
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConfined semiconducting polymers with boosted NIR light-triggered H2O2 production for hypoxia-tolerant persistent photodynamic therapy†
Feng
Lu
 a,
Lili
Li
a,
Meng
Zhang
a,
Chengwu
Yu
a,
Yonghui
Pan
a,
Fangfang
Cheng
b,
Wenbo
Hu
c,
Xiaomei
Lu
de,
Qi
Wang
*a and
Quli
Fan
*a
a,
Lili
Li
a,
Meng
Zhang
a,
Chengwu
Yu
a,
Yonghui
Pan
a,
Fangfang
Cheng
b,
Wenbo
Hu
c,
Xiaomei
Lu
de,
Qi
Wang
*a and
Quli
Fan
*a
aState Key Laboratory of Organic Electronics and Information Displays, Jiangsu Key Laboratory for Biosensors, Institute of Advanced Materials (IAM), Nanjing University of Posts & Telecommunications, Nanjing 210023, China. E-mail: iamqwang@njupt.edu.cn; iamqlfan@njupt.edu.cn
bSchool of Pharmacy, Nanjing University of Chinese Medicine, Nanjing 210023, China
cInstitute of Flexible Electronics (IFE), Northwestern Polytechnical University, Xi'an 710072, China
dInstitute of Advanced Materials (IAM) & School of Flexible Electronics (Future Technologies), Nanjing Tech University, Nanjing, 211816, China
eZhengzhou Institute of Biomedical Engineering and Technology, Zhengzhou, 450001, China
First published on 4th July 2024
Abstract
Hypoxia featured in malignant tumors and the short lifespan of photo-induced reactive oxygen species (ROS) are two major issues that limit the efficiency of photodynamic therapy (PDT) in oncotherapy. Developing efficient type-I photosensitizers with long-term ˙OH generation ability provides a possible solution. Herein, a semiconducting polymer-based photosensitizer PCPDTBT was found to generate 1O2, ˙OH, and H2O2 through type-I/II PDT paths. After encapsulation within a mesoporous silica matrix, the NIR-II fluorescence and ROS generation are enhanced by 3–4 times compared with the traditional phase transfer method, which can be attributed to the excited-state lifetime being prolonged by one order of magnitude, resulting from restricted nonradiative decay channels, as confirmed by femtosecond spectroscopy. Notably, H2O2 production reaches 15.8 μM min−1 under a 730 nm laser (80 mW cm−2). Further adsorption of Fe2+ ions on mesoporous silica not only improves the loading capacity of the chemotherapy drug doxorubicin but also triggers a Fenton reaction with photo-generated H2O2in situ to produce ˙OH continuously after the termination of laser irradiation. Thus, semiconducting polymer-based nanocomposites enables NIR-II fluorescence imaging guided persistent PDT under hypoxic conditions. This work provides a promising paradigm to fabricate persistent photodynamic therapy platforms for hypoxia-tolerant phototheranostics.
Introduction
Benefitting from the high safety and great operability of light, photodynamic therapy (PDT) has been widely used for various diseases in the clinic, such as actinic keratosis, acne vulgaris, and pathological myopia.1,2 In particular, this highly selective therapeutic technique with negligible invasiveness has been widely investigated for the treatment of malignant tumors over the past few decades.3,4 Currently, there are two major problems which severely limit the anti-tumor efficacy of PDT: the hypoxic microenvironment in solid tumors and short lifespan of photo-induced reactive oxygen species (ROS).5,6 Most photosensitizers (PSs) work in a type-II path, which produce singlet oxygen (1O2) through energy transfer from their excited triplet state to O2. This process is highly dependent on the concentration of O2; thus, type-II PDT become less effective for hypoxic tumors. Some techniques, such as the perfluorocarbon-based O2 delivery method and in situ O2 generation by catalytic reactions have been proposed to relieve hypoxia.7,8 However, these methods made the PDT system complicated, and the O2 delivery/generation efficiency was unclear. Meanwhile, PDT was considered to be short-acting. The tissue oxygen would be rapidly consumed during type-II PDT; therefore, the generation of 1O2 would be hindered in a short period of time.9 In addition, PDT takes effect only under light exposure; the generation of ROS stops immediately after the termination of irradiation. Although sustained release of 1O2 was achieved by combining PSs with anthracene or 2-pyridone groups, which can reversibly capture and release 1O2,6,10 these strategies still did not solve the hypoxia problem.From the perspective of a photosensitizer, the design of novel PSs capable of type-I PDT, which works through an electron transfer mechanism, would be a reliable way to combat tumor hypoxia.11 Type-I PDT is less dependent on O2 and can produce ˙OH, O2˙−, and H2O2 under hypoxic conditions.12 Most reported type-I PSs were inorganic semiconducting nanoparticles, which were generally excited at short wavelength and exhibited weak fluorescence.5 Apparently, organic PSs were more favorable for biomedical applications because of their better biocompatibility, near-infrared (NIR, 700–900 nm) absorption, and strong photoluminescence (PL), which also enables deep tissue penetration and photo diagnosis. For instance, many organic PSs can be used for fluorescence imaging in the second near-infrared window (NIR-II, 1000–1700 nm), which enables high resolution and contrast imaging in vivo due to reduced autofluorescence and photo scattering.13 However, developing organic-based NIR-light-triggered type-I PSs was still tricky, and they generally showed low ROS generation efficiency.11,14,15 Indeed, the PL and PDT performances of organic materials are commonly significantly reduced when their energy bandgaps get into the NIR region, probably due to the enhanced intramolecular nonradiative decay,16 which inhibits emissive transitions and intersystem crossing (ISC). As a result, type-I PDT was usually combined with photothermal therapy because high laser power was necessary to generate enough ROS.17,18 Nevertheless, this elevated the risk of thermal damage to normal tissues, which was not favorable for clinical practice. Therefore, developing organic-based photosensitizers with strong NIR-II emission and efficient type-I PDT capacity under low-power NIR excitation was greatly needed for hypoxia-tolerant phototheranostics.
To date, investigation of type-I PSs has focused mainly on the generation of ˙OH and O2˙−, since these short-lived radicals are considered to be highly cytotoxic.11 Although metal peroxide-, glucose oxidase-, and nanozyme-based strategies have been widely adopted to improve the intracellular H2O2 level to enhance the anticancer effect,19–21 the photo-generation of H2O2 through type-I PDT was largely neglected previously, probably due to the low H2O2 production efficiency and weak cytotoxicity. Actually, an organic photocatalyst has recently been used for H2O2 production,22 and it can provide much better controllability than above-mentioned methods. Notably, the photo-generated H2O2 could be used to produce cytotoxic ˙OH continuously in the presence of some transition metal ions (Fe2+, Cu2+, and Ti3+) through a Fenton reaction even after termination of exposure.23 Unfortunately, NIR organic photosensitizers with efficient H2O2 production have rarely been reported, and type-I PDT has not been combined with a Fenton reaction to achieve persistent photodynamic therapy.
Herein, we report that the NIR-II emissive semiconducting polymer (SP) poly[2,6-(4,4-bis-(2-ethylhexyl)-4H-cyclopenta[2,1-b;3,4-b′]dithiophene)-alt-4,7(2,1,3-benzothiadiazole)] (PCPDTBT) can produce 1O2, ˙OH, and H2O2 under 730 nm laser irradiation through type-I and type-II PDT approaches. After encapsulation in a mesoporous silica matrix, the fluorescence and ROS generation can be enhanced by about 3–4 times. Femtosecond transient absorption (fs-TA) investigation revealed that the excited-state lifetime was prolonged by one order of magnitude (264.5 ps vs. 19.1 ps) compared to amphiphilic polymer F127-coated ones, and intermolecular and intramolecular nonradiative decay channels were largely restricted due to the confinement effect. Importantly, the photoluminescence quantum yield (PLQY) in the NIR-II region was increased to 4.6%, and the H2O2 production yield could reach 15.8 μM min−1 under 730 nm laser irradiation with low power density (80 mW cm−2). Further adsorption of the Fe2+ ions on mesoporous silica not only improves the loading capacity of the chemotherapy drug doxorubicin (DOX) but also enhances ˙OH generation through the Fenton reaction. The photo-generated H2O2 here is enough to trigger the reaction with the Fe2+ ions in situ to produce ˙OH continuously after termination of laser irradiation, which greatly prolongs the duration of PDT. The obtained SP-based nanoplatform (Scheme 1) can generate ROS in cancer cells under hypoxic conditions and inhibit cell growth by apoptosis and ferroptosis mechanisms. NIR-II fluorescence imaging indicated an outstanding tumor accumulation of obtained nanoparticles in vivo, and 4T1 tumors can be significantly inhibited with a combination of chemotherapy and persistent PDT under a safe power density. This work provides a promising paradigm to fabricate persistent photodynamic therapy platforms for hypoxia-tolerant phototheranostics.
