
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceQuantifying Siglec-sialylated ligand interactions: a versatile 19F-T2 CPMG filtered competitive NMR displacement assay†
Unai
Atxabal‡
a,
Andrea
Fernández‡
ab,
Maria Jesús
Moure
a,
Klaudia
Sobczak
a,
Corwin
Nycholat
c,
Verónica
Almeida-Marrero
de,
Iker
Oyenarte
a,
James C.
Paulson
c,
Andrés de la
Escosura
 de,
Tomás
Torres
def,
Niels C.
Reichardt
bg,
Jesús
Jiménez-Barbero
*ahij and
June
Ereño-Orbea
*ah
de,
Tomás
Torres
def,
Niels C.
Reichardt
bg,
Jesús
Jiménez-Barbero
*ahij and
June
Ereño-Orbea
*ah
aChemical Glycobiology Lab, Center for Cooperative Research in Biosciences (CIC bioGUNE), Basque Research and Technology Alliance (BRTA), 48160 Derio, Bizkaia, Spain. E-mail: jjbarbero@cicbiogune.es; jereno@cicbiogune.es
bGlycotechnology Laboratory, CIC biomaGUNE, Paseo Miramon 194, San Sebastian, 20014, Spain
cDepartments of Molecular Medicine and Immunology & Microbiology, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, California 92037, USA
dDepartment of Organic Chemistry, Universidad Autónoma de Madrid, C/Francisco Tomás y Valiente 7, 28049 Madrid, Spain
eInstitute for Advanced Research in Chemical Sciences (IAdChem), Universidad Autónoma de Madrid, C/Francisco Tomás y Valiente 7, 28049 Madrid, Spain
fInstituto Madrileño de Estudios Avanzados (IMDEA)-Nanociencia, C/Faraday 9, 28049 Madrid, Spain
gCIBER-BBN, Paseo Miramon 194, San Sebastian, 20014, Spain
hIkerbasque, Basque Foundation for Science, Bilbao, Spain
iDepartment of Organic & Inorganic Chemistry, Faculty of Science and Technology, University of the Basque Country, EHU-UPV, 48940 Leioa, Bizkaia, Spain
jCentro de Investigacion Biomedica en Red de Enfermedades Respiratorias, 28029 Madrid, Spain
First published on 7th June 2024
Abstract
Sialic-acid-binding immunoglobulin-like lectins (Siglecs) are integral cell surface proteins crucial for the regulation of immune responses and the maintenance of immune tolerance through interactions with sialic acids. Siglecs recognize sialic acid moieties, usually found at the end of N-glycan and O-glycan chains. However, the different Siglecs prefer diverse presentations of the recognized sialic acid, depending on the type of glycosidic linkage used to link to the contiguous Gal/GalNAc or sialic acid moieties. This fact, together with possible O- or N-substitutions at the recognized glycan epitope significantly influences their roles in various immune-related processes. Understanding the molecular details of Siglec–sialoglycan interactions is essential for unraveling their specificities and for the development of new molecules targeting these receptors. While traditional biophysical methods like isothermal titration calorimetry (ITC) have been utilized to measure binding between lectins and glycans, contemporary techniques such as surface plasmon resonance (SPR), microscale thermophoresis (MST), and biolayer interferometry (BLI) offer improved throughput. However, these methodologies require chemical modification and immobilization of at least one binding partner, which can interfere the recognition between the lectin and the ligand. Since Siglecs display a large range of dissociation constants, depending on the (bio)chemical nature of the interacting partner, a general and robust method that could monitor and quantify binding would be highly welcomed. Herein, we propose the application of an NMR-based a competitive displacement assay, grounded on 19F T2-relaxation NMR and on the design, synthesis, and use of a strategic spy molecule, to assess and quantify sialoside ligand binding to Siglecs. We show that the use of this specific approach allows the quantification of Siglec binding for natural and modified sialosides, multivalent sialosides, and sialylated glycoproteins in solution, which differ in binding affinities in more than two orders of magnitude, thus providing invaluable insights into sialoglycan-mediated interactions.
Introduction
Sialic-acid-binding immunoglobulin-like lectins (Siglecs)1 form a crucial family of cell surface proteins involved in immune response regulation and immune tolerance maintenance through interactions with sialic acids on host cells.2 This biology is mediated by the presence of either immunoreceptor tyrosine-based inhibitory motifs (ITIMs) or ITIM-like sequences in the cytoplasmic tails of most Siglecs.3 However, a minority of Siglecs have no such cytoplasmic motifs, but have a basic residue in the transmembrane region that promotes association with the immunoreceptor tyrosine-based activation motif (ITAM)-containing adaptor protein DAP12.4 Typically, inhibitory Siglecs suppress immune cell activation by recruiting SHP family phosphatases to their ITIM or ITIM-like domains, downregulating signaling pathways, while Siglecs that engage DAP12 transmit activating signals. Sialoadhesin, lacking ITIM sequences and DAP12 binding sites, likely plays a more prominent role in adhesion than signaling.Thus, most Siglec are associated with regulation of immune cell responses such as immune cell killing, pathogen clearance, and cytokine production, which are linked to various inflammatory diseases and phenotypes. Structurally, the extracellular domain (ECD) consists of an amino-terminal variable (V)-set immunoglobulin (Ig)-like domain binding sialylated glycans, followed by several constant (C)-set Ig domains. The V-Ig domain is composed of antiparallel β sheets, and contains a shallow binding pocket for the glycan, as illustrated by the solved structures of Siglecs-1 (Sialoadhesin), -2 (CD22), -3 (CD33), -4 (MAG), -5, -7, -8, -15.5–10 The C–C′ loop adjacent to the pocket provides additional specificity for glycan recognition, and a conserved essential arginine residue forms a key molecular interaction with the sialic acid carboxylate.11,12
In eukaryotes, sialic acid (N-acetylneuraminic acid or Neu5Ac) can be linked to the underlying glycan in various ways, with Siglecs exhibiting selectivity in recognizing the presentation of the sialic acid moiety.13–15 For instance, CD22 strongly prefers α2,6-linked sialosides,6 while MAG favours α2-3-linked ones.15 Some Siglecs, such as Siglec-9, display greater promiscuity, binding multiple linkages.16 The monomeric KD values of Siglecs for their sialic acid-containing ligands span from high micromolar to low millimolar,17 driving substantial efforts to enhance affinity and selectivity through the development of chemically modified sialoside ligands.18
Given the therapeutic significance of the sialic acid–Siglec axis, investigating and quantifying interactions between Siglecs and sialosides is imperative. Traditionally, isothermal titration calorimetry (ITC) has been a staple for obtaining thermodynamic information and glycan–protein interaction valency.19 However, its widespread use has been limited by the necessity for relatively large protein (>10 mg) and glycan quantities, always depending on the KD value, to conduct multiple experiments. In recent years, other techniques such as surface plasmon resonance (SPR),20 microscale thermophoresis (MST),21 and biolayer interferometry (BLI)22 have emerged, offering enhanced throughput, less sample consumption, and expanded sample profiling capabilities. However, SPR and BLI require protein or ligand immobilization, which not only increases the number of steps to obtain the samples but also may affect the conformational and translational/rotational entropies and thus the association rate.23 Moreover, given the key role of presentation of the glycan and receptor partners in the outcome of the recognition event,24 special care should be taken to design the immobilization protocol. On the other hand, MST cannot be used for obtaining kinetic information as association and dissociation rates.25 In any case, the use of alternative and robust methodologies to obtain accurate binding affinities is essential to advance in understanding glycan-mediated interactions.
