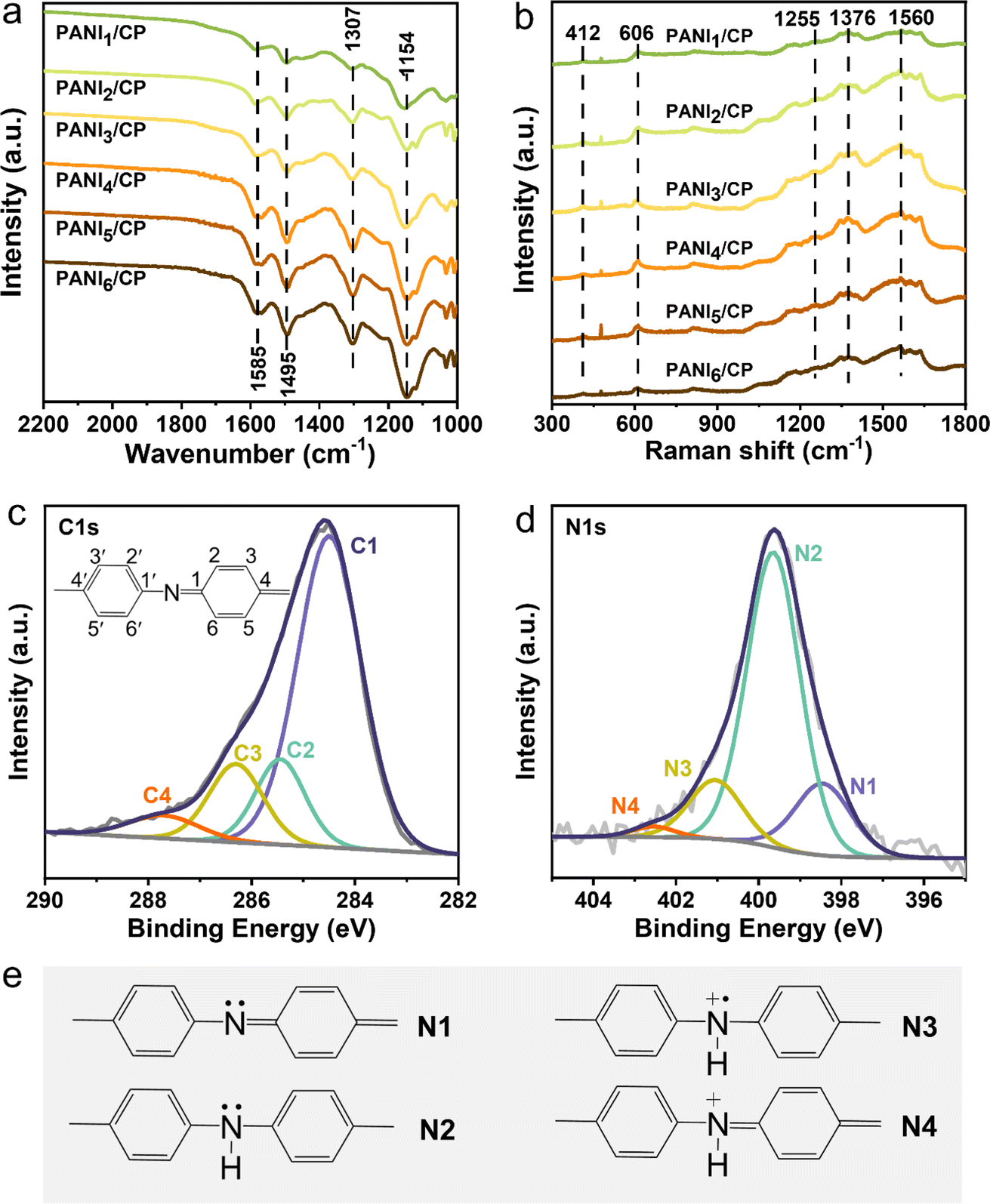
The characteristic FT-IR and Raman signals belonging to PANI could be observed as shown in Fig. 2a and b. The FT-IR spectrum shows vibration bands of N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) QN (Q represents the quinoid ring, 1154 cm−1), stretching mode of C–N (1307 cm−1), stretching vibration of CC in benzenoid rings (1495 cm−1), and stretching vibration of CC in quinoid rings (1585 cm−1) (Table S1†), and Raman spectra display out of plane C–H wag in polaronic structures (412 cm−1), benzenoid ring in-plane deformation vibration (606 cm−1), C–N stretching in polaronic units (1255 cm−1), C–N+˙ stretching of radical cations in semi quinoid rings (1376 cm−1), and C–C stretching of quinoid rings (1560 cm−1) (Table S2†), indicating the successful synthesis of PANI/CP catalysts,20–22 which have shown a variety of nitrogen functional groups on their surface.
QN (Q represents the quinoid ring, 1154 cm−1), stretching mode of C–N (1307 cm−1), stretching vibration of CC in benzenoid rings (1495 cm−1), and stretching vibration of CC in quinoid rings (1585 cm−1) (Table S1†), and Raman spectra display out of plane C–H wag in polaronic structures (412 cm−1), benzenoid ring in-plane deformation vibration (606 cm−1), C–N stretching in polaronic units (1255 cm−1), C–N+˙ stretching of radical cations in semi quinoid rings (1376 cm−1), and C–C stretching of quinoid rings (1560 cm−1) (Table S2†), indicating the successful synthesis of PANI/CP catalysts,20–22 which have shown a variety of nitrogen functional groups on their surface.
