
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotocarrier tunneling triggering CO2 photocatalysis†
Xian
Yan
a,
Meng
Yuan
a,
Ya-Long
Yuan
a,
Peng
Su
a,
Qing
Chen
a and
Fang-Xing
Xiao
 *ab
*ab
aCollege of Materials Science and Engineering, Fuzhou University, New Campus, Fujian Province 350108, China
bState Key Laboratory of Structural Chemistry, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Fuzhou, Fujian 350002, PR China. E-mail: fxxiao@fzu.edu.cn
First published on 3rd June 2024
Abstract
Solar CO2 reduction to renewable hydrocarbon fuels offers a promising pathway to carbon neutrality, but it is retarded by tough CO2 activation, complicated mechanisms, sluggish charge transport kinetics, and a scarcity of strategies for precise tuning of charge transport pathways. Herein, we first conceptually design a novel insulating polymer-mediated electron-tunneling artificial photosystem via progressive interface configuration regulation, wherein tailor-made Ag@citrate nanocrystals (NCs) are controllably self-assembled on transition metal chalcogenides (TMCs) assisted by an ultrathin insulating polymer interim layer, i.e., poly(allylamine hydrochloride) (PAH). In this multilayered nano-architecture, a solid ultra-thin insulating PAH interim layer serves as an unexpected charge tunneling mediator to stimulate smooth electron transfer from the TMC substrate to the terminal electron reservoirs of Ag@citrate NCs, engendering the tandem charge transfer route and significantly boosting the visible-light-driven photocatalytic CO2-to-syngas conversion performances. Furthermore, we have ascertained that such TMC–insulating polymer–metal NC tunneling photosystems are universal. This study would spark new inspiration for unleashing the long-term neglected charge tunneling capability of insulating polymers and diversifying non-conjugated polymer-based artificial photosystems for solar-to-fuel energy conversion.
1. Introduction
Solar CO2 conversion to syngas has been considered an alternative, sustainable and emerging strategy to provide renewable energy and achieve carbon neutrality in the near future.1,2 However, it is hampered by the chemical inertness of CO2 molecules, sluggish charge transfer kinetics and difficulty in accurately mediating the charge transport pathway.3,4 In addition, the CO2 reduction reaction involves multi-step electron transfer, representing a complex reaction mechanism. To surmount these obstacles, multifarious strategies have thus been carried out. Thus far, the majority of researchers have been devoted to exploring high-performance CO2 reduction photocatalysts that promote the adsorption and activation of inert CO2.5 Despite the advancement, there is a lack of studies that focus on the precise tuning of carrier transport behaviors by crafting spatially controllable charge transfer pathways at the micro-level in photocatalytic CO2 reduction reactions. The endeavor in this field would significantly unleash the correlation between charge transfer characteristics and the photocatalytic CO2 reduction pathway, which will without doubt reinforce our fundamental understanding of the photocatalytic mechanism of this emerging CO2 conversion technique.The harmonious interface design has been established as the essential premise for dictating spatially regulated vectorial charge transport pathways in artificial photosystems.6 Fundamentally, elegant interface design should rationally consider the following crucial factors: intimate integration of building blocks for crafting a clean interface, applicable interfacial co-catalyst engineering, and tunable optimization of interface configuration.7 To this end, construction of semiconductor–insulator–metal (SIM) photosystems requires precise interface configuration regulation for triggering spatially directional photocarrier migration via the electron tunneling effect and accessing charge separation.8 Noteworthily, as for the conventional SIM electron-tunneling photosystems, insulating layers are merely confined to oxides such as Al2O3 and TiO2, while exploration of non-conjugated insulating polymers as the intermediate layer has not yet been reported.9 This leads us to consider whether solid non-conjugated insulating polymers, which account for a vast majority of polymers, can be harnessed to mediate the electron tunneling process and what specific influence they would exert on the charge transfer mechanism in heterogeneous photocatalysis.
Inspired by the above discussion, we choose transition metal chalcogenides (TMCs) with excellent light-harvesting capability as substrates, solid non-conjugated insulating polymer as an interim layer, and tailor-made metal nanocrystals (NCs) as the terminal electron reservoirs for smartly constructing novel TMC–insulating polymer–metal NCs electron tunneling photosystems. Inspired by the charge properties of the ligand capped on the metal NCs surface, we speculate that the metal NCs can be accurately self-assembled on TMC substrates modified with an oppositely charged insulating polymer that serves as “molecular glue” via electrostatic interaction, which would produce well-defined TMC/insulating polymer/metal NC heterostructures.10,11 Considering the Schottky junction formed between TMCs and metal NCs, we believe spontaneous charge transfer would be stimulated once electron tunneling is triggered in such TMC–insulating polymer–metal NC photosystems.12 In this way, photoexcited electrons from the TMCs will effectively tunnel through the ultrathin insulating polymer interim layer and directionally migrate to the terminal electroreservoirs of metal NCs, engendering the tandem charge transfer route.
