
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceKey-to-lock halogen bond-based tetragonal pyramidal association of iodonium cations with the lacune rims of beta-octamolybdate†
Natalia S.
Soldatova
 a,
Amirbek D.
Radzhabov
a,
Daniil M.
Ivanov
ab,
Sergi
Burguera
c,
Antonio
Frontera
c,
Pavel A.
Abramov
*ad,
Pavel S.
Postnikov
*ae and
Vadim Yu.
Kukushkin
*bf
a,
Amirbek D.
Radzhabov
a,
Daniil M.
Ivanov
ab,
Sergi
Burguera
c,
Antonio
Frontera
c,
Pavel A.
Abramov
*ad,
Pavel S.
Postnikov
*ae and
Vadim Yu.
Kukushkin
*bf
aResearch School of Chemistry and Applied Biomedical Sciences, Tomsk Polytechnic University, Tomsk 634050, Russian Federation. E-mail: postnikov@tpu.ru
bInstitute of Chemistry, Saint Petersburg State University, Universitetskaya Nab. 7/9, Saint Petersburg 199034, Russian Federation. E-mail: v.kukushkin@spbu.ru
cDepartment of Chemistry, Universitat de les Illes Balears, Crta. de Valldemossa km 7.5, Palma de Mallorca (Baleares) 07122, Spain
dNikolaev Institute of Inorganic Chemistry SB RAS, 3 Acad. Lavrentiev Av., Novosibirsk 630090, Russian Federation. E-mail: abramov@niic.nsc.ru
eDepartment of Solid State Engineering, University of Chemical Technology, Prague 16628, Czech Republic
fInstitute of Chemistry and Pharmaceutical Technologies, Altai State University, Barnaul 656049, Russian Federation
First published on 24th June 2024
Abstract
The structure-directing “key-to-lock” interaction of double σ-(IIII)-hole donating iodonium cations with the O-flanked pseudo-lacune rims of [β-Mo8O26]4− gives halogen-bonded iodonium-beta-octamolybate supramolecular associates. In the occurrence of their tetragonal pyramidal motifs, deep and broad σ-(IIII)-holes of a cation recognize the molybdate backbone, which provides an electronic pool localized around the two lacunae. The halogen-bonded I⋯O linkages in the structures were thoroughly studied computationally and classified as two-center, three-center bifurcated, and unconventional “orthogonal” I⋯O halogen bonds. In the latter, the O-atom approaches orthogonally the C–IIII–C plane of an iodonium cation and this geometry diverge from the IUPAC criteria for the identification of the halogen bond.
1. Introduction
The specific molecular recognition represents an important driving force of a great number of processes spanning from supramolecular chemistry to biology and medicinal chemistry.1–8 The recognition parameters between “key” and “lock”, such as geometry, size, type of interactions and their number, are the main points to achieve proper conditions for the supramolecular assembly.9 In terms of bonding between coformers, the recognition pathway could involve a number of energetically differentiated steps. Inside this energetically scaled pool, noncovalent interactions (abbreviated as NCIs; for general reviews on NCIs see ref. 10–17) play a structure-directing role for various supramolecular associations or recognition processes between the “key” and “lock” coformers to accomplish, e.g. host–guest complexes18 or clathrates.19 A special attention, in this context, has been drawn to the utilization of polynuclear transition metal oxocomplexes (polyoxometalates, POM; for reviews on POMs see ref. 20–25) as building blocks for the design of new smart materials for energy conversion, storage and transfer systems.26–32 Despite the wide applicability of POMs, their involvement in “key-to-lock” recognition systems is poorly studied. A rare example, for instance, demonstrates site-specific anchoring of organo-boronic acids to the metal-functionalized Dawson anions [M3P2W15O62]9− (M = Ta, Nb).33In view of our interest in applications of a particular class of POMs, namely polyoxomolybdates (for recent relevant studies see ref. 34–39), we40–44 and other groups45–47 previously employed these species as receptors in the recognition systems. In the chemistry of polyoxomolybdates, the [β-Mo8O26]4− anion is a widely used tecton for crystal engineering and design of new compounds;38,48–53 it contains two lacunae that can function as “locks” for various electrophilic “keys” (Fig. 1A).
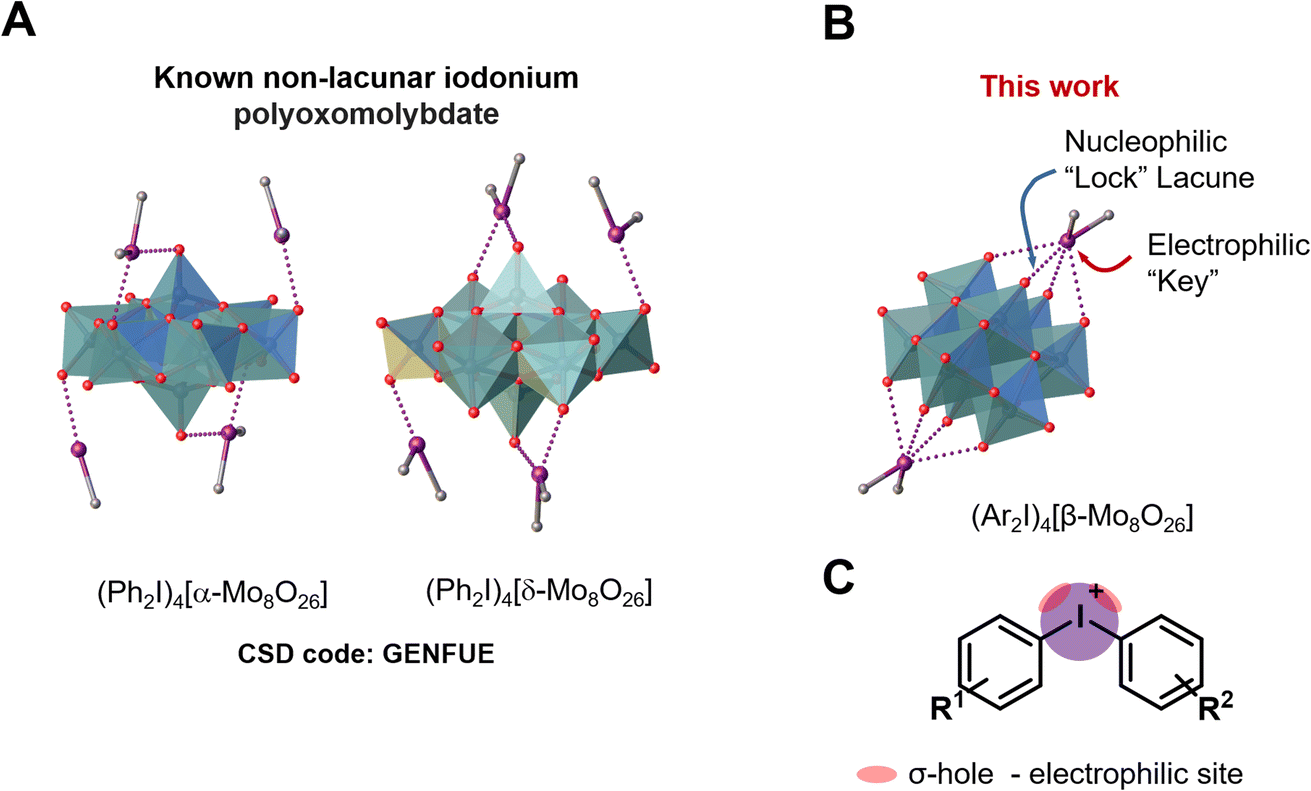 | ||
| Fig. 1 Structure of the iodonium α- and δ-octamolybdate (A); structure of [β-Mo8O26]4− anion (B) and graphical representation of iodonium cations highlighting σ-holes (C). | ||
Particularly, we reported on the employment of beta-octamolybdate anion (Fig. 1) as a ligand or “lock” for Ag+, which function as “key”.40–42 This “key-to-lock” recognition was achieved due to the presence of two pseudo-lacunae, in the structure of the POM anion. These lacunae of the [β-Mo8O26]4− anion also exhibited an ability to interact with hydrogen bond (abbreviated as HB) donors, like O–H, N–H, or even C–H groups – all acting as electrophilic coformers of NCIs.43 The ability of the lacunae to function as HB acceptor in the “key-to-lock” recognition stimulated our further studies focused on the employment of [β-Mo8O26]4− as nucleophilic partner toward such unconventional types of NCIs (sometimes refereed to unorthodox54,55 or exotic56) as halogen bonding (abbreviated as XB). According to with the IUPAC definition of XB,57 “a halogen bond… is a net attractive interaction between an electrophilic region associated with a halogen atom in a molecular entity and a nucleophilic region in another, or the same, molecular entity”. For general reviews on XB see ref. 58–62.