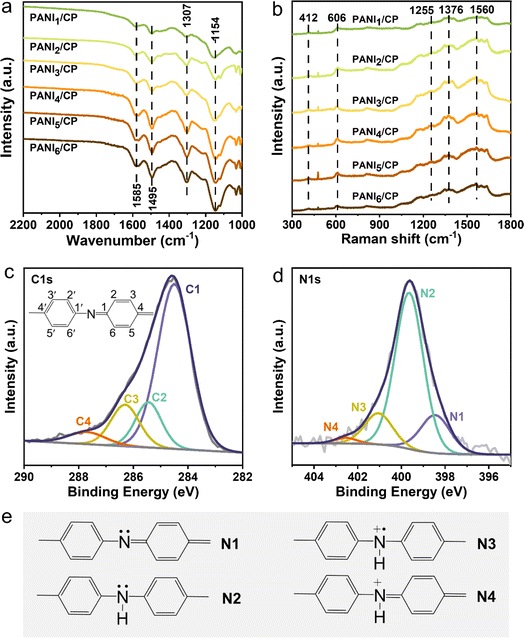 |
| | Fig. 2 Chemical structure characterization. (a) FT-IR and (b) Raman spectra of PANI/CP samples. (c) Deconvoluted C 1s XPS and (d) N 1s XPS spectra of PANI1/CP samples. (e) Structures of four nitrogen species. | |
The C ls X-ray photoelectron spectroscopy (XPS) spectra of PANI/CP samples are shown in Fig. 2c and S4,† which can be deconvoluted into four distinct peaks centered at 284.5, 285.4, 286.4 and 287.4 eV, respectively.23,24 The first peak at the lowest binding energy is attributed to the neutral C–C or C–H bonds, and all carbon atoms excluding those at the 1, 1′ and 4, 4′ positions in the polymer unit should correspond to this C1 species.25 C2 at ∼285.4 eV should be attributed to the carbon bonded with the neutral nitrogen atoms. Any increase in the positive charge on the carbon-bonded nitrogen atoms would result in a corresponding positive shift in the binding energy of the C ls line.25 The carbon atoms that are bonded to the polaronic-type nitrogen atoms could be attributed to the C3 species, and the carbon atoms bonded to the positively charged nitrogen atoms are assigned as the C4 species.26 The N 1s XPS spectra of all PANI/CP catalysts can be deconvoluted into four peaks (Fig. 2d), and these peaks correspond to quinoid imine nitrogen (N1), benzenoid amine nitrogen (N2), polaronic-type nitrogen (N3) and positively charged nitrogen species (N4) centered at 398.5, 399.6, 401.1 and 402.6 eV, respectively (Fig. 2e).27,28 It could be observed that the relative content of each N species in the PANI catalysts could be adjusted via alternating the electro-polymerization potentials (Fig. S5†), which will inevitably affect the intrinsic redox activity of the catalyst.28 In a word, combining the SEM, FT-IR, Raman and XPS results, the basic chemical structure of PANI/CP catalysts could be summarized as the PANI spheres that are firmly attached on carbon fiber, and the chemical structure diagram of PANI is determined as shown in Fig. S6.† There are four different types of N-containing species randomly distributed in the prepared PANI chain, and the relative content of these nitrogen species could be adjusted via tuning the electro-polymerization potential.