Herein, an efficient, easily accessible, and controllable electron-tunneling-mediated tandem charge transfer pathway is designed over TMC@insulating polymer/metal NC photosystems by an electrostatic self-assembly tactic, for which tailor-made negatively charged Ag@citrate NCs are accurately self-assembled on the positively charged ultrathin insulating poly(allylamine hydrochloride) (PAH) encapsulated CdS substrate at ambient conditions. Directional charge transfer is unanticipatedly stimulated over the CdS@PAH/Ag@citrate NCs heterostructure by electron tunneling penetrating the intermediate ultrathin insulating polymer layer, synergistically endowing the CdS@PAH/Ag@citrate NCs photosystem with excellent photoactivity toward CO2-to-syngas conversion under visible light irradiation. Furthermore, we ascertain that the insulating polymer-mediated electron tunneling photosystem is universal. Our study represents the construction of novel insulating polymer-based electron tunneling artificial photosystems for controllable charge transport modulation toward solar-to-fuel energy conversion.
2. Experimental section
2.1 Materials
All materials were of analytical grade and used as received without further purification, and detailed information is provided in the ESI.†2.2 Preparation of photocatalysts
Experimental procedures for preparing different photocatalysts in this study are provided in the ESI.† CdS/Ag@citrate NCs 1% was labeled as C/A1. CdS@PAH 1%/Ag@citrate NCs 1% was labeled as C@P1/A1.2.3 Characterization
Specific details are provided in the ESI.†2.4 Photocatalytic CO2 reduction
Photocatalytic CO2 reduction products were analyzed by gas chromatography (SHIMADZU, GC-2014C). The specific operational information is provided in the ESI.†2.5 Selective photocatalytic reduction performance
Photocatalytic reduction of nitroaromatics to amino compounds was performed in an anaerobic environment (inert atmosphere N2), along with the addition of a hole scavenger. Photoreduction products were analyzed by a UV-vis absorption spectrophotometer (Thermal Genesis). Specific operational information is provided in the ESI.†2.6 Photoelectrochemical (PEC) measurements
PEC measurements were carried out on an electrochemical workstation (CHI660E, CHI Shanghai, Inc.) with a conventional three-electrode system, and a 0.5 M Na2SO4 aqueous solution (pH = 6.69) was utilized as the electrolyte. Specific operational information is provided in the ESI.†3. Results and discussion
Scheme 1 depicts the flowchart for electrostatic self-assembly of CdS@PAH/Ag@citrate NCs (C@P/A) heterostructure under ambient conditions. Specifically, CdS nano-leaves are first closely encapsulated with an ultrathin PAH layer, which endows CdS nano-leaves with a positively charged surface (Fig. S1†). In contrast, Ag@citrate NCs are prepared by the reduction of silver precursors with citrate, thus presenting a carboxylic acid-capped negatively charged surface (Fig. S1†). Eventually, driven by electrostatic force, Ag@citrate NCs can be spontaneously and uniformly anchored on the positively charged CdS@PAH nano-leaves framework, forming the multilayered CdS@PAH/Ag@citrate NCs (C@P/A) ternary heterostructure.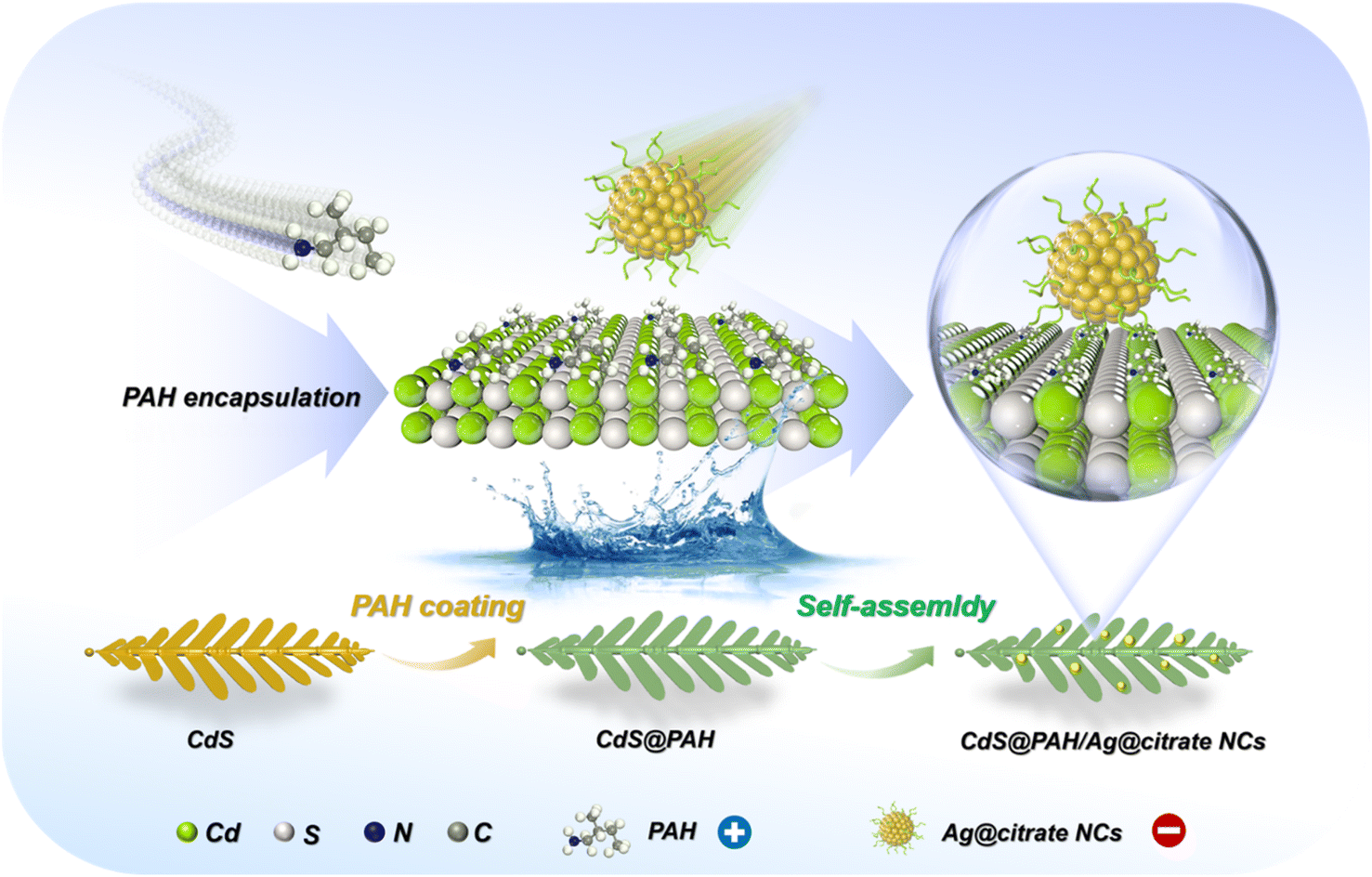 | ||
| Scheme 1 Schematic flowchart for fabrication of CdS@PAH/Ag@citrate NC heterostructures. | ||
3.1 Structural characterization
Fig. 1a shows the X-ray diffraction (XRD) patterns of samples, peaks at 24.90°, 26.52°, 28.28°, 36.72°, 43.83°, 47.94° and 51.97° are indexed to the (100), (002), (101), (102), (110), (103) and (112) crystal planes of hexagonal CdS (PDF# 6-314). Apparently, encapsulating PAH on CdS does not cause the crystal phase change. No peaks attributable to Ag@citrate NCs can be observed in the XRD patterns of C/A1 and C@P1/A1 heterostructures, which is mainly attributed to the low loading amount of Ag@citrate NCs. An analogous result is observed in Raman results (Fig. S2†), in which the peaks at 300 (Eg) and 602 cm−1 (Eg) for all these samples are attributed to CdS, and no characteristic peaks of Ag@citrate NCs and PAH are observed, consistent with XRD results. As demonstrated in Fig. 1b and Table S1,† the FTIR spectrum of CdS exhibits two bands at 3470 and 1665 cm−1, which correspond to the surface hydroxyl (–OH) group.13 In comparison with CdS, two bands at 2955 and 2882 cm−1 are seen in the FTIR spectra of C@P1 and C@P1/A1, which arise from the stretching vibration mode of –CH2– groups from PAH.14 Intriguingly, the band at ca. 1665 cm−1 attributed to –NH2– functional groups in the FTIR spectra of C@P1 and C@P1/A1 exhibits a more enhanced intensity compared to other counterparts, corroborating the successful attachment of the PAH layer to the CdS substrate.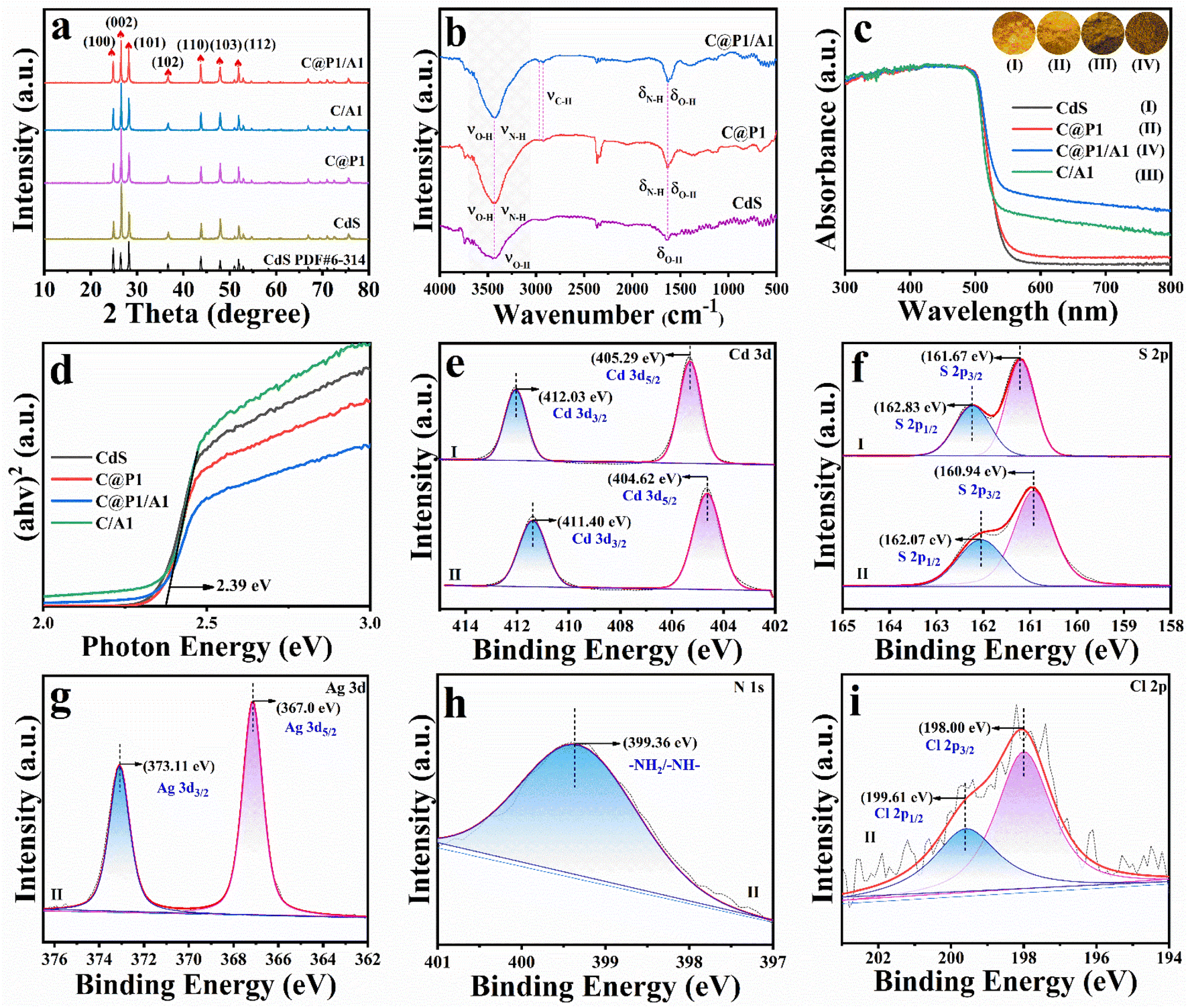 | ||
| Fig. 1 (a) XRD patterns of CdS, C@P1, C/A1 and C@P1/A1; (b) FTIR spectra of CdS, C@P1 and C@P1/A1; (c) DRS results of CdS, C@P1, C/A1 and C@P1/A1 with (d) transformed plots based on the Kubelka–Munk function vs. energy of light. High-resolution (e) Cd 3d, (f) S 2p, (g) Ag 3d, (h) N 1s and (i) Cl 2p spectra of (I) pristine CdS and (II) C@P1/A1. | ||
Optical properties of the samples were probed by UV-vis diffuse reflectance spectroscopy (DRS). As shown in Fig. 1c, all the samples exhibit a light absorption band edge at ca. 520 nm, and this stems from the inherent bandgap photoexcitation of the CdS substrate. It is worth noting that C@P1 and CdS exhibit an almost identical absorption band edge, implying PAH encapsulation exerts no influence on the light absorption of the CdS substrate, thus ruling out the photosensitization effect of PAH (Fig. S3†). Similarly, citrate does not possess photosensitivity (Fig. S4†). In addition, compared with pristine CdS, C/A and C@P1/A1 heterostructures present enhanced light absorption in the visible region ranging from 500 to 800 nm, verifying that integration of Ag@citrate NCs with the CdS substrate can enhance the light absorption capacity (Fig. S5†). According to the Kubelka–Munk function, the determination of the bandgap (Eg) is displayed in Fig. 1d, whereby Eg of CdS is determined to be ca. 2.39 eV, and it is revealed that PAH and Ag@citrate NCs deposition fails to alter the optical properties of the CdS substrate.