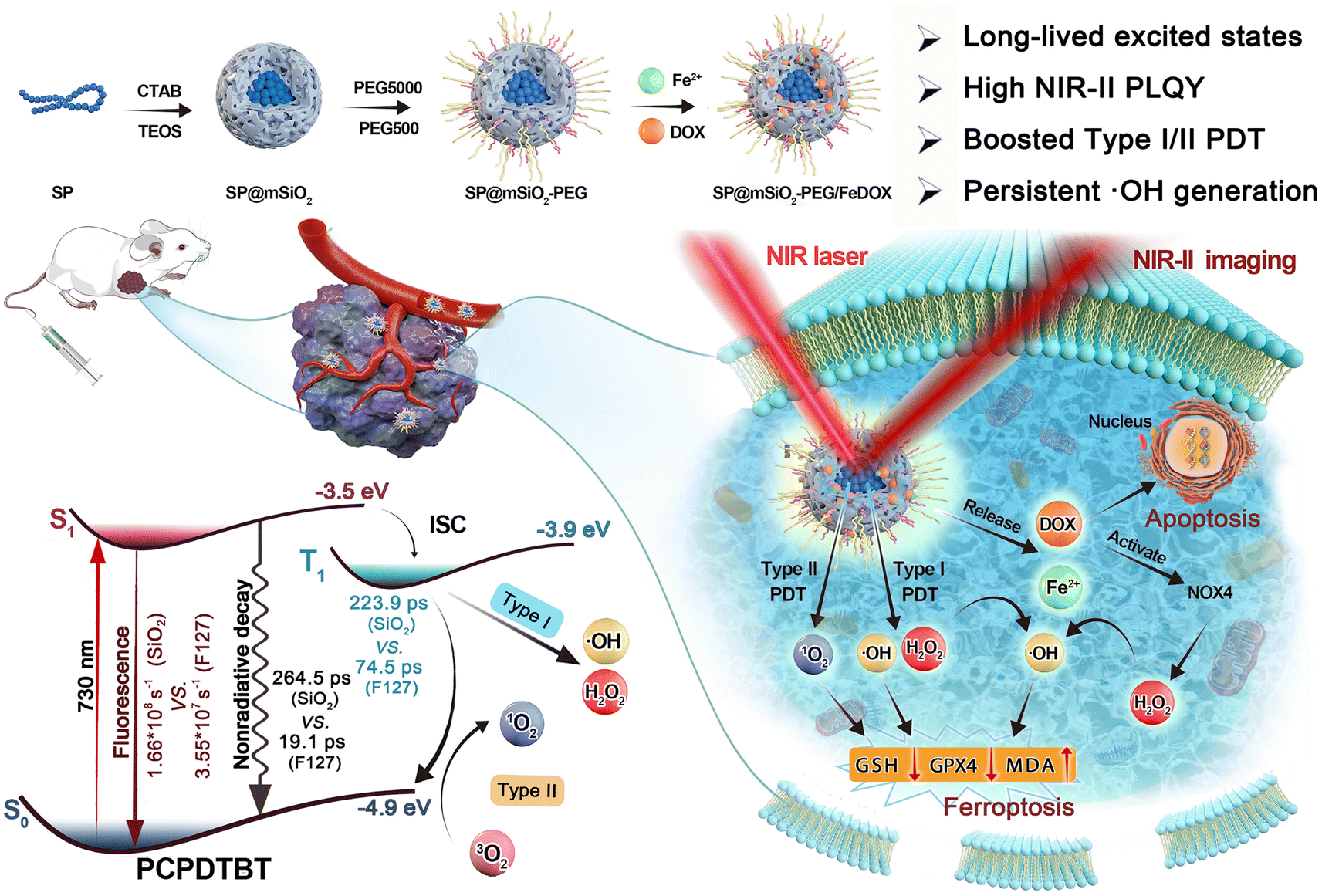 | ||
| Scheme 1 Schematic illustration of the synthetic process for the semiconducting polymer-based nanoplatform and its applications in phototheranostics. | ||
Results and discussion
Synthesis and characterization of SP@mSiO2-PEG
The semiconducting polymer PCPDTBT (Fig. 1A) was synthesized through a traditional Stille coupling reaction according to a previous report (Fig. S1†).24 Density functional theory calculation shows a planar skeleton structure for the polymer (Fig. 1B). Predicted frontier molecular orbitals indicate strong intramolecular charge transfer (ICT) and significant π-orbital overlap for the polymer (Fig. 1C). The electron density of the HOMO was located mainly on the dithienocyclopenta unit (donor), whereas it transferred to the benzothiadiazole unit (acceptor) for the LUMO. The π-orbital overlap originating from the planar skeleton guarantees strong NIR absorption and remarkable emission in the NIR-II region. The advantages of SPs for NIR-II imaging and PDT are summarized in Table S1.† Water-soluble PCPDTBT-based nanoparticles with uniform morphology can be obtained by F127 encapsulation (SP@F127, 16 nm) and PEGylated mesoporous silica coating (SP@mSiO2-PEG, 35.5 nm), as shown in Fig. 1D and E. Silica coating was achieved through CTAB-mediated phase transfer and subsequently the hydrolysis of tetraethyl orthosilicate.25 The porous structure was apparent from the enlarged transmission electron microscopy (TEM) image (Fig. S2†). The hydrodynamic diameter of SP@F127 was around 46.2 nm (Fig. S3†), which was similar to that of SP@mSiO2-PEG (∼49.5 nm, Fig. 1F). The larger hydrodynamic diameters of the nanoparticles compared with their TEM images were related to the extended PEG corona in solution.26 Here, two mPEG-silanes with different molecular weights were used to passivate the surface of silica, which can enhance the PEG coverage density on the nanoparticle.27 The salting out of SP@mSiO2-PEG from aqueous solution into chloroform strongly demonstrates this assumption (Fig. 1F). The efficient PEG modification not only improves the colloidal stability of the nanoparticle but also reduces protein adsorption and immune recognition, thereby largely improving the serum half-life in vivo.28 The absorption peak of SP@F127 was blue-shifted compared with PCPDTBT in organic solvent (Fig. 1G). This phenomenon can be attributed to conformational changes and inter-chain interactions accompanied by the aggregation of semiconducting polymers during the formation of the nanoparticles.29,30 These intramolecular and intermolecular interactions could shorten effective conjugation lengths and cause delocalization of π-electrons over polymeric chains, which all lead to the blue-shift of absorption spectra.31 Besides, these strong interactions lead to a significant decrease in photoluminescence, as shown in Fig. 1H. Here, it is interesting to find that the absorption of the polymer was less affected after silica coating (Fig. 1G), and the emission intensity of SP@mSiO2-PEG was much stronger than that of SP@F127 (Fig. 1H).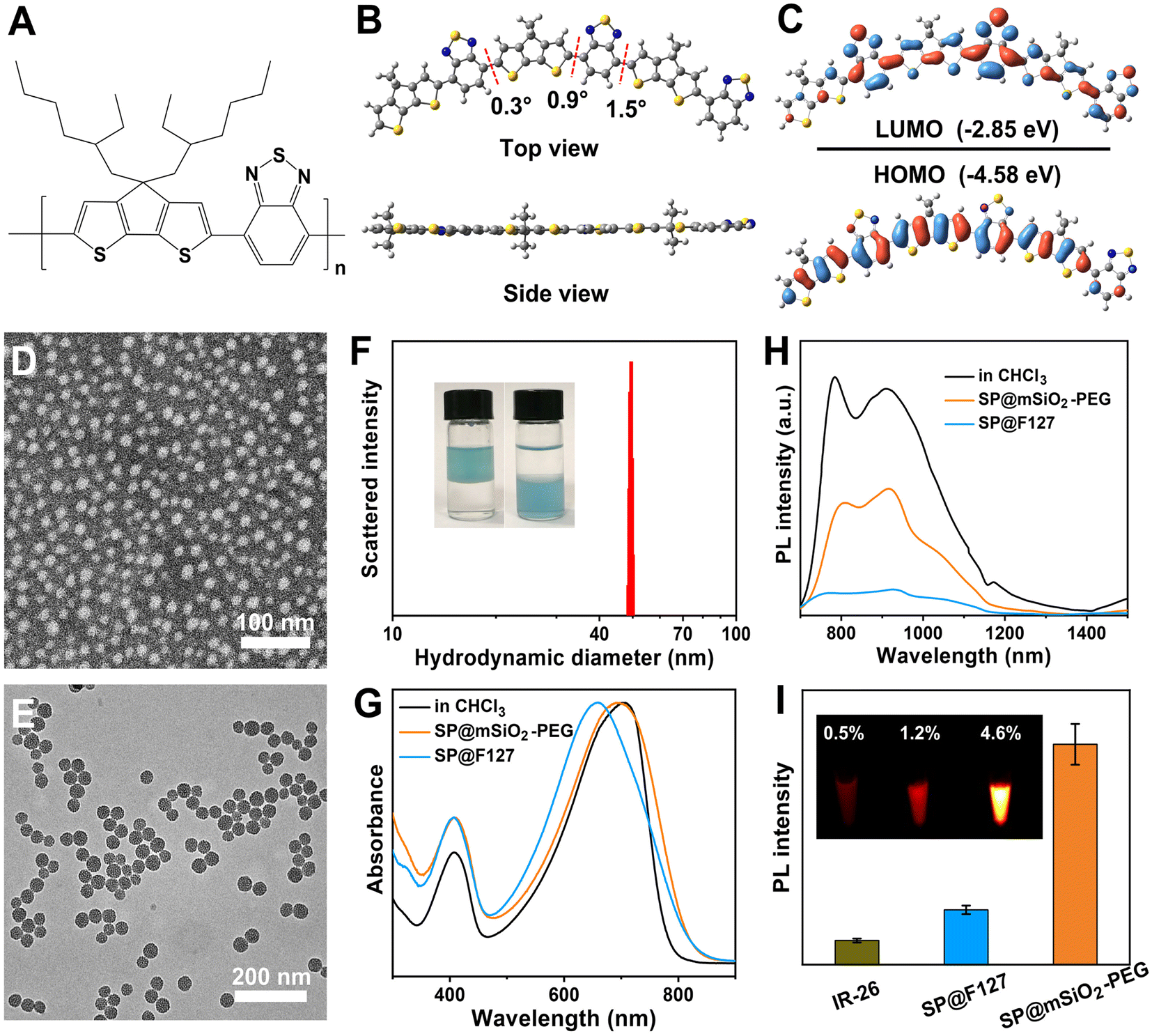 | ||
| Fig. 1 (A) Molecular structure, (B) optimized ground-state geometry, and (C) calculated molecular orbitals of PCPDTBT. (D) Negative-stained TEM image of SP@F127. (E) TEM image of SP@mSiO2-PEG. (F) Hydrodynamic diameter of SP@mSiO2-PEG. The inset shows photographs of the salting out of SP@mSiO2-PEG nanoparticles from water into chloroform. (G) Normalized absorption spectra of PCPDTBT in chloroform and encapsulated with F127 and mesoporous silica. (H) Emission spectra of PCPDTBT in chloroform and encapsulated with F127 and mesoporous silica, excited at 635 nm. (I) Average fluorescence intensities of IR-26 in 1,2-dichloroethane, SP@F127 and the SP@mSiO2-PEG nanoparticles in water. The inset shows corresponding NIR-II fluorescence images and the quantum yields. | ||
PCPDTBT has been heavily used for traditional NIR-I imaging with a strong emission around 800 nm,32 while its emission shoulder beyond 900 nm was neglected due to the low detection efficiency of silicon-based detectors (Fig. S4†). This is similar to many cyanine dyes, which exhibit strong emission tails in the NIR-II region.33,34 Photoluminescence quantum yields (PLQY) for SP@F127 and SP@mSiO2-PEG in the NIR-II region were determined to be 1.2% and 4.6%, respectively, using IR-26 (0.5% in 1,2-dichloroethane) as a reference (Fig. 1I and S5†), indicating their great potential for NIR-II imaging, especially for SP@mSiO2-PEG. Besides, these silica-based nanoparticles exhibited high stability: the absorption spectra did not change after 28 d of storage at room temperature (Fig. S6†) and can be stored for at least one year without any precipitation (Fig. S7†).
