
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReal-time observation of sub-100-fs charge and energy transfer processes in DNA dinucleotides†
Vasilis
Petropoulos
 a,
Lara
Martinez-Fernandez
*b,
Lorenzo
Uboldi
a,
Margherita
Maiuri
a,
Giulio
Cerullo
*ac,
Evangelos
Balanikas
d and
Dimitra
Markovitsi
*e
a,
Lara
Martinez-Fernandez
*b,
Lorenzo
Uboldi
a,
Margherita
Maiuri
a,
Giulio
Cerullo
*ac,
Evangelos
Balanikas
d and
Dimitra
Markovitsi
*e
aDipartimento di Fisica, Politecnico di Milano, Piazza Leonardo da Vinci 32, I-20133 Milano, Italy. E-mail: giulio.cerullo@polimi.it
bInstituto de Química Física Blas Cabrera, Consejo Superior de Investigaciones Científicas, Calle Serrano 119, Madrid, 28006, Spain. E-mail: lmartinez@iqf.csic.es
cIstituto di Fotonica e Nanotecnologie-CNR, Piazza Leonardo da Vinci 32, I-20133 Milano, Italy
dDepartment of Physical Chemistry, University of Geneva, CH-1211 Geneva-4, Switzerland
eUniversité Paris-Saclay, CNRS, Institut de Chimie Physique, UMR8000, 91405 Orsay, France. E-mail: dimitra.markovitsi@universite-paris-saclay.fr
First published on 27th June 2024
Abstract
Using as showcase the DNA dinucleotide 5′-dTpdG-3′, in which the thymine (T) is located at the 5′ end with respect to the guanine (G), we study the photoinduced electronic relaxation of coupled chromophores in solution with an unprecedented refinement. On the one hand, transient absorption spectra are recorded from 20 fs to 45 ps over the 330–650 nm range with a temporal resolution of 30 fs; on the other hand, quantum chemistry calculations determine the ground state geometry of the 4 possible conformers with stacked nucleobases, the associated Franck–Condon states, and map the relaxation pathways leading to excited state minima. Important spectral changes occurring before 100 fs are correlated with concomitant G+ → T− charge transfer and T → G energy transfer processes. The lifetime of the excited charge transfer state is only 5 ps and the absorption spectrum of a long-lived nπ*T state is detected. Our experimental results match the transient spectral properties computed for the anti–syn conformer of 5′-dTpdG-3′, which is characterized by the lowest ground state energy and differs from that encountered in B-form duplexes.
Introduction
Excited state relaxation in DNA is studied in relation with its damage induced by absorption of UV radiation1 and in view of future developments of DNA-based label-free optoelectronic devices.2,3 In both cases, charge and energy transfer processes play a key role. For example, the formation of the carcinogenic (6–4) photo-adducts,4 as well as charge carrier generation by low-energy photons,5 involve the population of an excited charge transfer (CT) state,6 in which an atomic charge has been transferred between two stacked nucleobases. Similarly, excitation energy transfer (ET) among nucleobases has been shown to provoke remote photodamage.7Numerous transient absorption (TA) studies have characterized the decay of CT states in various types of DNA multimers, ranging from dinucleotides and single strands to duplexes and guanine quadruplexes (G-Quadruplexes).8–22 However, the formation of these states from the excited states initially populated upon photon absorption (Franck–Condon states) has not been observed in real time. Likewise, ET was inferred from the fluorescence anisotropy values detected once the process has occurred.23 The main reason for the lack of direct experimental observation is that these processes are faster than the time resolution of the ultrafast spectroscopy setups used to date in these studies, which is typically in the 150–300 fs range.
A second, albeit equally important, difficulty is conceptual. So far, the data derived from TA experiments have been analysed using multiexponential functions, and time constants were correlated with monomer excited states and CT states (often referred as exciplexes/excimers). However, this approach becomes problematic when wave packets evolve along potential energy surfaces (PES). The latter do not fulfil the requirements for exponential decays, which are valid when the interactions of the chromophore with its environment are uniform in the three-dimensional space. They fall instead in the category of restricted geometries, in which dynamics has been discussed for a large variety of processes and systems.24–29
Here, we report the first comprehensive study monitoring the progression of electronic excitations from the Franck–Condon states to the PES minima in DNA multimers. We use as a showcase the DNA dinucleotide 5′-dTpdG-3′, abbreviated as TG, in which the thymine (T) is located at the 5′ end and the guanine (G) at the 3′ end. The investigation of dinucleotides by TA8–14 spectroscopy has proved helpful in disentangling processes in larger systems, in which a multitude of interactions are operative. In addition to being a convenient model system, the 5′-dTpdG-3′ sequence is frequently encountered in G-Quadruplexes. Thymines located at the 5′ position with respect to the G-core have important implications on the geometry adopted by these four-stranded structures.30
Our TA experiments are performed with an unprecedented combination of temporal resolution and spectral coverage;31TA spectra (TAS) following resonant photoexcitation by ≈25 fs pulses at 266 nm are recorded from 20 fs to 45 ps over the 330–650 nm range with a temporal resolution of ≈30 fs, as discussed in Section 1 of the ESI.† In parallel, quantum chemistry calculations determine the associated Franck–Condon excited states of the four possible conformers with stacked nucleobases, and map the relaxation pathways leading to excited state minima for all of them. The TAS corresponding to the most stable conformer are also computed. We show that the evolution from the Franck–Condon states toward these minima is manifested by an ultrafast spectral shift occurring with half-time (τ1/2) of 50 fs. Moreover, along with the formation of the G+ → T−CT state, we detect an ultrafast T → G ET. In a more general way, this is the first comprehensive study of photoinduced processes on bichromophoric systems performed with such refinement, i.e. detecting several transient species over a broad spectral range at early times and identifying them via the shapes of their TAS.