The surface elemental composition and chemical states of the samples were analyzed by X-ray photoelectron spectroscopy (XPS). As revealed in Fig. 1e(II), the high-resolution Cd 3d spectrum of the C@P1/A1 heterostructure with peaks situated at 404.62 (Cd 3d5/2) and 411.40 eV (Cd 3d3/2) is attributed to Cd2+ species.15 The high-resolution S 2p spectrum of the C@P1/A1 heterostructure exhibits two featured peaks at 160.94 (S 2p3/2) and 162.07 eV (S 2p1/2), ascribed to the S2− species (Fig. 1f(II)).16 Intriguingly, compared with pristine CdS, the Cd 3d and S 2p peaks of the C@P1/A1 heterostructure are shifted to a lower binding energy. This indicates a strong interaction, resulting in the overlap of the charge transfer state or electron wave function between CdS and Ag@citrate NCs.17 As displayed in Fig. 1g, the high-resolution Ag 3d spectrum of C@P1/A1 shows two peaks at 373.11 (Ag 3d3/2) and 367.0 eV (Ag 3d5/2), which correspond to Ag0 and Ag+ species,18 respectively, confirming the Ag@citrate NCs deposition on C@P1. The high-resolution N 1s (Fig. 1h) spectrum of C@P1/A1 exhibits a peak at 399.36 eV, which is assigned to nitrogen-containing (–NH2–) species from PAH.19 As depicted in Fig. 1i, the high-resolution Cl 2p spectrum of C@P1/A1 with two peaks located at 198.0 (Cl 2p3/2) and 199.61 eV (Cl 2p1/2) originates from the Cl− species of PAH.20 Perspicuously, the distinct elemental signals of Cd, S, Ag, N, and Cl unequivocally validate the successful fabrication of the C@P1/A1 heterostructure (Table S2†).
The morphology and textural structure of the samples were characterized by FESEM and TEM measurements. As shown in Fig. 2a and d, pristine CdS exhibits a leaf-like morphology, characterized by a pronounced trunk and meticulously arranged nano-leaves ranging in size from 4 to 6 μm. These unique nano-leaves structures not only expose a high density of active sites but also effectively reduce the vertical travel distance of photogenerated carriers, thereby suppressing charge recombination over CdS.21 The morphology of CdS encapsulated with an ultrathin PAH layer does not show the apparent change (Fig. 2b), primarily due to the low deposition amount of PAH. When Ag@citrate NCs are anchored on CdS@PAH by electrostatic self-assembly, as revealed in Fig. 2c, the morphology of CdS@PAH/Ag NCs is not markedly altered compared with CdS@PAH, which is due to the relatively small size of Ag@citrate NCs [Fig. S6,†ca. 11.27 nm], rendering clear differentiation of them merely from SEM images rather difficult. Furthermore, the approximate specific surface of CdS (4.325 m2 g−1) and C@P15@Au7 (3.587 m2 g−1) confirms the above statement (Fig. S7 and Table S3†). Fig. 2e shows the HRTEM image of pristine CdS, whose designated area exhibits a clear crystalline structure with a distinguishable lattice fringe of ca. 0.336 nm, corresponding to the (002) crystal facet of hexagonal CdS. Moreover, as shown in Fig. 2f, it is apparent that an amorphous PAH layer is closely and uniformly tethered to the outermost surface of CdS. To more intuitively understand the integration mode of building blocks in C@P1/A1, TEM and HRTEM images were probed. As displayed in Fig. 2g and h, various discernible Ag@citrate NCs are evenly distributed on the C@P1 nano-leaves substrate. HRTEM images of the C@P1/A1 heterostructure (Fig. 2i) show the lattice fringes of ca. 0.336 and 0.237 nm, matching well with the (002) and (111) crystal facets of CdS and Ag@citrate NCs, respectively. In addition, it can be observed that an ultrathin amorphous polymer layer tightly encases the edge of the CdS substrate, strongly confirming the existence of an elegant interface (C@P1/A1). Moreover, the PAH layer encapsulation on CdS can be corroborated by energy-dispersive X-ray spectroscopy (EDX) and elemental mapping results, as displayed in Fig. S8a† and 2j. Among them, N and Cl signals originating from PAH are distributed along the nano-leaves skeleton, verifying that the PAH layer is seamlessly coated on the whole CdS substrate. EDS (Fig. S8b†) and TEM elemental mapping (Fig. 2k1–6) results revealed the co-existence of Cd, S, C, Ag, N, and Cl elements, implying that CdS, PDDA and Au@citrate NCs are concurrently integrated in the C@P1/A1 heterostructure. Similarly, EDS and FESEM mapping results of C@P1/A1 (Fig. S9†) also point to an analogous conclusion.
 | ||
| Fig. 2 FESEM images of (a) CdS, (b) C@P1 and (c) C@P1/A1 with schematic models in insets; TEM images of (d) CdS and (g and h) C@P1/A1; HRTEM images of (e) CdS, (f) C@P1, (i) C@P1/A1; and elemental mapping results of (j) C@P1 and (k) C@P1/A1. | ||
3.2 Photocatalytic reduction activities
Photocatalytic CO2 reduction was carried out to evaluate the photoactivities of the C@P/A heterostructure under visible light irradiation (λ > 420 nm). After testing, it was determined that syngas (CO and H2) is the primary product of photocatalytic CO2 reduction reactions.22Fig. 3a illustrates the trend of syngas evolution over the C@P heterostructure as the PAH amount increases, based on which C@P1 is determined as the optimal encapsulation ratio. The excessive PAH encapsulation leads to a decrease in the photoactivity of C@P, which stems from the inherent insulating properties of PAH. Note that the molecular structure of PAH does not possess the π-conjugated structure, indicating it is an insulating polymer in a solid state (Fig. S10a†). Thus, encapsulation of CdS with PAH should remarkably increase the interfacial charge transfer resistance (Fig. S10b†) and even completely cut off the interfacial charge transfer, giving rise to reduced photoactivity. Nonetheless, it should be particularly stressed that the photoactivities of C@P are comparable to those of pristine CdS with minute fluctuation, implying that the insulating PAH coating in our case actually