For years, we have been engaged in the application of NMR methods to study glycan–receptor interactions,25 using diverse NMR-active nuclei (1H, 13C, 15N, 19F)26 to monitor these molecular recognition events, from the ligand and/or receptor perspectives. While NMR is a well-established method for monitoring glycan–lectin interactions,27 its low sensitivity and demand for substantial protein quantities (also 15N-isotopically labeled) have constrained its application for affinity determination, especially when using receptor-based NMR experiments. In fact, while receptor-based methods provide key information and may also allow disentangling the lectin's binding site, they usually require the use of labelled protein and a significant effort to assign the NMR signals for each specific amino acid through a combination heteronuclear experiments. The use of ligand-based experiments has expanded the applicability of NMR methods,28 with special mention to the STD NMR29 and 19F-NMR approaches.30 However, the determination of binding affinities by STD-NMR may be tedious and prone to significant errors.31 Moreover, STD-NMR may not work for systems that display high affinity, in the medium micromolar range or stronger.32 In those cases, 19F-NMR can be the method of choice33 and indeed, it has been widely employed for this task,34 also to monitor glycan interactions.35–37 and to guide the design of glycomimetic ligands.38 Nevertheless, from a general perspective, within the ligand-based NMR methods, an effective alternative involves the strategic use of competition ligand-observed NMR experiments39 employing a competitive displacement assay where signals from a competitor or “spy molecule” report on the interaction. The known binding affinity of the spy molecule enables the estimation of the competing ligands binding strength based on the extent of induced displacement.28 In this context, it has been demonstrated that 19F NMR competition experiments offer additional advantages compared with other nuclei, such as the ubiquitous 1H. Fluorine, an exogenous nucleus in biomolecules and uncommon in standard solvents or buffer components, provides high sensitivity, and the pronounced chemical shift anisotropy (CSA) inherent to 19F nuclei yields clear and distinct responses to changes in the chemical environment, ensuring exceptional sensitivity in detecting binding interactions.40 This methodology has been demonstrated to be particularly useful to screen libraries of molecular fragments (especially) or ligands and to estimate affinities.29 The key for success is the selection of the spy molecule, which should warrant the detection of binders within a wide scale of binding affinities.29 This is the challenge for Siglecs, which display a huge range of dissociation constants, depending on the (bio)chemical nature of the interacting partner. In this study, and based on those concepts, we have designed a novel spy molecule for monitoring ligand binding to Siglecs and used it within a competitive displacement assay, based on 19F T2-relaxation NMR, to assess and quantify sialoside ligand binding to Siglecs. The spy molecule has been designed on the one hand, to take advantage of the frequently found sugar–aromatic stacking interactions in lectin complexes.41 On the other hand, to combine unique 1H and 19F NMR features to allow its use in a variety of conditions and in the presence of diverse receptors. We show that the use of this innovative approach facilitates the quantification of binding for natural and modified sialoside, multivalent sialosides, and sialylated glycoproteins in solution, which differ in binding affinities in more than two orders of magnitude, thus providing invaluable insights into sialoglycan-mediated interactions, utilizing moderate amounts of the interacting partners. In particular, Siglec concentrations from 15 to 25 μM, with 10 equivalents of the difluorinated spy molecule have been used, although the lectin concentration can be safely decreased below than 1 μM, if required.42
Results and discussion
Design and synthesis of the spy molecule: α2,6SLN
An adequate spy molecule should mimic sialic acid (Neu5Ac), show intermediate binding affinity (high μM) and significant disparity in its transverse relaxation rate values between the unbound and bound states.43 The rationale for the design of the spy molecule was based on two ideas: First, given that sugar acetamide moieties are frequently involved in CH–π stacking interactions with the aromatic amino acid side chains and that polarization of the C–H bond enhances the strength of this interaction, we guessed that a CHF2 substitution would not decrease the affinity respect to the natural molecule and eventually, in some cases (as shown below), could improve it. Moreover, from the 1H NMR perspective, the CHF2 proton, being flanked by the two fluorine atoms, shows a very particular shape (a triplet, due to its coupling to the two 19F nuclei) chemical shift (δ 6.15 ppm, see Fig S2,† for instance), in a spectral region devoid of other signals. This fact makes this CHF2 fragment particularly suitable to monitor interactions of the spy molecule by both 1H and 19F NMR methods, both nuclei can be employed to monitor interactions (different of the CF3, with no protons and therefore without any possibility to provide CH–π interactions). This is why we preferred CHF2 over CF3 or CFH2 (less polarization and complex 1H NMR spectra for the diastereotopic geminal protons). Thus, we opted to introduce two fluorine atoms (CF2) at the acetamide group of Neu5Ac. In fact, it has been reported that the presence of the CF2 moiety induces polarization at the contiguous C–H bonds, thus enhancing the strength of the corresponding stacking CH–π interaction with aromatic residues.44,45 Indeed, the three-dimensional structures of Siglecs (CD22 (PDB ID: 5VKJ), MAG (PDB ID: 5LFU), CD33 (PDB ID: 5IHB), Siglec-5 (PDB ID: 2ZG2), Siglec-8 (PDB ID: 7QU6), Siglec-7 (PDB ID: 1O7S), and Siglec-15 (PDB ID: 7ZOZ)) show that most accessible aromatic amino acids on the sialic acid-binding pocket in the V-Ig domain are located in the G strand and B′–C loop, which contact with the N-acetyl group of sialic acids. Hence, we hypothesized that the incorporation of CF2 within the N-acetamide moiety of Neu5Ac of α2,6 sialyl lactosamine (SLN) to give the diF α2,6SLN analogue (Fig. 1A), could boost the affinity by promoting favorable CH–π interactions with the nearby aromatic residues located at the mentioned region.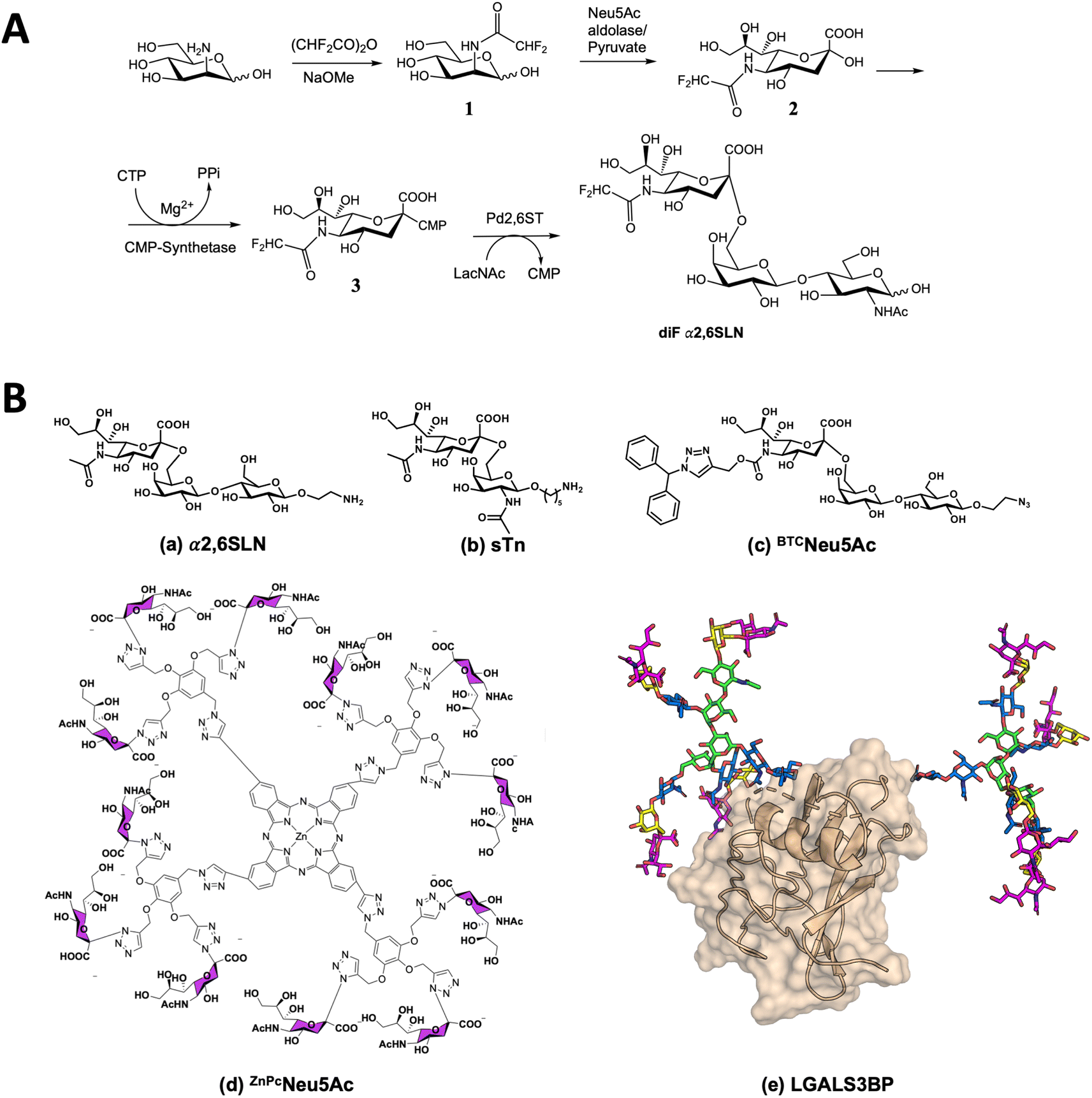 | ||
| Fig. 1 Synthesis and binding of diF α2,6SLN to Siglecs. (A) Scheme of the synthetic route for the preparation of diF α2,6SLN. (B) Sialoside ligands to target Siglec-9 or Siglec-15 and compete with diF α2,6SLN: (a) α2,6 sialyllactosamine, (b) a derivative of sialyl Tn antigen (sTn), (c) BTCNeu5Ac ligand, specific for Siglec-9, (d) a phthalocyanine-based multivalent ligand (ZnPcNeu5Ac) and (e) the intact Galectin-3 binding glycoprotein (LGALS3BP). | ||
The chemical synthesis of diF α2,6SLN was carried out using mannosamine as the starting material, which underwent N-acetylation using difluoroacetic anhydride under basic conditions. Following this, three chemoenzymatic reactions were conducted to produce the target N-difluoro acetamide neuraminic acid derivative and facilitate its transfer onto the N-acetyl lactosamine scaffold (Fig. 1A).