Results and discussion
Ground state properties
Fig. 1a shows the steady-state absorption spectrum of TG together with that of an equimolar mixture of the mononucleosides thymidine (dT) and deoxyguanosine (dG), their intensity being normalized at the peak wavelength of the laser pulse (266 nm). At this wavelength the molar absorption coefficients of dT and dG are the same.32 The maximum of the TG spectrum is slightly blue-shifted with respect to that of monomers (254 vs. 257 nm) and its intensity is somewhat higher, suggesting chromophore coupling. Base stacking in DNA is commonly detected by changes in the molar absorption coefficient upon heating (hypochromism/hyperchromism). This temperature dependence is explained by the fact that orbital overlap among stacked nucleobases leads to coupling between ππ* and CT transitions.33,34 Such changes are indeed observed in the difference of the absorption spectra recorded for TG at 95 °C and 23 °C (Fig. 1b), confirming base stacking. As reported in the literature,9,14 these changes in dinucleotides do not exceed a few percent.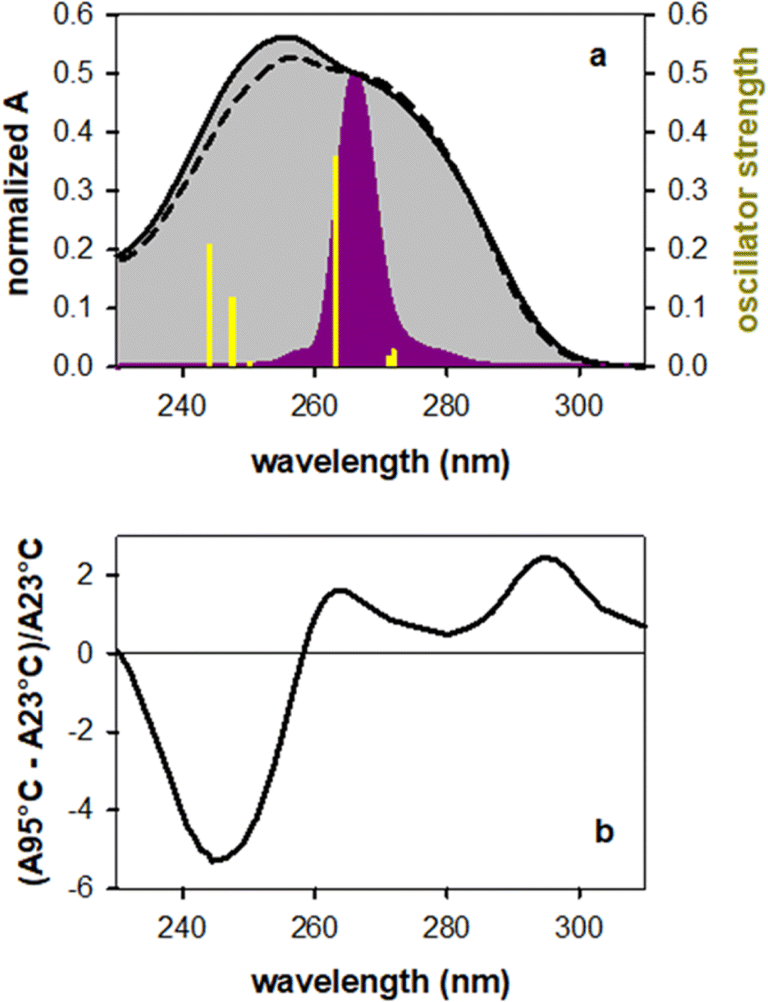 | ||
| Fig. 1 (a) Steady-state absorption spectra recorded for TG (solid lines) and an equimolar mixture of dT and dG (dashed lines) normalized at the peak wavelength (266 nm) of the exciting laser pulse (violet); yellow sticks correspond to the electronic transitions computed for the anti–synTG conformer. (b) Difference between the steady-state absorption spectra recorded for TG at 95° (A95 °C) and at 23 °C (A23 °C), divided by (A23 °C). | ||
In order to determine the stacking patterns, we optimized the four possible stacking geometries, anti–anti, anti–syn, syn–anti and syn–syn (Fig. 2), where anti and syn refer to the position of each nucleobase (in the order T-G) with respect to the angle of the glycosidic bond (Fig. S2†), associated with the deoxyribose moiety.
 | ||
| Fig. 2 Ground state structures of the four possible conformers with stacked nucleobases computed for TG and their relative energies (ΔG, in red). Thymine in blue; guanine in green. The dihedral angles that determine the syn and anti conformations are indicated in black. | ||
The energy of the anti–syn conformer is lower, respectively, by 0.06, 0.19 and 0.17 eV than those of the three other conformers (Fig. 2 and Table S1†). These values are higher than the thermal energy at room temperature (0.025 eV), making anti–synTG the most stable conformer. Therefore, we show detailed results for this conformer. However, in order to get a global picture, we also compute the Franck–Condon excited states and the associated relaxation pathways for the syn–anti and syn–syn conformers, and refine those of the anti–antiTG, whose properties were reported in ref. 35.
The properties of the 6 Franck–Condon states of anti–synTG are shown in Table 1 and the difference in the electronic density between their ground and excited state in Fig. S3.† Among them, S3, S5 and S6, having an oscillator strength higher than 0.1, are most likely to be populated by the exciting laser pulse. S3 is a ππ*T state, encompassing a very weak (0.08 a.u.) G+ → T−CT. A larger CT (0.57 a.u.) characterizes the S5 state, while S6 results from a mixing of the ππ*G (La) state with weak CT (0.20 a.u.). The omnipresence of the CT is understandable, because G and dT are the nucleobases with the highest oxidation and the lowest reduction potential, respectively.36 All 6 states are represented as sticks in Fig. 1a. We note that the energy of the S5 state coincides with that of the ππ*(Lb) state in the free guanosine chromophore, which is readily excited at 267 nm.32,37–39 Interestingly, no ππ*G(Lb) was identified for anti–synTG in the examined spectral range.
| State | Character | VAE (eV) | f | δ (a.u.) |
|---|---|---|---|---|
| S1 | nπ*(T) + ππ*G(La) | 5.21 | 0.0295 | 0.01 |
| S2 | ππ*G(La) + nπ*(T) | 5.22 | 0.0192 | 0.02 |
| S3 | ππ*T + G → T CT | 5.36 | 0.3573 | 0.08 |
| S4 | nπ*G(La) | 5.61 | 0.0093 | 0.01 |
| S5 | G → T CT | 5.66 | 0.1188 | 0.57 |
| S6 | ππ*G(La) + G → T CT | 5.73 | 0.2099 | 0.20 |
The properties of the Franck–Condon states of the three other conformers are presented in Tables S2, S3 and S4.† By comparing these states determined for all four conformers, we remark that the CT character is favoured when at least one base presents syn orientation. Indeed, no state with significant (>0.3 a.u.) CT is encountered below the S10 for the anti–anti conformer, whereas for the anti–syn and syn–syn conformers the S5/S6 states are characterized by a CT contribution. This trend is more pronounced in the case of the syn–anti conformer, for which the lowest-lying bright ππ* states (S2 and S4) exhibit modest (0.3–0.4 a.u.) CT character.