fails to retard the interfacial charge transfer and must function as an electron tunneling mediator to enable electron flow by penetrating through the PAH layer. In other words, the electrons photoexcited over CdS can still migrate to the outermost surface of the insulating PAH layer to participate in the photocatalytic CO2 reduction reaction. Inspired by this, we selected Ag@citrate NCs as the terminal electron reservoirs and homogeneously anchored them on the C@P1 heterostructure using a self-assembly strategy with a view to constructing a cascade charge transport chain given the Schottky junction endowed by NCs deposition. As shown in Fig. 3b, the CO evolution activity of C@P/A heterostructures gradually enhances with increasing the Ag@citrate NCs amount, and C@P1/A1 reaches the optimal photoactivity. The strong host–guest interaction between C@P1 and Ag@citrate NCs can facilitate the conversion of CO2 into CO. Obviously, driven by the Schottky barrier, the C@P/A heterostructure bursts out the more powerful potential of electron tunneling.23 The excessive loading of Ag@citrate NCs leads to the occupation of a majority of the active sites by H+, thus limiting the contact of CO2 with the active sites and facilitating the generation of H2.24 Since the photocatalytic syngas evolution process is rigorous and complicated, which involves the adsorption and activation of CO2 (ref. 25) and vectorial charge transport, the yield of CO, which accounts for the important ingredient of syngas, can reflect the CO2 conversion efficiency. Therefore, great attention is paid to the conversion of CO2 to CO in our current work. Undoubtedly, the optimal interface configuration is observed over C@P1/A1. It is clear that the syngas production rate of C@P1/A1 (CO: 52.60 μmol g−1 h−1, H2: 270.54 μmol g−1 h−1) is significantly higher than that of CdS (CO: 18 μmol g−1 h−1, H2: 38.241 μmol g−1 h−1), confirming the importance of harmonious interface configuration design. Noteworthily, the C/A1 heterostructure, which is fabricated by directly depositing the same amount of Ag@citrate NCs on the CdS substrate, exhibits a slight enhancement in CO production rate (CO: 23.22 μmol g−1 h−1) relative to the CdS substrate, but it is far inferior to the C@P1/A1 heterostructure under same conditions. The enhancement of syngas production rate over the C@P1/A1 heterostructure relative to the C/A1 heterostructure reveals the crucial role of the insulating PAH interim layer as an electron tunneling mediator in boosting the syngas evolution activities, i.e., PAH induces the photoelectron tunneling of CdS, which triggers the cascade electron transport and thus accelerates the interfacial charge separation.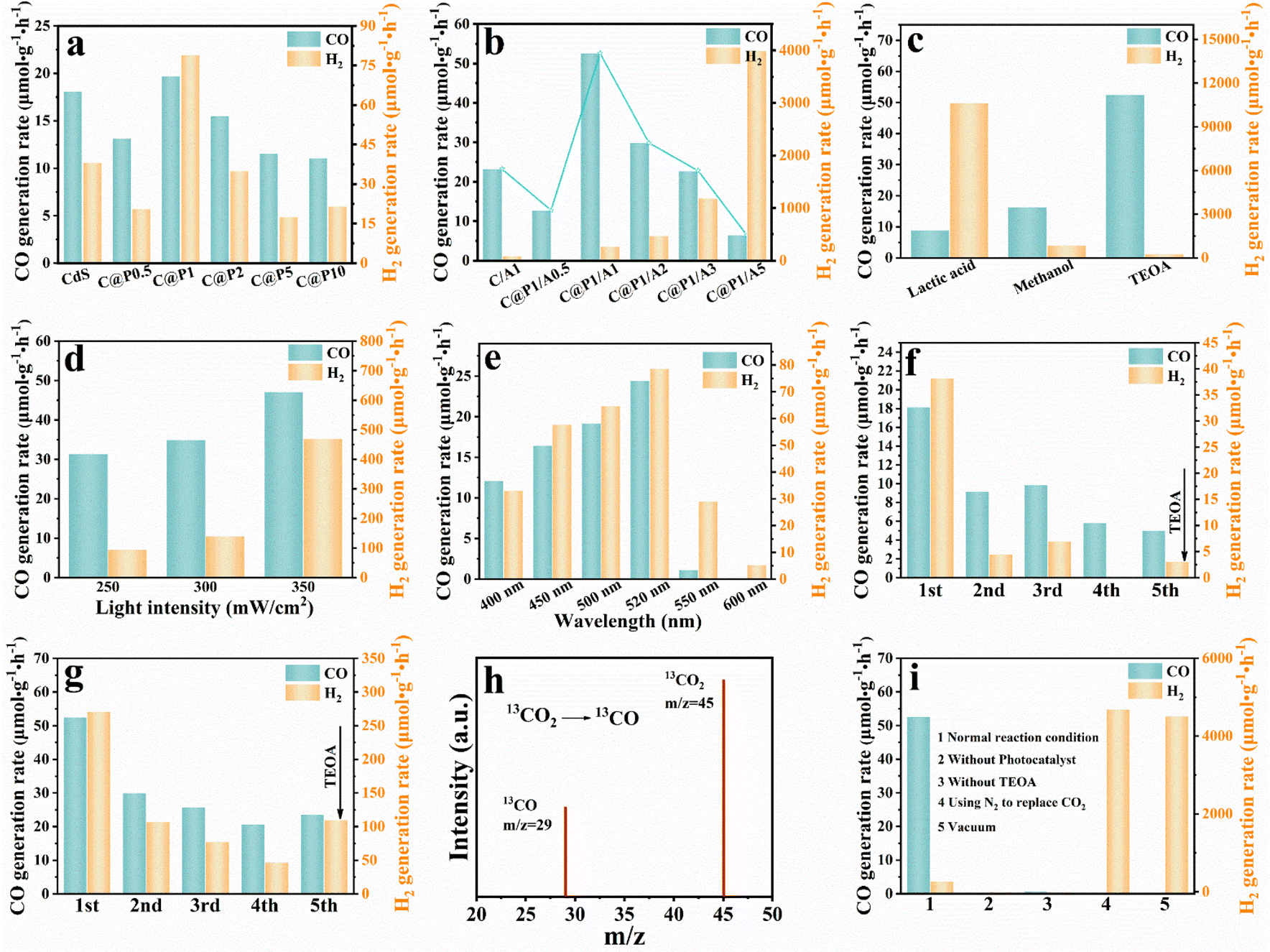 | ||
| Fig. 3 (a) Photocatalytic CO2 reduction performances of CdS and C@P-X% (X: 0.5, 1, 2, 5, and 10) with different mass ratios of PAH under visible light (λ > 420 nm) irradiation; (b) photocatalytic CO2 reduction performances of C/A1 and C@P1/A-X% (X: 0.5, 1, 2, 3, and 5) heterostructures with different mass ratios of Ag@citrate NCs; photoactivities of C@P1/A1 (c) using varying sacrificial reagents, (d) under different light intensity, (e) under different monochromatic light irradiation; cyclic photocatalytic reactions of (f) blank CdS and (g) C@P1/A1; (h) CO product generated from the 13CO2 isotope experiment; and (i) CO2 photoreduction performances of C@P1/A1 under different reaction conditions. | ||
Fig. 3c manifests that the sacrificial reagent category substantially influences the photoactivity of the C@P1/A1 heterostructure, among which TEOA demonstrates the optimal efficiency. Compared with TEOA, methanol and lactic acid have higher oxidation potentials, which means they need more energy during the oxidation process, resulting in a weaker ability to capture holes and slowing down the efficiency of electron–hole separation. Additionally, dissociation of lactic acid in the photoreaction leads to an increase in the concentration of protons, thereby accelerating the production of H2 and forming a competitive reaction with the evolution of CO. As revealed in Fig. 3d, the photoactivity of the C@P1/A1 heterostructure increases with increasing light intensity, indicating the photocatalytic syngas evolution reaction is indeed a photocatalytic process. Fig. 3e shows the syngas evolution rates of the C@P1/A1 heterostructure at different excitation wavelengths, which reveals the optimal syngas evolution activity at an incident wavelength of 520 nm. Note that the action spectrum is in faithful agreement with the DRS result, implying the predominant role of bandgap photoexcitation of the CdS substrate in triggering the photocatalytic CO2 reduction reaction. Apart from their outstanding photoactivity, stability is also an important indicator for evaluating the performance of the photocatalysts. As displayed in Fig. 3f and g, the reusability tests suggest that the photoactivities of CdS and the C@P1/A1 heterostructure show varying decay degrees after several sets of cyclic reactions, with CdS showing the most severe photoactivity decay. Disruption of nano-leaf morphology or dissociation of sodium citrate may cause a decrease in catalyst activity (Fig. S11†). However, the decrease in stability mainly arises from the generic photo-corrosion of the CdS substrate. C@P1/A1 achieves the directional charge transport modulation with the assistance of electron tunneling, which reduces the electron–hole pair recombination and retards the detrimental photo-corrosion of CdS. Furthermore, XRD (Fig. S12†), FTIR (Fig. S13†), Raman spectroscopy (Fig. S14†), and XPS (Fig. S15†) results of C@P1/A1 after cyclic reaction verify its relatively favorable photostability. To prove the true carbon source of CO, 13CO2 labeling experiments were carried out using 13CO2 as the reactant. Mass spectrum signals revealed that only 13CO (m/z = 29) is probed (Fig. 3h), corroborating that the CO product indeed originates from the photocatalytic reduction of CO2 reactant.26 Several control experiments were also conducted for the C@P1/A1 heterostructure, including testing with and without catalyst, TEOA, or CO2 under otherwise same reaction conditions (Fig. 3i). The results suggest that the design of a solar-driven CO2-to-syngas conversion photocatalytic system in our work is reasonable and efficient.