Among various NCIs, XB can be considered as one of the most suitable alternatives to HB for NCI-involving crystal engineering. It is well-documented58,59,63–65 that XB is significantly more directional force than HB and this structure-determining property has been actively used in the targeted construction of XB-based linear or orthogonal assemblies.58 In the vast majority of studies, monovalent halogen-containing organic compounds were implemented as XB donors.66 A relatively new trend in XB-involving crystal engineering and catalysis (explored mostly by Resnati,62,67 Huber,68–71 and some of us, see ref. 72–74 and references therein) is the utilization of hypervalent iodine compounds, particularly iodonium salts,75 where IIII sites function as efficient double σ-hole donors76 (biaxial σ-hole donors in another terminology;68Fig. 1B). Cationic nature of these iodonium species allows the rational design of new supramolecular architectures72–74 based on charge-supported XB that is conventionally stronger than XB, which is associated with coformers featuring a iodine(I) site. Thus, iodonium cations can function as a close-to-perfect “key” toward a lacune of POM which act as a “lock”. Notably, examples of nonlacunar iodonium POMs (e.g., [α- and δ-Mo8O26]4−)77 were reported (Fig. 1B). In contrast to the lacunar [β-Mo8O26]4−, the assemblies do not include any specified site of [α- and δ-Mo8O26]4− that will be bound to iodine(III) center.
In line with the previous studies focused on the [β-Mo8O26]4−-based recognition systems utilizing H⋯OPOM43,78–81 and AgI⋯OPOM40–42 bonding, we now report on the structure-directing “key-to-lock” XB-involving interaction of double σ-hole donating iodonium cations with the lacune rims of [β-Mo8O26]4− to give halogen-bonded supramolecular associates. In the occurrence of these assemblies, deep and broad σ-(IIII)-holes of the cation recognize the molybdate backbone which provides an electronic pool localized around the two lacunae.
2. Results and discussion
2.1. Halogen-bonded POMs
In overwhelming majority of the solid halogen-bonded POMs, monovalent halogen-containing compounds were used as XB donors,44,82–86 while pairing between POMs and iodonium salts, exhibiting double σ-(IIII)-hole site, is very little studied and only two relevant reports appeared in the literature. The first one has been focused on UV induced polymerization, in which the added POMs assisted the generation of phenyl radicals from corresponding iodonium salts.87–90 Although, the formed intermediate was isolated as the solid, it was not studied by single-crystal X-ray diffraction (SCXRD) and, consequently, its structure was not reliably established. In the second study, (Ph2I)4[Mo8O26]·3DMF·H2O was used for the design of iodine-doped molybdenum carbide nanocomposite for an electrochemical hydrogen evolution reaction.77 In the crystal structure of (Ph2I)4[Mo8O26]·3DMF·H2O, α- and δ-isomers of the octamolybdate ion are bound to the iodonium cations via the O-atom of the Mo![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group. However, XB features (even unconventional orthogonal XBs; Section 2.3.4) in the crystal structure organization have not been discussed. Notably, α- and δ-isomers of [Mo8O26]4− do not contain “lock” lacunae to accept the “key” site of R2I+ and it seemed that the use of [β-Mo8O26]4− is a prerequisite for the targeted assembly. Our MEP surface calculation data for α, β, γ, and δ-[Mo8O26]4− isomeric anions, clearly showing the availability of the lacunae in [β-Mo8O26]4− and their absence in α, γ, and δ-isomers, are given in the ESI.†
O group. However, XB features (even unconventional orthogonal XBs; Section 2.3.4) in the crystal structure organization have not been discussed. Notably, α- and δ-isomers of [Mo8O26]4− do not contain “lock” lacunae to accept the “key” site of R2I+ and it seemed that the use of [β-Mo8O26]4− is a prerequisite for the targeted assembly. Our MEP surface calculation data for α, β, γ, and δ-[Mo8O26]4− isomeric anions, clearly showing the availability of the lacunae in [β-Mo8O26]4− and their absence in α, γ, and δ-isomers, are given in the ESI.†
As electrophilic coformers for this study, we addressed diaryliodonium cations shown in Table 1. The corresponding iodonium salts are easy-to-hand stable substrates that can be prepared from elemental iodine, aromatic hydrocarbons, and such practical oxidant system as Oxone®-H2SO4.91 The anion metathesis of these salts with (n-Bu4N)4[β-Mo8O26] provides iodonium beta-octamolybdate associates detailed in the next section.
| Complex | Iodonium cation | Composition |
|---|---|---|
| 1 |

|
(Mes2I)4[Mo8O26]·2.6DMF |
| 2 |
|
(Ph2I)4[Mo8O26]·2DMSO |
| 3 | (Ph2I)4[Mo8O26]·2DMF | |
| 4 |

|
(Xyl2I)2(n-Bu4N)2[Mo8O26]·1.7DMSO |
| 5 | (Xyl2I)2(n-Bu4N)2[Mo8O26]·Et2O |
2.2. Synthesis and structures
Associates 1–5 were obtained as the solid by a conventional40,41,44 approach that includes vapor diffusion of Et2O (1, 3, 5) or i-PrOH (2, 4) into solutions of (n-Bu4N)4[β-Mo8O26] in DMF (1, 3,5) or DMSO (2, 4) (Table 1); for synthetic details see Experimental section. Complexes 1–5 were studied by SCXRD; the phase purity of 1, 2, and 4 was confirmed by powder X-ray diffraction. The SCXRD structures of 3 and 5 are well-refined and suitable for the analysis of NCIs. It is noteworthy, however, that the crystals of 3 and 5 contained phase admixtures; all our attempts to isolate phase pure complexes failed.Association mode of the iodonium cations with [β-Mo8O26]4− depends on the identity of Ar2I+. In the solid 1–3, all n-Bu4N+ cations from the starting material were fully replaced by the iodonium cations, while the association involving Xyl2I+ gave the mixed Xyl2I+/n-Bu4N+ counter-ion system observed in the structures of 4 and 5 (Table 1). The complete replacement of the n-Bu4N+ cations in 4 and 5 was not achieved even with 4 equiv. of (Xyl2I)OTf. Complex 2 contains two independent iodonium POM arrays, in the text referred to 2I and 2II.
Crystal packings of 1–5 are shown in Fig. S8–S13 (Section S5, the ESI†). The contacts between corresponding cations and the anion include C–H⋯OPOM H-bonds (1–5), π–π interactions between iodonium cations (2, 3), and C–H⋯π interactions (1, 2, 4, 5).
2.3. NCIs with the lacune rims
 | ||
| Fig. 2 XB-based linkage patterns of 1–3: (A) 1; (B) 2I; (C) 2II; (D) 3. | ||
 | ||
| Fig. 3 XB-based structural arrays of 4 (A) and 5 (B). | ||
Geometric parameters of NCIs involving the iodine(III) centers are summarized in Table 2. Consideration of chalcogen bond involving DMSO in the structure of 2II is given in the ESI (Section S1†) as these NCIs are out of scope of this study.
| a The following color code is used in table: purple (two-center XB), blue (bifurcated XB; bXB), and green (orthogonal XB). b The normalized contact (Nc) is defined as the ratio between the separation observed in the crystal and the sum of Bondi vdW radii of interacting atoms: Nc = d/ΣvdW; ΣvdW I + O = 3.50 Å. |
|---|

|
In the structures of 1–3, the I⋯O distances between an I-atom and all O-atoms of the lacune rim (namely, O1–O4) are smaller (2.74–3.44 Å) than the sum of Bondi vdW radii (ΣvdW I + O = 3.50 Å; Table 2). The systems of NCIs occurred between any one of the iodonium cations as “key” and the lacune rims as “lock” exhibit tetragonal pyramidal arrangement (Fig. 2). Notably, the IUPAC distance criterion for identification of XB57 is not obeyed for the I⋯O4 separation in the structure of 4 (Nc for I1⋯O4 = 1.01) and for the I⋯O3 and I⋯O4 contacts in 5 (Nc for I1⋯O3 = 1.13 and Nc for I1⋯O4 = 1.17). The structures of 4 and 5 are not tetragonal pyramidal, but can be considered as multi-center XB (three-center for 4 and four-center for 5, Fig. 3).