Transient absorption spectra and dynamics
The experimental TAS recorded for TG, presented in Fig. 3, consist of a broad photoinduced absorption (PA) band whose shape and intensity evolve with time. Their comparison with the TAS of an equimolar mixture of dT and dG at selected times reveals important differences (Fig. 3a), further confirming the existence of significant electronic coupling between the nucleobases, in line with the steady-state spectra in Fig. 1. In particular, the negative signal, stemming from stimulated emission (SE) of the thymidine chromophore (Fig. S4†), already reported in the literature,11,40 is completely absent from the TAS of the dinucleotide. Thus, it appears that the contribution of the ππ*T state, either correlated with unstacked nucleobases or present as a localized state within stacked ones, is significantly lower than what is expected for a solution containing 50% dT chromophores. However, we cannot rule out the presence of a weak SE, peaking below 400 nm, which may overlap with the UV PA band and decrease its intensity. Consequently, at early times, we focus mainly on the spectral changes in the visible spectral range.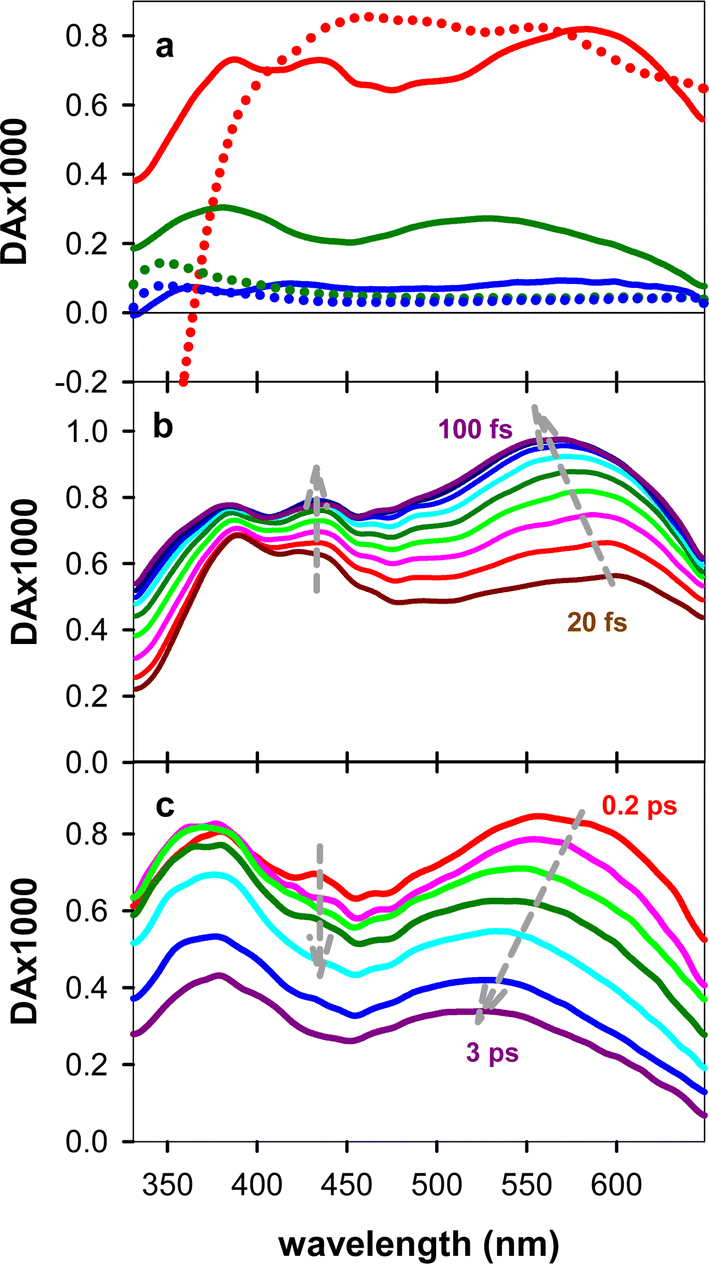 | ||
| Fig. 3 TAS recorded for TG (solid lines) and an equimolar mixture of dT and dG (dots). (a) 50 fs (red), 5 ps (green) and 40 ps (blue). (b) From 20 to 100 fs with 10 fs steps. (c) 0.2 ps (red), 0.3 ps (pink), 0.4 ps (light green), 0.6 ps (dark green), 1 ps (cyan), 2 ps (blue) and 3 ps (violet). | ||
Focusing on the evolution of the TGTAS, we observe that, at early times (Fig. 3b), their intensity increases with the time, while the maximum of the low-energy band moves from 602 to 565 nm. The dynamics of the shift can be approximated by a power law αt−0.04 (Fig. 4a), the τ1/2 being 50 fs. After 100 fs, the intensity in the visible starts decreasing slowly, while the shift toward shorter wavelengths persists, albeit at a significantly slower rate (Fig. 3c). In this case, the spectral shift can be described by a mono-exponential function with a time constant of 0.61 ± 0.08 ps (Fig. 4b). The maximum of the low-energy band is located at 530 nm at 3 ps. After this time, when the monomer signals have practically disappeared, the decay of the low-energy band is also described by a mono-exponential function with a time constant of 5.44 ± 0.03 ps (Fig. 4c). Following this decay, a long-lived background PA remains (Fig. 3a).
 | ||
| Fig. 4 Photo-induced dynamics (red circles) observed for TG over 3 timescales. (a) and (b) peak position (λmax) of the low-energy band. (c) Decay of the TA signal at 570 nm. Green lines correspond to fits with a power law (a) and mono-exponential (b and c) functions. The blue line in (a) indicates τ1/2. | ||
Computed relaxation paths
In order to get an insight into the origin of the above-described spectral evolution, we computationally mapped the deactivation pathways for the first 5 low-energy Franck–Condon states, plus the one exhibiting the largest charge transfer. For anti–synTG, we identified three minima in the PES of the first excited states: min-ππ*G (La), min-CT and min-nπ*T (Table S5†). The pathway leading from S3, which has predominantly ππ*T character, to min-ππ*G corresponds to a T → G ET. S5 leads to min-CT, where an electric charge of 0.81 a.u. has been transferred from G to T. The min-nπ*T is reached following internal conversion from the S2 and S4 states, and, since the former state is not completely dark, we also expect weak population of this minimum.The various relaxation pathways are schematically illustrated in Fig. 5; details are given in Table S5.† We stress that during the optimization procedure more than one reaction coordinate was varied. However, for two of the minima, one coordinate undergoes larger modifications than the others: the min-ππ*G strongly distorts the C2–NH2 coordinate whereas the min-nπ*T moves the O4 out of the plane (atom labelling in Fig. S5†). In the case of the min-CT there is a global rearrangement of the inter-base distance and displacements, plus some modifications in the thymine aromatic ring.
 | ||
| Fig. 5 Schematic representation of the PES associated with the 6 Franck–Condon states of anti–synTG leading to three minima, whose structure and relative energy ΔE (eV) are also shown. | ||
Similar calculations were performed for the three other conformers. The results are summarized in Tables S6, S7 and S8.† For anti–antiTG, three minima, min-ππ*G (La), min-ππ*T and min-nπ*T, were also determined. In agreement with the higher stability of the CT states at the Franck–Condon region, mentioned above for the “syn” conformers, a min-CT was optimized for the syn–anti conformer, while for syn–synTG a conical intersection between the first excited and the ground state exhibiting CT character was found.
The TAS computed for the Franck–Condon states of anti–synTG with the highest oscillator strength (Tables 1, S3, S5 and S6†) are presented in Fig. S6† together with the corresponding minima. We remark a clear trend: the spectra of the minima are all blue-shifted with respect to those of the initial states.