Laser-induced ROS generation
PCPDTBT can also produce abundant singlet oxygen under laser irradiation.35,36 The band gap between its T1 and S0 states is around 1.0 eV,37 which allows 1O2 generation under laser irradiation and enables type-II photodynamic therapy.38 Since the energy level for the S0 state of the SP is around −4.9 eV using the vacuum level as reference,39 the energy level for the T1 state can be calculated to be −3.9 eV, which is higher than the redox potential of O2 (−4.17 eV),40 suggesting its potential for type-I PDT (Fig. S8†). Using 1,3-diphenylisobenzofuran (DPBF), 3,3′,5,5′-tetramethylbenzidine (TMB) and Amplex Red as 1O2 (Fig. 2A–C), ˙OH (Fig. 2E–G) and H2O2 (Fig. 2I–K) indicators,41,42 photo-induced ROS generation was carefully investigated. It is clear that type-I and type-II processes exist for PCPDBT-based nanoparticles, and the PDT performance for SP@mSiO2-PEG was much better than that for SP@F127. The generation of 1O2 (Fig. 2D) and ˙OH (Fig. 2H) were further confirmed by electron spin resonance (ESR) characterization, which revealed a result consistent with the optical analysis. The production of ROS under normoxic and hypoxic conditions were thereafter compared (Fig. S9†). Obviously, the generation of 1O2 was largely inhibited under the hypoxic situation; nevertheless, the generation of ˙OH and H2O2 was less influenced, indicating the less oxygen consuming feature of type-I PDT. Note that the light-driven H2O2 production for SP@mSiO2-PEG was very efficient here with a yield of 15.8 μM min−1 under a power density of 80 mW cm−2 (Fig. S10†). This value was even higher than those of some photocatalysts for H2O2 synthesis.22,43 In general, silica coating provides around 3–4 fold enhancement in fluorescence emission and ROS generation, compared with the commonly used F127 encapsulation method (Fig. 2L). Although fluorescence amplification of organic dyes after silica coating was relatively common,44,45 enhancement in their PDT performance was rarely reported, which is actually more important for phototheranostics.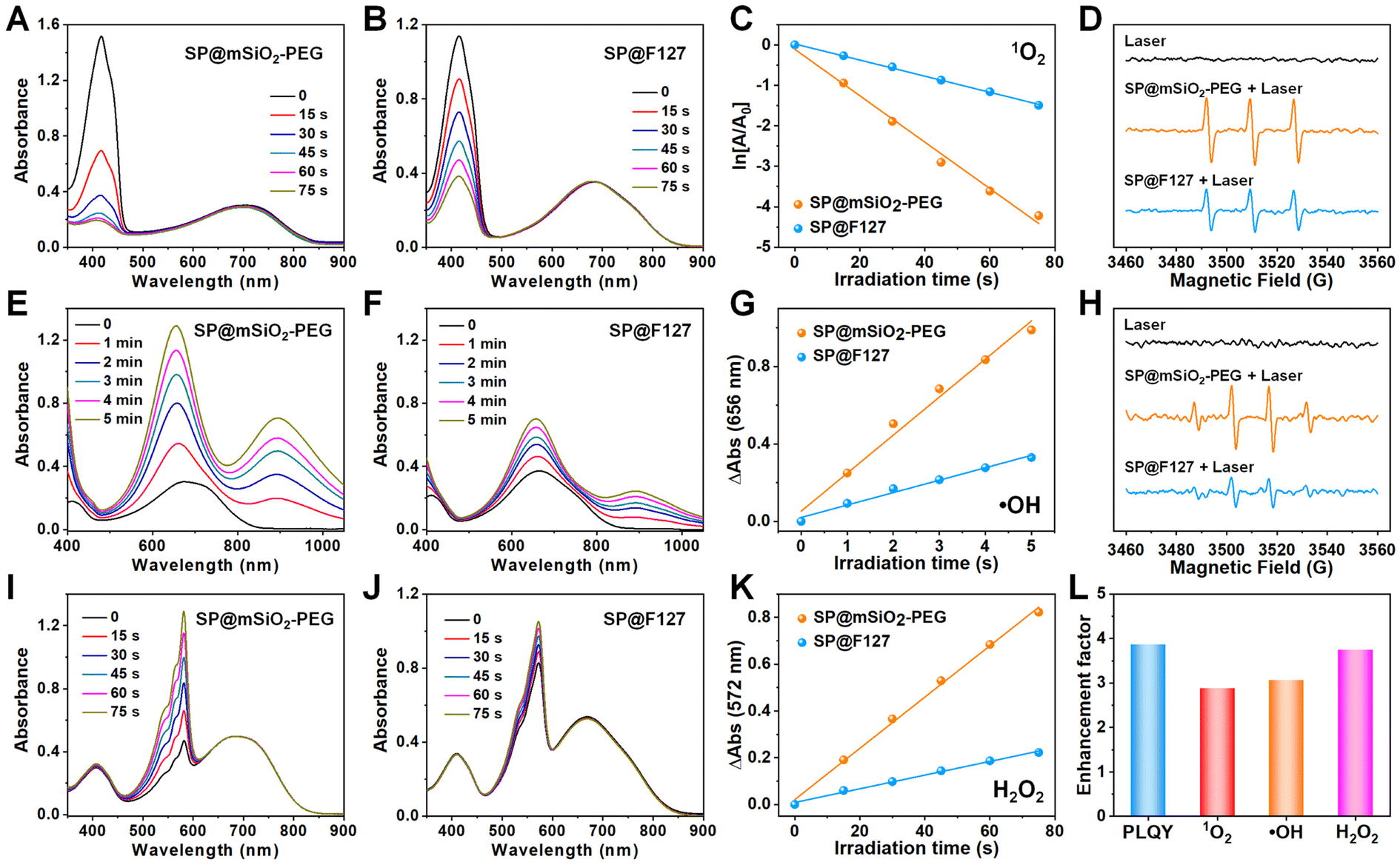 | ||
| Fig. 2 Time-dependent decay of DPBF by (A) SP@mSiO2-PEG and (B) SP@F127 under 730 nm laser irradiation. (C) Comparison of 1O2 generation by SP@mSiO2-PEG and SP@F127 under laser irradiation. (D) EPR spectra demonstrating the photo-triggered 1O2 generation with SP@mSiO2-PEG and SP@F127. Time-dependent oxidation of TMB by (E) SP@mSiO2-PEG and (F) SP@F127 under 730 nm laser irradiation. (G) Comparison of ˙OH generation by SP@mSiO2-PEG and SP@F127 under laser irradiation. (H) EPR spectra demonstrating the photo-triggered ˙OH generation with SP@mSiO2-PEG and SP@F127. Time-dependent evolution of Amplex Red by (I) SP@mSiO2-PEG and (J) SP@F127 under 730 nm laser irradiation. (K) Comparison of H2O2 generation by SP@mSiO2-PEG and SP@F127 under laser irradiation. The power density of the laser was 80 mW cm−2. (L) Enhancement factors of the NIR-II PLQY and ROS generation by mesoporous silica encapsulation. | ||
Excited-state dynamics study
The fluorescence enhancement of a semiconducting polymer was commonly attributed to isolation from water after silica coating.45 However, this assumption was inapplicable here due to the porous structure and hydrophilicity of mesoporous silica. In addition, a mesoporous silica coating for inorganic nanoparticles, such as rare-earth-doped nanoparticles (NaYF4:Yb0.05,Nd0.2@NaYF4) or Ag2S quantum dots, showed no evidence of amplified emission (Fig. S11†). These observations indicate that the characteristics of the SP play an important role here. To unravel the underlying mechanism for the enhancement in fluorescence and ROS generation, femtosecond transient absorption (fs-TA) spectroscopy was carried out to explore their excited-state dynamics (Fig. 3A, D and S12†).46 Positive signals around 465 nm were observed for both samples, and this excited-state absorption (ESA) band was commonly related to the photoinduced triplet state.47,48 Negative signals above 550 nm can be assigned to the combination of ground-state bleaching (GSB) and stimulated emission, as their plots are consistent with their steady-state absorption and fluorescence spectra.49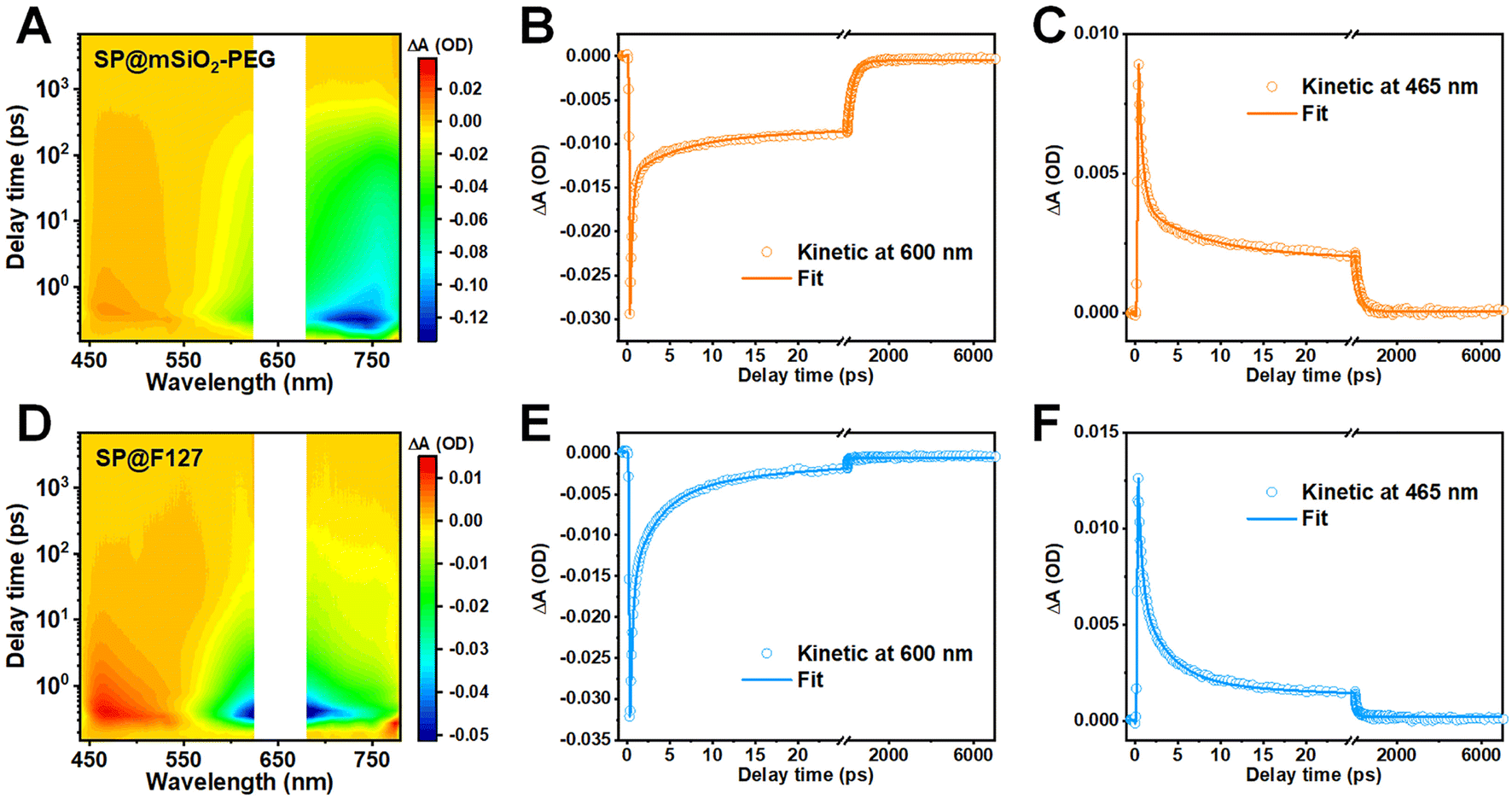 | ||
| Fig. 3 Investigation of excited state dynamics. 2D pseudocolor fs-TA mapping of (A) SP@mSiO2-PEG, and (D) SP@F127 excited with 660 nm laser pulse. Kinetic curves and fitting lines of SP@mSiO2-PEG at (B) 600 nm and (C) 465 nm. Kinetic curves and fitting lines of SP@F127 at (E) 600 nm and (F) 465 nm. | ||