Although the I⋯O distance fulfill the IUPAC criteria for XB,57 some I⋯O contacts cannot be unambiguously attributed to XB because of significant deviations of some ∠C–I⋯O angles (up to 79°) from linearity (Table 2).
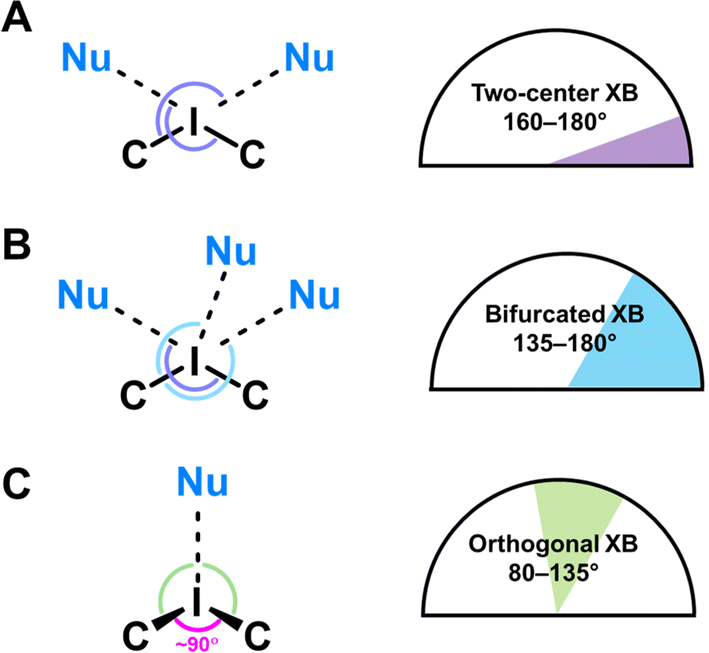 | ||
| Fig. 4 Classification of the NCIs in 1–5. (A) Two-center XB; (B) bifurcated XB; (C) orthogonal XB. | ||
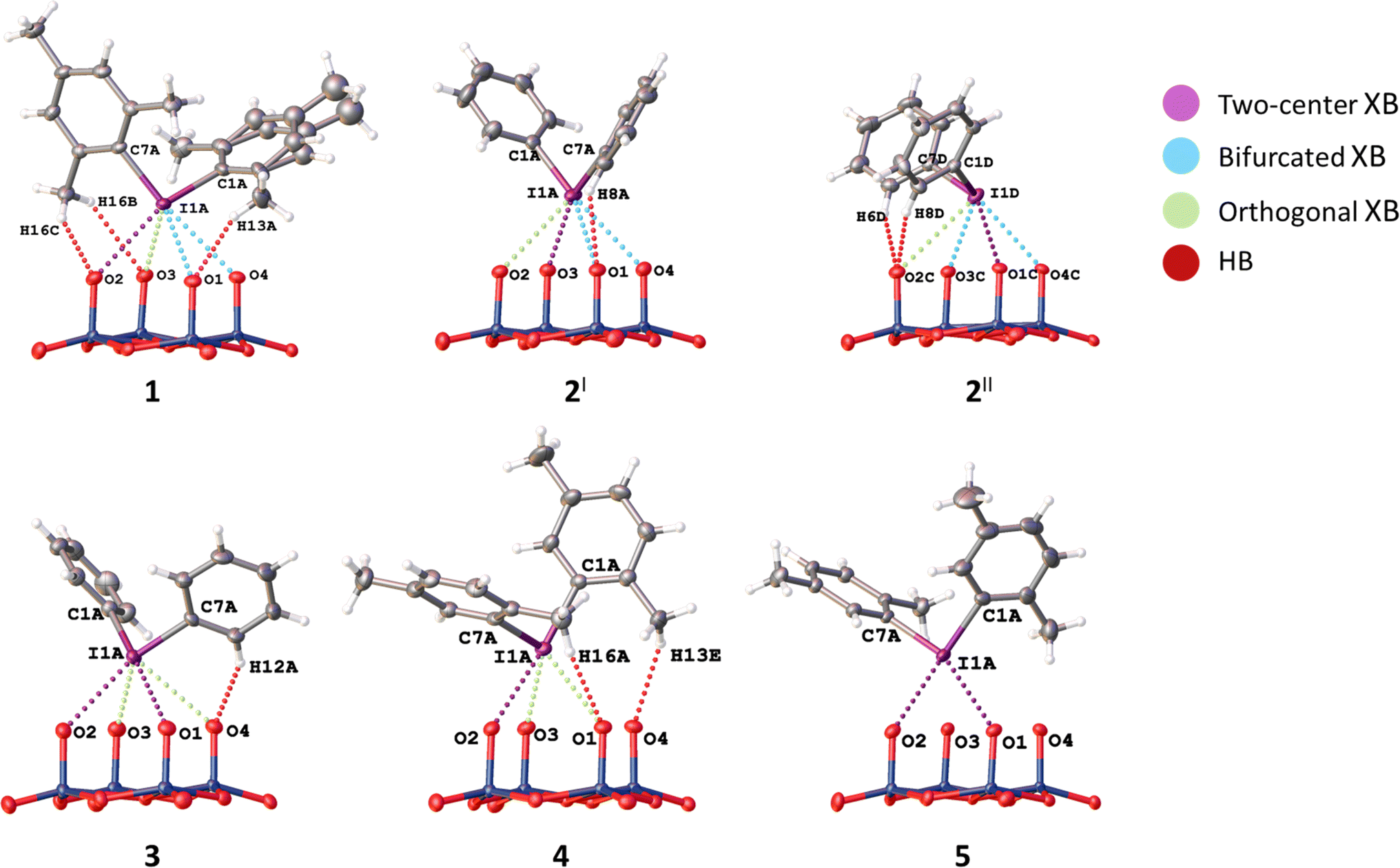 | ||
| Fig. 5 Classified NCIs with lacune rims in 1–5. | ||
To describe these specific interactions, we adopted the term “orthogonal XB”, which is used to facilitate a clearer understanding of these particular NCIs. The orthogonal XBs, as found in this study, do not involve interactions between the O-atoms and the σ*(C–I) orbital, signifying that these are not related to the σ-hole region of the I-atom. Notably, the average Nc value for the orthogonal XBs is 0.94, the highest among all XB types associated with the lacune rim. Based on this classification, in the structures of 1–3, at least one NCI with the lacune rim qualifies as an orthogonal XB (Table 2).
The molecular structure of compound 4 showcases a combination of two-center XB (I1⋯O2) and two orthogonal XBs (I1⋯O3 and I1⋯O2; Fig. 3). Interestingly, the IUPAC criteria for XB distance57 are not fully met in certain interactions within this structure. Thus, the I⋯O4 separation in 4 (Nc 1.01) and the I⋯O3 and I⋯O4 contacts in 5 (Nc 1.13 and 1.17, respectively) do not align with the standard XB identification parameters.57 Furthermore, our analysis of the structure of 5 confirmed the absence of orthogonal XB.
In certain instances, orthogonal XBs are observed to synergistically93 coexist with hydrogen bonding94 involving the O-atom of the POM and the ortho-H or ortho-CH3 groups of the iodonium cation (Fig. 5). An extensive review of the CSD data, conforming to our classification criteria for the orthogonal XBs (d(I⋯O) < ΣvdW, ∠C–I⋯X 70–130°), revealed over 100 iodonium structures featuring the orthogonal XBs. Remarkably, only about a third of these structures exhibit a synergistic combination of orthogonal XB and HBs; this synergistic occurrence typically results in ∠C–I⋯X approaching 90°.