3.2 HMFOR activity and reaction pathway analysis
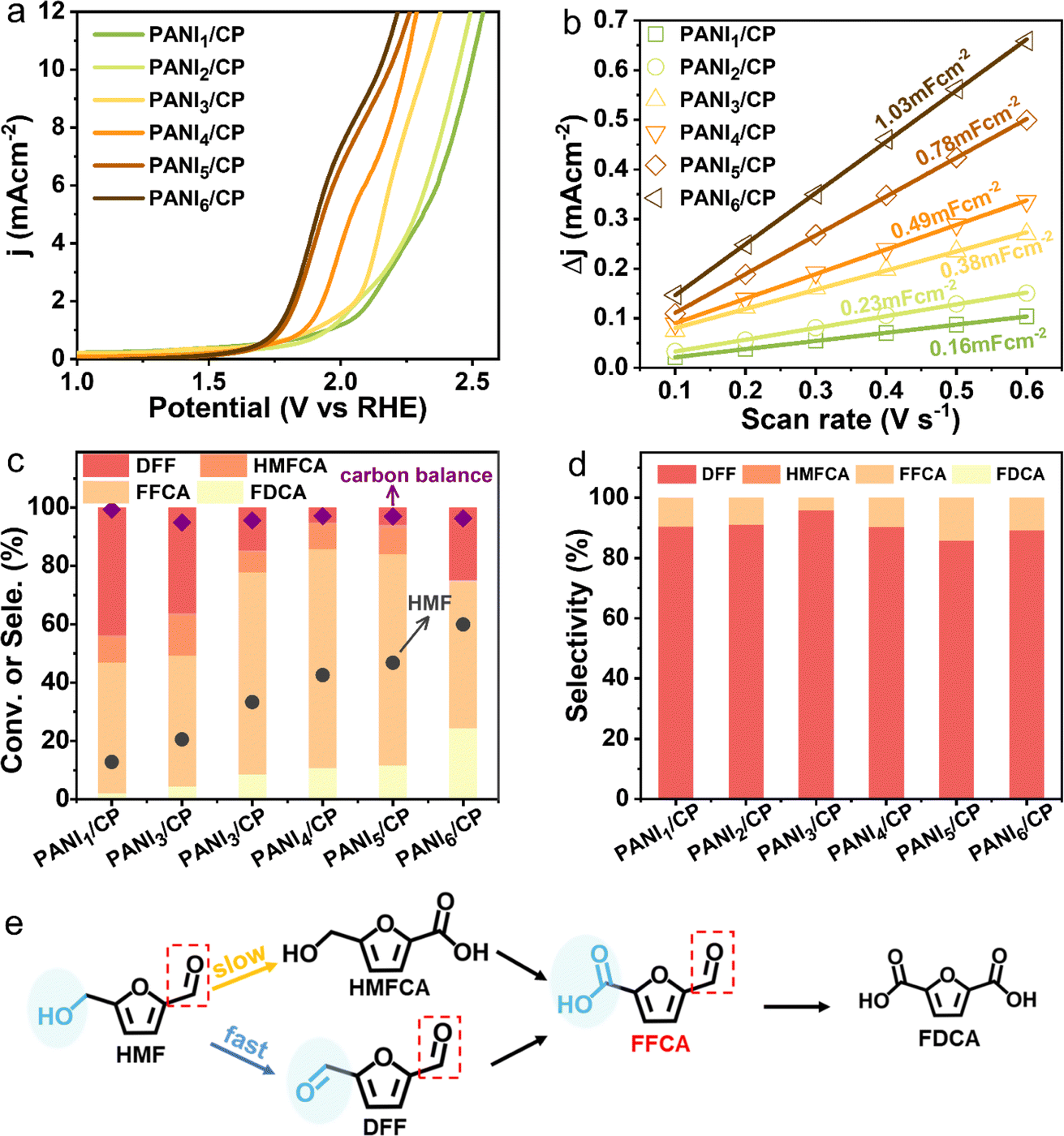
The HMFOR activity of PANI/CP catalysts was tested in an H-type electrochemical cell, and a commercial Pt sheet and Hg/HgO were applied as the counter and reference electrode, respectively. The oxygen evolution reaction (OER) is the major competing reaction for the HMFOR. Linear sweep voltammetry (LSV) exhibits a decrease in the onset potential after adding 10 mM HMF into the electrolyte (Fig. S7†), suggesting that the HMFOR is more favorable than the OER over PANI/CP from the viewpoint of thermodynamics.29–31 Moreover, the onset potential of the HMFOR decreases gradually at 10 mA cm−2 (Fig. 3a), and the electrochemical specific surface area (ECSA) increases gradually from PANI1/CP to PANI6/CP catalysts (Fig. 3b and S8†), indicating that the adsorption/activation capacity of the catalysts for HMF improves with the increasing deposition potential, which could be directly related to the chemical structure of the catalyst, namely the relative content of different nitrogen species. Conversion of HMF and corresponding product distributions and the carbon balance of the reaction system are shown in Fig. 3c. It needs to be mentioned that these reaction results are obtained by chronoamperometry until 86 C electrons are transferred at a potential of 1.96 VRHE, where the reaction reaches the stoichiometric number for complete oxidation of HMF to FDCA. However, it is surprising to observe that FFCA is the main oxidation product, and only a small amount of FDCA can be detected on these carbon catalysts (Fig. S9†). An optimal FFCA selectivity of 76% for PANI4/CP and a corresponding HER faradaic efficiency (FE) of ∼100% could be achieved as shown in Fig. S10.† For comparison, we prepared NiO, CuO, polypyrrole and polythiophene catalysts and applied them in the electrooxidation reaction of HMF under alkaline conditions. The relevant experimental results are summarized in Table S3.† The results showed that the main product was FDCA for the transition metal catalysts NiO and CuO due to its strong oxidation ability. The ability of polythiophene and polypyrrole to catalyze HMF oxidation was very poor, and the conversion rate of HMF was less than 20%. Therefore, the N atom between benzene rings in PANI may be the main active site. In addition, DFF was the main product rather than FFCA, indicating that PANI/CP as a non-metallic catalyst with mild oxidation ability can promote the selective production of intermediate product FFCA, which is totally different from the results from metal-based catalysts reported in most literature studies that FDCA is the main oxidation product for the HMFOR (ESI Table S4†).9–11,14,32–46 In addition, when DFF or FFCA as the only reactant was added into the electrolyte, the conversion rates of DFF were faster than that of FFCA for all PANI/CP samples (Fig. S11†), which resulted in the accumulation of FFCA. Moreover, the stability test of the catalyst showed that the conversion of HMF and the selectivity of FFCA remained at about 43% and 74% after 6 cycles (Fig. S12†). In addition, the substrate concentration was expanded to 0.5 M and the electrooxidation reaction of HMF was carried out in a flowing system and the FFCA selectivity reached 72% after 24 hours, indicating that the catalyst has excellent durability.