Assignment of the observed spectral evolution
Next, we search the fingerprints of the above-described relaxation pathways in the experimental TAS, starting from the long times. We also discuss these data in comparison with a TA study on 5′-dGpdT-3′dinucleotide,11 whose polarity, i.e. the order according to which two nucleobases are connected via the phosphodiester backbone, is opposite than that of TG.The longest living transient species detected in 5′-dGpdT-3′ was attributed to the thymine triplet state (3ππ*T).11 However, the spectral shape of 3ππ*T, studied by ns flash photolysis and peaking at 350 nm,41 is very different than our TAS at 40 ps (Fig. 6a), exhibiting three well-defined absorption bands in the probed wavelength range (see Fig. S7a† for a comparison). These bands are absent from the 40 ps TAS determined for an equimolar mixture of dG and dT, shown on an appropriate scale in Fig. S8,† and from the 3ππ*G(La) spectrum, peaking at 370 nm.42 The 40 ps TAS (Fig. 6a) resembles instead the spectrum computed for the min-nπ*T state (Fig. 6b), which also exhibits three bands. The lifetime of the nπ* state in thymidine monophosphate is 130 ps,43 explaining why it appears as a constant background in our measurements. We have not found any reference in the literature on the broad band TA spectra of nπ*T or nπ*G(La) states.
 | ||
| Fig. 6 Comparison between experimental TAS recorded for TG (a) with those computed for anti–synTG (b); (a) TAS at 40 ps (blue) and 5 ps (red); the dashed red line corresponds to the difference between the TAS at 5 and 40 ps. (b) min-nπ*T (blue) and min-CT (red). | ||
We assign the peak at 530 nm decaying with a time constant of 5.4 ps to a CT state. To obtain a rough evaluation of the CT state spectrum, we can consider an equimolar mixture of the guanosine radical cation and the thymidine radical anion, whose TAS have been obtained, respectively, by ns flash photolysis44 and pulse radiolysis.45 The resulting TAS, shown in Fig. S7b,† presents an absorption band around 505 nm. The overlap with that of the CT state of TG is not expected to be perfect for three reasons. (i) Not an entire atomic charge is transferred in the CT state (Table S5†), and as the amount of the transferred charge increases, the spectrum of the CT state shifts toward shorter wavelengths (Fig. S6b†). (ii) In a previous experimental and theoretical study, we reported that the spectral shape of the CT state in dinucleotides is affected by their polarity.13 (iii) In pulse radiolysis experiments electrons give rise to additional transient species absorbing in the UV. The spectrum computed for the min-CT of anti–synTG is shown in Fig. 6b, while the experimental TAS at 5 ps, when the contribution from localized ππ* states has practically vanished (Fig. 3a) in Fig. 6a; the comparison between the two spectra is improved after subtraction of the long-lived background PA at 40 ps. The 5 ps TAS spectrum exhibits an intense band at 380 nm and a second one in the visible, in good agreement with the computed min-CT spectrum (Fig. 6b).
The measured lifetime of 5.44 ± 0.03 ps is about half of that reported for 5′-dGpdT-3′ (10–14 ps).11,12 This is not surprising because the DNA polarity affects the excited state relaxation.13 Thus, the lifetime of the CT state in 5′-dApdG-3′ (112 ps) was found to be shorter than that in 5′-dGpdA-3′ (170 ps).13
The build-up of a small PA band at ∼445 nm observed in Fig. 3b and its disappearance within 2–3 ps (Fig. 3c) are assigned, respectively, to T → G ET and to the decay of the ππ*G(La) state. Indeed, the dT TAS does not exhibit such a band (Fig. S4†), while it is present in the 50 fs TAS of dG. A previous theoretical study assigned this spectral feature to a minimum in the PES of the first excited state of dG.46 Although a build-up at this wavelength is also observed for the free dG, it is significantly smaller compared to that found for TG (Fig. 7a). Moreover, the maximum intensity is reached within 60 fs for dG, as compared to 90 fs for TG. These build-up times correspond to relaxation processes and are not limited by the time resolution, as attested by Fig. S1.†
 | ||
| Fig. 7 (a) Rise of the TA signals at 445 nm recorded for TG (red) and a dG solution with equivalent concentration (green) at 445 nm; the temporal profile of the laser pulse is shown in violet. (b) Comparison of the dynamics observed for the shift of the low-energy band in the TGTAS (red circles) with the decays of dG (green) and dT (blue), averaged over the spectral range of the shift (530–560 nm). | ||
According to our calculations, population of the min-CT, whose PA bands are located at shorter wavelengths with respect to those of the initial Franck–Condon state S5 (Fig. S6b†), is expected to induce a blue shift, in line with what is observed in Fig. 3b. The associated non-exponential dynamics (Fig. 4a), is explained by the fact that the process takes place within the restricted geometry defined by the PES. We remark that, in these spectral and temporal ranges, the TAS recorded for an equimolar mixture of mononucleotides do not exhibit any significant change either in the position or in the intensity of their maximum (Fig. S9†).
The slower blue shift of the low-energy band (Fig. 3c and 4b) is explained by the decay of the ππ*G(La) state. This is better illustrated by overlaying the position of λmax between 0.2 and 2.5 ps with the dG decay averaged within the considered spectral range, 530–560 nm (Fig. 7b). It is further supported by the similar time constants derived by fits with mono-exponential functions of the spectral shift (0.61 ± 0.08 ps; Fig. 4b) and the dG decay (0.64 ± 0.03 ps). We stress that the latter value is not representative of the entire dG dynamics, which is very complex;37–39,46,47 it simply reflects its behaviour within the spectral and temporal limits corresponding to Fig. 7b, where it is also clear that the dT decay follows a different pattern.
So far, we discussed the experimental TAS in the light of the relaxation pathways computed for anti–synTG. Now we examine whether the other less stable conformers could be responsible for the three excited state PES minima that have been associated with the observed spectral evolution. The min-nπ*T is reached in all of them. The min-CT is encountered only for syn–antiTG. The pathway corresponding to T → G ET is detected for the syn–syn conformer.
Although anti–syn is the most stable TG conformer in the ground state and the fingerprints corresponding to the minima of its PES are detected in the experimental TAS, we cannot rule out that the other conformers are also present in solution, albeit with a much lower concentration. Similarly, it is quite possible that we have a contribution from unstacked nucleobases, mainly characterized by localized excited states; yet the lack of a negative signal due to the SE of ππ*T, which does not correspond to a PES minimum in anti–synTG, indicates that this contribution is rather low.
Global analysis
The multitude of the above discussed processes, associated with 3 minima in the 3 PES of each conformer and 2 for the unstacked nucleobases, giving rise to a complex network of excited states, is one reason why we have not based our analysis on global fits. The second and most important reason for avoiding analysis with exponential functions over the entire time domain explored in our experiments is related with the physics of the processes. As mentioned in the introduction, an excited state decays exponentially when its interactions with the environment are uniform within the three-dimensional space. In low-dimensional spaces the dynamics tend to be slower, described, for example, by stretched exponentials or exhibiting power-law patterns.25,48 To correctly describe the dynamics in a given system, specific theoretical analysis and/or numerical simulations are required. We stress that the power-law function used to fit the spectral shift in Fig. 4a is a phenomenological approach, simply showing that the dynamics of the process is not exponential.In any case, we did perform a global analysis within the framework of sequentially evolving species. Fig. S10† reports the evolution associated spectra (EAS) for an equimolar mixture of dT and dG. Five time constants (120 fs, 250 fs, 800 fs, 2.1 ps and 2 ns) are needed to fit the data, numerically close to those reported by Kwok and co-workers for dT (150 fs and 760 fs)40 and dG (200 fs, 790 fs and 2.0 ps)38 using a lower temporal resolution. However, the similar time evolution of the individual nucleosides does not allow for their spectral discrimination within the studied equimolar mixture. As a result, instead of identifying distinct spectral signatures for each nucleoside, an average spectrum is obtained.