Kinetic curves within the GSB region could reflect the nonradiative decay of excited states, and the fractions of depopulation through different channels can be evaluated with corresponding relative amplitudes.50 The kinetic plots and fitting curves of SP@mSiO2-PEG and SP@F127 are shown in Fig. 3B and E. A triexponential function was employed to achieve a satisfactory fit, and the corresponding fraction and relative amplitudes are summarized in Table 1. The kinetic curves demonstrate significantly accelerated nonradiative decay for SP@F127, which favors thermal deactivation and commonly leads to a decrease in fluorescence and intersystem crossing.51 According to a previous dynamic investigation into semiconducting polymers,16 comparable femtosecond components of 0.31 ps in SP@mSiO2-PEG and 0.32 ps in SP@F127 can be ascribed to intermolecular nonradiative decay, while the time constants of 5.9 ps for SP@mSiO2-PEG and 3.1 ps for SP@F127 can be ascribed to intramolecular nonradiative decay. Apparently, these short-lived components were largely inhibited by silica coating relative to F127 encapsulation (0.2% vs. 3.9% for the intermolecular nonradiative decay, 1.0% vs. 25.7% for the intramolecular nonradiative decay), which leads to the excited states of PCPDTBT living longer by an order of magnitude (264.5 ps vs. 19.1 ps). Due to the planar conjugated structure (Fig. 1B), aromatic interactions and π–π stacking can take place after F127 encapsulation,52 leading to severe aggregation of the SPs, causing strong intermolecular and intramolecular interactions. For SP@mSiO2-PEG, cationic surfactant CTAB-mediated phase transfer and subsequent rigid silica coating were considered to prevent these interactions by electrostatic repulsion and steric hindrance. Besides, intramolecular rotation and vibration of molecules can be inhibited within the silica matrix.44,53 The decay of the photoinduced triplet state follows the same trend (Fig. 3C and F). The average lifetime for the ESA band of SP@mSiO2-PEG was 223.9 ps, which was much longer than that of SP@F127 (74.5 ps). The long-lived triplet state would improve energy or electron transfer to oxygen and amplify the ROS generation.54,55
| Wavelength | τ 1 (ps) | A 1 | τ 2 (ps) | A 2 | τ 3 (ps) | A 3 | τ avg (ps) | |
|---|---|---|---|---|---|---|---|---|
| a A 1, A2, and A3 represent the fractions of associated components. | ||||||||
| SP@mSiO2-PEG | 600 nm | 0.31 | 0.2% | 5.9 | 1.0% | 264.6 | 98.8% | 264.5 |
| 465 nm | 0.49 | 0.5% | 6.5 | 2.2% | 224.0 | 97.3% | 223.8 | |
| SP@F127 | 600 nm | 0.32 | 3.9% | 3.1 | 25.7% | 20.0 | 70.4% | 19.1 |
| 465 nm | 0.46 | 1.8% | 3.5 | 10.9% | 75.0 | 87.3% | 74.5 | |
Combining the PLQY and fluorescent lifetime (Fig. S13†), the photophysical properties of both the nanoparticles can be obtained (Table S2†) according to previous reports.56,57 Total deexcitation rates, including the rate of fluorescence (Kf), rate of intersystem crossing (Kisc), and rate of nonradiative pathways (Knr), were close for SP@mSiO2-PEG (8.77 × 108 s−1) and SP@F127 (8.47 × 108 s−1). Nevertheless, Kf for SP@mSiO2-PEG was around 3.7 times faster than that of SP@F127. In addition, Kisc for SP@mSiO2-PEG should be largely enhanced due to the improved ROS generation and similar deexcitation rates.58 Therefore, the rate of nonradiative decay (Knr) of SP@mSiO2-PEG was slowed down here.
On the basis of above results, we conclude that the confinement effect of the rigid mesoporous silica matrix significantly reduces the aggregation of PCPDTBT and restricts the intermolecular and intramolecular interactions, which inhibits the nonradiative decay rate, prolongs the lifetime of the excited states and enables strong fluorescent emission and abundant ROS generation under photon excitation.
Drug loading and persistent ˙OH generation
The drug loading capacity of mesoporous silica can be seriously influenced after high-density PEG modification.59 Fortunately, we found that the loading capacity of DOX can be dramatically enhanced with the decoration of metal ions on silica (Fig. 4A and B), which may be related to chelation between metals and phenolic hydroxyl groups on DOX (Fig. S14†).60 The loading capacity for Fe2+ was determined to be 0.51% using 1.10-phenanthroline as a probe (Fig. S15†). From high resolution transmission electron microscopy (HRTEM) images, the SPs were found to be in the core of the nanocomposites, and the Fe2+ ions were most likely located in the mesoporous silica shell (Fig. S16†). Since the Fe2+ ions can be oxidized at high temperature, it is better to store the nanoparticles in a refrigerator (Fig. S17†). Fe2+-ion-modified SP@mSiO2-PEG (SP@mSiO2-PEG/Fe) exhibited the highest DOX loading capacity (16.2%) among all the samples. DOX-loaded nanoparticles (SP@mSiO2-PEG/FeDOX) presented a grey color (Fig. 4A) with the highest characteristic absorption of DOX at around 500 nm (Fig. 4B).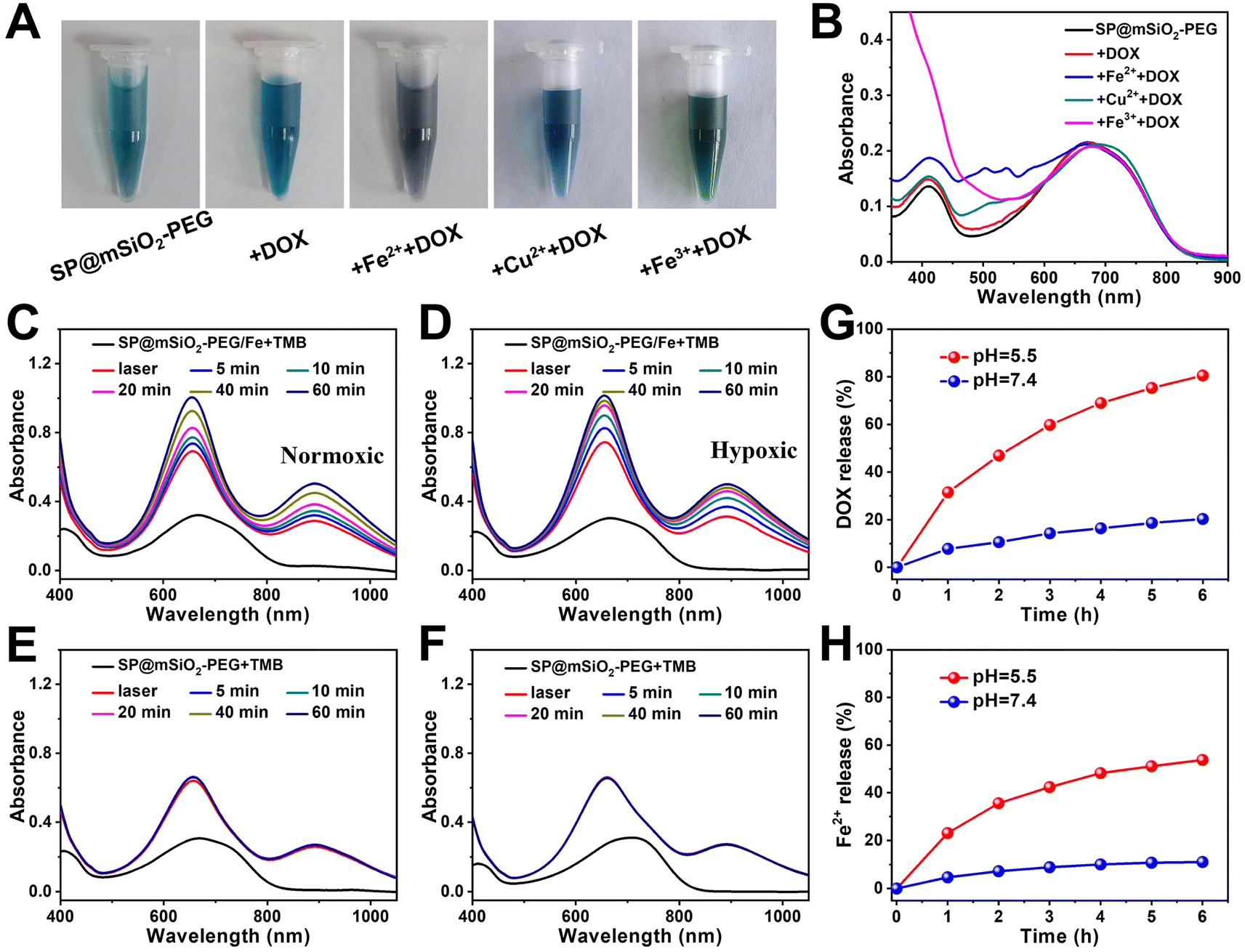 | ||
| Fig. 4 (A) Photographs and (B) corresponding absorption spectra of SP@mSiO2-PEG before and after loading with DOX, Fe2++DOX, Cu2++DOX, and Fe3++DOX. Persistent ˙OH generation from SP@mSiO2-PEG/Fe after 730 nm laser irradiation under (C) normoxic and (D) hypoxic conditions. Generation of ˙OH from SP@mSiO2-PEG after 730 nm laser irradiation under (E) normoxic and (F) hypoxic conditions. Cumulative release of (G) DOX and (H) the Fe2+ ions from SP@mSiO2-PEG/FeDOX at different pH values. | ||