Binding of diF α2,6SLN to Siglecs-9 and -15 measured by 1D 19F CPMG NMR experiments
The transverse relaxation rate (R2) of diF α2,6SLN was determined when free in solution and in the presence of Siglec-9 (full extracellular domain (ECD) with human IgG1 Fc (Siglec-9d1–d3-FC)) and Siglec-15 (full ECD with mVENUS46 (Siglec-15d1–d2-mVENUS)) by performing 1D 19F Carr–Purcell–Meiboom–Gill (CPMG) experiments, with diverse CPMG filter times (0 s, 0.05 s, 0.1 s, 0.2 s, 0.35 s, and 0.5 s). The CF2 signal intensity of the diF α2,6SLN exhibited a significant decrease in the presence of both Siglec-9 and -15 (10 molar equivalents of ligand versus Siglec). In particular, the use of a 200 ms CPMG filter provided the largest intensity difference between the unbound and bound forms (Fig. 2). As a control, the binding of diF α2,6SLN to Siglecs-9 and -15 with abrogated capacity to bind to sialic acid (Siglec-9d1–d3-Fc R120A and Siglec-15d1–d2-mVENUS R143A mutants) was also tested under the same experimental conditions, using the 0.2 s CPMG filter time (Fig. S1†). As expected, the CF2 signal did not undergo changes in intensity, demonstrating that binding of the diF α2,6SLN to Siglecs takes place at the canonical sialic acid binding pocket.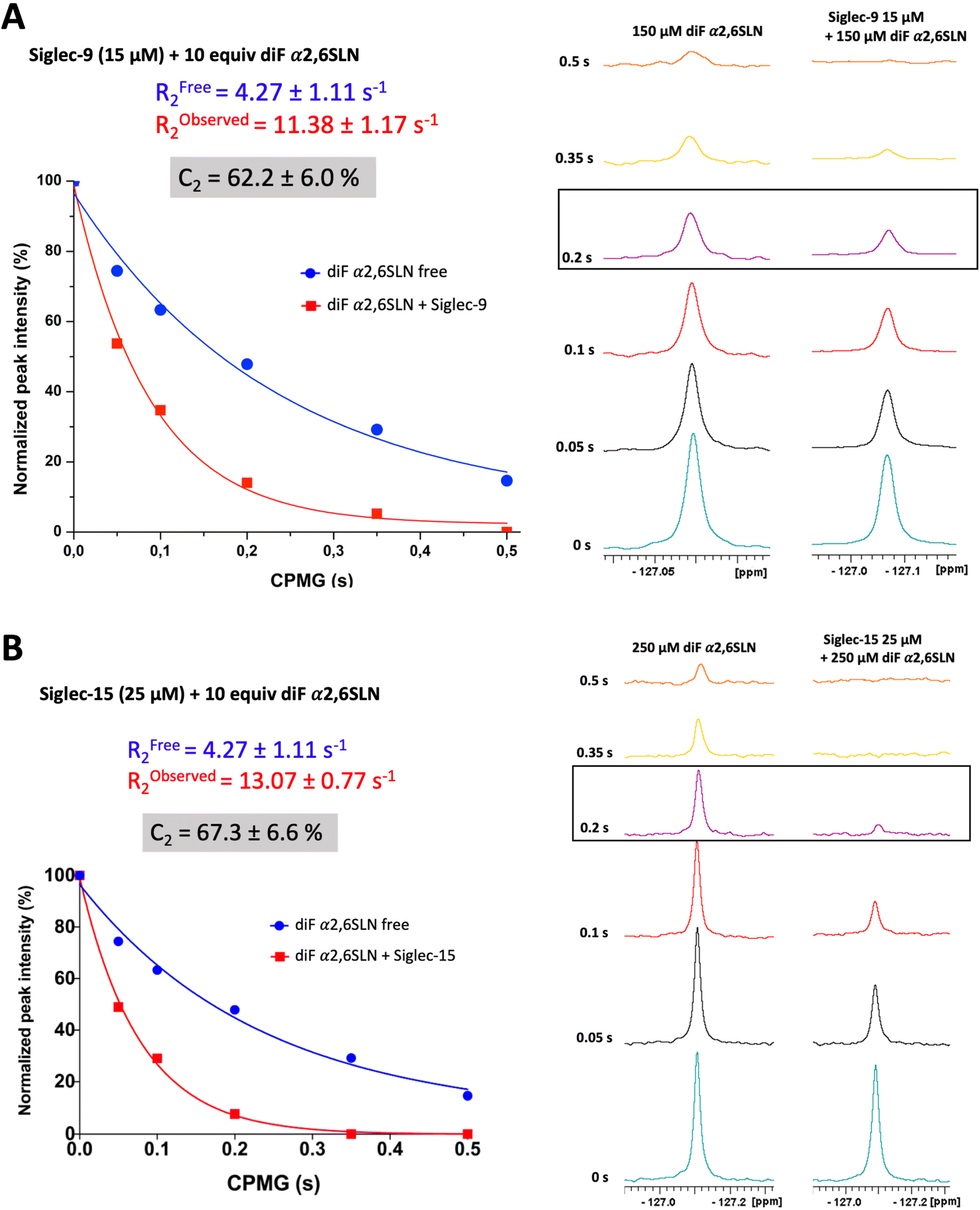 | ||
| Fig. 2 Characterization of diF α2,6SLN by 19F NMR. (A) Measurement of the transverse relaxation rate of diF α2,6SLN (150 μM) in the absence (blue) and presence (red) of Siglec-9 (15 μM). (B) Measurement of the transverse relaxation rate of diF α2,6SLN (250 μM) in the absence (blue) and presence (red) of Siglec-15 (25 μM). | ||
Remarkably, the decay of the difluoro acetamide 19F-NMR signal intensity of the spy molecule was significantly faster in the presence of Siglec-9 and Siglec-15 compared to its unbound state in solution (Fig. 2). To quantitatively elucidate the extent of this difference in rate, the so-called contrast factor (C2)47,48 was calculated (eqn (1)).
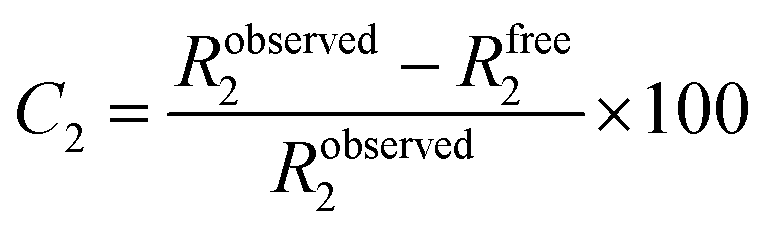 | (1) |
Calculation of C2 (transversal relaxation rate contrast). Robserved2 stands for the transversal relaxation rate of diF α2,6SLN in the presence of the protein while Rfree2 stands for the transversal relaxation rate of the ligand free in solution.
This factor highlights the extent of divergence in R2 relaxation rates between the bound and free states. Notably, the C2 value for diF α2,6SLN in the case of Siglec-9 was 62 ± 6%, while for Siglec-15 was 67 ± 7%. These results indicate that the designed diF α2,6SLN entity is indeed an effective reporter molecule for both Siglec receptors. The large increase in the relaxation rate R2 in the presence of the lectin allows for a large assay window for competition experiments.