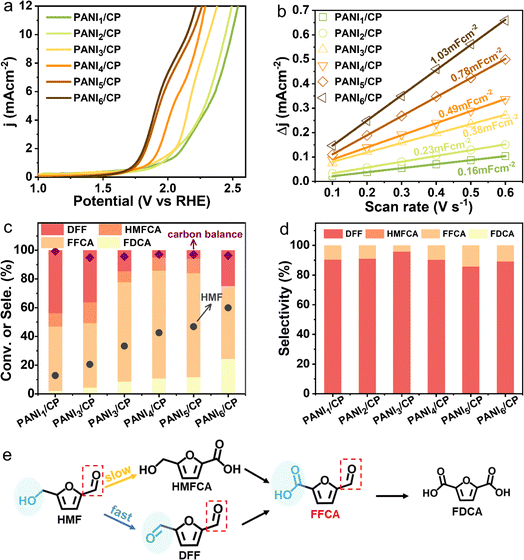 |
| | Fig. 3 Electrochemical performances and reaction pathway. (a) Linear sweep voltammetry curves of all PANI/CP catalysts in 0.1 M KOH solution (pH 13) with 10 mM HMF at a scan rate of 10 mV s−1. (b) Scan rate as a function of current density for all PANI/CP catalysts. (c) Conversion of HMF, selectivity of DFF, HMFCA, FFCA and FDCA and carbon balance of all PANI/CP samples until transfer of 86 C electrons at a potential of 1.96 VRHE. (d) Product distribution for all PANI/CP catalysts at a potential of 1.96 VRHE in 0.1 M KOH after transferring 10 C electrons. (e) Schematic reaction pathways in the process of the HMFOR. | |
Fig. 3d exhibits the HMFOR product distribution on PANI/CP catalysts at a fixed passing electron of 10 C, where the HMF conversion is below 1%, and these data may exclude the effect of substrate concentration variation and reflect the intrinsic selectivity and reaction pathways.14 As shown in Fig. 3d, DFF is the primary product and 5-hydroxymethyl-2-furancarboxylic acid (HMFCA) could not be detected for all PANI/CP catalysts. Furthermore, the conversion rate of HMF is higher than that of DFF via the PANI4/CP catalyst (Fig. S13†), indicating that the oxidation of the hydroxyl (–OH) group is faster than that of the aldehyde (–CO) functionality for PANI/CP catalysts (Fig. 3e). Therefore, the HMFOR process is more inclined to follow the HMF-DFF-FFCA reaction path (Fig. 3e), which is different from the HMF-HMFCA-FFCA path under the catalysis of metallic materials in alkaline solutions reported in most of the literature studies.32–46 This unique behavior of non-metallic PANI catalysts may effectively protect the aldehyde group (–CO) in HMF from oxidation via altering the reaction path, leading to the highly selective HMFOR process yielding FFCA.
3.3 Identification and quantification of the active sites and structure–function relation analysis
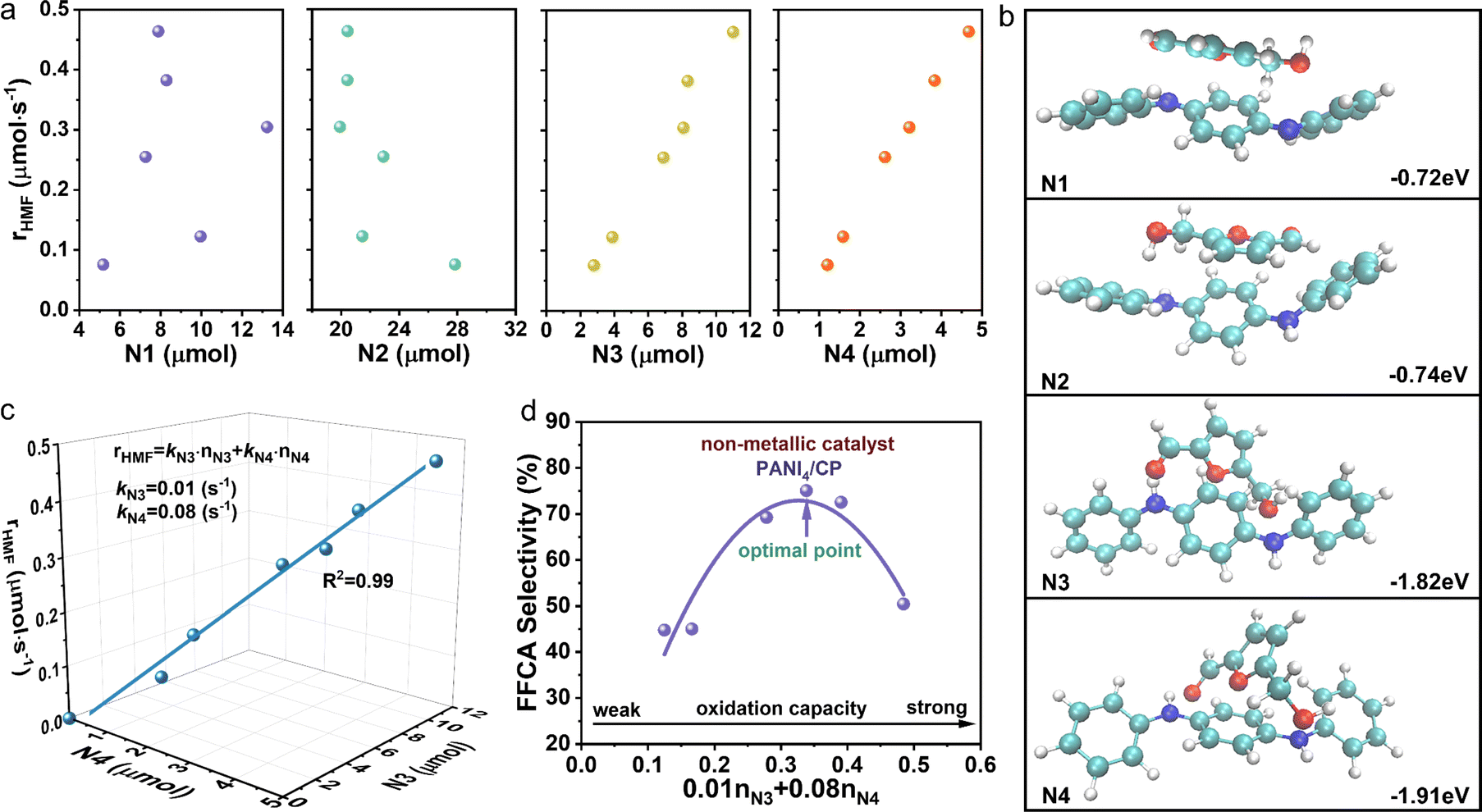
As introduced above, the relative content of nitrogen functional groups in PANI/CP catalysts can be adjusted by changing the electro-polymerization potential. The concentration of N1, N2, N3 and N4 could be determined from the peak area ratio of N1s XPS bands (Fig. S14†),2,27,28 and the detailed values of the concentrations for various nitrogen functional groups for all PANI/CP catalysts after the HMFOR are listed in ESI Table S5† (the detailed calculation method could be found in Note S1†). In this section, we will link the intrinsic HMFOR catalytic activity to the chemical structure of the proposed PANI/CP catalysts to reveal the basic structure–function relationship for this reaction process. It is known that the relative content of nitrogen species will affect the