In addition, it is exciting to see that the absorbance from TMB at 656 nm and 900 nm continues to increases for at least 1 h after removing the laser (Fig. 4C), indicating the persistent generation of ˙OH from SP@mSiO2-PEG/Fe, even under hypoxic conditions (Fig. 4D). For SP@mSiO2-PEG, in contrast, the absorption spectra remain almost unchanged after turning off the laser (Fig. 4E and F). The Fe2+ ions themselves also cannot change the absorbance of TMB (Fig. S18†). This means the photo-generated H2O2 was enough to trigger the Fenton reaction with Fe2+ loaded on the nanoparticles to produce ˙OH in situ. This strategy with the photo-generated H2O2 would be advantageous for precise tumor therapy, because it provides high precision and controllability. In comparison, endogenous H2O2, H2O2 generated by the glucose oxidase catalytic reaction and other H2O2 delivery methods, were relatively less controllable.20,61
The release of Fe2+ and DOX from SP@mSiO2-PEG/FeDOX exhibited a pH-dependent manner (Fig. 4H). Both of them are released much faster at lower pH, which can be beneficial for tumor therapy due to the acidic tumor microenvironment. One interesting aspect is that the simultaneous release of Fe2+ and DOX could further trigger ferroptosis to improve the therapeutic outcome.62
In vitro cytotoxic investigation
Intracellular ROS generation of SP@mSiO2-PEG in cancer cells under 730 nm laser irradiation under normoxic and hypoxic conditions was further investigated. The hypoxic environment was created with a Mitsubishi™ AnaeroPack, and intracellular hypoxia was confirmed with the hypoxia indicator ROS-ID (Fig. 5A). A 5.2-fold increase in fluorescence was observed after hypoxic treatment (Fig. 5B), indicating the successful creation of intracellular hypoxia. Different probes were applied for the selective imaging of corresponding ROS (Fig. 5C). The probes emitted weak fluorescence without laser irradiation under normoxic conditions. Their fluorescence became dramatically enhanced after exposure to a 730 nm laser (230 mW cm−2, 10 min), while under hypoxic conditions, the fluorescence from SOSG became much weaker, indicating that the generation of 1O2 was inhibited. The other probes still revealed obvious emission, demonstrating that the generation of ˙OH and H2O2 through a type-I process was still effective under hypoxic conditions. The SP@mSiO2-PEG nanoparticles were generally biocompatible without light. The cytotoxicity increased with Fe2+ modification and became even higher after DOX loading (Fig. 5D), indicating a chemotherapy effect. Under laser irradiation, SP@mSiO2-PEG exhibited remarkable cell toxicity due to the efficient PDT (Fig. 5E). The cytotoxicity was only slightly decreased under hypoxic conditions, proving the effectiveness of type-I PDT. SP@mSiO2-PEG/Fe and SP@mSiO2-PEG/FeDOX follow same trends, but the cell inhibition efficiency for tumor cells was enhanced due to the combination of photodynamic therapy and chemotherapy.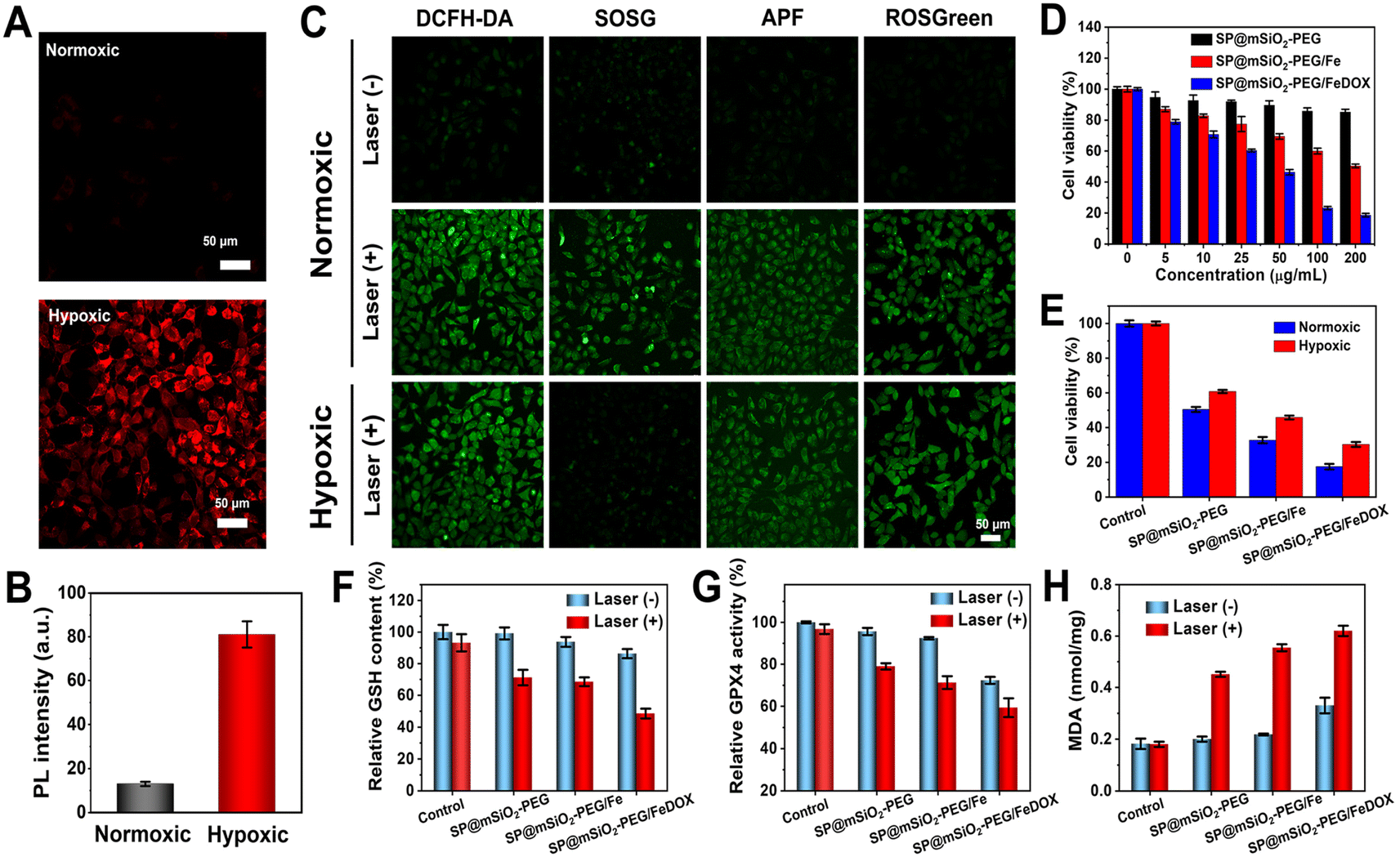 | ||
| Fig. 5 (A) Intracellular hypoxia state imaging with hypoxia indicator ROS-ID and (B) quantitative fluorescence intensities. (C) ROS detection under normoxic and hypoxic conditions with DCFH-DA, SOSG, APF, and ROSGreen as total ROS, 1O2, ˙OH, and H2O2 fluorescence probes, respectively. (D) Cell viabilities of 4T1 cells after incubation with the nanoparticles at different concentrations. (E) Cell viabilities after incubation with nanoparticles (25 μg mL−1) and exposure to a 730 nm laser (230 mW cm−2, 10 min). Intracellular (F) GSH content, (G) GPX4 activity and (H) MDA amount assays after different treatments. | ||
The mechanism for tumor cell inhibition was further studied. As shown in Fig. S19–S21,† DOX can not only induce the apoptosis of cancer cells through DNA damage but also up-regulate the intracellular H2O2 level by activating intracellular NADPH oxidase 4 (NOX4).62 The semiconducting polymer here can produce 1O2 through type-II PDT and ˙OH and H2O2 through type-I PDT. Thus, the high level of H2O2 generated from DOX and type-I PDT could further react with ferrous ions to continually produce highly cytotoxic ˙OH (Scheme 1). The up-regulated total ROS concentration leads to oxidative damage to cancer cells and initiates ferroptosis.63 Iron-loaded silica nanoparticles were also reported to induce ferroptosis in cancer cells.64 To confirm the ferroptosis mechanism, glutathione (GSH) depletion, glutathione peroxidase 4 (GPX4) down-regulation, and lipid peroxidation (LPO) were further investigated, which are considered three crucial events during ferroptosis.65 The intracellular GSH concentration in SP@mSiO2-PEG/FeDOX-treated cells under laser irradiation dropped obviously compared with other groups (Fig. 5F). Similarly, GSH-associated GPX4 was largely deactivated after incubation with SP@mSiO2-PEG/FeDOX and laser treatment (Fig. 5G). The deactivation of GPX4 can suppress the repair function of lipid antioxidants and block LPO elimination.66 Malonaldehyde (MDA) was the final product of LPO under oxidative damage.67 The MDA level for SP@mSiO2-PEG/FeDOX and laser-treated cells was also the highest among all the groups (Fig. 5H and S22†). Therefore, SP@mSiO2-PEG/FeDOX could inhibit the viability of cancer cells through apoptosis and ferroptosis mechanisms with a combination of chemotherapy and photodynamic therapy.