STD-NMR experiments for affinity calculation and epitope mapping of diF α2,6SLN to Siglecs-9 and -15
The use of the spy molecule for estimating the binding affinities of the competitor molecules requires the knowledge of its KD value versus the target Siglecs. Therefore, as first step, 1H STD-NMR titration experiments were carried out to determine the affinity of diF α2,6SLN towards Siglec-9d1–d3-Fc and Siglec-15d1–d2-mVENUS respectively. Following the methodology developed by Angulo and coworkers,33 several STD NMR spectra were measured using different saturation times (1 s, 2 s, 4 s and 6 s) for each diF α2,6SLN concentration (ranging from 0.05 to 0.7 mM for Siglec-9d1–d3-Fc and from 0.05 to 0.5 mM for Siglec-15d1–d2-mVENUS) (Tables S1 and S3†). Since the 1H NMR signal for the difluoroacetamide moiety is fairly isolated at δ 6.1 ppm (Fig. S2†), it was used to estimate the normalized STD-AF0 values (Tables S2 and S4†), which were obtained from plotting the STD-AF vs. the saturation time at the STD build-up curves.29 Thus, the normalized STD-AF0 values were calculated for every point of the titration and then plotted versus the ligand concentration (Fig. 3A). The fitting using a Langmuir isotherm (Hill equation) allowed estimating the KD. Interestingly, the obtained KD values for the diF α2,6SLN spy molecule were in the micromolar range (285 ± 64 μM for Siglecs-9 and 469 ± 77 μM for Siglec-15, Fig. 3A). These affinity values are definitively stronger than those estimated for the natural non-fluorinated analogue, which had been previously estimated to be above 5 mM for 15N-labelled Siglec-9 using the receptor-based NMR approach, following a chemical shift perturbation analysis.49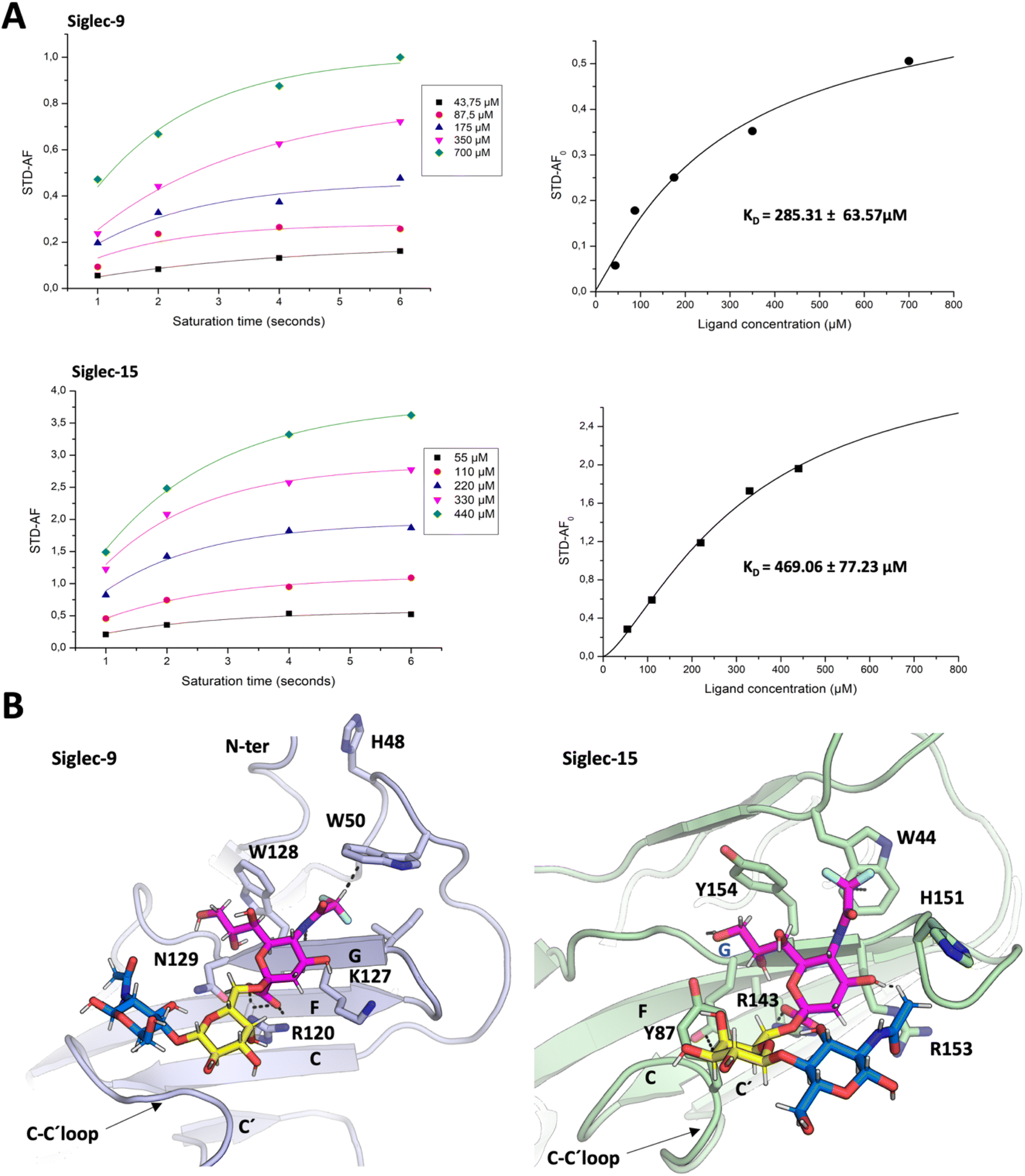 | ||
| Fig. 3 Binding of diF α2,6SLN to Siglec-9 and Siglec-15. (A) Estimation of KD for the binding process of the diF α2,6SLN spy molecule to Siglec-9 and Siglec-15. (Right) STD-AF intensities of the diF-NHAc Neu5Ac of the spy molecule recorded at different saturation times, using increasing concentrations of the ligand, in the presence of 17.5 μM of Siglec-9 or 25 μM of Siglec-15. To obtain the STD AF normalized values, for each concentration, the curves were fitted to the equation STD AF (tsat) = STD AFmax [1 − e−ksattsat]. The STD AF0 values were obtained by applying the equation STD AF0 = STD AFmaxksat. (Left) STD AF0 values are plotted against the spy molecule concentration and fitted to the Langmuir isotherm (Hill equation) y = Bmaxx/(KD + x) to obtain the KD. (B) The possible structure of the complexes: representation of the ligand–lectin interactions in the binding sites, deduced from representative frames derived from 100 ns MD simulation of diF α2,6SLN bound to Siglec-9 and Siglec-15. | ||
The glycan epitope map for the diF α2,6SLN molecule was also determined from the STD NMR experiments. For both Siglec-9 and Siglec-15, the highest STD intensity peak corresponded to the key N-difluoroacetamide proton mentioned above (Fig. S2, Tables S5 and S6†), followed by other protons at the Neu5Ac moiety. The protons at the Gal moiety showed reduced intensity, as well as the NHAc group at the GlcNAc moiety. In both cases, the epitope map of diF α2,6SLN was comparable to that presented by α2,6SLN in the presence of Siglec-9 (ref. 50) and Siglec-15.10
Regarding the three-dimensional structure of the complexes, molecular dynamics (MD) simulations were carried out to generate a model geometry. The binding modes of the non-fluorinated “natural” α2,6SLN ligand have been previously determined both for Siglec-9 (ref. 50) and for Siglec-15.10 Thus, since the STD profile for the difluoro α2,6SLN and the natural α2,6SLN were indeed very similar, the starting geometries for the MD simulations with the fluorinated analogue were built from the binding pose deduced for the “natural” molecule. The MD results showed that the V-set Ig domains of Siglec-9 (generated with AlphaFold51) and Siglec-15 (from the crystal structure (PDB ID: 7ZOZ)) nicely accommodate diF α2,6SLN at the canonical sialic acid binding pocket, by establishing the essential salt bridge between the C1 carboxylate and the conserved Arg (Fig. 3B). The diF α2,6SLN molecule remained stable within the binding site throughout the entire simulation (100 ns) in both cases. For Siglec-9, the H3ax and H4 of Neu5Ac sited on top of the aliphatic side chain of the K127. Additionally, the H9proR establishes a key CH–π interaction with the W128 indole ring, which further stabilizes the Neu5Ac scaffold within the binding cleft.
For Siglec-15, the obtained binding mode of the spy was also similar to that reported for α2,6SLN.10 The carboxylic acid moiety of the Neu5Ac establishes the key ion–pair interaction with R143, while H3ax and H4 make van der Waals contacts with the aliphatic side chain of R153. The described CH–π interaction between H9proR and Y154 was also maintained through the simulation. Moreover, the aromatic side chain of Y87 stabilizes the Gal moiety through CH–π interactions. Herein, the polarized hydrogen of the N-difluoroacetamide group establishes a CH–π interaction with the indole ring of W44, which was kept throughout the entire simulation as it is reflected within the trajectory in Fig. S3B.† Additionally, for both Siglec-9 and Siglec-15, the N-difluoroacetamide group fits within a hydrophobic cleft, composed by aliphatic and aromatic residues (Fig. 3B). Besides the key CH–π contacts, the presence of two fluorine atoms increases the hydrophobicity of this ligand region and thus establishes stronger hydrophobic contacts with the surrounding aromatic and aliphatic amino acids compared to the non-fluorinated N-acetamide group. In conclusion, the CH–π interaction and the increased hydrophobicity provide the impetus for the improved affinity of the spy molecule.
19F T2 filtered NMR competitive displacement assays to determine dissociation constants
Once the KD values for the spy molecule for both Siglecs were determined, 19F T2 filtered NMR competitive displacement assays were performed to determine the dissociation constants for a variety of sialoside ligands of different chemical nature: the natural α2,6SLN ligand itself, sialyl Tn antigen (sTn), a modified sialoglycan BTCNeu5Ac specific for Siglec-9, a multivalent ligand (ZnPcNeu5Ac)52 and a sialylated glycoprotein, galectin-3 binding protein (LGALS3BP)53 (Fig. 1B). Since the largest intensity differences between the free and bound forms of the spy molecule were observed using a CPMG filter time of 0.2 s (Fig. 2), this value was selected as filter time for all the competitive displacement assays. 0.1 mM of TFA was also added in every sample as a reference signal.Siglec-15 binding to a standard sialyl Tn antigen (sTn) derivative54 was also tested. However, as for α2,6SLN, despite adding 340 equivalents of sTn versus the 10 equivalents of spy molecule, only 30% was displaced (Fig. S4C†). The displacement achieved with sTn compared to that of α2,6SLN using the same equivalents was somehow larger (29% vs. 22%), corresponding to a lower KD limit of 4 mM, suggesting that Siglec-15 has a comparable, yet slightly better affinity for sTn relative to α2,6SLN, still in the mM range.