In the case of TG, the use of 4 exponential functions (50 fs, 380 fs, 4.5 ps and 2 ns) provides acceptable fits (see EAS in Fig. S11†). The spectral features corresponding to the time constants of 4.5 ps and 2 ns resemble those attributed to min-CT and min-nπ*T (Fig. S12†) but the evolution on shorter times, in particular, that corresponding to the complex decay of ππ*G(La), is not correctly resolved.
Conclusion
This article reports an important breakthrough in the study of photoinduced excited state relaxation in coupled nucleobases in solution. The novelties concern both the approach and the obtained results. Two main methodological innovations need to be underlined. On the one hand, this is the first observation of transient absorption spectra and dynamics on the sub-100-fs timescale. It was rendered possible by the exceptional combination of sensitivity and temporal resolution of our femtosecond TA setup. On the other hand, the interpretation of the results has been based on a battery of quantum chemistry calculations on the most stable geometry in the ground state, the Franck–Condon states, their evolution toward the excited state minima and the corresponding TAS. In this way, we circumvented the global fits with exponential functions, which are inadequate to describe evolution along potential energy surfaces.Several compelling results emerged from our investigation.
(i) Our computations showed that the most stable conformation of TG involving stacked nucleobases (anti–syn) differs from that adopted by the T/G step in B-form duplexes (anti–anti), which has been implicitly considered for the interpretation of previous femtosecond measurements on dinucleotides.11,13 The anti–syn conformation is encountered in G-Quadruplexes.30
(ii) The minima corresponding to relaxation pathways computed for the anti–synTG, summarized in Fig. 8, are detected in the experimental TAS.
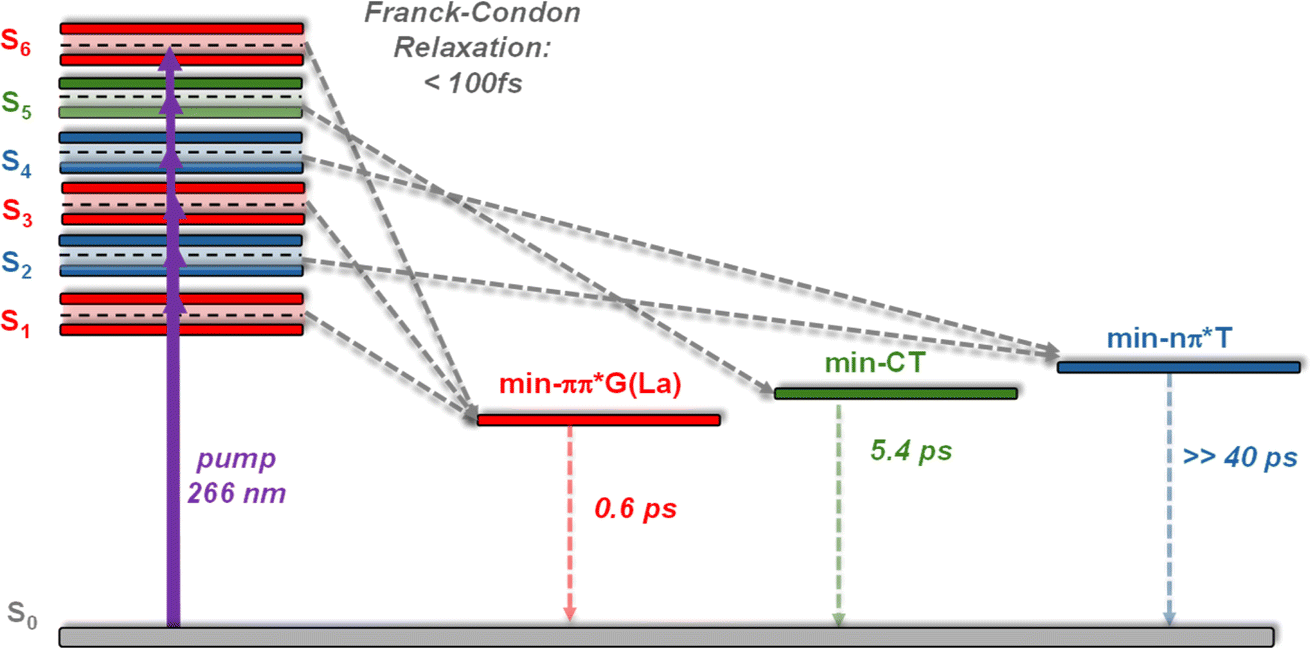 | ||
| Fig. 8 Schematic drawing of the Franck–Condon excited states and the corresponding minima computed in the PES of anti–synTG. In italics: dynamics determined from the TAS evolution. The relaxation time leading to min-nπ*T has not been observed, because the associated signal is too weak and blurred by those corresponding to other excited states; the 0.6 ps indicated for the decay of min-ππ*G(La) corresponds only to the minimum absorbing in the red.37–39,47 | ||
(iii) According to TA measurements, the CT state is reached within 100 fs, following non-exponential dynamics and decays with a lifetime of 5.4 ps. This the shortest lifetime ever determined for DNA dinucleotides,8–14 and, probably, for any donor–acceptor pair of aromatic chromophores.
(iv) The PES leading to min-ππ*G(La) corresponds to T → G ET. The associated spectral evolution occurs within 90 fs. Such an ultrafast ET could be facilitated by a very small CT between the nucleobases characterizing the initial Franck–Condon state (Table 1). So far, ultrafast ET in DNA was considered to proceed exclusively via exciton states.23
(v) Several detailed studies, dealing with monomeric pyrimidines, reported population of nπ* states.43,49,50 But so far, no experimental studies identified such states in dinucleotides or larger DNA multimeric systems composed of the major nucleobases.
The use of quantum chemistry calculations allowed a qualitative assignment of the dominant processes. In this way, we circumvented the global fits with exponential functions which, although they bring precious information on longer timescales, are inadequate to describe wave packet evolution along PES and/or systems involving a large number of transient species. Yet, the question of dynamical models that are appropriate to describe these processes remains open and requires specific work. We hope that our experimental results will inspire the future development of less time-consuming methods than those currently available that can be applied on large molecular systems.