Apart from the photocatalytic CO2 reduction, aromatic nitro compounds (6 electrons) were chosen for selective photoreduction to verify the universality of the PAH-mediated electron tunneling effect. Photoactivities of C/A1 and C@P1/A1 heterostructures were evaluated by anaerobic photocatalytic reduction of aromatic nitro compounds to amino derivatives under visible light irradiation (λ > 420 nm) with the addition of ammonium formate as a hole quencher and N2 purging under ambient conditions. As shown in Fig. 4a, the C@P1/A1 heterostructure can complete the photoreduction of nitrobenzene to aminobenzene within 100 s, with a conversion rate as high as nearly 85%, far exceeding that of C/A1. More intriguingly, as displayed in Fig. 4b–f and Table S5,† analogous results are visualized in the photoreduction of other nitro-aromatics, including 4-nitrotoluene, 4-nitroanisole, 4-nitroaniline (4-NA), 3-nitroaniline (3-NA) and o-nitroacetophenone. Based on the above analysis, it can be concluded that the PAH-mediated electron tunneling photosystem is general for diverse photo-reduction reactions apart from photocatalytic CO2 reduction reactions.
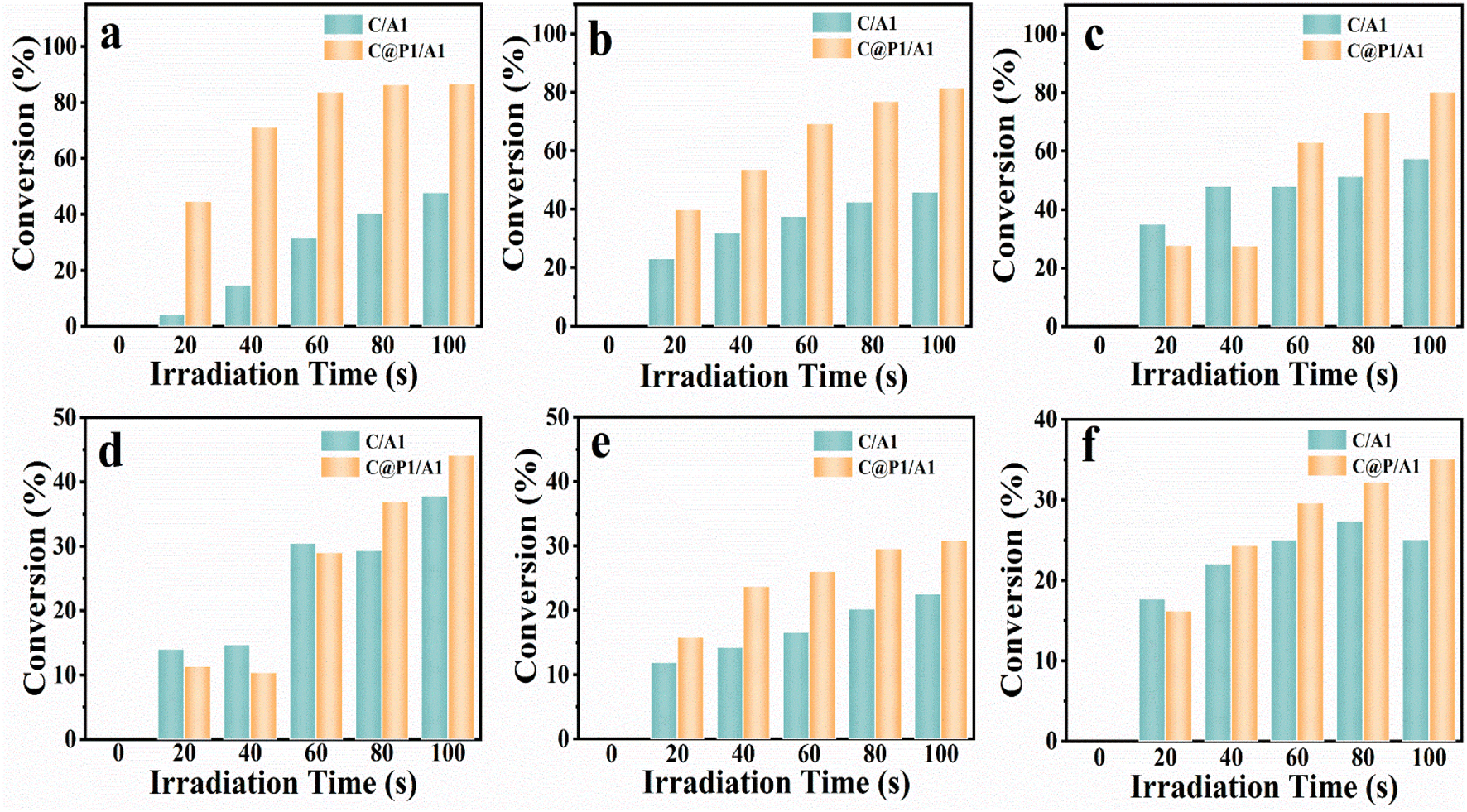 | ||
| Fig. 4 Photocatalytic performances of C/A1 and C@P1/A1 heterostructures toward selective reduction of nitroaromatics under visible light irradiation (λ > 420 nm) with the addition of ammonium formate as a hole scavenger and N2 bubbling at ambient conditions, including (a) nitrobenzene, (b) 4-nitrotoluene, (c) 4-nitroanisole, (d) 4-NA, (e) 3-NA and (f) o-nitroacetophenone. | ||
3.3 PEC and spectroscopic analysis
Photoelectrochemical (PEC) measurements were performed to disclose the interfacial charge separation efficiency of the electron tunneling photosystem. Fig. 5a shows the transient photocurrent response (I–t) of C/A1 and C@P1/A1 photoelectrodes under intermittent visible light (λ > 420 nm) irradiation, wherein the photocurrent density of C@P1/A is significantly higher than that of C/A1. The apparent photocurrent density difference between above-mentioned photoelectrodes provides compelling evidence that the PAH-mediated electron tunneling photosystem shows higher charge separation and transport efficiency. Consistently, as shown in Fig. 5b, the open-circuit photovoltage decay (OCVD) test exhibits the analogous result. Electrochemical impedance spectroscopy (EIS) was utilized to evaluate the interfacial charge transfer resistance between the photoelectrodes and electrolytes under visible light irradiation.27 In the Nyquist plots of EIS results, the semicircle arc radius represents the charge transfer resistance between the photoelectrode and electrolyte, with a smaller arc radius indicating lower charge transfer resistance. As exhibited in Fig. 5c and Table S4,† compared with C/A1 (19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 510 ohm), C@P1/A1 (5159 ohm) exhibits a smaller charge transfer resistance (Rct), manifesting its faster interfacial charge transfer rate. Additionally, to further evaluate the efficiency of interfacial charge separation, the charge carrier density (ND) of the photoelectrodes was determined by analyzing Mott–Schottky (M–S) results (Fig. 5d and e). The charge carrier density (ND) of the photoelectrode is calculated using the following equation:
510 ohm), C@P1/A1 (5159 ohm) exhibits a smaller charge transfer resistance (Rct), manifesting its faster interfacial charge transfer rate. Additionally, to further evaluate the efficiency of interfacial charge separation, the charge carrier density (ND) of the photoelectrodes was determined by analyzing Mott–Schottky (M–S) results (Fig. 5d and e). The charge carrier density (ND) of the photoelectrode is calculated using the following equation: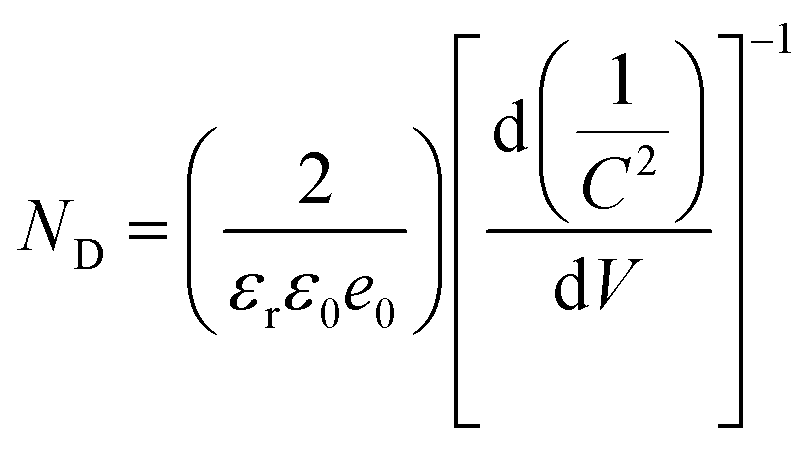 | (1) |
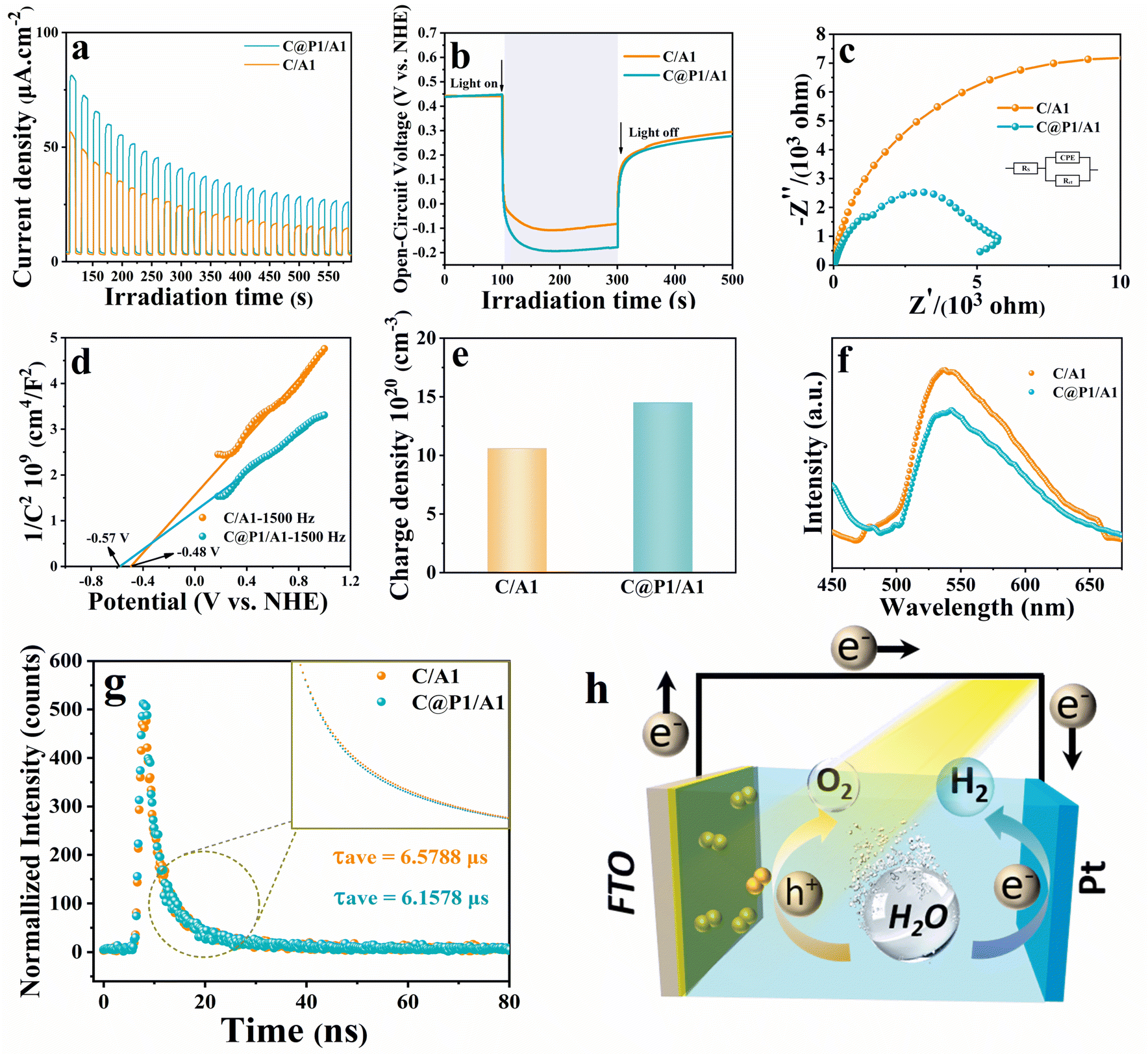 | ||
| Fig. 5 (a) Photocurrent, (b) open-circuit potential decay, and (c) EIS Nyquist plots of C@A1 and C@P1/A1 under visible light (λ > 420 nm) irradiation in an aqueous Na2SO4 solution (0.5 M, pH = 6.69); (d) Mott–Schottky plots, (e) charge carrier densities, (f) PL spectra, (g) time-resolved transient PL decay of C@A1 and C@P1/A1 heterostructures, and (h) schematic illustration of the PEC mechanism. | ||
3.4 Photocatalytic mechanism
To highlight the influence of the ultra-thin insulating PAH interim layer on the electron-tunneling effect, insulating SiO2 was encapsulated on the C@P1 surface for blocking electron transport, and the thus prepared control sample was labeled as C@P1@S/A1 (Fig. S16–18† and 6a). Another control sample named C@P1/A1-700 °C is simultaneously prepared, for which the C@P1/A1 heterostructure underwent high-temperature calcination in an inert atmosphere to eliminate the PAH layer (Fig. S19† and 6b). Compared with pristine C@P1/A1, C@P1/A1 after high-temperature calcination in a nitrogen atmosphere showed slight fragmentation at the edge of its nano-leaves, but still retained the nano-leaves skeleton (Fig. S20†). As shown in Fig. 6c, the photoactivities of the samples towards photoreduction of nitrobenzene follow the order of C@P1/A1 > C@P1/A1-700 °C > C@P1@S/A1. Unsurprisingly, interfacial charge transfer between C@P1 and Ag@citrate NCs is blocked by the SiO2 interim layer. In other