2.4. NCIs with out of the lacune rim oxo-ligands
In the structures of 1–3, iodonium cations also interact with out of the lacune rim oxoligands (Fig. 2), while 4 and 5 do not display such interactions due to incomplete anion metathesis (Section 2.3.). For the structures of 1 and 2I XB involves the μ2-O ligands, while for 2II and 3 the terminal MoO functionality functions as XB acceptor. All these I⋯O distances are smaller than Bondi ΣvdW (3.50 Å; Nc = 0.76–0.92). Contacts with the bridging μ2-O ligands are rather short (Nc 0.78 and 0.81) in comparison with XBs occurred between the IIII site and the MoO group (Nc 0.82 and 0.84). In addition, the structure of 1 exhibits NCI with the O-atom of the Mo–O–Mo linkage. The latter is characterized by a rather small Nc value (0.92) and low linearity (∠C7B–I1B⋯O11 = 134.39(10)°); this deviation is apparently caused by steric effects of iodonium cations and the anion. In 2–3, the iodonium cations form XB with O-atoms out of the lacune rim and also O-atoms of the crystallization solvent (DMF or DMSO) and such I⋯O XBs are almost linear ∠C–I1⋯O = 161–171° (Table 3). Notably, all XBs with O-atoms of the solvent are shorter that XB with any oxo-ligands (Table 3); a similar situation was observed in the structures of the previously reported solvates of a iodonium cation with DMSO.95,96
| Iodonium POM motif | XB | d(C–I⋯O), Å/Nca | ∠(C–I1⋯O), ° |
|---|---|---|---|
| a The normalized contact (Nc) is defined as the ratio between the separation observed in the crystal and the sum of Bondi vdW radii of interacting atoms: Nc = d/ΣvdW; ΣvdW I + O = 3.50 Å | |||
| 1 | C1B–I1B⋯O6 | 2.733(2)/0.78 | 174.14(9) |
| C7B–I1B⋯O11 | 3.237(3)/0.92 | 134.39(10) | |
| 2 I | C1B–I1B⋯O8 | 2.838(3)/0.81 | 162.57(14) |
| C7B–I1B⋯O1S | 2.745(3)/0.78 | 161.80(13) | |
| 2 II | C1E–I1E⋯O5C | 2.855(3)/0.82 | 169.87(14) |
| C7E–I1E⋯O2S | 2.646(5)/0.76 | 163.25(16) | |
| C7E–I1E⋯O3S | 2.783(15)/0.80 | 171.2(4) | |
| 3 | C1B–I1B⋯O5 | 2.956(4)/0.84 | 164.51(17) |
| C7B–I1B⋯O1S | 2.830(5)/0.81 | 161.2(2) | |
2.5. DFT calculations
First, MEP surfaces of the cations and the anion were performed to rationalize the assemblies described above and the existence of multi-center XBs. The MEP surfaces of the cations are given in Fig. 6, showing the expected pair of σ-holes on each I-atom (marked with asterisks in Fig. 6) opposite to the I–C bonds. Their relative intensity behaves as expected considering the number of electron donor methyl groups, being the most positive ones those of the Ph2I+ moiety without additional Me-groups (Fig. 6b). Since the iodoniums are the cationic species, the MEP values are positive all over the van der Waals surface. However, an anisotropy of the electron density is observed at the I-atom with two regions where the MEP is less positive (local minima) above and below the C2I plane (at the expected location of iodine's LPs). | ||
| Fig. 6 MEP surfaces (isodensity 0.001 a.u.) of Mes2I+ (a), Ph2I+ (b), and Xyl2I+ (c). The MEP maximum, minimum values are given in kcal mol−1. Moreover, the MEP values at the local minimum located over the I-atom are also indicated (Vs,LP(I)) in kcal mol−1. | ||
The MEP surface of the anion is represented in Fig. 7, where the MEP values are very large and negative all over the van der Waals surface due to the tetra-anionic character of the polyoxometalate. Notably, it can be observed that the existence of two symmetrically equivalent MEP minima located at the lacune rim that is under the influence of four O-atoms (only one minimum is shown in Fig. 7). The maxima are located at two opposite vertices. This explains the occurrence of the tetragonal pyramidal arrangement, since the positive I-atom of the cation will tend to interact with the most negative part of the anion, under the effect of four O-atoms. Once two cations occupy the two minima, other regions with less negative MEP values are available, which are under the influence of three monocoordinated O-atoms, also in line with the occurrence of four-center XBs.
 | ||
| Fig. 7 MEP surface (isodensity 0.001 a.u.) of β-[Mo8O26]4−. The MEP maximum, minimum values are given in kcal mol−1. | ||
We analyzed neutral pentameric assemblies (the anion surrounded by four cations) of all compounds reported herein using a combination of QTAIM and NCIplot computational tools to study the contacts established between the cations and the anions. The assemblies of 1 and 3 are commented below (Fig. 8 and 9, respectively) and the rest are provided in the ESI (Fig. S4 and S5, Section S2 of the ESI†).
 | ||
| Fig. 8 Combined QTAIM (BCPs in red and bond path as solid orange lines) and NCIplot analysis (RDG = 0.5, ρcut-off = 0.04, color range −0.04 a.u. ≤ (signλ2)*ρ ≤ 0.04 a.u. of the pentameric assembly of 1. The interaction energy is also indicated. The total contributions of the CH⋯O and I⋯O derived from the Vb values are indicated. Moreover, the individual contributions of the XBs are indicated in blue adjacent to the BCPs. Only intermolecular interactions are represented. | ||
 | ||
| Fig. 9 Combined QTAIM (BCPs in red and bond path as solid orange lines) and NCIplot analysis (RDG = 0.5, ρcut-off = 0.04, color range −0.04 a.u. ≤ (signλ2)*ρ ≤ 0.04 a.u. of the pentameric assembly of 3. The interaction energy is also indicated. The total contributions of the CH⋯O and I⋯O derived from the Vb values are indicated. Moreover, the individual contributions of the XBs are indicated in blue adjacent to the BCPs. Only intermolecular interactions are represented. | ||
For the pentameric assembly of 1, the QTAIM/NCIplot analysis evidences the existence of a multitude CH⋯O contacts formed due to the electron-donating effect of the methyl substituents. Each contact is characterized by a bond critical point (BCP) and bond path connecting the H and O-atoms. The XBs are highlighted at the bottom of the Fig. 8 where the ancillary CH⋯O were omitted for the sake of clarity. It can be observed that for one pair of cations, the I-atom is linked to four O-atoms of the anion, establishing a tetragonal pyramidal assembly. This agrees well with the MEP analysis, since this iodine is located at the MEP minimum of the anion, thus maximizing the electrostatic attraction. Each XB is characterized by a BCP, bond path and green reduced density gradient (RDG) isosurface, thus disclosing the attractive nature of these XB contacts. For the other pair of cations, the I-atom is connected to three O-atoms of the anion by three BCPs and bond paths. By examining the color of the RDG isosurfaces, it can be deduced that in this case one of the XBs is stronger (blue isosurface) than the other two (green isosurface), also in agreement with the geometric features of the XBs (Section 2.3.).
The formation energy of this pentameric assembly is very large (−836.7 kcal mol−1) due to the pure coulombic attraction between a tetra-anionic species and four surrounding cations. In this case, it is more convenient to discuss the energetic features of the assemblies using the interaction energies derived from the QTAIM analysis (using the Vb values), since they are free from the influence of pure electrostatic effects. The energetic contribution of all CH⋯O contacts observed in the pentamer is indicated in Fig. 8 (see also Table 4). This contribution is quite significant (−16.1 kcal mol−1) due to the presence of numerous CH⋯O contacts. The contribution of the XBs is also indicated in Fig. 8 and it is much larger (−53.4 kcal mol−1), evidencing that it is the dominant interaction. The individual energies of the XBs are indicated in blue close to the BCPs that characterize the XBs. It is interesting to emphasize that for the XB in the tetragonal pyramidal arrangement, all energies are similar (ranging −2.5 to −3.6 kcal mol−1), thus suggesting that the location of the I-atom in this cation is likely dominated by the nondirectional electrostatic attraction. However, in the other binding mode (four-center XBs), one XB is very strong and directional (−9.1 kcal mol−1) and the other two are much weaker ancillary XBs. Hence, in this case the directional XB dominates over the effect of the simple electrostatic attraction.
| Compound | E | E HB | E XB |
|---|---|---|---|
| 1 | −836.7 | −16.1 | −52.2 |
| 2 | −816.3 | −6.7 | −38.6 |
| 3 | −825.0 | −8.4 | −46.0 |
| 4 | −837.6 | −32.3 (−5.6) | −29.4 |
| 5 | −822.2 | −26.7 (−5.0) | −31.6 |
A parallel analysis for 3 is provided in Fig. 9, where the main difference with 1 is the reduced number of CH⋯O contacts due to the absence of methyl groups. In case of the XBs, the main difference with 1 is that in one pair of symmetrically equivalent cations, the I-atoms form three-center XBs instead of four-center XB. Therefore, the total contribution of the I⋯O contacts is smaller in 3. For the tetragonal pyramidal arrangement that is highlighted in the bottom part of Fig. 9 there are two XBs that are clearly stronger than the other two. This is not observed in 1 and a likely explanation is that in 3 a better adjustment of the geometry of the cation on the surface of the anion is possible due to the absence of the CH3⋯O contacts, facilitating the occurrence of more directional and shorter XBs.