Finally, an interesting perspective is to examine whether the ultrafast T → G energy transfer, detected in TG, also occurs in G-Quadruplexes. If this were the case, positioning peripheral thymines at the 5′ end with respect to the guanines of the G-core should further enhance their fluorescence. Such an enhancement is important because the intrinsic fluorescence of G-Quadruplexes is currently scrutinized in view of biosensors development.3,51,52
Methods
Transient absorption setup
Ultrafast TA experiments were conducted using a custom-built setup, employing an amplified Ti:Sapphire laser (Coherent Libra) that generates 100 fs pulses at a central wavelength of 800 nm, operating at a repetition rate of 1 kHz.31,53 A fraction of the fundamental laser beam is first frequency-doubled and then directed to drive a broadband visible non-collinear optical parametric amplifier (NOPA). The output of the NOPA consists of broadband visible pulses, which are subsequently compressed using chirped dielectric mirrors. These compressed pulses are further frequency-doubled in a 20-μm-thick β-barium borate crystal, resulting in broadband UV pump pulses that can be tuned within the range of 250–300 nm. The generated UV pump pulses, fully characterized through two-dimensional spectral interferometry, are compressed to 24 fs (FWHM) duration with the help of a prism pair54 (Fig. S13†). For the purpose of the experiment, the UV pump pulses are tuned to 266 nm. Broadband probe pulses, with spectrum spanning from 320 nm to 650 nm, were obtained by white-light continuum generation, focusing a portion of the laser fundamental beam onto a slowly moving 2-mm-thick CaF2 plate. Pump and probe pulses are non-collinearly focused on the sample under study, with spot sizes of 180 μm and 95 μm, respectively. The relative pump and probe polarizations are adjusted to the magic angle (54.7°). The pump fluence is kept at 100 μJ cm−2, resulting in differential absorption (DA) signals lower than 10−3; in this way, the coherent artefact and solvated electrons stemming from two-photon ionization of the solvent are minimized. The transmitted probe spectrum is recorded by an optical multichannel analyser, operating at the full laser repetition rate. The temporal resolution is estimated to be sub-30-fs. At 20 fs, although spectral intensities are not fully developed, spectral shapes in the visible are meaningful. During the experiment 6 mL of the solution kept flowing through a 1-mm-thick quartz cell controlled by a pump. For a detailed discussion of the temporal resolution in the UV TA experiments, see Section 1 in the ESI.†Key experimental points
Dinucleotides are difficult to purify and residual mononucleotides may “contaminate” their proprieties. Therefore, purchased from Eurogentec, TG was purified by reverse phase HPLC and tested by MALDI-TOF (Fig. S14†). It was dissolved in phosphate buffer (∼0.12 mol L−1, pH 7.0). The concentration of absorbed photons (8 × 10−6 mol L−1) was much lower than the dinucleotide concentration (∼4 × 10−3 mol L−1), precluding two-photon absorption. Experiments on the monomeric chromophores were performed using the mononucleosides dT and dG in water, instead of the corresponding mononucleotides, which have a great propensity to aggregate in solutions containing salts.55Computational details
We used quantum mechanical (QM) calculations, based on the Density Functional Theory (DFT) with the M052X functional,56–58 the 6-31G(d) basis set. The solvent effect was taken into account via an implicit polarizable continuum model (PCM),59 which was shown to reproduce experimental trends in a satisfactory way.60,61 In our computations we included one Na+ ion per dinucleotide so as to keep the system electrically neutral.The vertical absorption energies of the different excited states and the corresponding PES were characterized using the above-described methodologies, but resorting to the time-dependent version (TD-DFT). The charge transfer character was computed by a simple Mulliken population analysis in terms of δq, i.e., the difference between the charges in the excited state and in the ground state.
For the TA spectra, the transition dipole moments between the excited states were obtained by a multifunctional analyser (multiwfn program).62 For comparison with the experimental spectra, the computed ones were shifted by −0.65 eV. Such a shift, typical for this level of calculation, matches the difference between the energy computed for the lowest bright state (La) of dG in water and the corresponding value derived from deconvolution of the experimental spectrum (Fig. 2 in ref. 63). Subsequently, we applied a phenomenological broadening via a Gaussian function with half width at half maximum of 0.2 eV.
Data availability
The data that support the findings of this study are available in the ESI† and the on: https://doi.org/10.5281/zenodo.11453876. They are also available upon request from the corresponding authors.Author contributions
Conceptualization: LMF, GC, DM; data curation: VP, LMF; formal analysis: VP, LMF, EB, DM; funding acquisition: GC, MM, DM; investigation: VP, LMF, LU; methodology: LMF, GC, MM; supervision: GC, MM; validation: all authors; visualization: VP, LMF, EB, DM; writing original draft: VP, LMF, DM; writing – review & editing: VP, LMF, GC, EB, DM.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has received funding from the European Union’s Horizon 2020 research and innovation programme under the Marie Sklodowska-Curie ITN programme (grant No. 765266 – LightDyNAmics and grant No. 812992 – MUSIQ). G.C. acknowledges financial support by the European Union's NextGenerationEU Programme with the I-PHOQS Infrastructure [IR0000016, ID D2B8D520, CUP B53C22001750006] “Integrated infrastructure initiative in Photonic and Quantum Sciences”. L. M.-F. is indebted to the Galician Supercomputing Center for the access granted to its supercomputing infrastructure.References
- DNA Photodamage: From Light Absorption to Cellular Responses and Skin Cancer, ed. R. Improta and T. Douki, RSC, Cambridge, 2021 Search PubMed.
- M. E. Barbinta-Patrascu and S. M. Iordache, DNA - the fascinating biomacromolecule in optoelectronics and photonics applications, J. Optoelectron. Adv. Mater., 2022, 24, 563–575 CAS.
- D. Markovitsi, Processes triggered in guanine quadruplexes by direct absorption of UV radiation: From fundamental studies toward optoelectronic biosensors, Photochem. Photobiol., 2024, 100, 262–274 CrossRef CAS PubMed.
- J. Cadet and T. Douki, Formation of UV-induced DNA damage contributing to skin cancer development, Photochem. Photobiol. Sci., 2018, 17, 1816–1841 CrossRef CAS PubMed.
- E. Balanikas, A. Banyasz, T. Douki, G. Baldacchino and D. Markovitsi, Guanine Radicals Induced in DNA by Low-Energy Photoionization, Acc. Chem. Res., 2020, 53, 1511–1519 CrossRef CAS PubMed.
- L. Martinez Fernandez and R. Improrta, in DNA Photodamage: From Light Absorption to Cellular Responses and Skin Cancer, ed. R. Improta and T. Douki, RSC, Cambridge, 2021, doi: 10.1039/9781839165580, ch. 2, pp. 17–36.
- H. A. Wagenknecht, Remote Photodamaging of DNA by Photoinduced Energy Transport, Chembiochem, 2022, 23, e202100265 CrossRef CAS PubMed.
- I. Buchvarov, Q. Wang, M. Raytchev, A. Trifonov and T. Fiebig, Electronic energy delocalization and dissipation in single- and double-stranded DNA, Proc. Natl. Acad. Sci. U.S.A., 2007, 104, 4794–4797 CrossRef CAS PubMed.
- M. C. Stuhldreier and F. Temps, Ultrafast photo-initiated molecular quantum dynamics in the DNA dinucleotide d(ApG) revealed by broadband transient absorption spectroscopy, Faraday Discuss, 2013, 163, 173–188 RSC.
- J. Chen and B. Kohler, Base Stacking in Adenosine Dimers Revealed by Femtosecond Transient Absorption Spectroscopy, J. Am. Chem. Soc., 2014, 136, 6362–6372 CrossRef CAS PubMed.
- M. Duchi, M. P. O'Hagan, R. Kumar, S. J. Bennie, M. C. Galan, B. F. E. Curchod and T. A. A. Oliver, Exploring ultraviolet photoinduced charge-transfer dynamics in a model dinucleotide of guanine and thymine, Phys. Chem. Chem. Phys., 2019, 21, 14407–14417 RSC.