In vivo NIR-II phototheranostics
With their distinguished NIR-II fluorescent property, mesoporous silica-coated SP nanoparticles could be excellent in vivo imaging agents. Healthy BALB/c mouse were intravenously injected with the SP@mSiO2-PEG nanoparticles, and fluorescence images were obtained with an InGaAs camera under 808 nm laser irradiation (∼60 mW cm−2). The nanoparticles can circulate with the blood flow, and the whole-body blood vessels were unambiguously visualized (Fig. 6A). A capillary of 0.17 mm at the abdomen can be discerned (Fig. 6B), indicating a high resolution of NIR-II imaging. The feasibility of using SP@mSiO2-PEG as tumor imaging agents was then validated with 4T1 tumor bearing mice. The tumor was already visible in fluorescence images 4 h after injection, and the emission intensity continued to increase with post-injection time (Fig. 6C). At 48 h post-injection, the fluorescence intensity at the tumor site did not change too much (Fig. 6D). Note that the fluorescence intensity at the tumor was comparable with the liver at this time, indicating an efficient tumor accumulation of the nanoparticles, which was difficult to achieve for amphiphilic polymer-encapsulated fluorophores.68 We believe the high tumor accumulation was related to the long blood circulation time and passive targeting ability of the nanoparticles due to the dense PEG grafting. To confirm this point of view, the fluorescence intensities of the liver and major blood vessel at the hind limb were monitored (Fig. S23†). The liver exhibited the highest fluorescence at 48 h, which then decreased gradually. The blood vessel was still apparent at 24 h; it became fuzzy at 36 h and completely invisible after 48 h. These data were consistent with Fig. 6D. The blood circulation half-life for silica-coated nanoparticles reached 11.4 h (Fig. S23B†), which was superior for phototheranostics applications in vivo. The mice were sacrificed after 96 h, and ex vivo images confirm the accumulation of the nanoparticles in the tumor (Fig. 6E). Besides, the obvious accumulation in the liver and spleen (Fig. 6F) indicated a metabolic pathway based on the hepatobiliary system.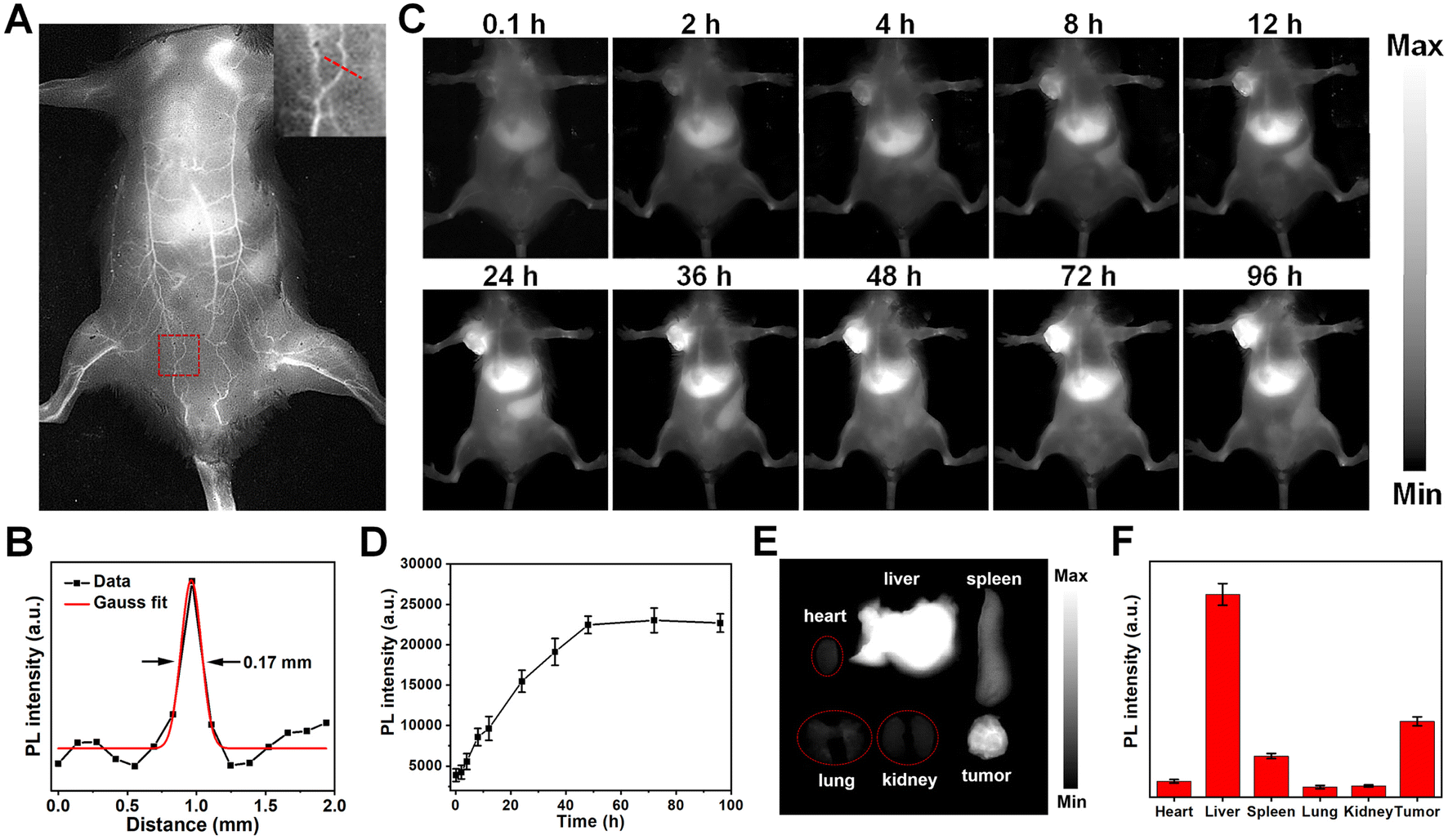 | ||
| Fig. 6 (A) In vivo NIR-II image of a mouse 20 min after intravenous injection of the SP@mSiO2-PEG nanoparticles. (B) Cross-sectional emission intensity profile along the red line in (A) and fitted to a Gaussian function. (C) Time-dependent NIR-II fluorescence imaging of a 4T1 tumor in a living mouse. (D) NIR-II fluorescence intensity of the tumor region at different time points. (E) Ex vivo fluorescence image and (F) corresponding quantitative analysis of major organs 96 h after intravenous injection. 808 nm laser with a power density of ∼60 mW cm−2 was used as the excitation source. | ||
Encouraged by the abundant accumulation of the nanoparticles in the tumor, tumor inhibition experiments were carried out with these nanoparticles in vivo. 4T1 tumor-bearing mice were divided into five groups and treated with different formulae: (i) Saline, (ii) SP@mSiO2-PEG, (iii) SP@mSiO2-PEG/FeDOX, (iv) SP@mSiO2-PEG + laser, and (v) SP@mSiO2-PEG/FeDOX + laser. Laser irradiation was performed at 48 h post-injection. The power density of the 730 nm-laser was adjusted to 230 mW cm−2 according to the maximum permissible exposure (MPE) for skin (ANSI Z136.1-2014). From Fig. 7A, the SP@mSiO2-PEG nanoparticles (group ii) did not provide any therapeutic effect, while SP@mSiO2-PEG/FeDOX (group iii) exhibited obvious anti-tumor ability due to the chemotherapy from the combination of Fe2+ and DOX. In comparison, the SP@mSiO2-PEG nanoparticles can provide a slightly better tumor inhibition ratio under laser exposure (group iv). At the adopted power density, the temperature of the tumors increased to only around 38.5 °C for group iv (Fig. S24†), which was below the temperature required for photothermal therapy.35 The poor photothermal effect can be related to the inhibited nonradiative decay and low power density of the laser. Therefore, the anti-tumor effect of SP@mSiO2-PEG observed here results mainly from the PDT effect rather than the photothermal effect. Under same conditions, SP@mSiO2-PEG/FeDOX revealed the highest anti-tumor activity with laser irradiation, which can be attributed to the rational combination of chemotherapy and persistent photodynamic therapy. The ex vivo tumor images (Fig. 7B) after treatment displayed same results, and a tumor inhibition ratio of ∼85% was obtained for group v (Fig. 7C). Histopathological tissue analysis of tumor tissues also confirmed severe cell apoptosis and necrosis after treatment (Fig. 7E). The high anti-tumor effect was believed to be related to the effective photosensitization under hypoxic conditions and subsequent persistent ˙OH generation, which enhanced apoptosis and ferroptosis. The body weights of the mice during treatment did not show a significant difference for each group (Fig. 7D), indicating limited side effects for the fabricated nanoplatform. In addition, hematoxylin and eosin (H&E) staining showed no sign of damage in the main organs of the mice after treatment (Fig. S25†), demonstrating the safety of semiconducting polymer-based nanoparticles and utilization of the laser. These results show convincingly that mesoporous silica-coated semiconducting polymers with restricted conformation are a safe and efficient nanoplatform for tumor phototheranostics.