The energetic and QTAIM/NCIplot analyses of 2, 4, and 5 are given in the ESI,† disclosing similar results. Moreover, the energetic results for all compounds are summarized in Table 4, evidencing that in 1–3 the XB is more relevant than the HB. In the cases of 4 and 5, since two cations of the pentameric assemblies are tetrabutylammonium cations instead of iodonium cations, the contribution of CH⋯O interactions significantly increases and that of the I⋯O XBs decreases. For 4 and 5, the energies given in parenthesis in the EHB column correspond to the CH⋯O contribution of the Xyl2I+ cations. It can be observed that it is smaller than half of EHB of 1 but larger than half of EHB in 2 and 3, in line with the absence of methyl groups in the latter two compounds.
The natural bond orbital (NBO) analysis of the assemblies under consideration was performed to verify the orbitals involved in the donor–acceptor interactions and to confirm the σ-hole nature of the XB contacts by the implication of the antibonding σ*(C–I) orbital. The results for 1 and 3 are shown in Fig. 10 and 11 and those of 2, 4, and 5 in the ESI (Fig. S6 and S7†). In the case of 1, for the tetragonal pyramidal binding mode (Fig. 10a and b), we observed the electron donation from LPs located at three O-atoms of the anion to the antibonding σ*(C–I) orbitals of the cation. The I⋯O contact of the tetragonal pyramidal binding mode that does not exhibit orbital mediated charge transfer corresponds to the longest I⋯O contact, where the overlap of the orbitals is likely too small (smaller than the threshold used by the program). In any case, the fact that, for three out of four contacts, the LP(O) → σ*(C–I) charge transfer is observed strongly suggests the σ-hole nature of the interaction. In the case of the four-center interaction (Fig. 10c), the NBO discloses the existence of a single LP(O) → σ*(C–I) charge transfer that corresponds to the strongest XB, in line with the QTAIM/NCIplot analysis data.
 | ||
| Fig. 10 NBOs involved in the XBs observed in 1 for the tetragonal pyramidal (a and b) and four-center (c) binding modes. The second order perturbation analysis energies are also given, E(2). | ||
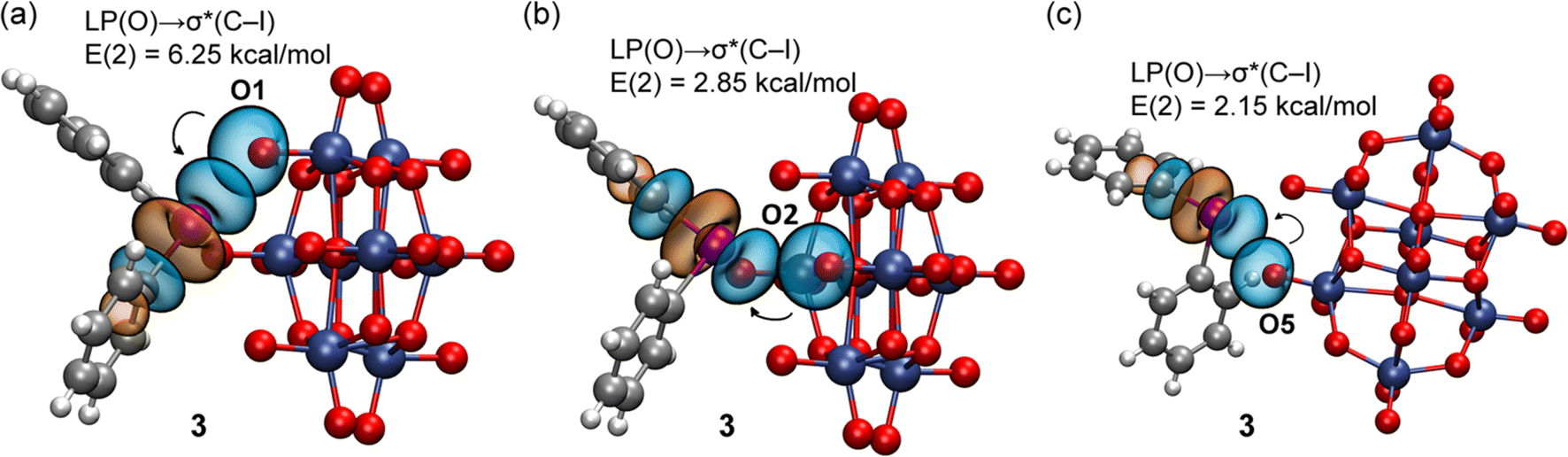 | ||
| Fig. 11 NBOs involved in the XBs observed in 3 for the tetragonal pyramidal (a and b) and three-center (c) binding modes. The second order perturbation analysis energies are also given, E(2). | ||
The NBOs corresponding to the XBs in 3 are given in Fig. 11. As also observed in 1, for the tetragonal pyramidal assembly, three LP(O) → σ*(C–I) donor–acceptor interactions are observed and for the three-center XB only one, corresponding to the strongest and more directional XB. No other orbital donor–acceptor interactions were identified, thus confirming that only the antibonding σ*(C–I) orbital of the cation is involved in the binding mechanism. The results for 2, 4, and 5 (the ESI†) demonstrate a similar behavior.
3. Conclusions
The results of this study can be considered from at least two main perspectives. Firstly, we observed that iodonium cations interact with the lacune rim of the beta-octamolybate anion, [β-Mo8O26]4−, to form halogen-bonded assemblies, exhibiting the tetragonal pyramidal motif. This association belongs to the “key-to-lock” category (as it was defined by Lehn9) when the IIII-site recognizes O-atoms of the rim, namely deep and broad σ-(IIII)-holes of the cation interact with the molybdate backbone which provides an electronic pool localized around two pseudo-cavities. The iodonium cation acts as a “key”, and the anion acts as a “lock”: these partners form “well-defined noncovalent interaction, geometrical and interactional complementarity and energy gain”.9 It is noteworthy that α- and δ-[Mo8O26]4− do not exhibit “lock” lacunae to accept the “key” R2I+ and the use of the lacunar cluster [β-Mo8O26]4− is a prerequisite for the XB-based assembly.In a broader sense, we found a novel type of supramolecular organization of POMs, which is based on XB. Consideration of the previous data on hydrogen-bond43,78–81 and silver(I)40–42 recognitions of the O-flanked lacune of beta-octamolybate and also this study focused on XB-involving assembly – all together – give a hint that similar recognition for the targeted supramolecular assembly could also be achieved using other powerful σ-hole donors. Furthermore, this “key-to-lock” binding could be applied to various lacunary POMs such as, for instance, [H3PW11O39]4− (ref. 97) or [γ-SiW10O34(H2O)2]4− (ref. 98–101) and also other double-σ-hole donors such as, for instance, the chalcogen bond donating compounds R2EWGCh (Ch = Se, Te).102
Secondly, examination of the occurrence of the XB-based tetragonal pyramidal moiety and analysis of appropriate NCIs provides an illustration of how different types of XB (namely, the two-center, bifurcated, and orthogonal XB; Fig. 4) could complement each other in the recognition system. The identification of the unusual orthogonal XB (which does not correspond to the IUPAC criteria for the recognition of XB) could be of interest for the crystal engineering community as this force might provide novel opportunities for targeted halogen-bond driven supramolecular assembly.
Finally, iodonium salts of α-[SiMo12O40]4− and [W10O32]4− are applied as efficient initiators of radical photopolymerization of acrylates, cationic photopolymerization of epoxides, and photopolymerization of epoxy/acrylate blends.88,89,103 These POMs are also used as POM-containing photocomposites exhibiting high catalytic activity toward photodecomposition (and decoloration) of dyes in aqueous solutions.103 The achieved results of this study should be useful for the rational design of the iodonium-modified POM catalysts and final POM-based photocomposites. Works in this direction are underway in our group.