words, the electrons photoexcited over C@P1 cannot tunnel through the insulating SiO2 layer to Ag@citrate NCs, thus resulting in markedly decreased photoactivity. Consistently, as shown in Fig. 6d and e, compared with C@P1/A1, C@P1@S/A1 exhibits the lower photocurrent density and photovoltage, confirming that the SiO2 interim layer indeed cuts off the interfacial electron transport chain between C@P1 and Ag@citrate NCs. Unlike the SiO2 insulating layer, the insulating PAH interim layer exhibits excellent electron tunneling capability, facilitating interfacial electron transport. This speculation is also rationalized by the in situ XPS measurement, which was generally used to determine interfacial charge migration paths. As shown in Fig. 6f–h, compared with C@P1/A1 in the dark, the binding energies of Cd 3d and S 2p for C@P1/A1 under visible light irradiation markedly move to the higher energy level, while the binding energy of Ag 3d obviously shifts to the lower energy level. The variation of the elemental binding energy directly reflects the change in electron density, where a positive shift in the binding energy of the core element suggests an electron density reduction and vice versa.29 This implies that the electron density around Cd and S atoms is depleted, and the electrons pour into Ag atoms. This result strongly confirms that photoelectrons flow from CdS to Ag@citrate NCs in the C@P1/A1 heterostructure under the PAH-mediated electron tunneling effect.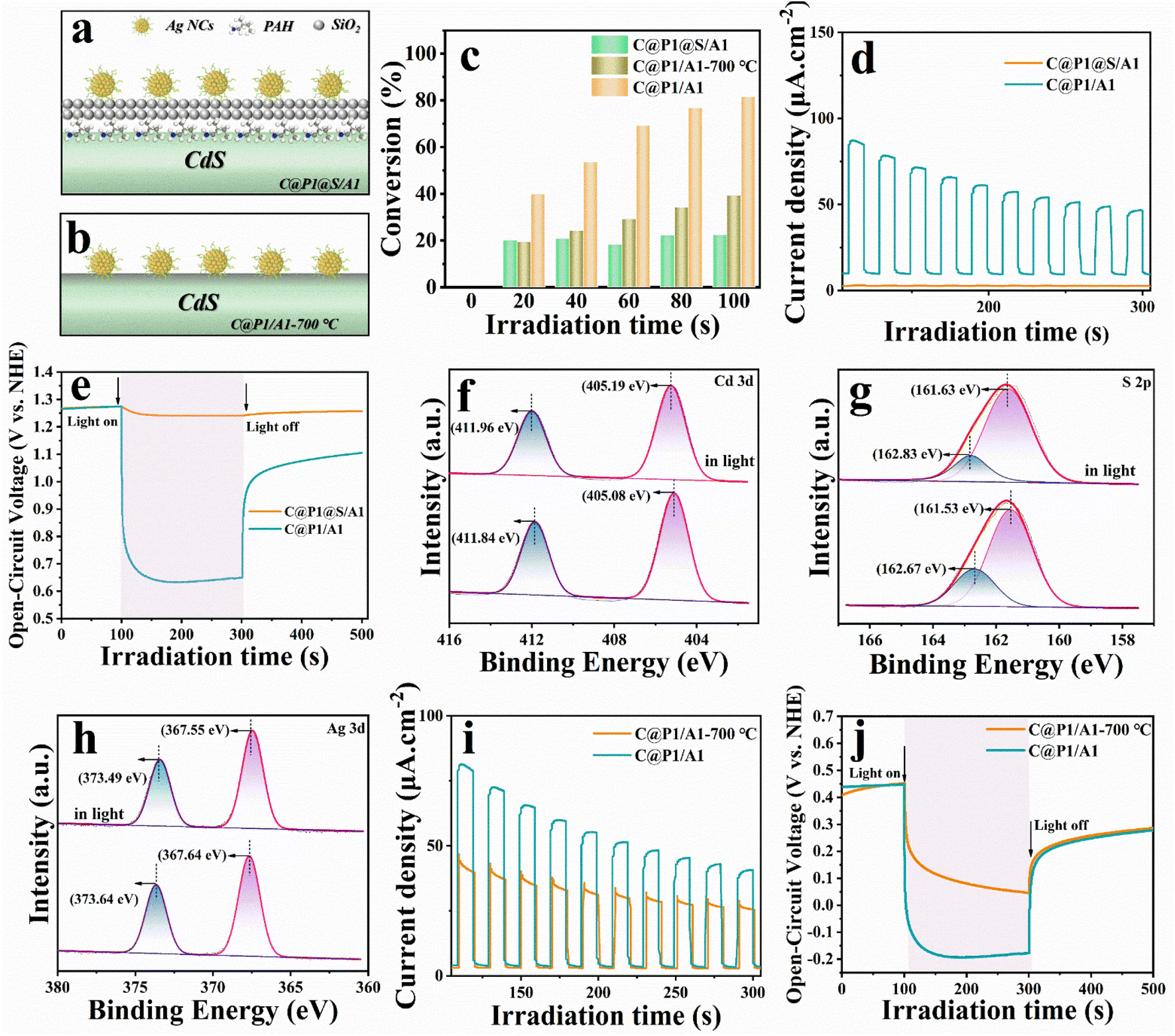 | ||
| Fig. 6 Schematic models of (a) C@P1@S/A1 and (b) C@P1/A1-700 °C; (c) photoactivities of C@P1/A1, C@P1@S/A1, and C@P1/A1-700 °C toward nitrobenzene reduction; (d and i) photocurrents and (e and j) open-circuit potential decay results of C@P1/A1, C@P1/A1-700 °C, and C@P1@S/A1; in situ irradiated high-resolution (f) Cd 3d, (g) S 2p and (h) Ag 3d spectra of C@P1/A1. | ||
Although it is generally believed that high-temperature calcination can facilitate the interfacial integration of building blocks in heterostructure and effectively shorten the interfacial carrier transport distance,30 the reality is that the photoactivity of C@P1/A1-700 °C is far inferior to that of C@P1/A1. This is ascribed to the destruction of the molecular structure of PAH, rendering the electron tunneling effect disappear. Undoubtedly, introducing an ultra-thin PAH interim layer in the C@P1/A1 heterostructure benefits interfacial electron transport. Consistently, transient photocurrent density and OCVD measurement results further confirm that the carrier separation efficiency of C@P1/A1 is much higher than that of C@P1/A1-700 °C (Fig. 6i and j). Based on the above analysis, we ascertain that the PAH interim layer can harmoniously modulate the charge migration by triggering the tunneling effect. Analogous results have also been observed in the photoreduction of other nitro-aromatics, including 4-nitrotoluene, 4-nitroanisole, 4-nitroaniline (4-NA), 3-nitroaniline (3-NA), 2-nitrophenol (2-NP) and o-nitroacetophenone, over C@P1/A1, C@P1@S/A1, and C@P1/A1-700 °C (Fig. S21 and Table S6†), and the results point to the same conclusion, thereby signifying the universal role of PAH as electron tunneling mediator.
In situ diffuse reflectance infrared Fourier-transform spectroscopy (DRIFTS) was used to reveal the evolution of intermediates during the CO2 photocatalytic reduction process. As shown in Fig. 7(a, c and e), the distinct peak at 1510 cm−1 could be attributed to monodentate carbonate (m-CO32−) groups,31 while the band at 1340 cm−1 corresponds to the asymmetric O–C–O stretches of b-CO32− groups,32 which originated from the interaction of H2O and CO2. The peak at 1224 cm−1 is assigned to HCO3−.33 The appearance of carbonate species suggested that CO2 could be efficiently absorbed and activated on the surface of catalysts.34 Importantly, a new peak at ca. 1547 cm−1 arises and is gradually strengthened when the irradiation time is prolonged, and the peak could be assigned to COOH* species,35 which represents key intermediates during photoreduction of CO2 to CO. The result affirms that CO2 is reduced to CO through the COOH* intermediate under light irradiation in our current work. It is noteworthy that the peak intensity of COOH* is particularly substantial for the DRIFTS spectrum of the C@P1/A1 heterostructure, showing the pivotal role of Ag@citrate NCs to adsorb, activate and convert CO2, which is also consistent with the photocatalytic CO2 reduction performance aforementioned. In addition, strong peaks centered at 1723 cm−1 are attributed to the bending vibration of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, which is derived from the accumulation of CO.36,37Fig. 7(b, d and f) display the adsorption FTIR spectra of CdS, C@P1, and C@P1/A1 under similar experimental conditions. Noteworthily, C@P1 and C@P1/A1 exhibit similar CO2 adsorption capacities, but both of them are lower than those of the CdS substrate. This result also confirms that the introduction of an ultra-thin insulating PAH interim layer does not increase the CO2 adsorption capacity; thus, the efficient evolution of CO over the C@P1/A1 heterostructure can be attributed to the PAH-mediated electron tunneling effect.
O, which is derived from the accumulation of CO.36,37Fig. 7(b, d and f) display the adsorption FTIR spectra of CdS, C@P1, and C@P1/A1 under similar experimental conditions. Noteworthily, C@P1 and C@P1/A1 exhibit similar CO2 adsorption capacities, but both of them are lower than those of the CdS substrate. This result also confirms that the introduction of an ultra-thin insulating PAH interim layer does not increase the CO2 adsorption capacity; thus, the efficient evolution of CO over the C@P1/A1 heterostructure can be attributed to the PAH-mediated electron tunneling effect.