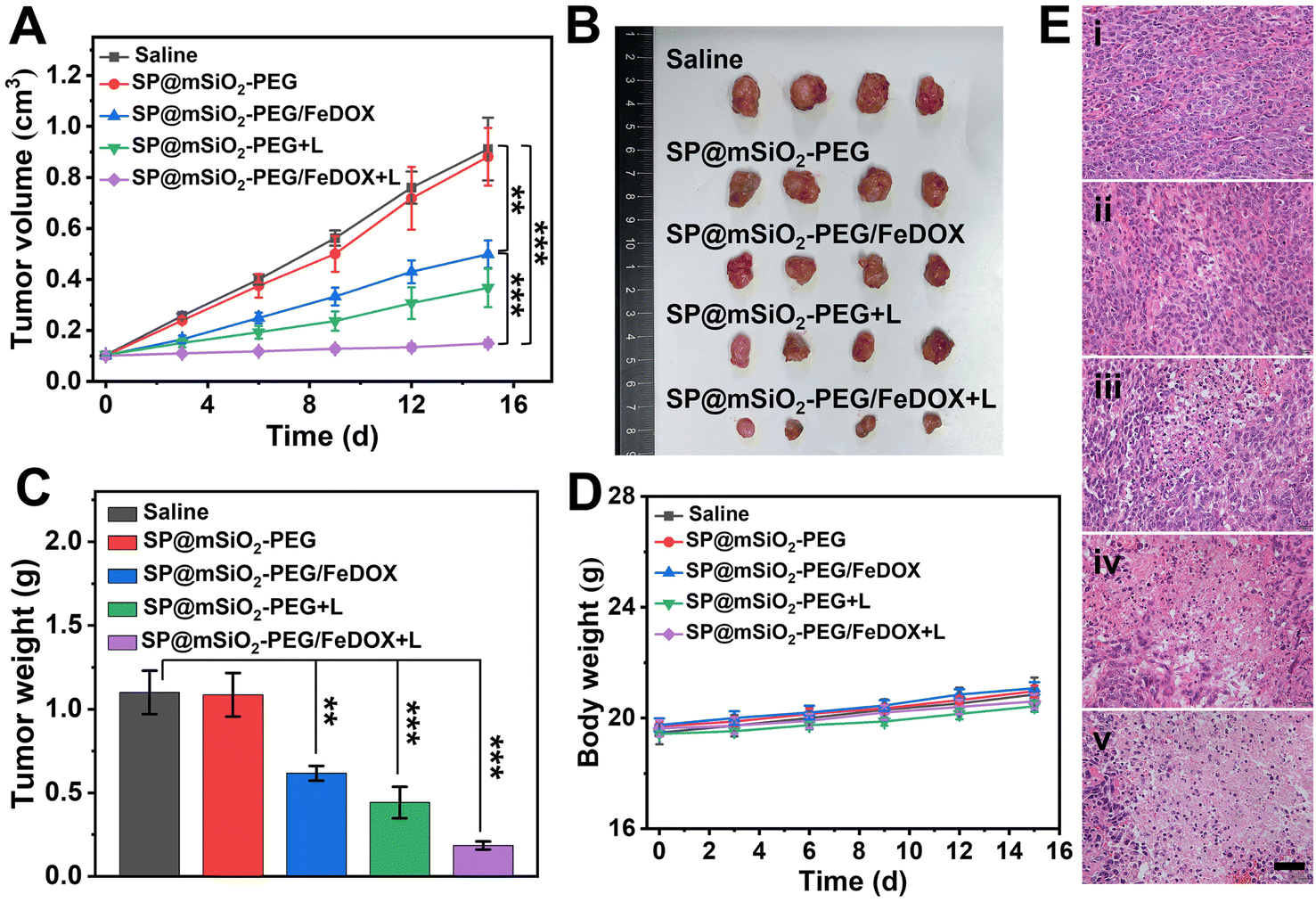 | ||
| Fig. 7 (A) Time-dependent tumor volumes of 4T1 tumor-bearing mice after different treatments: (i) saline, (ii) SP@mSiO2-PEG, (iii) SP@mSiO2-PEG/FeDOX, (iv) SP@mSiO2-PEG + laser, and (v) SP@mSiO2-PEG/FeDOX + laser. (B) Photographs and (C) weights of isolated tumors after treatment. (D) Changes in the body weights of the mice during treatment. (E) H&E staining of tumor slides after treatment. Scale bars: 50 μm. (**p < 0.01 and ***p < 0.001). | ||
Conclusions
In summary, we have reported the encapsulation of semiconducting polymers within a mesoporous silica matrix, which leads to 3–5 fold enhancement in NIR-II fluorescence quantum yield and ROS generation. A femtosecond spectroscopy study revealed that the excited-state lifetime of PCPDTBT in the silica matrix was one order of magnitude longer than that in a F127 micelle, which resulted from significantly restricted intermolecular and intramolecular interactions of the confined SP. Chemotherapy drug DOX was efficiently loaded onto the nanoparticles after modification with the Fe2+ ions. The photo-induced ˙OH and H2O2 through type-I PDT here can be more competent against tumor hypoxia. Meanwhile, the photo-generated H2O2 was enough to react with the Fe2+ ions in situ to produce ˙OH persistently after 730 nm laser irradiation. Thus, SP@mSiO2-PEG/FeDOX nanoparticles can be used for NIR-II imaging guided chemotherapy and persistent photodynamic therapy. This work not only provides a general approach to fabricating semiconducting polymer-based phototheranostic nanoplatforms but also inspires the use of the photo-generated H2O2 for persistent tumor therapy.Data availability
The data supporting this article have been included in the ESI.†Author contributions
F. Lu, Q. Wang, and Q. Fan conceived and designed the study. L. Li, M. Zhang, C. Yu, and F. Cheng performed the materials synthesis, in vivo and in vitro experiments. Y. Pan and W. Hu performed the femtosecond spectroscopy characterizations. X. Lu provided technical support. F. Lu contributed to the writing of the manuscript. Q. Wang and Q. Fan revised the manuscript. All the authors discussed the results.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 21975131, 22175098, and 52373142), China Postdoctoral Science Foundation (2023M741804), Natural Science Foundation of Nanjing University of Posts and Telecommunications (NY223100), and Synergetic Innovation Center for Organic Electronics and Information Displays.Notes and references
- M. Kolarikova, B. Hosikova, H. Dilenko, K. Barton-Tomankova, L. Valkova, R. Bajgar, L. Malina and H. Kolarova, Med. Res. Rev., 2023, 43, 717 CrossRef CAS PubMed.
- J. H. Correia, J. A. Rodrigues, S. Pimenta, T. Dong and Z. C. Yang, Pharmaceutics, 2021, 13, 1332 CrossRef CAS PubMed.
- M. Overchuk, R. A. Weersink, B. C. Wilson and G. Zheng, ACS Nano, 2023, 17, 7979 CrossRef CAS PubMed.
- X. S. Li, J. F. Lovell, J. Yoon and X. Y. Chen, Nat. Rev. Clin. Oncol., 2020, 17, 657 CrossRef PubMed.
- L. Q. Li, C. Shao, T. Liu, Z. C. Chao, H. L. Chen, F. Xiao, H. M. He, Z. X. Wei, Y. L. Zhu, H. Wang, X. D. Zhang, Y. T. Wen, B. Yang, F. He and L. L. Tian, Adv. Mater., 2020, 32, 2003471 CrossRef CAS PubMed.
- J. H. Zou, L. Li, J. W. Zhu, X. C. Li, Z. Yang, W. Huang and X. Y. Chen, Adv. Mater., 2021, 33, 2103627 CrossRef CAS PubMed.
- W. Tang, Z. Yang, L. C. He, L. M. Deng, P. Fathi, S. J. Zhu, L. Li, B. Shen, Z. T. Wang, O. Jacobson, J. B. Song, J. H. Zou, P. Hu, M. Wang, J. Mu, Y. Y. Cheng, Y. Y. Ma, L. G. Tang, W. P. Fan and X. Y. Chen, Nat. Commun., 2021, 12, 523 CrossRef CAS PubMed.
- Y. Zhu, D. Jin, M. M. Liu, Y. Dai, L. Li, X. W. Zheng, L. L. Wang, A. Z. Shen, J. N. Yu, S. S. Wu, Y. Wu, K. Zhong, J. J. Cheng and Y. Z. Liu, Small, 2022, 18, 2200116 CrossRef CAS PubMed.
- S. J. Wang, P. Huang, L. M. Nie, R. J. Xing, D. B. Liu, Z. Wang, J. Lin, S. H. Chen, G. Niu, G. M. Lu and X. Y. Chen, Adv. Mater., 2013, 25, 3055 CrossRef CAS PubMed.
- J. H. Zou, J. W. Zhu, Z. Yang, L. Li, W. P. Fan, L. C. He, W. Tang, L. M. Deng, J. Mu, Y. Y. Ma, Y. Y. Cheng, W. Huang, X. C. Dong and X. Y. Chen, Angew. Chem., Int. Ed., 2020, 59, 8833 CrossRef CAS PubMed.
- Y. Gu, H. J. Lai, Z. Y. Chen, Y. L. Zhu, Z. Z. Sun, X. Lai, H. T. Wang, Z. X. Wei, L. Chen, L. M. Huang, Y. Z. Zhang, F. He and L. L. Tian, Angew. Chem., Int. Ed., 2023, 62, e202303476 CrossRef CAS PubMed.
- T. K. Luo, K. Y. Ni, A. Culbert, G. X. Lan, Z. Li, X. M. Jiang, M. Kaufmann and W. B. Lin, J. Am. Chem. Soc., 2020, 142, 7334 CrossRef CAS PubMed.
- G. Hong, Y. Zou, A. L. Antaris, S. Diao, D. Wu, K. Cheng, X. Zhang, C. Chen, B. Liu, Y. He, J. Z. Wu, J. Yuan, B. Zhang, Z. Tao, C. Fukunaga and H. Dai, Nat. Commun., 2014, 5, 4206 CrossRef CAS PubMed.
- Y. W. Yu, S. Wu, L. Zhang, S. D. Xu, C. H. Dai, S. M. Gan, G. F. Xie, G. X. Feng and B. Z. Tang, Biomaterials, 2022, 280, 121255 CrossRef CAS PubMed.
- H. Y. Huang, W. S. Xie, Q. Wan, L. C. Mao, D. N. Hu, H. Sun, X. Y. Zhang and Y. Wei, Adv. Sci., 2022, 9, 2104101 CrossRef CAS PubMed.
- X. F. Miao, W. Y. Yao, R. Z. Chen, M. X. Jia, C. Ren, H. Zhao, T. C. He, Q. L. Fan and W. B. Hu, Adv. Mater., 2023, 35, 2301739 CrossRef CAS PubMed.
- K. K. Wen, H. Tan, Q. Peng, H. Chen, H. Ma, L. Wang, A. D. Peng, Q. Q. Shi, X. D. Cai and H. Huang, Adv. Mater., 2022, 34, 2108146 CrossRef CAS PubMed.
- K. Yang, F. Long, W. Liu, Z. Q. Zhang, S. J. Zhao, B. H. Wang, Y. P. Zou, M. H. Lan, J. Yuan, X. Z. Song and C. W. Lin, ACS Appl. Mater. Interfaces, 2022, 14, 18043 CrossRef CAS PubMed.
- S. Lei, J. Zhang, N. T. Blum, M. Li, D. Y. Zhang, W. M. Yin, F. Zhao, J. Lin and P. Huang, Nat. Commun., 2022, 13, 1298 CrossRef CAS PubMed.
- L. S. Lin, T. Huang, J. B. Song, X. Y. Ou, Z. T. Wang, H. Z. Deng, R. Tian, Y. J. Liu, J. F. Wang, Y. Liu, G. C. Yu, Z. J. Zhou, S. Wang, G. Niu, H. H. Yang and X. Y. Chen, J. Am. Chem. Soc., 2019, 141, 9937 CrossRef CAS PubMed.
- C. Liu, J. Xing, O. U. Akakuru, L. J. Luo, S. Sun, R. F. Zou, Z. S. Yu, Q. L. Fang and A. G. Wu, Nano Lett., 2019, 19, 5674 CrossRef CAS PubMed.
- Y. N. Zhang, C. S. Pan, G. M. Bian, J. Xu, Y. M. Dong, Y. Zhang, Y. Lou, W. X. Liu and Y. F. Zhu, Nat. Energy, 2023, 8, 361 CrossRef CAS.
- Y. D. Wang, F. C. Gao, X. F. Li, G. M. Niu, Y. F. Yang, H. Li and Y. Y. Jiang, J. Nanobiotechnol., 2022, 20, 69 CrossRef CAS PubMed.