intrinsic redox capacity of PANI.47 As shown in Fig. 4a, the conversion rate of HMF (rHMF) increases with an increase in content of N3 and N4 species, suggesting that N3 and N4 are likely to be the active sites for the HMFOR. This result is well proven using the theoretical calculation results. In a typical procedure, the theoretical calculation models were constructed for these four nitrogen species in PANI, and the adsorption energies of HMF on N1, N2, N3 and N4 were calculated to be −0.72, −0.74, −1.82 and −1.91 eV, respectively (Fig. 4b), which indicates a very weak interaction (e.g., van der Waals forces) between N1 and N2 species and HMF molecules, but a relative strong chemisorption exists between N3 and N4 groups and HMF reactants. It could also be observed that the adsorption energy of N4 is higher than that of N3, suggesting its higher activity to activate HMF.
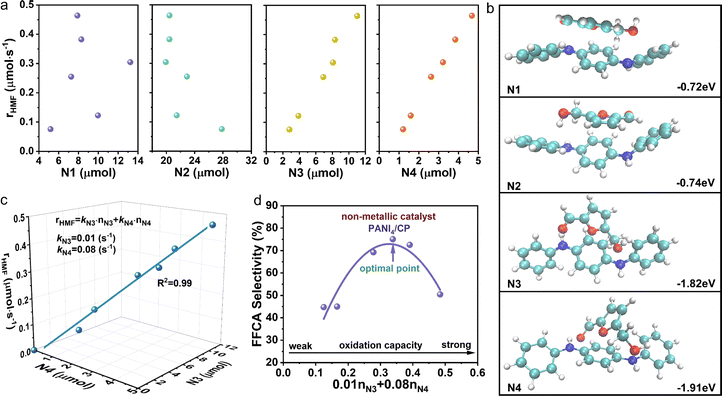 |
| | Fig. 4 Identification and quantification analysis of active sites. (a) Amount of HMF transformation as a function of content of N1, N2, N3 and N4, respectively. (b) Optimized structure and adsorption energies of HMF adsorbed on N1, N2, N3 and N4 of PANI, respectively. (c) Content of N3 and N4 as a function of conversion rate of HMF. (d) Contribution of all active sites (oxidation capacity) of PANI catalysts as a function of selectivity of FFCA. | |
A simple kinetic analysis was further performed to further quantify the intrinsic catalytic activity and the contribution of N3 and N4 active sites for the HMFOR. We assume that these two active sites work independently, and the apparent conversion rate of HMF (rHMF) can reflect oxidation capacity of all active sites on the catalyst surface, which could be expressed as the summary of the conversion rate on each site as shown in eqn (5):
| | | rHMF = kN3·nN3 + kN4·nN4 | (5) |
where
k and
n represent the intrinsic catalytic activity (turn over frequency, namely the reaction rate normalized by the number of active sites in the unit of s
−1) and the number (in the unit of μmol) of N3 and N4 active sites, respectively. The values of
kN3 and
kN4 could be calculated
via linear regression of
rHMF and
n, which could be obtained experimentally from activity measurement and structural characterization results. The intrinsic catalytic activity for N4 (
kN4 = 0.08 s
−1) is about 8 times higher than that of N3 (
kN3 = 0.01 s
−1), which is consistent with the theoretical calculation results that N4 type nitrogen species exhibit higher activity than N3 species. If we consider both the intrinsic activity and the number of active sites, the contribution of N4 is about 4 times greater than that of N3 (
e.g., 0.26
vs. 0.06 μmol s
−1 for PANI
4/CP) as shown in Fig. S15.