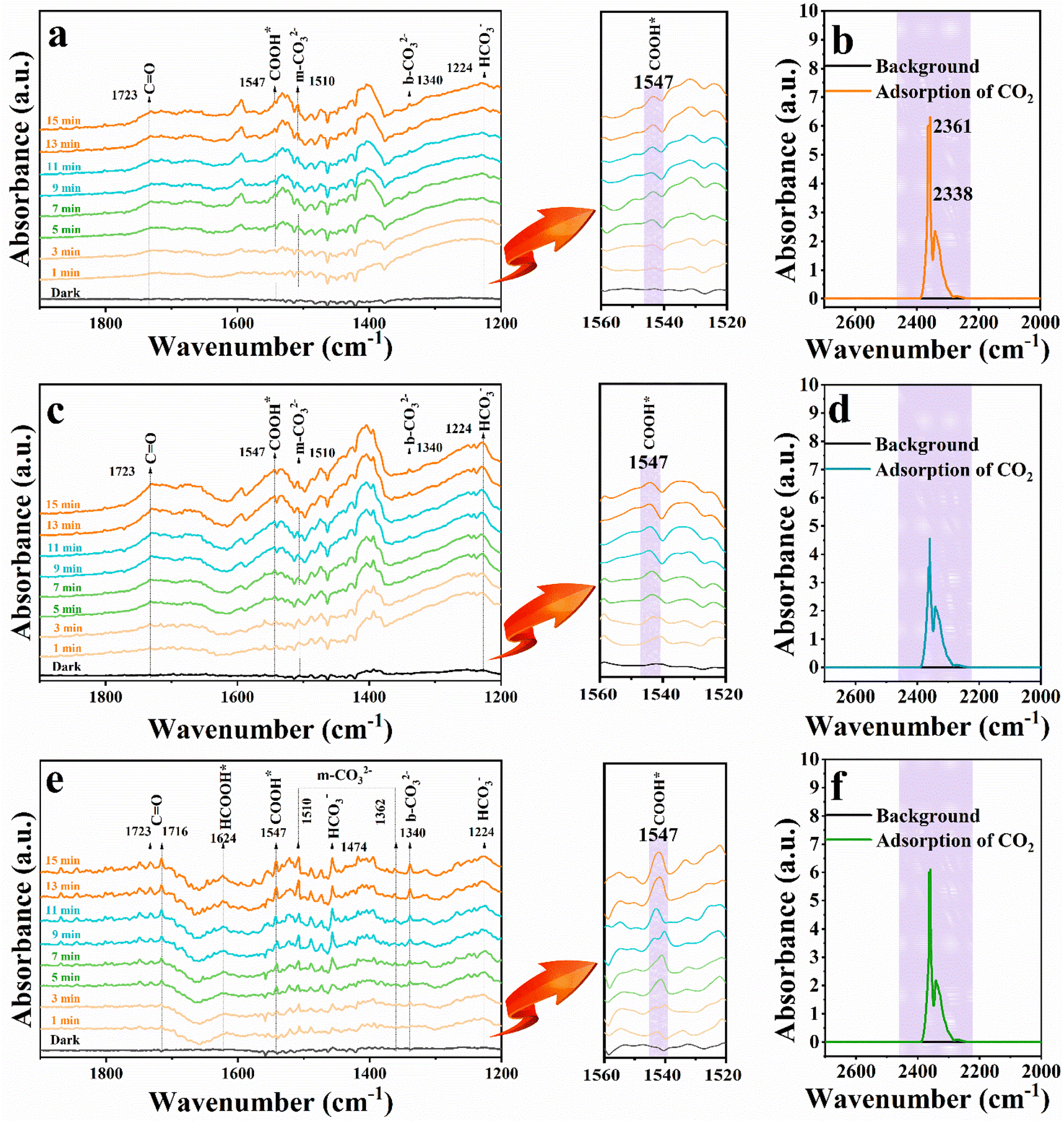 | ||
| Fig. 7 In situ DRIFTS analysis of the photocatalytic CO2 reduction over (a) CdS, (c) C@P1 and (e) C@P1/A1. The sorption equilibrium spectra of (b) CdS, (d) C@P1 and (f) C@P1/A1 after CO2 adsorption in the dark for 5 min. | ||
According to the DRS and M–S results, the conduction band (CB) and valence band (VB) positions of the CdS NC substrate are determined to be −0.7 and 1.69 V vs. NHE, respectively (Fig. S22†). Based on the above analysis, CO2 photoreduction over the C@P/A heterostructure may undergo the following steps (Scheme 2). Upon the irradiation of visible light, CdS is photoexcited to produce electron–hole pairs, with electrons flowing to the CB and leaving holes in the VB. Subsequently, the ultra-thin insulating PAH layer intercalated between CdS and Ag@citrate NCs functions as an efficient electron tunneling modulator to stimulate the electron transport from CdS to Ag@citrate NCs. Ag@citrate NCs are characterized by their high conductivity, which enables the accumulation of more photoelectrons. At the same time, adsorbed 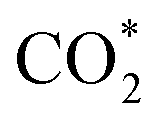 species over Ag@citrate NCs capture the electrons to form the carboxyl intermediate (COOH*) with the assistance of the protons
species over Ag@citrate NCs capture the electrons to form the carboxyl intermediate (COOH*) with the assistance of the protons 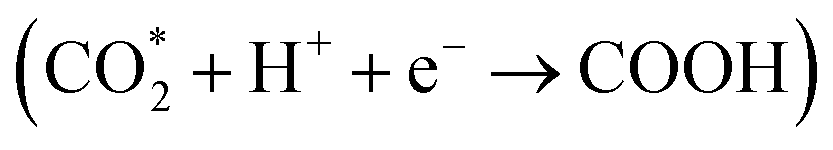 .38 Through a further COOH* protonation process, CO* molecules are yielded progressively (COOH* + H+ + e− → CO* + H2O), which are eventually transformed into a free CO product (CO* → CO). Simultaneously, holes remaining in the VB of CdS are quenched by electron donors (TEOA). In addition, hydrogen ions in the water can inevitably react with photoelectrons to produce H2, ultimately fulfilling the photoreduction of CO2 to syngas.
.38 Through a further COOH* protonation process, CO* molecules are yielded progressively (COOH* + H+ + e− → CO* + H2O), which are eventually transformed into a free CO product (CO* → CO). Simultaneously, holes remaining in the VB of CdS are quenched by electron donors (TEOA). In addition, hydrogen ions in the water can inevitably react with photoelectrons to produce H2, ultimately fulfilling the photoreduction of CO2 to syngas.
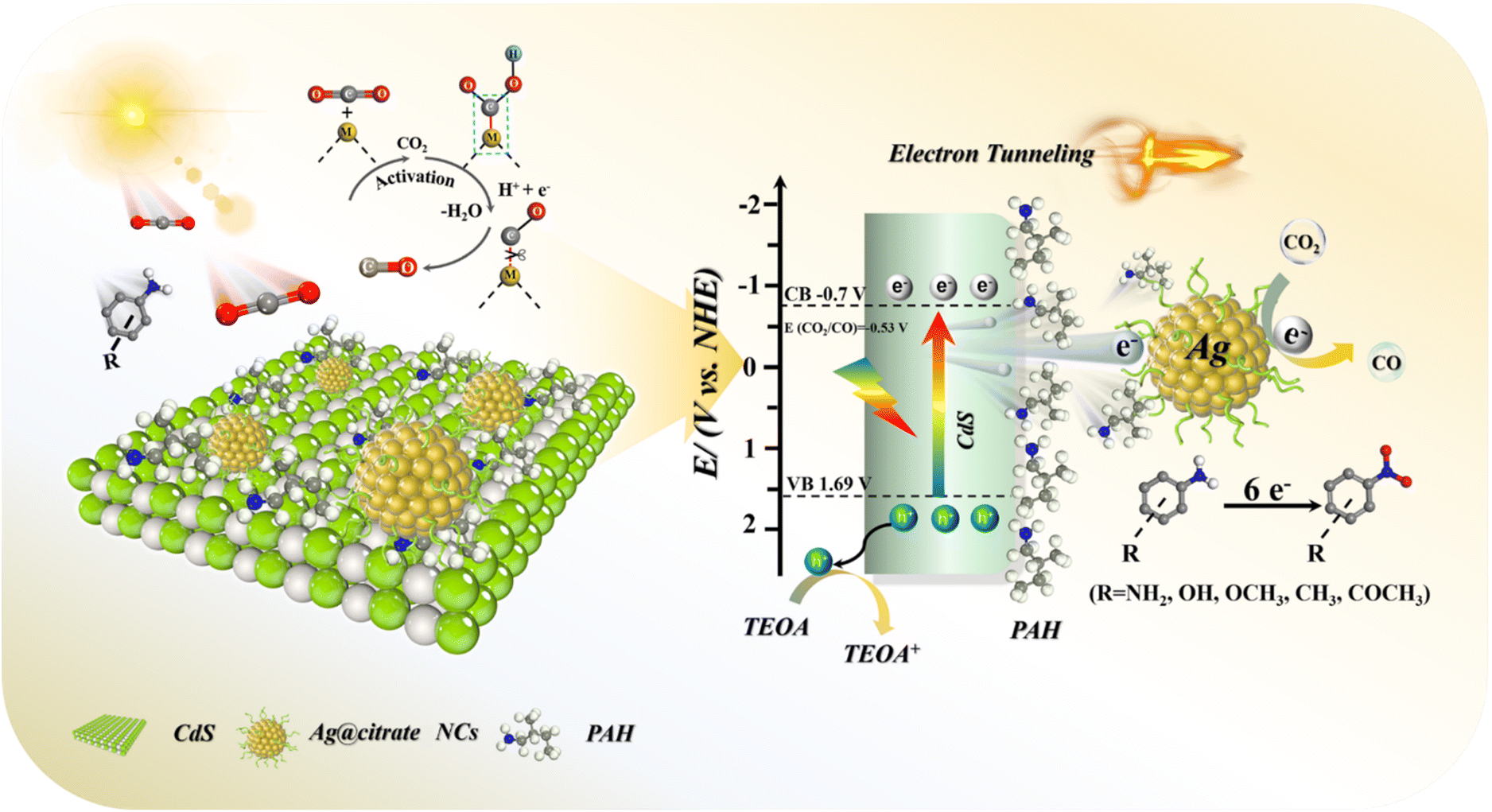 | ||
| Scheme 2 Schematic illustration of the photocatalytic mechanisms of the C@P/A heterostructure (metal site M: Ag). | ||
4. Conclusions
In summary, tandem charge transfer was stimulated over a C@P/A heterostructure, assisted by the electron tunneling effect endowed by an ultra-thin insulating polymer interim layer. In this well-defined heterostructure nano-architecture, the CdS substrate functions as a light-harvesting antenna for generating electron–hole pairs. The ultra-thin insulating PAH interim layer induces electron tunneling, accelerating the migration of photoelectrons from CdS to the terminal electron reservoir of Ag@citrate NCs, forming the electron relay. Benefiting from this synergistic effect, the C@P/A heterostructure demonstrates significantly enhanced photocatalytic CO2-to-syngas conversion performances under visible light irradiation. Our work offers new inspiration for finely modulating the charge transfer via novel non-conjugated insulating polymer-mediated electron-tunneling photosystems towards solar-to-fuel conversion.Data availability
All data supporting the findings of this study are available within the paper and its ESI† files. Source data are provided with this paper.Author contributions
Xian Yan performed the experiments, analyzed all data, and drafted the manuscript. Meng Yuan and Ya-Long Yuan help to check the manuscript. Peng Su and Qing Chen carried out DRIFTS experiments. Fang-Xing Xiao guided this work and corrected the manuscript. All authors contributed to a critical discussion of the data and manuscript.Conflicts of interest
The authors declare no competing interests.Acknowledgements
The support provided by the award program for Minjiang scholar professorship is greatly acknowledged. This work was financially supported by the National Natural Science Foundation of China (No. 21703038 and 22072025). The financial support from the State Key Laboratory of Structural Chemistry, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Science, is acknowledged (No. 20240018).References
- H. Li, Q. Song, S. Wan, C. W. Tung, C. Liu, Y. Pan, G. Luo, H. M. Chen, S. Cao, J. Yu and L. Zhang, Small, 2023, 19, 2301711 CrossRef CAS PubMed.