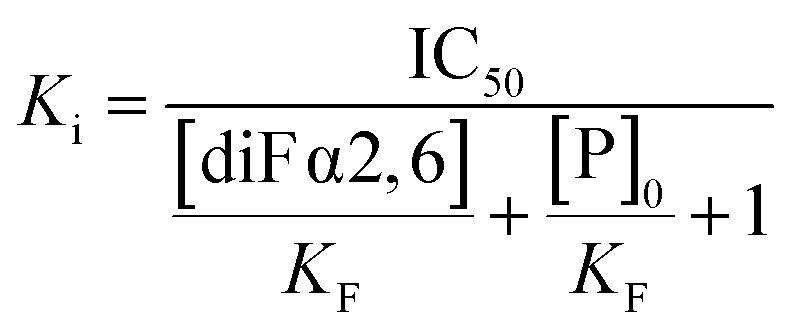 | (2) |
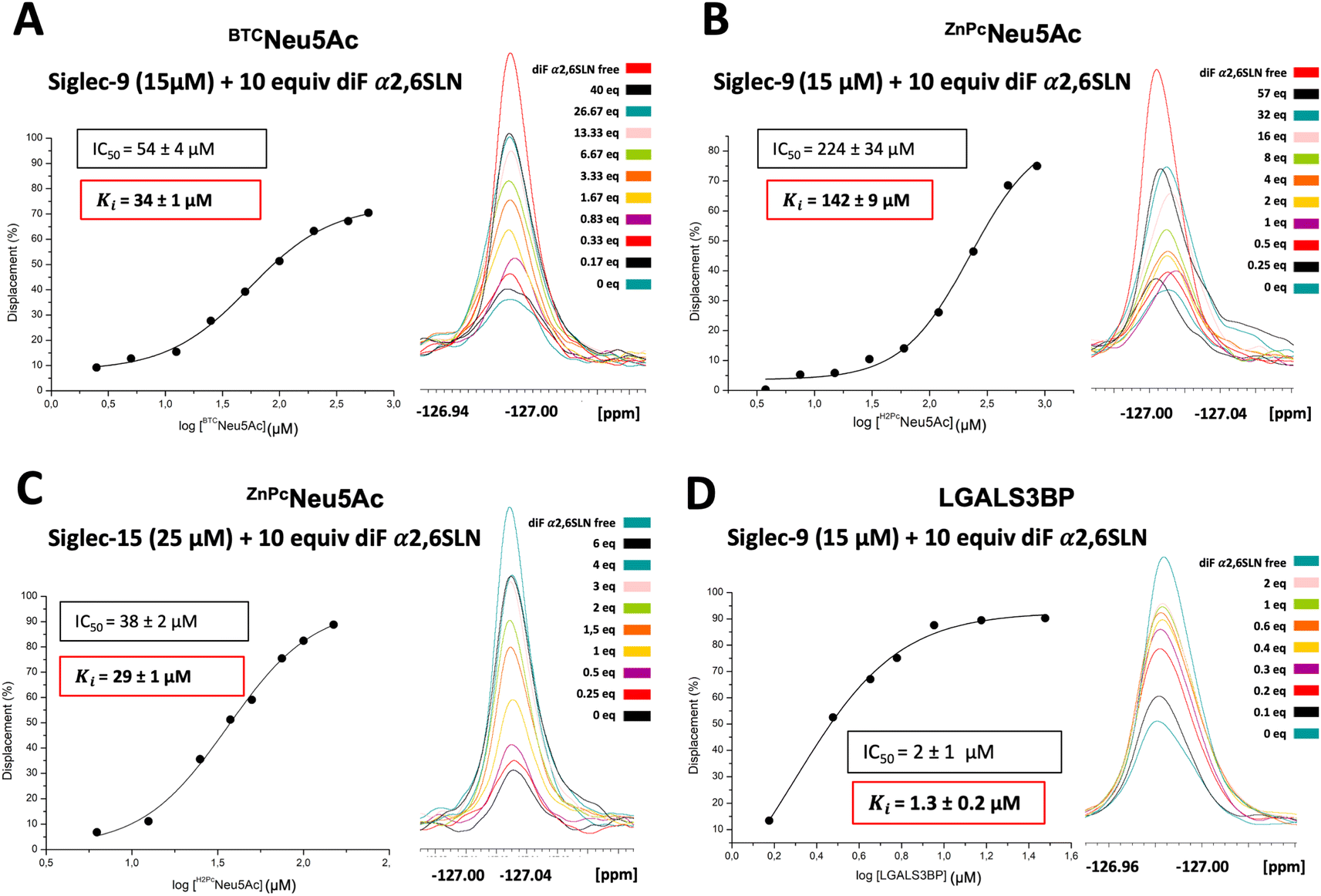 | ||
| Fig. 4 19F CPMG filtered NMR competitive displacement assay with BTCNeu5Ac glycan mimetic, ZnPcNeu5Ac and LGALS3BP glycoprotein in the presence of Siglec-9. (A) A dose response plot was fitted to estimate IC50 of BTCNeu5Ac. From this value, the Cheng–Prussof equation allowed estimating Ki. (B) Competitive displacement with the multivalent ligand ZnPcNeu5Ac. Dose response plot to calculate IC50 from the displacement of the spy with the multivalent competitor in Siglec-9. (C) Competitive displacement with the multivalent ligand ZnPcNeu5Ac. Dose response plot to calculate IC50 from the displacement of the spy with the multivalent competitor in Siglec-15. (D) 19F T2 NMR filtered competitive displacement assays with the intact glycoprotein SRCR domain of LGALS3BP expressed in HEK293F mammalian cells bearing two sialylated N-linked glycans. | ||
Calculation of the Ki of the competitor using the Cheng–Prusoff equation. IC50 stands for the concentration of the competitor required to displace the 50% of the spy molecule. KF is the dissociation constant of the fluorinated spy molecule. [diF α2,6SLN] is the concentration employed of the spy molecule, while [P]0 is the employed protein concentration.
Systematic additions of the glycoprotein produced in HEK293F cells, up to 2 equivalents, resulted in a significant signal displacement of 90%. The dose–response curve allowed deriving an IC50 value of 2 ± 1 μM. By applying the Cheng–Prusoff equation, the dissociation constant between LGALS3BP and Siglec-9 was calculated to be 1.3 ± 0.2 μM (Fig. 4D). The significant displacement of the 19F-labelled spy molecule indicates the glycan-mediated nature of the interaction between Siglec-9 and LGALS3BP. In contrast, no intensity changes were observed in the presence of LGALS3BP expressed in HEK293S (Fig. S5B†), indicating that Siglec-9 does not interact with LGALS3BP when decorated with high-Man N-glycans. Additionally, to check the selectivity of LGALS3BP towards Siglec-9, Siglec-15, which does not belong to the CD33-type Siglec subfamily was tested as well. Interestingly, even in the presence of 4 equivalents of LGALS3BP from HEK293F, there was hardly any displacement of the diF α2,6SLN spy from Siglec-15 (Fig. S6†). The structural details behind this preference of LGALS3BP for Siglec-9 remain elusive. It has also been previously reported61 that LGALS3BP shows predilection towards Siglec-9 over other Siglecs. We may speculate that LGALS3BP contains specific glycan epitopes, such as sulfated GlcNAc moieties, which might boost the preference for Siglec-9.
Materials and methods
Ligands synthesis
Chemicals were purchased from Sigma-Aldrich or Acros Organics and were used without further purification. Thin layer chromatography was carried out using Merck aluminum sheets silica gel 60 F254 and visualized by UV irradiation (254 nm) or by staining with vanillin solution. Purifications of compounds were performed on: SampliQ high performance graphitized carbon cartridges (1 mL) from Agilent Technologies, C18 Sep-Pak Cartridges (1 mL) from Waters (Milford), biogel columns packed with BioGelP-4 (Biorad) and flash chromatography using Merck 62 Å 230–400 mesh silica gel. All aqueous solutions were prepared from nanopure water produced with a Diamond UV water purification system (Branstead International). All organic solvents were concentrated using rotary evaporation. NMR spectra were acquired on a Bruker 500 MHz spectrometer. High-resolution mass spectra were acquired on a Waters LCT Premier XE instrument, (Waters) equipped with a standard ESI source by direct injection. The instrument was operated with a capillary voltage of 1.0 kV and a cone voltage of 200 V. Cone and desolvation gas flow were set to 50 and 600 L h−1, respectively; source and desolvation temperatures were 100 °C. MALDI-TOF mass analyses were performed on an Ultraflextreme III time-of-flight mass spectrometer equipped with a pulsed N2 laser (337 nm) and controlled by FlexControl 3.3 software (Bruker Daltonics). CMP-NeuAc Synthetase from N. meningitidis and Sialic acid aldolase from Escherichia coli K12 were purchased from Sigma-Aldrich and P. damselae α2,6 sialyltransferase (Pd2,6ST) was expressed and purified as previously reported.63The synthesis and characterization of the multivalent ligand ZnPcNeu5Ac has been previously described in ref. 39. The same batch has been utilized in the present study. The synthesis of glycomimetic BTCNeu5Ac has already been described.55 α2,6SLN (6′-sialyl N-acetyl-D-lactosamine aminoethylglycoside) was purchased from Asparia Glycomics. DiF α2,6SLN and sTn derivative have been synthesized as described in ESI Materials and methods.†
The synthesis of BTCNeu5Ac has already been described in ref. 43.