4. Experimental section
4.1. General information
(n-Bu4N)4[β-Mo8O26]104 and the iodonium salts91 were prepared according to the literature data. Other reagents were of commercial quality (Sigma-Aldrich) and were used without additional purification. Elemental analyses were carried out on a MICRO Cube CHN analyzer. IR spectra were recorded on a Bruker Vertex 60 FT-IR spectrometer (Section S8, the ESI†). Electrospray ionization (ESI) mass spectra were obtained on a Bruker maXis spectrometer equipped with an ESI source. The instrument was operated in positive ion mode using an m/z range 50–2600 (Section S9, the ESI†). The nebulizer gas flow was 0.4 bar, and the drying gas flow was 4.0 L min−1.4.2. Synthetic work and crystal growth
For details see Section S6, the ESI.†4.3. X-ray single-crystal diffraction studies
Crystallographic data and refinement details are given in Table S2.† The diffraction data for 1, 3, and 4 were collected on a New Xcalibur (Agilent Technologies) diffractometer with MoKα radiation (λ = 0.71073) by doing φ scans of 0.5° frames at 150 K. Absorption correction was done empirically using SCALE3 ABSPACK (CrysAlisPro, Agilent Technologies, Version 1.171.37.35 (release 13-08-2014 CrysAlis171.NET)). The diffraction data for 2 and 5 were collected on a Bruker D8 Venture diffractometer with a CMOS PHOTON III detector and IμS 3.0 source (Mo Kα radiation, λ = 0.71073 Å) at 150 K. The φ- and ω-scan techniques were employed. Absorption correction was applied by SADABS (Bruker Apex3 software suite: Apex3, SADABS-2016/2 and SAINT, version 2018.7-2; Bruker AXS Inc.: Madison, WI, 2017). The structures were solved by SHELXT105 and refined by full-matrix least-squares treatment against |F|2 in anisotropic approximation with SHELX 2014/7 (ref. 106) in ShelXle program.107 H-atoms were refined in geometrically calculated positions. In the crystal structure of 5, DMSO molecules of crystallization demonstrate complicated orientation disorder with low occupancies. These molecules were treated by SQUEEZE procedure108 of PLATON program.109 This gives 74e per void which can be assigned as 1.7 DMSO per formula unit.4.4. X-ray powder diffraction
X-ray powder diffraction patterns were measured on a Bruker D8 Advance diffractometer using LynxEye XE T discriminated CuKα radiation. Samples were layered on a flat plastic specimen holder. For details see Section S7, the ESI.†4.5. Theoretical methods
The calculations reported herein were performed using the Turbomole 7.2 program.110 The level of theory used for the calculations was PBE0 (ref. 111)-D3 (ref. 112)/def2-TZVP.113,114 For iodine, this basis set includes effective core potentials (ECP) and takes into consideration relativistic effects for the inner electrons.114 The MEP surface plots of the anions and cations were generated using the 0.001 a.u. isosurface and the same level of theory. The topological analysis of the electron density was carried out according to the quantum theory of atoms in molecules (QTAIM) and noncovalent interaction plot index (NCIplot) methods proposed by Bader115 and W. Yang et al.,116 respectively. For clarity, only intermolecular critical points were represented in the QTAIM analyses. Both were represented using the VMD program.117 The individual XB energies were evaluated using the equations proposed by Bartashevich and Tsirelson118 for XB and Espinosa et al.119 for HB. The NBO analysis120 was performed using the same level of theory and the NBO7.0 program.121Data availability
Supplementary crystallographic data for this paper have been deposited at Cambridge Crystallographic Data Centre (CCDC 2334360–2334364) and can be obtained viahttps://www.ccdc.cam.ac.uk/data_request/cif. Other data available within the article or its ESI† and can be provided in raw format upon request.Author contributions
Natalia S. Soldatova – methodology, investigation, funding acquisition, writing – original draft; Amirbek D. Radzhabov – investigation, formal analysis; Daniil M. Ivanov – conceptualization, methodology, data curation; Sergi Burguera – investigation, formal analysis; Antonio Frontera – methodology, resources, writing – original draft; Pavel A. Abramov – conceptualization, methodology, writing – original draft; Pavel S. Postnikov – conceptualization, methodology, supervision, writing – review & editing; Vadim Yu. Kukushkin – conceptualization, supervision, project administration, writing – review & editing.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the Russian Science Foundation, project 23-73-10091. P. A. A. thanks the Ministry of Science and Higher Education of the Russian Federation for the access SCXRD facilities of NIIC SB RAS. Authors are grateful to the Center for Chemical Analysis and Materials Research (Saint Petersburg State University) for the physicochemical measurements.References
- E. Persch, O. Dumele and F. Diederich, Angew. Chem., Int. Ed., 2015, 54, 3290–3327 CrossRef CAS PubMed
.
- X. Wu, A. M. Gilchrist and P. A. Gale, Chem, 2020, 6, 1296–1309 CAS
- L. Escobar and P. Ballester, Chem. Rev., 2021, 121, 2445–2514 CrossRef CAS PubMed
- J. Pancholi and P. D. Beer, Coord. Chem. Rev., 2020, 416, 213281 CrossRef CAS
- N. Biot and D. Bonifazi, Coord. Chem. Rev., 2020, 413, 213243 CrossRef CAS
- X. Zhang, Z. An, J. An and X. Tian, Coord. Chem. Rev., 2024, 502, 215601 CrossRef CAS
- Y. Fan, J. He, S. Guo and H. Jiang, ChemPlusChem, 2024, e202300536 CrossRef PubMed
- B. B. Patnaik, S. Baliarsingh, A. Sarkar, A. S. S. Hameed, Y. S. Lee, Y. H. Jo, Y. S. Han and J. Mohanty, Rev. Aquac., 2024, 16, 190–233 CrossRef
-
Perspectives in Supramolecular Chemistry, ed. J. Behr, Wiley, 1994, vol. 1 Search PubMed
- I. Alkorta, J. Elguero and A. Frontera, Crystals, 2020, 10, 180 CrossRef CAS
- S. Scheiner, J. Chem. Phys., 2020, 153, 140901 CrossRef PubMed
- P. Molina, F. Zapata and A. Caballero, Chem. Rev., 2017, 117, 9907–9972 CrossRef CAS PubMed
- A. S. Mahadevi and G. N. Sastry, Chem. Rev., 2016, 116, 2775–2825 CrossRef CAS PubMed
- L. Brammer, A. Peuronen and T. M. Roseveare, Acta Crystallogr., Sect. C: Struct. Chem., 2023, 79, 204–216 CrossRef CAS PubMed
- Y. Cornaton and J.-P. Djukic, Acc. Chem. Res., 2021, 54, 3828–3840 CrossRef CAS PubMed
- G. Resnati and P. Metrangolo, Coord. Chem. Rev., 2020, 420, 213409 CrossRef CAS
- M. Savastano, Dalton Trans., 2024, 53, 1373–1392 RSC
- H. Schneider, J. Phys. Org. Chem., 2022, 35, e4340 CrossRef CAS
- N. N. Nguyen, R. Berger, M. Wagner, J. Thiel, H.-J. Butt and R. Graf, J. Phys. Chem. C, 2021, 125, 15751–15757 CrossRef CAS
- M. I. S. Veríssimo, D. V. Evtuguin and M. T. S. R. Gomes, Front. Chem., 2022, 10, 840657 CrossRef PubMed
- N. I. Gumerova and A. Rompel, Nat. Rev. Chem., 2018, 2, 0112 CrossRef CAS
- B. D'Cruz, M. O. Amin and E. Al-Hetlani, Ind. Eng. Chem. Res., 2021, 60, 10960–10977 CrossRef
- N. I. Gumerova and A. Rompel, Chem. Soc. Rev., 2020, 49, 7568–7601 RSC
- Y. Gao, M. Choudhari, G. K. Such and C. Ritchie, Chem. Sci., 2022, 13, 2510–2527 RSC
- B. Yu, X. Zhao, J. Ni and F. Yang, ChemPhysMater, 2023, 2, 20–29 CrossRef
- Y. Zhang, J. Liu, S.-L. Li, Z.-M. Su and Y.-Q. Lan, EnergyChem, 2019, 1, 100021 CrossRef
- M. R. Horn, A. Singh, S. Alomari, S. Goberna-Ferrón, R. Benages-Vilau, N. Chodankar, N. Motta, K. (Ken) Ostrikov, J. MacLeod, P. Sonar, P. Gomez-Romero and D. Dubal, Energy Environ.