- C. L. Kufner, W. Zinth and D. B. Bucher, UV-Induced Charge-Transfer States in Short Guanosine-Containing DNA Oligonucleotides, Chembiochem, 2020, 21, 2306–2310 CrossRef CAS PubMed.
- V. Petropoulos, L. Uboldi, M. Maiuri, G. Cerullo, L. Martinez-Fernandez, E. Balanikas and D. Markovitsi, Effect of the DNA polarity on the relaxation of its electronic excited states, J. Phys. Chem. Lett., 2023, 14, 10219–10224 CrossRef CAS PubMed.
- T. Takaya, C. Su, K. de La Harpe, C. E. Crespo-Hernandez and B. Kohler, UV excitation of single DNA and RNA strands produces high yields of exciplex states between two stacked bases, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 10285–10290 CrossRef CAS PubMed.
- J. Chen, Y. Zhang and B. Kohler, Excited states in DNA strands investigated by ultrafast laser spectroscopy, Top. Curr. Chem., 2015, 356, 39–87 CrossRef CAS PubMed.
- W. J. Schreier, P. Gilch and W. Zinth, Early Events of DNA Photodamage, Annu. Rev. Phys. Chem., 2015, 66, 497–519 CrossRef CAS PubMed.
- W. M. Kwok, C. S. Ma and D. L. Phillips, “Bright” and “Dark” excited states of an alternating AT oligomer characterized by femtosecond broadband spectroscopy, J. Phys. Chem. B, 2009, 113, 11527–11534 CrossRef CAS PubMed.
- C. S. Ma, R. C.-T. Chan, C. T.-L. Chan, A. K.-W. Wong and W.-M. Kwok, Real-time Monitoring Excitation Dynamics of Human Telomeric Guanine Quadruplexes: Effect of Folding Topology, Metal Cation, and Confinement by Nanocavity Water Pool, J. Phys. Chem. Lett., 2019, 10, 7577–7585 CrossRef CAS PubMed.
- G. W. Doorley, D. A. McGovern, M. W. George, M. Towrie, A. W. Parker, J. M. Kelly and S. J. Quinn, Picosecond transient infrared study of the ultrafast deactivation processes of electronically excited B-DNA and Z-DNA forms of [poly(dG-dC)]2, Angew. Chem., Int. Ed., 2009, 48, 123–127 CrossRef CAS PubMed.
- G. W. Doorley, M. Wojdyla, G. W. Watson, M. Towrie, A. W. Parker, J. M. Kelly and S. J. Quinn, Tracking DNA Excited States by Picosecond-Time-Resolved Infrared Spectroscopy: Signature Band for a Charge-Transfer Excited State in Stacked Adenine-Thymine Systems, J. Phys. Chem. Lett., 2013, 4, 2739–2744 CrossRef CAS.
- P. M. Keane, M. Wojdyla, G. W. Doorley, J. M. Kelly, A. W. Parker, I. P. Clark, G. M. Greetham, M. Towrie, L. M. Magno and S. J. Quinn, Long-lived excited states in i-motif DNA studied by picosecond time-resolved IR spectroscopy, Chem. Commun., 2014, 50, 2990–2992 RSC.
- L. Martínez-Fernández, J. A. Green, L. Esposito, M. Y. Jouybari, Y. Y. Zhang, F. Santoro, B. Kohler and R. Improta, The photoactivated dynamics of dGpdC and dCpdG sequences in DNA: a comprehensive quantum mechanical study, Chem. Sci., 2024, 9676–9693 RSC.
- T. Gustavsson and D. Markovitsi, Fundamentals of the Intrinsic DNA Fluorescence, Acc. Chem. Res., 2021, 54, 1226–1235 CrossRef CAS PubMed.
- A. Blumen, J. Klafter and G. Zumofen, in Optical spectroscopy of glasses, ed. I. Zschokke, Springer, Dordecht, the Netherlands, 1986, pp. 199–265 Search PubMed.
- J. M. Drake, J. Klafter and P. Levitz, Chemical and Biological Microstructures as Probed by Dynamical Processes, Science, 1991, 251, 1574–1579 CrossRef CAS PubMed.
- D. Markovitsi, A. Germain, P. Millie, I. Lécuyer, L. Gallos, P. Argyrakis, H. Bengs and H. Ringsdorf, Triphenylene columnar liquid crystals: excited states and energy transfer, J. Phys. Chem., 1995, 99, 1005–1017 CrossRef CAS.
- K. Y. Guslienko, Magnetic vortex state stability, reversal and dynamics in restricted geometries, J. Nanosci. Nanotechnol., 2008, 8, 2745–2760 CrossRef CAS PubMed.
- O. Benichou, C. Chevalier, J. Klafter, B. Meyer and R. Voituriez, Geometry-controlled kinetics, Nat. Chem., 2010, 2, 472–477 CrossRef CAS PubMed.
- M. Zarea, R. Carmieli, M. A. Ratner and M. R. Wasielewski, Spin Dynamics of Radical Pairs with Restricted Geometries and Strong Exchange Coupling: The Role of Hyperfine Coupling, J. Phys. Chem. A, 2014, 118, 4249–4255 CrossRef CAS PubMed.
- J. L. Chen, M. P. Cheng, G. F. Salgado, P. Stadlbauer, X. B. Zhang, S. Amrane, A. Guédin, F. N. He, J. Sponer, H. X. Ju, J. L. Mergny and J. Zhou, The beginning and the end: flanking nucleotides induce a parallel G-quadruplex topology, Nucleic Acids Res., 2021, 49, 9548–9559 CrossRef CAS PubMed.
- R. Borrego-Varillas, L. Ganzer, G. Cerullo and C. Manzoni, Ultraviolet Transient Absorption Spectrometer with Sub-20-fs Time Resolution, Appl. Sci., 2018, 8, 989 CrossRef.
- D. Onidas, D. Markovitsi, S. Marguet, A. Sharonov and T. Gustavsson, Fluorescence properties of DNA nucleosides and nucleotides: a refined steady-state and femtosecond investigation, J. Phys. Chem. B, 2002, 106, 11367–11374 CrossRef CAS.
- D. Varsano, R. Di Felice, M. A. L. Marques and A. Rubio, A TDDFT study of the excited states of DNA bases and their assemblies, J. Phys. Chem. B, 2006, 110, 7129–7138 CrossRef CAS PubMed.
- F. Plasser, A. Aquino, H. Lischka and D. Nachtigallová, Electronic Excitation Processes in Single-Strand and Double-Strand DNA: A Computational Approach, Top. Curr. Chem., 2015, 356, 1–38 CAS.
- E. Balanikas, L. Martinez-Fernadez, R. Improta, P. Podbevsek, G. Baldacchino and D. Markovitsi, The Structural Duality of Nucleobases in Guanine Quadruplexes Controls Their Low-Energy Photoionization, J. Phys. Chem. Lett., 2021, 12, 8309–8313 CrossRef CAS PubMed.
- E. Palecek and M. Bartosik, Electrochemistry of Nucleic Acids, Chem. Rev., 2012, 112, 3427–3481 CrossRef CAS PubMed.
- V. Karunakaran, K. Kleinermanns, R. Improta and S. A. Kovalenko, Photoinduced dynamics of guanosine monophosphate in water from broad-band transient absorption spectroscopy and quantum-chemical calculations, J. Am. Chem. Soc., 2009, 131, 5839–5850 CrossRef CAS PubMed.