- L. Liu, Z. Wang, J. Zhang, O. Ruzimuradov, K. Dai and J. Low, Adv. Mater., 2023, 35, 2300643 CrossRef CAS PubMed.
- Z. Liu, J. Ma, M. Hong and R. Sun, ACS Catal., 2023, 13, 2106–2117 CrossRef CAS.
- Z. Zhao, Z. Wang, J. Zhang, C. Shao, K. Dai, K. Fan and C. Liang, Adv. Funct. Mater., 2023, 33, 2214470 CrossRef CAS.
- Z. Liu, Z. Chen, M. Li, J. Li, W. Zhuang, X. Yang, S. Wu and J. Zhang, ACS Catal., 2023, 13, 6630–6640 CrossRef CAS.
- Z.-Q. Wei, S. Hou, X. Lin, S. Xu, X.-C. Dai, Y.-H. Li, J.-Y. Li, F.-X. Xiao and Y.-J. Xu, J. Am. Chem. Soc., 2020, 142, 21899–21912 CrossRef CAS PubMed.
- Z.-Q. Wei, S. Hou, S.-C. Zhu, Y. Xiao, G. Wu and F.-X. Xiao, Adv. Funct. Mater., 2022, 32, 2106338 CrossRef CAS.
- T. Zhu and M. N. Chong, Nano Energy, 2015, 12, 347–373 CrossRef CAS.
- Y. W. Chen, J. D. Prange, S. Duehnen, Y. Park, M. Gunji, C. E. D. Chidsey and P. C. McIntyre, Nat. Mater., 2011, 10, 539–544 CrossRef CAS PubMed.
- S. Li, Q.-L. Mo, S.-C. Zhu, Z.-Q. Wei, B. Tang, B.-J. Liu, H. Liang, Y. Xiao, G. Wu, X.-Z. Ge and F.-X. Xiao, Adv. Funct. Mater., 2022, 32, 2110848 CrossRef CAS.
- B.-J. Liu, H. Liang, Q.-L. Mo, S. Li, B. Tang, S.-C. Zhu and F.-X. Xiao, Chem. Sci., 2022, 13, 497–509 RSC.
- K. Wang, X.-Z. Ge, Q.-L. Mo, X. Yan, Y. Xiao, G. Wu, S.-R. Xu, J.-L. Li, Z.-X. Chen and F.-X. Xiao, J. Catal., 2022, 416, 92–102 CrossRef CAS.
- S. Xu, H.-J. Lin, X. Lin, X.-Y. Fu, S. Hou, Z.-Q. Wei, Q.-L. Mo and F.-X. Xiao, J. Catal., 2021, 399, 150–161 CrossRef CAS.
- Q. Zhang, Q. An, X. Luan, H. Huang, X. Li, Z. Meng, W. Tong, X. Chen, P. K. Chu and Y. Zhang, Nanoscale, 2015, 7, 14002–14009 RSC.
- S. Rengaraj, S. Venkataraj, S. H. Jee, Y. Kim, C.-W. Tai, E. Repo, A. Koistinen, A. Ferancova and M. Sillanpaa, Langmuir, 2011, 27, 352–358 CrossRef CAS PubMed.
- K. Ojha, T. Debnath, P. Maity, M. Makkar, S. Nejati, K. V. Ramanujachary, P. K. Chowdhury, H. N. Ghosh and A. K. Ganguli, J. Phys. Chem. C, 2017, 121, 6581–6588 CrossRef CAS.
- L. Du, Y. Chen, Q. Wang, Y. Zhao, L. Li, X. Liu and G. Tian, Small, 2021, 17, 2100412 CrossRef CAS PubMed.
- H. Liang, B.-J. Liu, B. Tang, S.-C. Zhu, S. Li, X.-Z. Ge, J.-L. Li, J.-R. Zhu and F.-X. Xiao, ACS Catal., 2022, 12, 4216–4226 CrossRef CAS.
- W. Cheng, C. Yang, X. Ding, A. C. Engler, J. L. Hedrick and Y. Y. Yang, Biomacromolecules, 2015, 16, 1967–1977 CrossRef CAS PubMed.
- S. Zhao, Y. Zhang, Y. Zhou, C. Zhang, X. Sheng, J. Fang and M. Zhang, ACS Sustainable Chem. Eng., 2017, 5, 1416–1424 CrossRef CAS.
- B. Lin, H. Li, H. An, W. Hao, J. Wei, Y. Dai, C. Ma and G. Yang, Appl. Catal., B, 2018, 220, 542–552 CrossRef CAS.
- M.-Y. Qi and Y.-J. Xu, Angew. Chem., Int. Ed., 2023, 62, 202311731 CrossRef PubMed.
- X.-H. Li and M. Antonietti, Chem. Soc. Rev., 2013, 42, 6593–6604 RSC.
- Z. Liu, Z. Chen, M. Li, J. Li, W. Zhuang, X. Yang, S. Wu and J. Zhang, ACS Catal., 2023, 13, 6630–6640 CrossRef CAS.
- L. Yuan, M.-Y. Qi, Z.-R. Tang and Y.-J. Xu, Angew. Chem., Int. Ed., 2021, 60, 21150–21172 CrossRef CAS PubMed.
- C. Bie, B. Zhu, F. Xu, L. Zhang and J. Yu, Adv. Mater., 2019, 31, 1902868 CrossRef CAS PubMed.
- A. P. Singh, N. Kodan, B. R. Mehta, A. Held, L. Mayrhofer and M. Moseler, ACS Catal., 2016, 6, 5311–5318 CrossRef CAS.
- X. An, Q. Tang, H. Lan, H. Liu, X. Yu, J. Qu, H. Lin and J. Ye, Angew. Chem., Int. Ed., 2022, 61, 202212706 CrossRef PubMed.
- H. Li, Q. Song, S. Wan, C.-W. Tung, C. Liu, Y. Pan, G. Luo, H. M. Chen, S. Cao, J. Yu and L. Zhang, Small, 2023, 19, 2301711 CrossRef CAS PubMed.
- H.-J. Lin, T. Li, M.-H. Huang, X.-C. Dai, Y.-B. Li and F.-X. Xiao, J. Phys. Chem. C, 2019, 123, 28066–28080 CrossRef CAS.
- X. Jiao, X. Li, X. Jin, Y. Sun, J. Xu, L. Liang, H. Ju, J. Zhu, Y. Pan, W. Yan, Y. Lin and Y. Xie, J. Am. Chem. Soc., 2017, 139, 18044–18051 CrossRef CAS PubMed.
- J. Di, C. Zhu, M. Ji, M. Duan, R. Long, C. Yan, K. Gu, J. Xiong, Y. She, J. Xia, H. Li and Z. Liu, Angew. Chem., Int. Ed., 2018, 57, 14847–14851 CrossRef CAS PubMed.
- H. Jiang, M. Xu, X. Zhao, H. Wang, Q. Liu, Z. Liu, Q. Liu, G. Yang and P. Huo, Inorg. Chem., 2022, 61, 11207–11217 CrossRef CAS PubMed.
- X. Wu, W. Zhang, J. Li, Q. Xiang, Z. Liu and B. Liu, Angew. Chem., Int. Ed., 2023, 62, 202213124 CrossRef PubMed.
- L. Liang, X. Li, J. Zhang, P. Ling, Y. Sun, C. Wang, Q. Zhang, Y. Pan, Q. Xu, J. Zhu, Y. Luo and Y. Xie, Nano Energy, 2020, 69, 104421 CrossRef CAS.
- S. Karmakar, S. Barman, F. A. Rahimi, D. Rambabu, S. Nath and T. K. Maji, Nat. Commun., 2023, 14, 4508 CrossRef CAS PubMed.
- G. Wang, Y. Wu, Z. Li, Z. Lou, Q. Chen, Y. Li, D. Wang and J. Mao, Angew. Chem., Int. Ed., 2023, 62, 202218460 CrossRef PubMed.
- E. Gong, S. Ali, C. B. Hiragond, H. S. Kim, N. S. Powar, D. Kim, H. Kim and S.-I. In, Energy Environ. Sci., 2022, 15, 880–937 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc02313g |
| This journal is © The Royal Society of Chemistry 2024 |