Sci., 2021, 14, 1652–1700 RSC
- F. M. B. Gusmão, D. Mladenović, K. Radinović, D. M. F. Santos and B. Šljukić, Energies, 2022, 15, 9021 CrossRef
- H. Hu, L. Lian, X. Ji, W.-L. Zhao, H. Li, W. Chen, H. N. Miras and Y.-F. Song, Coord. Chem. Rev., 2024, 503, 215640 CrossRef CAS
- T. Ma, R. Yan, X. Wu, M. Wang, B. Yin, S. Li, C. Cheng and A. Thomas, Adv. Mater., 2024, 36, 2310283 CrossRef CAS PubMed
- L. Li, Y.-T. Yu, N.-N. Zhang, S.-H. Li, J.-G. Zeng, Y. Hua and H. Zhang, Coord. Chem. Rev., 2024, 500, 215526 CrossRef CAS
- C. Wang, B. Wang, H. Yang, Y. Wan, H. Fang, W. Bao, W. Wang, N. Wang and Y. Lu, Chem. Eng. J., 2024, 483, 149143 CrossRef CAS
- S. Li, Y. Zhou, N. Ma, J. Zhang, Z. Zheng, C. Streb and X. Chen, Angew. Chem., Int. Ed., 2020, 59, 8537–8540 CrossRef CAS PubMed
- M. Zhu, S. Han, J. Liu, M. Tan, W. Wang, K. Suzuki, P. Yin, D. Xia and X. Fang, Angew. Chem., Int. Ed., 2022, 61, e202213910 CrossRef CAS PubMed
- E. G. Ribó, N. L. Bell, D. Long and L. Cronin, Angew. Chem., Int. Ed., 2022, 61, e202201672 CrossRef PubMed
- M. T.-K. Ng, N. L. Bell, D.-L. Long and L. Cronin, J. Am. Chem. Soc., 2021, 143, 20059–20063 CrossRef CAS PubMed
- S. She, W. Xuan, N. L. Bell, R. Pow, E. G. Ribo, Z. Sinclair, D.-L. Long and L. Cronin, Chem. Sci., 2021, 12, 2427–2432 RSC
- D. Li, X. Zhang, J. Lv, P. Cai, Y. Sun, C. Sun and S. Zheng, Angew. Chem., Int. Ed., 2023, 62, e202312706 CrossRef CAS PubMed
- A. Jimbo, C. Li, K. Yonesato, T. Ushiyama, K. Yamaguchi and K. Suzuki, Chem. Sci., 2023, 14, 10280–10284 RSC
- V. I. Komlyagina, N. F. Romashev, V. V. Kokovkin, A. L. Gushchin, E. Benassi, M. N. Sokolov and P. A. Abramov, Molecules, 2022, 27, 6961 CrossRef CAS PubMed
- P. A. Abramov, V. Y. Komarov, D. A. Pischur, V. S. Sulyaeva, E. Benassi and M. N. Sokolov, CrystEngComm, 2021, 23, 8527–8537 RSC
- P. A. Abramov, J. Struct. Chem., 2022, 63, 2068–2082 CrossRef CAS
- V. V. Volchek, N. B. Kompankov, M. N. Sokolov and P. A. Abramov, Molecules, 2022, 27, 8368 CrossRef CAS PubMed
- A. V. Chupina, V. Shayapov, A. S. Novikov, V. V. Volchek, E. Benassi, P. A. Abramov and M. N. Sokolov, Dalton Trans., 2020, 49, 1522–1530 RSC
- Q. Wu, J. Wang, L. Zhang, A. Hong and J. Ren, Angew. Chem., 2005, 117, 4116–4120 CrossRef
- A. Tian, Y. Ning, Y. Yang, X. Hou, J. Ying, G. Liu, J. Zhang and X. Wang, Dalton Trans., 2015, 44, 16486–16493 RSC
- X.-Y. Wang, W.-C. Chen, K.-Z. Shao, X.-L. Wang, L. Zhao and Z.-M. Su, Chem. Commun., 2021, 57, 1042–1045 RSC
- N. N. Harmalkar, B. R. Srinivasan and S. N. Dhuri, Z. Naturforsch., B: J. Chem. Sci., 2022, 77, 245–252 CrossRef CAS
- Y.-Q. Lan, J.-F. Ma, J. Yang, X.-H. Wang and Z.-M. Su, Inorg. Chem., 2007, 46, 8283–8290 CrossRef PubMed
- P. Bolle, H. Serier-Brault, A. Boulmier, M. Puget, C. Menet, O. Oms, J. Marrot, P. Mialane, A. Dolbecq and R. Dessapt, Cryst. Growth Des., 2018, 18, 7426–7434 CrossRef CAS
- J. Ying, C. Sun, L. Jin, A. Tian and X. Wang, CrystEngComm, 2021, 23, 5385–5396 RSC
- S. Yue, W. Song, S. Pan, D. Liu, C. Li, J. Zang, J. Nan and J. Gui, Energy Fuels, 2023, 37, 8988–8998 CrossRef CAS
- Q. Lu, J. Ying, A. Tian and X. Wang, Inorg. Chem., 2023, 62, 16617–16626 CrossRef CAS PubMed
- S. Benz, A. I. Poblador-Bahamonde, N. Low-Ders and S. Matile, Angew. Chem., 2018, 130, 5506–5510 CrossRef
- Y. Zhao, Y. Cotelle, N. Sakai and S. Matile, J. Am. Chem. Soc., 2016, 138, 4270–4277 CrossRef CAS PubMed
- A. Docker, A. J. Martínez Martínez, H. Kuhn and P. D. Beer, Chem. Commun., 2022, 58, 3318–3321 RSC
- G. R. Desiraju, P. S. Ho, L. Kloo, A. C. Legon, R. Marquardt, P. Metrangolo, P. Politzer, G. Resnati and K. Rissanen, Pure Appl. Chem., 2013, 85, 1711–1713 CrossRef CAS
- B. K. Saha, R. V. P. Veluthaparambath and V. G. Krishna, Chem.–Asian J., 2023, 18, e202300067 CrossRef CAS PubMed
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS PubMed
- R. Tepper and U. S. Schubert, Angew. Chem., Int. Ed., 2018, 57, 6004–6016 CrossRef CAS PubMed
- L. C. Gilday, S. W. Robinson, T. A. Barendt, M. J. Langton, B. R. Mullaney and P. D. Beer, Chem. Rev., 2015, 115, 7118–7195 CrossRef CAS PubMed
-
L. Catalano, G. Cavallo, P. Metrangolo, G. Resnati and G. Terraneo, in Topics in Current Chemistry, Springer Verlag, 2016, vol. 373, pp. 289–309 Search PubMed
- A. J. Peloquin, S. C. Hill, H. D. Arman, C. D. McMillen, D. Rabinovich and W. T. Pennington, J. Chem. Crystallogr., 2021, 52, 62–72 CrossRef
- A. Mukherjee, S. Tothadi and G. R. Desiraju, Acc. Chem. Res., 2014, 47, 2514–2524 CrossRef CAS PubMed
- D. M. Ivanov, A. S. Novikov, G. L. Starova, M. Haukka and V. Y. Kukushkin, CrystEngComm, 2016, 18, 5278–5286 RSC
- C. B. Aakeröy, M. Baldrighi, J. Desper, P. Metrangolo and G. Resnati, Chem.–Eur. J., 2013, 19, 16240–16247 CrossRef PubMed
- G. Cavallo, J. S. Murray, P. Politzer, T. Pilati, M. Ursini and G. Resnati, IUCrJ, 2017, 4, 411–419 CrossRef CAS PubMed
- F. Heinen, E. Engelage, C. J. Cramer and S. M. Huber, J. Am. Chem. Soc., 2020, 142, 8633–8640 CrossRef CAS PubMed
- R. L. Sutar and S. M. Huber, ACS Catal., 2019, 9, 9622–9639 CrossRef CAS
- D. L. Reinhard, D. Kutzinski, M. Hatta, E. Engelage and S. M. Huber, Synlett, 2024, 35, 209–214 CrossRef CAS
- R. Robidas, D. L. Reinhard, C. Y. Legault and S. M. Huber, Chem. Rec., 2021, 21, 1912–1927 CrossRef CAS PubMed
- N. S. Soldatova, P. S. Postnikov, D. M. Ivanov, O. V. Semyonov, O. S. Kukurina, O. Guselnikova, Y. Yamauchi, T. Wirth, V. V. Zhdankin, M. S. Yusubov, R. M. Gomila, A. Frontera, G. Resnati and V. Y. Kukushkin, Chem. Sci., 2022, 13, 5650–5658 RSC