† Therefore, N4 functionality, which contributes over 80% of overall activity, is considered the main active site to adsorb and activate HMF.
48Fig. 4d shows the plot of the selectivity of FFCA as a function of the total oxidation capacity of all active sites on the catalyst surface showing a volcanic type of relation, and this result suggests that a moderate oxidation capacity of the catalyst is the key for highly selective electrochemical synthesis of FFCA. It is worth noting that the prepared PANI
4/CP catalysts with proper oxidation capacity could achieve a relatively high FFCA selectivity of 76%
via adjusting the number of active sites. To our best knowledge, this is the best performance for electrochemical synthesis of FFCA
49,50 (Table S4
†), shedding light on the potential applications of non-metallic catalysts in related systems.
4. Conclusions
In summary, we have developed a new reaction route for electrochemical synthesis of FFCA as a high value-added product via selective oxidation of HMF using PANI/CP catalysts. Optimized FFCA selectivity up to 76% in alkaline media is exhibited, which has reached the best performance in reported literature studies. Identification and quantification of active sites are further realized via linking the activity to structural compositions of PANI; both polaronic-type nitrogen (N3) and positively charged nitrogen (N4) species are proved responsible for adsorption and activation of HMF, and the intrinsic activity of N4 is higher than that of N3. In this study, the structure–function relationship of the HMFOR on PANI was proposed, which laid the foundation for further development of non-metallic applications in the field of efficient utilization of biomass.
Data availability
All data (experimental procedures and characterization) that support the findings of this study are available within the article and its ESI.†
Author contributions
All authors contributed to the planning of the work. X. L. performed the synthesis and characterization of the catalysts, the catalytic experiments, and the analysis of the characterization results. K. Q. performed the SEM analysis of the catalysts. X. D. and Y. L. performed the TEM analysis of the catalysts. D. W. and J. D. contributed to the synthesis of catalysts. W. Q. led the research and provided guidance for experiments and analysis.
Conflicts of interest
The authors declare no competing interests.
Acknowledgements
The authors acknowledge the financial support from the National Natural Science Foundation of China (22072163 and U23A20545), Shccig-Qinling Program, China Baowu Low Carbon Metallurgy Innovation Foundation-BWLCF202113, and IMR Innovation Fund (2023-PY13).
References
- M. Asif and T. Muneer, Energy supply, its demand and security issues for developed and emerging economies, Renewable Sustainable Energy Rev., 2007, 11, 1388–1413 CrossRef
 .
.
- A. N. Menegaki and K. P. Tsagarakis, Rich enough to go renewable, but too early to leave fossil energy, Renewable Sustainable Energy Rev., 2015, 41, 1465–1477 CrossRef .
- G. H. Wang, Platinum-cobalt bimetallic nanoparticles in hollow carbon nanospheres for hydrogenolysis of 5-hydroxymethylfurfural, Nat. Mater., 2014, 13, 294–301 Search PubMed .
- C. Y. Zhang, Highly efficient and selective production of FFCA from CotA-TJ102 laccase-catalyzed oxidation of 5-HMF, Int. J. Biol. Macromol., 2019, 128, 132–139 CrossRef CAS PubMed .
- F. Mafakheri and F. Nasiri, Modeling of biomass-to-energy supply chain operations: Applications, challenges and research directions, Energy Policy, 2014, 67, 116–126 CrossRef .
- L. F. T. Novaes, Electrocatalysis as an enabling technology for organic synthesis, Chem. Soc. Rev., 2021, 50, 7941–8002 RSC .
- C. Tang, Y. Zheng, M. Jaroniec and S. Z. Qiao, Electrocatalytic refinery for sustainable production of fuels and chemicals, Angew. Chem., Int. Ed., 2021, 60, 19572–19590 CrossRef CAS PubMed .
- M. Zhang, Trimetallic NiCoFe-layered double hydroxides nanosheets efficient for oxygen evolution and highly selective oxidation of biomass-derived 5-hydroxymethylfurfural, ACS Catal., 2020, 10, 5179–5189 CrossRef CAS .