- C. C.-W. Cheng, C. Ma, C. T.-L. Chan, K. Y.-F. Ho and W.-M. Kwok, The solvent effect and identification of a weakly emissive state in nonradiative dynamics of guanine nucleosides and nucleotides - a combined femtosecond broadband time-resolved fluorescence and transient absorption study, Photochem. Photobiol. Sci., 2013, 12, 1351–1365 CrossRef CAS PubMed.
- S. E. Krul, S. J. Hoehn, K. J. Feierabend and C. E. Crespo-Hernández, Excited state dynamics of 7-deazaguanosine and guanosine 5′-monophosphate, J. Chem. Phys., 2021, 154, 075103 CrossRef CAS PubMed.
- W. M. Kwok, C. Ma and D. L. Phillips, A doorway state leads to photostability or triplet photodamage in thymine DNA, J. Am. Chem. Soc., 2008, 130, 5131–5139 CrossRef CAS PubMed.
- S. Marguet and D. Markovitsi, Time-resolved study of thymine dimer formation, J. Am. Chem. Soc., 2005, 127, 5780–5781 CrossRef CAS PubMed.
- P. D. Wood and R. Redmond, Triplet state interactions between nucleic acid bases in solution at room temperature: intermolecular energy and electron transfer, J. Am. Chem. Soc., 1996, 118, 4256–4263 CrossRef CAS.
- P. M. Hare, C. Crespo-Hernández and B. Kohler, Internal conversion to electronic ground state occurs via two distinct pathways for pyrimidine bases in aqueous solution, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 435–440 CrossRef CAS PubMed.
- L. P. Candeias and S. Steenken, Ionization of purine nucleosides and nucleotides and their components by 193-nm laser photolysis in aqueous solution: model studies for oxidative damage of DNA, J. Am. Chem. Soc., 1992, 114, 699–704 CrossRef CAS.
- R. Yamagami, K. Kobayashi and S. Tagawa, Formation of Spectral Intermediate G-C and A-T Anion Complex in Duplex DNA Studied by Pulse Radiolysis, J. Am. Chem. Soc., 2008, 130, 14772–14777 CrossRef CAS PubMed.
- J. Ortín-Fernández, J. González-Vázquez, L. Martínez-Fernández and I. Corral, Molecular Identification of the Transient Species Mediating the Deactivation Dynamics of Solvated Guanosine and Deazaguanosine, Molecules, 2022, 27, 989 CrossRef PubMed.
- Y. Zhang, R. Improta and B. Kohler, Mode-specific vibrational relaxation of photoexcited guanosine 5'-monophosphate and its acid form: a femtosecond broadband mid-IR transient absorption and theoretical study, Phys. Chem. Chem. Phys., 2014, 16, 1487–1499 RSC.
- M. F. Shlessinger and A. W. Montroll, On the Willams-Watts function of dielectric relaxation, Proc. Natl. Acad. Sci. U. S. A., 1984, 81, 1280–1283 CrossRef PubMed.
- Y. Miura, Y. Yamamoto, S. Karashima, N. Orimo, A. Hara, K. Fukuoka, T. Ishiyama and T. Suzuki, Formation of Long-Lived Dark States during Electronic Relaxation of Pyrimidine Nucleobases Studied Using Extreme Ultraviolet Time-Resolved Photoelectron Spectroscopy, J. Am. Chem. Soc., 2023, 145, 3369–3381 CrossRef CAS PubMed.
- S. J. Hoehn, S. E. Krul, M. M. Pogharian, E. Mao and C. E. Crespo-Hernández, Photochemical Stability of 5-Methylcytidine Relative to Cytidine: Photophysical Insight for mRNA Therapeutic Applications, J. Phys. Chem. Lett., 2023, 14, 10856–10862 CrossRef CAS PubMed.
- C. Lu, A. Lopez, J. K. Zheng and J. W. Liu, Using the Intrinsic Fluorescence of DNA to Characterize Aptamer Binding, Molecules, 2022, 27, 7809 CrossRef CAS PubMed.
- A. Lopez and J. Liu, Probing metal-dependent G-quadruplexes using the intrinsic fluorescence of DNA, Chem. Commun., 2022, 58, 10225–10228 RSC.
- D. Polli, L. Luer and G. Cerullo, High-time-resolution pump-probe system with broadband detection for the study of time-domain vibrational dynamics, Rev. Sci. Instrum., 2007, 78, 103108 CrossRef PubMed.
- R. Borrego-Varillas, A. Oriana, F. Branchi, S. De Silvestri, G. Cerullo and C. Manzoni, Optimized ancillae generation for ultra-broadband two-dimensional spectral-shearing interferometry, J. Opt. Soc. Am. B, 2015, 32, 1851–1855 CrossRef CAS.
- E. Baldassarri, M. G. Ortore, F. Spinozzi, A. Round, C. Ferrero and P. Mariani, K+vs. Na+ Effects on the Self-Assembly of Guanosine 5′-Monophosphate: A Solution SAXS Structural Study, Nanomaterials, 2020, 10, 629 CrossRef CAS PubMed.
- Y. Zhao, N. E. Schultz and D. G. Truhlar, Design of density functionals by combining the method of constraint satisfaction with parametrization for thermochemistry, thermochemical kinetics, and noncovalent interactions, J. Chem. Theory Comput., 2006, 2, 364–382 CrossRef PubMed.
- Y. Zhao and D. G. Truhlar, Density functionals with broad applicability in chemistry, Acc. Chem. Res., 2008, 41, 157–167 CrossRef CAS PubMed.
- R. Improta, F. Santoro and L. Blancafort, Quantum Mechanical Studies on the Photophysics and the Photochemistry of Nucleic Acids and Nucleobases, Chem. Rev., 2016, 116, 3540–3593 CrossRef CAS PubMed.
- J. Tomasi, B. Mennucci and R. Cammi, Quantum mechanical continuum solvation models, Chem. Rev., 2005, 105, 2999–3093 CrossRef CAS PubMed.
- L. Martinez-Fernandez, A. J. Pepino, J. Segarra-Marti, A. Banyasz, M. Garavelli and R. Improta, Computing the Absorption and Emission Spectra of 5-Methylcytidine in Different Solvents: A Test-Case for Different Solvation Models, J. Chem. Theor. Comput., 2016, 12, 4430–4439 CrossRef CAS PubMed.
- L. Martinez-Fernandez, F. Santoro and R. Improta, Nucleic Acids as a Playground for the Computational Study of the Photophysics and Photochemistry of Multichromophore Assemblies, Acc. Chem. Res., 2022, 55, 2077–2087 CrossRef CAS PubMed.
- T. Lu and F. W. Chen, Multiwfn: A multifunctional wavefunction analyzer, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- B. Bouvier, T. Gustavsson, D. Markovitsi and P. Millié, Dipolar coupling between electronic transitions of the DNA bases and its relevance to exciton states in double helices, Chem. Phys., 2002, 275, 75–92 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc02514h |
| This journal is © The Royal Society of Chemistry 2024 |