- A. D. Radzhabov, A. I. Ledneva, N. S. Soldatova, I. I. Fedorova, D. M. Ivanov, A. A. Ivanov, M. S. Yusubov, V. Y. Kukushkin and P. S. Postnikov, Int. J. Mol. Sci., 2023, 24, 14642 CrossRef CAS PubMed
- N. S. Soldatova, V. V. Suslonov, D. M. Ivanov, M. S. Yusubov, G. Resnati, P. S. Postnikov and V. Y. Kukushkin, Cryst. Growth Des., 2023, 23, 413–423 CrossRef CAS
- E. Merritt and B. Olofsson, Angew. Chem., Int. Ed., 2009, 48, 9052–9070 CrossRef CAS PubMed
- N. S. Soldatova, P. S. Postnikov, V. V. Suslonov, T. Y. Kissler, D. M. Ivanov, M. S. Yusubov, B. Galmés, A. Frontera and V. Y. Kukushkin, Org. Chem. Front., 2020, 7, 2230–2242 RSC
- J. Li, Y. Zhao, B. Huang, Y. Wang, Z. Xiong, B. Xiao, Y. Zhao, Z. Xiao and P. Wu, J. Cluster Sci., 2022, 33, 2375–2381 CrossRef CAS
- P. Politzer, J. S. Murray and T. Clark, Phys. Chem. Chem. Phys., 2013, 15, 11178 RSC
- J. S. Murray, P. Lane and P. Politzer, J. Mol. Model., 2009, 15, 723–729 CrossRef CAS PubMed
- H. Wang, W. Wang and W. J. Jin, Chem. Rev., 2016, 116, 5072–5104 CrossRef CAS PubMed
- T. Clark, M. Hennemann, J. S. Murray and P. Politzer, J. Mol. Model., 2007, 13, 291–296 CrossRef CAS PubMed
- L. Fotović, N. Bedeković and V. Stilinović, Cryst. Growth Des., 2023, 23, 3384–3392 CrossRef PubMed
- L.-S. Wang, Y. Lu, G. M. Ó. Máille, S. P. Anthony, D. Nolan and S. M. Draper, Inorg. Chem., 2016, 55, 9497–9500 CrossRef CAS PubMed
- D. Kuriakose and M. R. Prathapachandra Kurup, Inorg. Chim. Acta, 2020, 505, 119472 CrossRef CAS
- D. Kuriakose and M. R. P. Kurup, Polyhedron, 2019, 170, 749–761 CrossRef CAS
- M. Oszajca, Ľ. Smrčok and W. Łasocha, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2013, 69, 1367–1372 CrossRef CAS PubMed
- P. Xiao, F. Dumur, M.-A. Tehfe, B. Graff, J. P. Fouassier, D. Gigmes and J. Lalevée, Macromol. Chem. Phys., 2013, 214, 1749–1755 CrossRef CAS
- P. Xiao, C. Simonnet-Jégat, F. Dumur, G. Schrodj, M.-A. Tehfe, J. P. Fouassier, D. Gigmes and J. Lalevée, Polym. Chem., 2013, 4, 4526 RSC
- H. Mokbel, P. Xiao, C. Simonnet-Jégat, F. Dumur, D. Gigmes, J. Toufaily, T. Hamieh, J. P. Fouassier and J. Lalevée, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 981–989 CrossRef CAS
- F. Dumur, Eur. Polym. J., 2023, 195, 112193 CrossRef CAS
- N. Soldatova, P. Postnikov, O. Kukurina, V. V. V. Zhdankin, A. Yoshimura, T. Wirth and M. S. Yusubov, Beilstein J. Org. Chem., 2018, 14, 849–855 CrossRef CAS PubMed
- I. S. Aliyarova, D. M. Ivanov, N. S. Soldatova, A. S. Novikov, P. S. Postnikov, M. S. Yusubov and V. Y. Kukushkin, Cryst. Growth Des., 2021, 21, 1136–1147 CrossRef CAS
- I. I. Fedorova, N. S. Soldatova, D. M. Ivanov, K. Nikiforova, I. S. Aliyarova, M. S. Yusubov, P. M. Tolstoy, R. M. Gomila, A. Frontera, V. Y. Kukushkin, P. S. Postnikov and G. Resnati, Cryst. Growth Des., 2023, 23, 2661–2674 CrossRef CAS
- E. Arunan, G. R. Desiraju, R. A. Klein, J. Sadlej, S. Scheiner, I. Alkorta, D. C. Clary, R. H. Crabtree, J. J. Dannenberg, P. Hobza, H. G. Kjaergaard, A. C. Legon, B. Mennucci and D. J. Nesbitt, Pure Appl. Chem., 2011, 83, 1637–1641 CrossRef CAS
- D. D. DesMarteau, W. T. Pennington, V. Montanari, B. H. Thomas and J. Fluor, Chem, 2003, 122, 57–61 CAS
- G. Wu, P. J. Zheng, S. Z. Zhu and Q. Y. Chen, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1991, 47, 1227–1230 CrossRef
- E. Radkov and R. H. Beer, Polyhedron, 1995, 14, 2139–2143 CrossRef CAS
- K. Kamata, M. Kotani, K. Yamaguchi, S. Hikichi and N. Mizuno, Chem.–Eur. J., 2007, 13, 639–648 CrossRef CAS PubMed
- K. Kamata, Y. Nakagawa, K. Yamaguchi and N. Mizuno, J. Catal., 2004, 224, 224–228 CrossRef CAS
- T. Hirano, K. Uehara, K. Kamata and N. Mizuno, J. Am. Chem. Soc., 2012, 134, 6425–6433 CrossRef CAS PubMed
- V. Y. Evtushok, V. A. Lopatkin, O. Y. Podyacheva and O. A. Kholdeeva, Catalysts, 2022, 12, 472 CrossRef CAS
- A. V. Rozhkov, E. A. Katlenok, M. V. Zhmykhova, A. Y. Ivanov, M. L. Kuznetsov, N. A. Bokach and V. Y. Kukushkin, J. Am. Chem. Soc., 2021, 143, 15701–15710 CrossRef CAS PubMed
- M. Ghali, C. Brahmi, M. Benltifa, C. Vaulot, A. Airoudj, P. Fioux, F. Dumur, C. Simonnet-Jégat, F. Morlet-Savary, S. Jellali, L. Bousselmi and J. Lalevée, J. Polym. Sci., 2021, 59, 153–169 CrossRef CAS
-
W. G. Klemperer, Inorganic syntheses, 1990, pp. 74–85 Search PubMed
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed
- C. B. Hübschle, G. M. Sheldrick and B. Dittrich, J. Appl. Crystallogr., 2011, 44, 1281–1284 CrossRef PubMed
- A. L. Spek, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 9–18 CrossRef CAS PubMed
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS PubMed
- R. Ahlrichs, M. Bär, M. Häser, H. Horn and C. Kölmel, Chem. Phys. Lett., 1989, 162, 165–169 CrossRef CAS
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057 RSC
- R. F. W. Bader, Chem. Rev., 1991, 91, 893–928 CrossRef CAS
- J. Contreras-García, E. R. Johnson, S. Keinan, R. Chaudret, J.-P. Piquemal, D. N. Beratan and W. Yang, J. Chem. Theory Comput., 2011, 7, 625–632 CrossRef PubMed
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed
- E. V Bartashevich and V. G. Tsirelson, Russ. Chem. Rev., 2014, 83, 1181–1203 CrossRef
- E. Espinosa, E. Molins and C. Lecomte, Chem. Phys. Lett., 1998, 285, 170–173 CrossRef CAS
- E. D. Glendening, C. R. Landis and F. Weinhold, J. Comput. Chem., 2019, 40, 2234–2241 CrossRef CAS PubMed
-
E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales, P. Karafiloglou, C. R. Landis and F. Weinhold, Theor. Chem. Institute, Univ. Wisconsin, Madison, WI, 2018 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2334360–2334364. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc01695e |
| This journal is © The Royal Society of Chemistry 2024 |