- N. Zhang, Electrochemical oxidation of 5-hydroxymethylfurfural on nickel nitride/carbon nanosheets: Reaction pathway determined by in situ sum frequency generation vibrational spectroscopy, Angew. Chem., Int. Ed., 2019, 58, 15895–15903 CrossRef CAS PubMed .
- Y. X. Lu, Tuning the selective adsorption site of biomass on Co3O4 by ir single atoms for electrosynthesis, Adv. Mater., 2021, 33, 2007056 CrossRef CAS PubMed .
- X. Lu, Highly efficient electro-reforming of 5-hydroxymethylfurfural on vertically oriented nickel nanosheet/carbon hybrid catalysts: Structure-function relationships, Angew. Chem., Int. Ed., 2021, 60, 14528–14535 CrossRef CAS PubMed .
- Y. Zhu, Novel Application of g-C3N4/NaNbO3 composite for photocatalytic selective oxidation of biomass-derived HMF to FFCA under visible light irradiation, Adv. Powder Technol., 2020, 31, 1148–1159 CrossRef CAS .
- Z. H. Zhang and G. W. Huber, Catalytic oxidation of carbohydrates into organic acids and furan chemicals, Chem. Soc. Rev., 2018, 47, 1351–1390 RSC .
- R. Ge, Selective electrooxidation of biomass-derived alcohols to aldehydes in a neutral medium: Promoted water dissociation over a nickel-oxide-supported ruthenium single-atom catalyst, Angew. Chem., Int. Ed., 2022, 61, e202200211 CrossRef CAS PubMed .
- E. Hayashi, Effect of MnO2 crystal structure on aerobic oxidation of 5-hydroxymethylfurfural to 2,5-furandicarboxylic acid, J. Am. Chem. Soc., 2019, 141, 890–900 CrossRef CAS PubMed .
- F. Zhang, Y. Y. Feng and W. Feng, Three-dimensional interconnected networks for thermally conductive polymer composites: Design, preparation, properties, and mechanisms, Mater. Sci. Eng., R, 2020, 142, 34 CrossRef .
- Y. Shi, L. L. Peng, Y. Ding, Y. Zhao and G. H. Yu, Nanostructured conductive polymers for advanced energy storage, Chem. Soc. Rev., 2015, 44, 6684–6696 RSC .
- S. Bhadra, D. Khastgir, N. K. Singha and J. H. Lee, Progress in preparation, processing and applications of polyaniline, Prog. Polym. Sci., 2009, 34, 783–810 CrossRef CAS .
- Z. H. Xue, Tuning the adsorption energy of methanol molecules along Ni-N-doped carbon phase boundaries by the Mott-Schottky effect for gas-phase methanol dehydrogenation, Angew. Chem., Int. Ed., 2018, 57, 2697–2701 CrossRef CAS PubMed .
- Z. Zhang, M. Wan and Y. Wei, Highly crystalline polyaniline nanostructures doped with dicarboxylic acids, Adv. Funct. Mater., 2006, 16, 1100–1104 CrossRef CAS .
- H. S. O. Chan, Poly(meta-phenylenediamine)-synthesis and characterization by X-ray photoelectron-spectroscopy, Eur. Polym. J., 1991, 27, 1303–1308 CrossRef CAS .
- M. Jain and S. Annapoorni, Raman study of polyaniline nanofibers prepared by interfacial polymerization, Synth. Met., 2010, 160, 1727–1732 CrossRef CAS .
- W. Qi, W. Liu, X. Guo, R. Schlogl and D. Su, Oxidative dehydrogenation on danocarbon: Intrinsic catalytic activity and structure-function relationships, Angew. Chem., Int. Ed., 2015, 54, 13682–13685 CrossRef CAS PubMed .
- W. Qi, Oxidative Dehydrogenation on nanocarbon: Identification and quantification of active sites by chemical titration, Angew. Chem., Int. Ed., 2013, 52, 14224–14228 CrossRef CAS PubMed .
- S. Golczak, A. Kanciurzewska, M. Fahlman, K. Langer and J. Langer, Comparative XPS surface study of polyaniline thin films, Solid State Ionics, 2008, 179, 2234–2239 CrossRef CAS .
- X. L. Wei, M. Fahlman and K. J. Epstein, XPS study of highly sulfonated polyaniline, Macromolecules, 1999, 32, 3114–3117 CrossRef CAS .
- S. N. Kumar, F. Gaillard, G. Bouyssoux and A. Sartre, High-resolution XPS studies of electrochemically synthesized conducting polyaniline films, Synth. Met., 1990, 36, 111–127 CrossRef CAS .
- C. O. Baker, X. Huang, W. Nelson and R. B. Kaner, Polyaniline nanofibers: Broadening applications for conducting polymers, Chem. Soc. Rev., 2017, 46, 1510–1525 RSC .
- B. You, N. Jiang, X. Liu and Y. Sun, Simultaneous H2 generation and biomass upgrading in water by an efficient noble-metal-free bifunctional electrocatalyst, Angew. Chem., Int. Ed., 2016, 55, 9913–9917 CrossRef CAS PubMed .
- L. Gao, NiSe@NiOx Core-shell nanowires as a non-precious electrocatalyst for upgrading 5-hydroxymethylfurfural into 2,5-furandicarboxylic acid, Appl. Catal., B, 2020, 261, 118235 CrossRef .
- X.-J. Bai, W.-X. He, X.-Y. Lu, Y. Fu and W. Qi, Electrochemical oxidation of 5-hydroxymethylfurfural on ternary metal-organic framework nanoarrays: Enhancement from electronic structure modulation, J. Mater. Chem. A, 2021, 9, 14270–14275 RSC .
- D.-H. Nam, B. J. Taitt and K.-S. Choi, Copper-based catalytic anodes to produce 2,5-furandicarboxylic acid, a biomass-derived alternative to terephthalic acid, ACS Catal., 2018, 8, 1197–1206 CrossRef CAS .
- M. J. Kang, Electrocatalysis of 5-hydroxymethylfurfural at cobalt based spinel catalysts with filamentous nanoarchitecture in alkaline media, Appl. Catal., B, 2019, 242, 85–91 CrossRef CAS .
- R. Latsuzbaia, Continuous electrochemical oxidation of biomass derived 5-(hydroxymethyl)furfural into 2,5-furandicarboxylic acid, J. Appl. Electrochem., 2018, 48, 611–626 CrossRef CAS .
- Y. Lu, Tailoring competitive adsorption sites by oxygen-vacancy on cobalt oxides to enhance the electrooxidation of biomass, Adv. Mater., 2022, 34, e2107185 CrossRef PubMed .
- Q. Qin, Electrochemical fixation of nitrogen and its coupling with biomass valorization with a strongly adsorbing and defect optimized boron-carbon-nitrogen catalyst, ACS Appl. Energy Mater., 2019, 2, 8359–8365 CrossRef CAS .
- X. Huang, Enhancing the electrocatalytic activity of CoO for the oxidation of 5-hydroxymethylfurfural by introducing oxygen vacancies, Green Chem., 2020, 22, 843–849 RSC .
- Y. Lu, Identifying the geometric site dependence of spinel oxides for the electrooxidation of 5-hydroxymethylfurfural, Angew. Chem., Int. Ed., 2020, 59, 19215–19221 CrossRef CAS PubMed .
- B. You, X. Liu, N. Jiang and Y. Sun, A general strategy for decoupled hydrogen production from water splitting by integrating oxidative biomass valorization, J. Am. Chem. Soc., 2016, 138, 13639–13646 CrossRef CAS PubMed .
- G. Yang, Interfacial engineering of MoO2-FeP heterojunction for highly efficient hydrogen evolution coupled with biomass electrooxidation, Adv. Mater., 2020, 32, e2000455 CrossRef PubMed .
- K. Gu, Defect-rich high-entropy oxide nanosheets for efficient 5-hydroxymethylfurfural electrooxidation, Angew. Chem., Int. Ed., 2021, 60, 20253–20258 CrossRef CAS PubMed .
- M. Sun, Nitrogen-doped Co3O4 nanowires enable high-efficiency electrochemical oxidation of 5-hydroxymethylfurfural, Chin. Chem. Lett., 2022, 33, 385–389 CrossRef CAS .
- D. J. Chadderdon, Electrocatalytic oxidation of 5-hydroxymethylfurfural to 2,5-furandicarboxylic acid on supported Au and Pd bimetallic nanoparticles, Green Chem., 2014, 16, 3778–3786 RSC .
- B. Zhou, Platinum modulates redox properties and 5-hydroxymethylfurfural adsorption kinetics of Ni(OH)2 for biomass upgrading, Angew. Chem., Int. Ed., 2021, 60, 22908–22914 CrossRef CAS PubMed .
- X. J. Bai, Preparation of MOF film/aerogel composite catalysts via substrate-seeding secondary-growth for the oxygen evolution reaction and CO2 cycloaddition, Angew. Chem., Int. Ed., 2021, 60, 701–705 CrossRef CAS PubMed .
- X. Lu, K. Qi, D. Wang, X. Dai and W. Qi, The Highly efficient electrocatalytic oxidation of 5-hydroxymethylfurfural on copper nanocrystalline/carbon hybrid catalysts: Structure-function relations, Catal. Sci. Technol., 2022, 12, 6437–6443 RSC .
- A. A. E. Fakir, Engineering of new hydrogel beads based conducting polymers: Metal-free catalysis for highly organic pollutants degradation, Appl. Catal., B, 2021, 286, 119948 CrossRef .
- X. Lu, D. Wang, K.-H. Wu, X. Guo and W. Qi, Oxygen reduction to hydrogen peroxide on oxidized nanocarbon: Identification and quantification of active sites, J. Colloid Interface Sci., 2020, 573, 376–383 CrossRef CAS PubMed .
- S. Choi, Mechanistic Investigation of biomass oxidation using nickel oxide nanoparticles in a CO2-saturated electrolyte for paired electrolysis, J. Phys. Chem. Lett., 2020, 11, 2941–2948 CrossRef CAS PubMed .
- Y. Lu, Tailoring competitive adsorption sites by oxygen-vacancy on cobalt oxides to enhance the electrooxidation of biomass, Adv. Mater., 2022, 34, e2107185 CrossRef PubMed .
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab
*ab