Expression and purification of Siglec-9, Siglec-15 and LGALS3BP
The DNA encoding the full-length extracellular domain (d1–d2) of human Siglec-15 (UniprotKB Q6ZMC9, residues 20–263) fused to mVENUS and cloned into pHLsec64 vector was generated as previously described.10 The DNA encoding human Siglec-9d1–d3 (UniprotKB Q9NYZ4) (residues 18–336) wild-type and R120A mutant, fused to the human IgG1 Fc region (UniprotKB P01857, residues 99–330) and with a C-terminal 6× His tag, was subcloned between XbaI and AfeI restriction sites into pcDNA 3.4 (Invitrogen). SRCR LGALS3BP (UniprotKB K7ES75) (residues 20–133) was cloned into pHLsec vector64 between AgeI and KpnI restriction sites. All plasmids were codon optimized for expression in human cells and synthesized by Genscript. Siglec-15-mVENUS (WT and R143A mutant), Siglec-9-Fc (WT and R120A mutant) and SRCR LGALS3BP constructs were transiently transfected into HEK293 Free Style (F) (R79007, Thermo Fisher) HEK293S (CRL-3022, ATCC) suspension cells. Cells were split in 200 mL cultures at 1 × 106 cells mL−1. The DNA: FectoPRO transfection reagent solution (Polyplus) was then added directly to the cells, and cells were incubated at 37 °C, 130 rpm, 8% CO2 and 70% humidity for 6–7 days. Supernatants were passed through a HisTrap Ni-NTA (17528601, GE Healthcare) and then separated on a Superdex 200 Increase size exclusion column (GE28-9909-44, GE Healthcare) in 20 mM Tris pH 7.5 (PHG0002, Sigma-Aldrich), 300 mM NaCl (S9888, Sigma-Aldrich) buffer to achieve size homogeneity.Western blot
Input samples were separated by 4–15% Mini-PROTEAN TGX precast protein gel (4561083, BioRad) and transferred to a 0.2 μm PVDF membrane (1704156, BioRad) using a trans-blot turbo transfer system (BioRad). The membrane was blocked with 1× Carbo-free blocking 1% Tween-20 solution (SP-5040-125, Vectorlabs) for 1 h at RT. Membranes were then incubated for an hour with 1 μg mL−1 MAL II in the same carbo-free blocking solution. After 3 washes with carbo-free solution, membranes were incubated with streptavidin-HRP at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5000 in the same buffer. Chemiluminescence detection was performed using Clarity Max Western ECL Substrate (170506, BioRad) on an iBright CL1500 system (Invitrogen).
:5000 in the same buffer. Chemiluminescence detection was performed using Clarity Max Western ECL Substrate (170506, BioRad) on an iBright CL1500 system (Invitrogen).
NMR experiments
All 19F NMR-based spectra were recorded on the 600 MHz AVANCE 2 spectrometer, equipped with a 19F probe (19F, 1H SEF from Bruker) at 298 K. The 19F NMR experiments were carried out in 10% D2O/90% H2O saline buffer (20 mM Tris pH 8, 300 mM NaCl), using 100 μM of trifluoroacetic acid (TFA) as a control molecule.For the measurement of the transversal relaxation rate (R2) of the diF α2,6SLN spy molecule bound to the Siglec-9-Fc (15 μM) or Siglec-15-mVENUS (25 μM), a 1:10 protein/ligand ratio was used. For the R2 measurement of the free molecule, two different concentrations (150 μM and 250 μM) were employed. For each sample, an in house 19F CPMG pulse sequence (decoupled) was applied, where the total CPMG filter time was varied (0 ms, 50 ms, 100 ms, 200 ms, 350 ms and 500 ms). In order to minimize the R2exchange effect, the τ within CPMG pulse was kept very short (τ = 75 μs) in all cases. We preferred using this short inter-pulse delay to minimize the exchange and to be able to properly quantify the signal intensities of the spy molecule, avoiding a fast decrease in the intensity. Since the fragment contains two fluorine atoms, we also minimize the irreversible dephasing that could take place.
The 19F NMR peaks were integrated and plotted against the CPMG filter time. The R2 relaxation rates were obtained from the fitting to the corresponding exponential decay equation ((t) = (O) × e−R2t) by using the OriginLab software. For every point of the competition experiment, a single 19F NMR CPMG (decoupled) experiment was conducted for each sample, using a fixed CPMG delay (200 ms). For every competitive displacement assay, a given concentration of Siglec-9d1d3-Fc (15 μM) and Siglec-15-mVENUS (25 μM), was used, while 10 equivalents of diF α2,6SLN spy molecule were always present. Then, increasing equivalents of the competitor molecule were added.
Molecular dynamics simulations
The binding modes of the non-fluorinated “natural” α2,6SLN ligand have been previously determined both for Siglec-9 (ref. 50) and for Siglec-15.10 Thus, since the STD profile for the difluoro α2,6SLN and the natural α2,6SLN were indeed very similar, the starting geometries for the MD simulations with the fluorinated analogue were built from the binding pose deduced for the “natural” molecule. In particular, a homology 3D model of Siglec-9d1 was first constructed using AlphaFold.51 For Siglec-15d1 the PDB ID: 7ZOZ was employed. The fluorinated spy molecule was manually docked within the sialic acid binding site by superimposing the Siglec-9 homology model and Siglec-15 (PDB ID: 7ZOZ) with the co-crystallized X-ray structure of Siglec-8 with NSANeu5Ac (PDB ID: 7QUI). The corresponding ligand was superposed to the co-crystallized NSANeu5Ac, adopting the same geometry as the Neu5Ac of the glycan.MD simulations for the protein were then carried out with the AMBER 20 (ref. 65) package implemented with ff14SB66 for the fluorinated spy molecule diF α2,6SLN the general Amber force field (GAFF2)67 was used. The initial structures were neutralized with either Na+ or Cl− ions and set at the centre of a cubic TIP3P68 water box with a buffering distance between solute and box of 10 Å. For each system, a two-stage geometry optimization approach was followed: the first stage minimized only the positions of the solvent molecules and ions, while the second stage provided an unrestrained minimization of all the atoms in the simulation cell. The systems were then heated by incrementing the temperature from 0 to 300 K under a constant pressure of 1 atm and periodic boundary conditions. Harmonic restraints of 10 kcal mol−1 were applied to the solute, under the Andersen temperature coupling scheme.69 The time step was kept at 1 fs during the heating stages, allowing potential inhomogeneities to self-adjust. Water molecules were treated with the SHAKE70 algorithm such that the angle between the hydrogen atoms was kept fixed through the simulations. Long-range electrostatic effects were modelled using the particle mesh Ewald (PME) method.71 An 8 Å cut-off was applied to non-bonded interactions. Each system was equilibrated for 2 ns with a 2 fs time step at a constant volume and temperature of 300 K. Three independent production trajectories were then run for additional 100 ns under the same simulation conditions, leading to accumulated simulation times of 300 ns for each system.
Conclusions
An efficient and robust screening method based on competitive displacement of a fluorinated ligand detected by 19F NMR with a T2-filter has been developed for detecting and quantifying the binding events between Siglecs and diverse types of sialosides (Fig. 1B). A suitable fluorinated molecule has been designed and synthesized by replacing the N-acetamide group of Neu5Ac with a N-difluoroacetamide group. The two fluorine nuclei contribute to provide an intense signal in the 19F NMR spectrum (δ −127 ppm) and a separated triplet signal in a fairly isolated region of the 1H NMR spectrum (δ 6 ppm). Additionally, the difluoro substitution at the N-acetamide group of the Neu5Ac of the spy molecule significantly increases the binding affinity in at least one order of magnitude (versus the parent non fluorinated molecule) towards Siglec-9 and Siglec-15, likely due to the enhanced CH–π interaction stablished between the polarized hydrogen at the N-difluoroacetamide moiety and the neighboring aromatic residues in the Siglec. In this context, the difluoro substitution at the N-acetamide moiety of Neu5Ac could be used for the future design and development of other novel modified sialoglycans for targeting Siglecs.The 19F NMR based T2-filtered competitive binding assay has proven to be a valuable method to detect and quantify binding interactions between Siglecs and diverse ligands. For small monovalent glycans, binding affinities obtained by other methods were nearly identical to those obtained through 1H–15N HSQC NMR titrations. Moreover, this technique was equally useful to deduce and quantify the molecular recognition event between a high molecular weight (6.5 kDa) ZnPcNeu5Ac multivalent ligand with interesting photodynamic properties, containing 12 sialic acids, with both Siglec-9 and Siglec-15. The ligand displayed significant differences in the relative affinity, always in the μM range, and showed 5-fold better affinity for Siglec-15 than for Siglec-9. The ability to assess the relative affinity to synthetic multivalent ligands could be exploited in the future to generate novel multivalent ligands to selectively target a given Siglec.
An intact glycoprotein served as another very physiologically relevant ligand. The use of 1H–15N HSQC NMR for measuring KD values in large molecular weight systems is rather challenging, as the increased molecular weight of the formed complex can lead to a significant intensity decrease in the cross peaks due the fast T2 relaxation rate of the complex. For this reason, the competitive NMR method described herein, based on competitive displacement, exhibits versatility for application with any system since the monovalent spy ligand is used to measure the interaction. In this study, LGALS3BP was employed. This glycoprotein contains two multi-antennary N-linked glycans and plays a role in down-regulating the immune response by engaging with CD33-type Siglec receptors, particularly Siglec-9. Remarkably, the glycoprotein had a relatively high affinity for Siglec-9 (KD of 1.2 ± 0.2 μM), nearly 16000-fold higher than that for the monomeric α2,6SLN. The binding interaction is exclusively mediated by the sialylated N-linked glycans, since LGALS3BP produced with no sialic acids in HEK293S cells showed no engagement with Siglec-9. Regarding the increased affinity, it is notable that the Siglec-9 target protein was a bi-valent Fc chimeric protein (Siglec-9-Fc), allowing the potential for multivalent interactions between the multi-antennary N-linked glycans of LGALS3BP and the dimeric Siglec-9-Fc. Indeed, such large increases in affinity between monovalent ligands and multivalent N-linked glycan ligands have been seen for CD22 (Siglec-2).62 In contrast, LGALS3BP induced scarce displacements when Siglec-15 was used as a target.