- S. Albrecht, S. Janietz, W. Schindler, J. Frisch, J. Kurpiers, J. Kniepert, S. Inal, P. Pingel, K. Fostiropoulos, N. Koch and D. Neher, J. Am. Chem. Soc., 2012, 134, 14932 CrossRef CAS PubMed.
- F. Lu, L. Yang, Y. J. Ding and J. J. Zhu, Adv. Funct. Mater., 2016, 26, 4778 CrossRef CAS.
- F. Lu, T. L. Doane, J. J. Zhu and C. Burda, Chem. Commun., 2014, 50, 642 RSC.
- C. F. Wu, S. J. Hansen, Q. O. Hou, J. B. Yu, M. Zeigler, Y. H. Jin, D. R. Burnham, J. D. McNeill, J. M. Olson and D. T. Chiu, Angew. Chem., Int. Ed., 2011, 50, 3430 CrossRef CAS PubMed.
- J. A. Hubbell and A. Chilkoti, Science, 2012, 337, 303 CrossRef PubMed.
- T. F. Abelha, P. R. Neumann, J. Holthof, C. A. Dreiss, C. Alexander, M. Green and L. A. Dailey, J. Mater. Chem. B, 2019, 7, 5115 RSC.
- C. Szymanski, C. F. Wu, J. Hooper, M. A. Salazar, A. Perdomo, A. Dukes and J. McNeill, J. Phys. Chem. B, 2005, 109, 8543 CrossRef CAS PubMed.
- H. Tan, Y. Zhang, M. Wang, Z. X. Zhang, X. H. Zhang, A. M. Yong, S. Y. Wong, A. Y. C. Chang, Z. K. Chen, X. Li, M. Choolani and J. Wang, Biomaterials, 2012, 33, 237 CrossRef CAS PubMed.
- W. H. Zeng, L. Y. Wu, Y. D. Sun, Y. Q. Wang, J. F. Wang and D. J. Ye, Small, 2021, 17, 2101924 CrossRef CAS PubMed.
- S. J. Zhu, Z. B. Hu, R. Tian, B. C. Yung, Q. L. Yang, S. Zhao, D. O. Kiesewetter, G. Niu, H. T. Sun, A. L. Antaris and X. Y. Chen, Adv. Mater., 2018, 30, 1802546 CrossRef PubMed.
- Z. C. Cai, L. Zhu, M. Q. Wang, A. W. Roe, W. Xi and J. Qian, Theranostics, 2020, 10, 4265 CrossRef CAS PubMed.
- H. J. Zhu, J. C. Li, X. Y. Qi, P. Chen and K. Y. Pu, Nano Lett., 2018, 18, 586 CrossRef CAS PubMed.
- W. Zhou, Y. Chen, Y. T. Zhang, X. Y. Xin, R. T. Li, C. Xie and Q. L. Fan, Small, 2020, 16, 1905641 CrossRef CAS PubMed.
- D. Di Nuzzo, A. Aguirre, M. Shahid, V. S. Gevaerts, S. C. J. Meskers and R. A. J. Janssen, Adv. Mater., 2010, 22, 4321 CrossRef CAS PubMed.
- J. C. Li and K. Y. Pu, Acc. Chem. Res., 2020, 53, 752 CrossRef CAS PubMed.
- X. O. Gong, M. H. Tong, S. H. Park, M. Liu, A. Jen and A. J. Heeger, Sensors, 2010, 10, 6488 CrossRef CAS PubMed.
- Y. Nosaka and A. Y. Nosaka, Chem. Rev., 2017, 117, 11302 CrossRef CAS PubMed.
- F. Gong, L. Cheng, N. Yang, O. Betzer, L. Feng, Q. Zhou, Y. Li, R. Chen, R. Popovtzer and Z. Liu, Adv. Mater., 2019, 31, 1900730 CrossRef PubMed.
- Y. J. Sang, F. F. Cao, W. Li, L. Zhang, Y. W. You, Q. Q. Deng, K. Dong, J. S. Ren and X. G. Qu, J. Am. Chem. Soc., 2020, 142, 5177 CrossRef CAS PubMed.
- J. Ma, X. X. Peng, Z. X. Zhou, H. Yang, K. Q. Wu, Z. Z. Fang, D. Han, Y. F. Fang, S. Q. Liu, Y. F. Shen and Y. J. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202210856 CrossRef CAS PubMed.
- E. Herz, T. Marchincin, L. Connelly, D. Bonner, A. Burns, S. Switalski and U. Wiesner, J. Fluoresc., 2010, 20, 67 CrossRef CAS PubMed.
- H. Zhu, Y. Fang, X. Zhen, N. Wei, Y. Gao, K. Q. Luo, C. Xu, H. Duan, D. Ding, P. Chen and K. Pu, Chem. Sci., 2016, 7, 5118 RSC.
- M. Maiuri, M. Garavelli and G. Cerullo, J. Am. Chem. Soc., 2020, 142, 3 CrossRef CAS PubMed.
- B. Z. Zheng, D. N. Zhong, T. T. Xie, J. Zhou, W. L. Li, A. Ilyas, Y. H. Lu, M. Zhou and R. R. Deng, Chem, 2021, 7, 1615 CAS.
- B. Cai, H. W. Song, A. Brnovic, M. V. Pavliuk, L. Hammarstrom and H. N. Tian, J. Am. Chem. Soc., 2023, 145, 18687 CrossRef CAS PubMed.
- C. Yin, H. Zhang, B. Sun, S. Y. Chen, X. Y. Jiang, X. F. Miao, P. F. Sun, W. B. Hu, Q. L. Fan and W. Huang, Adv. Funct. Mater., 2021, 31, 2106575 CrossRef CAS.
- W. B. Hu, P. N. Prasad and W. Huang, Acc. Chem. Res., 2021, 54, 697 CrossRef CAS PubMed.
- Y. S. Liu, M. J. Gu, Q. H. Ding, Z. Y. Zhang, W. X. Gong, Y. C. Yuan, X. F. Miao, H. L. Ma, X. C. Hong, W. B. Hu and Y. L. Xiao, Angew. Chem., Int. Ed., 2023, 62, e202214875 CrossRef CAS PubMed.
- H. K. Zhang, Z. Zhao, A. T. Turley, L. Wang, P. R. McGonigal, Y. J. Tu, Y. Y. Li, Z. Y. Wang, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Adv. Mater., 2020, 32, 2001457 CrossRef CAS PubMed.
- J. Kang, S. Lhee, J. K. Lee, R. N. Zare and H. G. Nam, Sci. Rep., 2020, 10, 16859 CrossRef CAS PubMed.
- L. K. McKenzie, H. E. Bryant and J. A. Weinstein, Coord. Chem. Rev., 2019, 379, 2 CrossRef CAS.
- D. L. Sai, J. Lee, D. L. Nguyen and Y. P. Kim, Exp. Mol. Med., 2021, 53, 495 CrossRef CAS PubMed.
- A. Cravcenco, Y. Yu, F. Edhborg, J. F. Goebel, Z. Takacs, Y. Z. Yang, B. Albinsson and K. Börjesson, J. Am. Chem. Soc., 2021, 143, 19232 CrossRef CAS PubMed.
- P. Holzmeister, E. Pibiri, J. J. Schmied, T. Sen, G. P. Acuna and P. Tinnefeld, Nat. Commun., 2014, 5, 5356 CrossRef CAS PubMed.
- T. Liu, X. Y. Zhang, H. L. Zhang, H. Zhao, Z. G. Zhang and Y. Tian, Opt. Express, 2020, 28, 25757 CrossRef CAS PubMed.
- Y. S. Lin, N. Abadeer, K. R. Hurley and C. L. Haynes, J. Am. Chem. Soc., 2011, 133, 20444 CrossRef CAS PubMed.
- Z. P. Zhen, W. Tang, H. M. Chen, X. Lin, T. Todd, G. Wang, T. Cowger, X. Y. Chen and J. Xie, ACS Nano, 2013, 7, 4830 CrossRef CAS PubMed.
- L. H. Fu, Y. L. Wan, C. Qi, J. He, C. Y. Li, C. Yang, H. Xu, J. Lin and P. Huang, Adv. Mater., 2021, 33, 2006892 CrossRef CAS PubMed.
- C. C. Xue, M. H. Li, Y. Zhao, J. Zhou, Y. Hu, K. Y. Cai, Y. L. Zhao, S. H. Yu and Z. Luo, Sci. Adv., 2020, 6, eaax1346 CrossRef CAS PubMed.
- S. S. He, J. Yu, M. K. Xu, C. Zhang, C. Xu, P. H. Cheng and K. Y. Pu, Angew. Chem., Int. Ed., 2023, 62, e202310178 CrossRef CAS PubMed.
- S. E. Kim, L. Zhang, K. Ma, M. Riegman, F. Chen, I. Ingold, M. Conrad, M. Z. Turker, M. H. Gao, X. J. Jiang, S. Monette, M. Pauliah, M. Gonen, P. Zanzonico, T. Quinn, U. Wiesner, M. S. Bradbury and M. Overholtzer, Nat. Nanotechnol., 2016, 11, 977 CrossRef CAS PubMed.
- B. R. Stockwell, J. P. F. Angeli, H. Bayir, A. I. Bush, M. Conrad, S. J. Dixon, S. Fulda, S. Gascón, S. K. Hatzios, V. E. Kagan, K. Noel, X. J. Jiang, A. Linkermann, M. E. Murphy, M. Overholtzer, A. Oyagi, G. C. Pagnussat, J. Park, Q. Ran, C. S. Rosenfeld, K. Salnikow, D. L. Tang, F. M. Torti, S. V. Torti, S. Toyokuni, K. A. Woerpel and D. D. Zhang, Cell, 2017, 171, 273 CrossRef CAS PubMed.
- D. P. Huang, H. Q. Huang, M. L. Li, J. L. Fan, W. Sun, J. J. Du, S. R. Long and X. J. Peng, Adv. Funct. Mater., 2022, 32, 2208105 CrossRef CAS.
- J. H. Liu, W. Zhang, C. M. Zhou, M. M. Li, X. Wang, W. Zhang, Z. Z. Liu, L. L. Wu, T. D. James, P. Li and B. Tang, J. Am. Chem. Soc., 2022, 144, 13586 CrossRef CAS PubMed.
- J. W. Liu, Y. W. Xiong, Y. C. Gao, X. P. Xu, K. Chen, Q. M. Shen, W. Huang, Q. L. Fan and Q. Wang, Small, 2023, 19, 2205640 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc01609b |
| This journal is © The Royal Society of Chemistry 2024 |