In conclusion, an efficient and robust NMR method for assessing and quantifying molecular interactions between glycans and lectins (such as Siglecs) has been developed, based on 19F-T2 CPMG-filtered NMR experiments. Using this methodology, reliable Ki values within one (30–40 μM) and even two orders of magnitude (1.2 μM) respect to the Kd value (ca. 300 μM) of the spy molecule have been determined. The shapes of the measured displacement curves were different enough to allow for the determination of those values. Entering into the nM range could probably be difficult to quantify, although minute amounts of the titrating ligand can always be used to certify the existence of a very tight binding. On the other hand, the quantitative measurement of low affinity events (above 2 mM) remains elusive, yet detectable.
The dissociation constants for the screened molecules can be determined in a straightforward manner through competition experiments with a spy molecule that contains a fluorine tag (difluoroacetamide). The method allows determining the affinity values without the need of using labeled proteins or high quantities of protein. Moreover, it has been proven to be effective for monitoring interactions with complex systems, including multivalent presentations of glycans, either prepared by chemical synthesis or natural. In fact, using the intact glycoprotein, the method allows quantifying a glycan-mediated protein-glycoprotein interaction, which remains rather elusive by employing other experimental techniques.
Data availability
Detailed synthetic procedures, supporting experimental results and complete characterization data presented on this article can be found in the ESI.†Author contributions
Experimental conception and design: J. E.-O., J. J-B.; data acquisition: U. A., A. F., M. J. M., K. S., I. O., C. N.; and A. d. l. E.; analysis of data: U. A., A. F., J. E.-O., J. J-B.; drafting the article or revising it critically for important intellectual content: U. A., A. F., J. C. P., A. d. l. E., T. T., J. J-B.; J. E.-O.Conflicts of interest
There is no conflict of interest to declare.Acknowledgements
This work was supported by the European Research Council (788143-RECGLYCANMR to J. J.-B) and the Marie-Skłodowska-Curie actions (ITN Glytunes grant agreement no. 956758 to J. E.-O; and ITN BactiVax under grant agreement no. 860325 to U. A.). The NMR spectrometers are part of the National NMR Network (PTNMR) and are partially supported by Infrastructure Project No. 22161 (co-financed by FEDER through COMPETE 2020, POCI and PORL and FCT through PIDDAC). JJB acknowledge to the European commission for the COST Action 18132 – GLYCONANOPROBES and the GLYCOTWINNING project and CIBERES, an initiative of Instituto de Salud Carlos III (ISCIII, Madrid, Spain). We thank Agencia Estatal de Investigación of Spain for grants PID2019-107770RA-I00 (J. E.-O.), PID2020-119306GB-100 (A. d. l. E.), and the Severo Ochoa Center of Excellence Accreditation CEX2021-001136-S, all funded by MCIN/AEI/10.13039/501100011033. T. T. acknowledges financial support from the Spanish MCIN/AEI/10.13039/501100011033 (PID2020-116490GB-I00), the Comunidad de Madrid and the Spanish State through the Recovery, Transformation and Resilience Plan [“Materiales Disruptivos Bidimensionales (2D)” (MAD2D-CM) (UAM1)-MRR Materiales Avanzados], and the European Union through the Next Generation EU funds. IMDEA Nanociencia acknowledges support from the “Severo Ochoa” Programme for Centres of Excellence in R&D (MINECO, Grant SEV2016-0686).References
- M. P. Lenza, U. Atxabal, I. Oyenarte, J. Jiménez-Barbero and J. Ereño-Orbea, Cells, 2020, 9, 1–19 CrossRef PubMed.
- P. R. Crocker, J. C. Paulson and A. Varki, Nat. Rev. Immunol., 2007, 7, 255–266 CrossRef CAS PubMed.
- B. E. Tourdot, M. K. Brenner, K. C. Keough, T. Holyst, P. J. Newman and D. K. Newman, Biochemistry, 2013, 52, 2597–2608 CrossRef CAS PubMed.
- T. Angata, Adv. Exp. Med. Biol., 2020, 215–230 CrossRef CAS PubMed.
- A. P. May, R. C. Robinson, M. Vinson, P. R. Crocker and E. Y. Jones, Mol. Cell, 1998, 1, 719–728 CrossRef CAS PubMed.
- J. Ereño-Orbea, T. Sicard, H. Cui, M. T. Mazhab-Jafari, S. Benlekbir, A. Guarné, J. L. Rubinstein and J. P. Julien, Nat. Commun., 2017, 8(1), 764 CrossRef PubMed.
- M. A. Zhuravleva, K. Trandem and P. D. Sun, J. Mol. Biol., 2008, 375, 437–447 CrossRef CAS PubMed.
- M. S. Alphey, H. Attrill, P. R. Crocker and D. M. F. van Aalten, J. Biol. Chem., 2003, 278, 3372–3377 CrossRef CAS PubMed.
- M. P. Lenza, U. Atxabal, C. Nycholat, I. Oyenarte, A. Franconetti, J. I. Quintana, S. Delgado, R. Núñez-Franco, C. T. Garnica Marroquín, H. Coelho, L. Unione, G. Jiménez-Oses, F. Marcelo, M. Schubert, J. C. Paulson, J. Jiménez-Barbero and J. Ereño-Orbea, JACS Au, 2023, 3, 204–215 CrossRef CAS PubMed.
- M. P. Lenza, L. Egia-Mendikute, A. Antoñana-Vildosola, C. O. Soares, H. Coelho, F. Corzana, A. Bosch, P. Manisha, J. I. Quintana, I. Oyenarte, L. Unione, M. J. Moure, M. Azkargorta, U. Atxabal, K. Sobczak, F. Elortza, J. D. Sutherland, R. Barrio, F. Marcelo, J. Jiménez-Barbero, A. Palazon and J. Ereño-Orbea, Nat. Commun., 2023, 14, 3496 CrossRef CAS PubMed.
- W.-X. Shi, R. Chammas, N. M. Varki, L. Powell and A. Varki, J. Biol. Chem., 1996, 271, 31526–31532 CrossRef CAS PubMed.
- E. C. M. Brinkman-Van der Linden and A. Varki, J. Biol. Chem., 2000, 275, 8625–8632 CrossRef CAS PubMed.
- T. Yamaji, T. Teranishi, M. S. Alphey, P. R. Crocker and Y. Hashimoto, J. Biol. Chem., 2002, 277, 6324–6332 CrossRef CAS PubMed.
- L. D. Powell, D. Sgroi, E. R. Sjoberg, I. Stamenkovic and A. Varki, J. Biol. Chem., 1993, 268, 7019–7027 CrossRef CAS PubMed.
- K. Strenge, R. Schauer, N. Bovin, A. Hasegawa, H. Ishida, M. Kiso and S. Kelm, Eur. J. Biochem., 1998, 258, 677–685 CrossRef CAS PubMed.
- J. Q. Zhang, G. Nicoll, C. Jones and P. R. Crocker, J. Biol. Chem., 2000, 275, 22121–22126 CrossRef CAS PubMed.
- C. Büll, M. A. Stoel, M. H. den Brok and G. J. Adema, Cancer Res., 2014, 74, 3199–3204 CrossRef PubMed.
- R. Hevey, Pharmaceuticals, 2019, 12, 55 CrossRef CAS PubMed.
- H. Su and Y. Xu, Front. Pharmacol, 2018, 9, 413254 Search PubMed.
- H. Nguyen, J. Park, S. Kang and M. Kim, Sensors, 2015, 15, 10481–10510 CrossRef CAS PubMed.
- M. Jerabek-Willemsen, T. André, R. Wanner, H. M. Roth, S. Duhr, P. Baaske and D. Breitsprecher, J. Mol. Struct., 2014, 1077, 101–113 CrossRef CAS.
- E. Laigre, D. Goyard, C. Tiertant, J. Dejeu and O. Renaudet, Org. Biomol. Chem., 2018, 16, 8899–8903 RSC.
- S. Murali, R. Rustandi, X. Zheng, A. Payne and L. Shang, Viruses, 2022, 14, 717 CrossRef CAS PubMed.
- S. Bertuzzi, F. Peccati, S. Serna, R. Artschwager, S. Notova, M. Thépaut, G. Jiménez-Osés, F. Fieschi, N. C. Reichardt, J. Jiménez-Barbero and A. Ardá, ACS Cent. Sci., 2022, 8, 1415–1423 CrossRef CAS PubMed.
- R. P. Sparks, W. Lawless, A. S. Arango, E. Tajkhorshid and R. A. Fratti, J. Visualized Exp., 2022, 180, e60607 Search PubMed.
- P. Valverde, J. I. Quintana, J. I. Santos, A. Ardá and J. Jiménez-Barbero, ACS Omega, 2019, 4, 13618–13630 CrossRef CAS PubMed.
- A. Ardá and J. Jiménez-Barbero, Chem. Commun., 2018, 54, 4761–4769 RSC.
- C. Dalvit, I. Gmür, P. Rößler and A. D. Gossert, Prog. Nucl. Magn. Reson. Spectrosc., 2023, 138–139, 52–69 CrossRef CAS PubMed.
- J. Angulo, P. M. Enríquez-Navas and P. M. Nieto, Chem.–Eur. J., 2010, 16, 7803–7812 CrossRef CAS PubMed.
- C. Dalvit, P. E. Fagerness, D. T. A. Hadden, R. W. Sarver and B. J. Stockman, J. Am. Chem. Soc., 2003, 125, 7696–7703 CrossRef CAS PubMed.
- G. Rocha, J. Ramírez-Cárdenas, M. C. Padilla-Pérez, S. Walpole, R. Nepravishta, M. I. García-Moreno, E. M. Sánchez-Fernández, C. Ortiz Mellet, J. Angulo and J. C. Muñoz-García, Anal. Chem., 2024, 96, 615–619 CrossRef CAS PubMed.
- S. Walpole, S. Monaco, R. Nepravishta and J. Angulo, in Methods in Enzymology, Academic Press, 2019, vol. 615, pp. 423–451 Search PubMed.
- C. Dalvit and A. Vulpetti, J. Med. Chem., 2019, 62, 2218–2244 CrossRef CAS PubMed.
- C. Dalvit and S. Knapp, Magn. Reson. Chem., 2017, 55, 1091–1095 CrossRef CAS PubMed.
- T. Diercks, A. S. Infantino, L. Unione, J. Jiménez-Barbero, S. Oscarson and H. Gabius, Chem.–Eur. J., 2018, 24, 15761–15765 CrossRef CAS PubMed.
- J. D. Martínez, A. I. Manzano, E. Calviño, A. de Diego, B. Rodriguez de Francisco, C. Romanò, S. Oscarson, O. Millet, H.-J. Gabius, J. Jiménez-Barbero and F. J. Cañada, J. Org. Chem., 2020, 85, 16072–16081 CrossRef PubMed.
- B. Linclau, A. Ardá, N.-C. Reichardt, M. Sollogoub, L. Unione, S. P. Vincent and J. Jiménez-Barbero, Chem. Soc. Rev., 2020, 49, 3863–3888 RSC.
- E. C. Wamhoff, J. Hanske, L. Schnirch, J. Aretz, M. Grube, D. Varón Silva and C. Rademacher, ACS Chem. Biol., 2016, 11, 2407–2413 CrossRef CAS PubMed.
- C. Dalvit, M. Flocco, S. Knapp, M. Mostardini, R. Perego, B. J. Stockman, M. Veronesi and M. Varasi, J. Am. Chem. Soc., 2002, 124, 7702–7709 CrossRef CAS PubMed.
- C. Dalvit, M. Flocco, M. Veronesi and B. J. Stockman, Comb. Chem. High Throughput Screening, 2002, 5, 605–611 CrossRef CAS PubMed.
- J. L. Asensio, A. Ardá, F. J. Cañada and J. Jiménez-Barbero, Acc. Chem. Res., 2013, 46, 946–954 CrossRef CAS PubMed.
- G. V. de Castro and A. Ciulli, Chem. Commun., 2019, 55, 1482–1485 RSC.
- C. Dalvit, Prog. Nucl. Magn. Reson. Spectrosc., 2007, 4, 243–271 CrossRef.
- L. Unione, M. Alcalá, B. Echeverria, S. Serna, A. Ardá, A. Franconetti, F. J. Cañada, T. Diercks, N. Reichardt and J. Jiménez-Barbero, Chem.–Eur. J., 2017, 23, 3957–3965 CrossRef CAS PubMed.
- E. Jiménez-Moreno, A. M. Gómez, A. Bastida, F. Corzana, G. Jiménez-Oses, J. Jiménez-Barbero and J. L. Asensio, Angew. Chem., 2015, 127, 4418–4422 CrossRef.
- G.-J. Kremers, J. Goedhart, E. B. van Munster and T. W. J. Gadella, Biochemistry, 2006, 45, 6570–6580 CrossRef CAS PubMed.
- R. Buratto, D. Mammoli, E. Chiarparin, G. Williams and G. Bodenhausen, Angew. Chem., Int. Ed., 2014, 53, 11376–11380 CrossRef CAS PubMed.
- G. V. de Castro and A. Ciulli, Chem. Commun., 2019, 55, 1482–1485 RSC.
- U. Atxabal, C. Nycholat, J. M. Pröpster, A. Fernández, I. Oyenarte, M. P. Lenza, A. Franconetti, C. O. Soares, H. Coelho, F. Marcelo, M. Schubert, J. C. Paulson, J. Jiménez-Barbero and J. Ereño-Orbea, ACS Chem. Biol., 2024, 19, 483–496 CrossRef CAS PubMed.
- H. Yu, A. Gonzalez-Gil, Y. Wei, S. M. Fernandes, R. N. Porell, K. Vajn, J. C. Paulson, C. M. Nycholat and R. L. Schnaar, Glycobiology, 2017, 27, 657–668 CrossRef CAS PubMed.
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov, O. Ronneberger, K. Tunyasuvunakool, R. Bates, A. Žídek, A. Potapenko, A. Bridgland, C. Meyer, S. A. A. Kohl, A. J. Ballard, A. Cowie, B. Romera-Paredes, S. Nikolov, R. Jain, J. Adler, T. Back, S. Petersen, D. Reiman, E. Clancy, M. Zielinski, M. Steinegger, M. Pacholska, T. Berghammer, S. Bodenstein, D. Silver, O. Vinyals, A. W. Senior, K. Kavukcuoglu, P. Kohli and D. Hassabis, Nature, 2021, 596, 583–589 CrossRef CAS PubMed.
- V. Almeida-Marrero, M. Mascaraque, M. Jesús Vicente-Arana, Á. Juarranz, T. Torres and A. de la Escosura, Chem.–Eur. J., 2021, 27, 9634–9642 CrossRef CAS PubMed.
- V. Loimaranta, J. Hepojoki, O. Laaksoaho and A. T. Pulliainen, J. Leukocyte Biol., 2018, 104, 777–786 CrossRef CAS PubMed.
- J. Munkley, Int. J. Mol. Sci., 2016, 17, 275 CrossRef PubMed.
- C. D. Rillahan, E. Schwartz, R. McBride, V. V. Fokin and J. C. Paulson, Angew. Chem., Int. Ed., 2012, 51, 11014–11018 CrossRef CAS PubMed.
- C. Yung-Chi and W. H. Prusoff, Biochem. Pharmacol., 1973, 22, 3099–3108 CrossRef PubMed.
- L. L. Kiessling, T. Young, T. D. Gruber and K. H. Mortell, Glycoscience, Springer Berlin Heidelberg, Berlin, Heidelberg, 2008 Search PubMed.
- J. I. Quintana, U. Atxabal, L. Unione, A. Ardá and J. Jiménez-Barbero, Chem. Soc. Rev., 2023, 52, 1591–1613 RSC.
- V. Almeida-Marrero, F. Bethlehem, S. Longo, M. C. Bertolino, T. Torres, J. Huskens and A. de la Escosura, Angew. Chem., Int. Ed., 2022, 134(31), e202206900 CrossRef.
- E. Capone, S. Iacobelli and G. Sala, J. Transl. Med., 2021, 19, 405 CrossRef CAS PubMed.
- H. Läubli, F. Alisson-Silva, M. A. Stanczak, S. S. Siddiqui, L. Deng, A. Verhagen, N. Varki and A. Varki, J. Biol. Chem., 2014, 289, 33481–33491 CrossRef PubMed.
- W. Peng and J. C. Paulson, J. Am. Chem. Soc., 2017, 139, 12450–12458 CrossRef CAS PubMed.
- H. Yu, S. Huang, H. Chokhawala, M. Sun, H. Zheng and X. Chen, Angew. Chem., Int. Ed., 2006, 45, 3938–3944 CrossRef CAS PubMed.
- A. R. Aricescu, W. Lu and E. Y. Jones, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2006, 62, 1243–1250 CrossRef PubMed.
- D. A. Case, K. Belfon, I. Y. Ben-Shalom, S. R. Brozell, D. S. Cerutti, T. E. Cheatham III, V. W. D. Cruzeiro, T. A. Darden, R. E. Duke, G. Giambasu, M. K. Gilson, H. Gohlke, A. W. Goetz, R. Harris, S. Izadi, S. A. Iz-mailov, K. Kasavajhala, A. Kovalenko, R. Krasny, T. Kurtzman, T. S. Lee, S. LeGrand, P. Li, C. Lin, J. Liu, T. Luchko, R. Luo, V. Man, K. M. Merz, Y. Miao, O. Mikhailovskii, G. Monard, H. Nguyen, A. Onufriev, F. Pan, S. Pantano, R. Qi, D. R. Roe, A. Roitberg, C. Sagui, S. Schott-Verdugo, J. Shen, C. L. Simmerling, N. R. Skrynnikov, J. Smith, J. Swails, R. C. Walker, J. Wang, L. Wilson, R. M. Wolf, X. Wu, Y. Xiong, Y. Xue, D. M. York and P. A. Kollman, AMBER 2020, University of California, San Francisco, 2020 Search PubMed.
- J. A. Maier, C. Martinez, K. Kasavajhala, L. Wickstrom, K. E. Hauser and C. Simmerling, J. Chem. Theory Comput., 2015, 11, 3696–3713 CrossRef CAS PubMed.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- H. C. Andersen, J. Chem. Phys., 1980, 72, 2384–2393 CrossRef CAS.
- S. Miyamoto and P. A. Kollman, J. Comput. Chem., 1992, 13, 952–962 CrossRef CAS.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc01723d |
| ‡ Contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |