
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceControlling the regioselectivity of the bromolactonization reaction in HFIP†
Tuong Anh
To‡
 a,
Nhu T. A.
Phan‡
a,
Binh Khanh
Mai
*b and
Thanh Vinh
Nguyen
*a
a,
Nhu T. A.
Phan‡
a,
Binh Khanh
Mai
*b and
Thanh Vinh
Nguyen
*a
aSchool of Chemistry, University of New South Wales, Sydney, NSW 2052, Australia. E-mail: t.v.nguyen@unsw.edu.au
bDepartment of Chemistry, University of Pittsburgh, Pennsylvania 15260, USA. E-mail: binh.mai@pitt.edu
First published on 11th April 2024
Abstract
The halolactonization reaction provides rapid access to densely functionalized lactones from unsaturated carboxylic acids. The endo/exo regioselectivity of this cyclization reaction is primarily determined by the electronic stabilization of alkene substituents, thus making it inherently dependent on substrate structures. Therefore this method often affords one type of halolactone regioisomer only. Herein, we introduce a simple and efficient method for regioselectivity-switchable bromolactonization reactions mediated by HFIP solvent. Two sets of reaction conditions were developed, each forming endo-products or exo-products in excellent regioselectivity. A combination of computational and experimental mechanistic studies not only confirmed the crucial role of HFIP, but also revealed the formation of endo-products under kinetic control and exo-products under thermodynamic control. This study paves the way for future work on the use of perfluorinated solvents to dictate reaction outcomes in organic synthesis.
Introduction
Halonium-promoted addition of nucleophiles to alkenes is one of the most fundamental reactions in organic chemistry, which offers widespread applications in organic synthesis.1 The intramolecular variant of this transformation is a powerful tool to construct molecular complexity by not only creating a new ring and stereogenic centres but also introducing a halide group for subsequent functionalizations.2 Halolactonization is a typical example of this transformation, offering rapid access to densely functionalized lactones from acyclic unsaturated carboxylic acids. A wide range of valuable lactone analogues varying in both structures and stereochemistry could be obtained through this protocol by controlling the diastereoselectivity, enatioselectivity, and regioselectivity of the reaction (Scheme 1a).3 Traditionally, diastereoselective and enantioselective halolactonizations are usually directed by substrate structures.3a,b Over the past two decades, there have been also developments of reagent-controlled enantioselective halolactonization, relying on chiral electrophilic halogenating reagents which in turn can be generated in situ through the coordination of chiral catalysts to halogenating reagents.4 On the other hand, regioselectivity of halolactonization has predominantly been dictated by the electronic effects of alkene substituents, with carboxylate groups intercepting halonium intermediates at the position where positive charge stabilization is most favorable. To the best of our knowledge, methods to alter this inherent regioselectivity of halolactonization or offer controllable formation of both regioisomers from one single unsaturated carboxylic acid substrate are scarce in the literature.5 | ||
| Scheme 1 (a) Halolactonization reaction; (b) HFIP-mediated halocyclization; and (c) this work: HFIP-mediated regioselectivity-switchable bromolactonization. | ||
In the last decade, hexafluoroisopropanol (HFIP) has attracted increasing attention as a reaction solvent due to its strong hydrogen-bond donating ability, low nucleophilicity, stability under redox conditions and most importantly the unique capacity to stabilize ionic reaction intermediates.6 HFIP has also demonstrated its ability to facilitate a wide range of difunctionalization reactions.7 In relevant context to this work, there have been seminal contributions from the Gulder group8 on HFIP-mediated halocyclization of terpenes, and the Lebœuf and Gandon groups9 on HFIP-mediated haloamidation and halolactonization of alkenes with excellent reaction outcomes (Scheme 1b). Building upon these research studies and our prior investigations on halide-promoted addition reaction to alkenes,10 acid-promoted cyclization reactions11 and HFIP-assisted Brønsted acid-catalyzed chemistry,12 we envisaged that HFIP can be used to activate bromonium sources such as NBS and promote endo-bromolactonization of readily available conjugated unsaturated carboxylic acids13 (Scheme 1c, upper route). On the other hand, the other bromolactonization regioisomers with smaller ring sizes are inherently more thermodynamically stable but require higher activation energies due to the lack of electronic stabilization from the aromatic ring. We believed that the presence of HFIP-activated Brønsted acid catalysts can potentially activate the reaction substrates to overcome these higher activation energy barriers, leading to the formation of the exo-cyclization product (Scheme 1c, lower route). This novel regioselectivity-switchable protocol would enable rapid regioselective synthesis of two different analogues of densely functionalized lactones from readily available precursors.
Results and discussion
We elected to focus on bromolactonization in this work, as brominating reagents and their corresponding products possess balanced reactivity and stability compared to chloro or iodo counterparts. We initiated this study by choosing 1a, which was efficiently obtained by ring-opening olefination of cyclopentanone ketal according to our recent work,13 as the model substrate and N-bromosuccinimide (NBS) as a brominating reagent, which can generate ε-caprolactone 2a and δ-valerolactone 3a as endo-cyclization and exo-cyclization products respectively (Table 1). The bromolactonization of 1a in HFIP happened smoothly at room temperature, selectively forming endo-product 2a in 91% yield with a 13/1 regioisomer ratio (entry 1, Table 1). Both reactivity and selectivity of this HFIP-mediated endo-bromolactonization are in accordance with Lebœuf and Gandon work.9 Lowering the reaction temperature to 0 °C, with a slight decrease in yield and longer reaction time, resulted in absolute selectivity to endo-product 2a (entry 2). This observation is in agreement with our initial hypothesis that endo-products of bromolactonization reactions are kinetically favorable while exo-products are thermodynamically favorable. For a simple reaction setup and better reaction efficiency, we chose to carry out the reaction at room temperature in subsequent studies.|
|
|||||
|---|---|---|---|---|---|
| Entrya | Catalyst (mol%) | Solvent (v/v) | Yield of 2ab | Yield of 3ab | 2a/3a ratiob |
| a Reaction conditions: 1a (0.1 mmol), NBS (1.1 equiv.), catalyst, and solvent (v/v, 0.1 M) were stirred at room temperature for 1 h. b Yields and regioisomer ratios were determined by 1H NMR using methyl benzoate as an internal standard. See pages S4–S5 in the ESI for full optimization studies. ND = not detected. c Reaction was carried out at 0 °C for 2 h. | |||||
| 1 | HFIP | 91% | 7% | 13/1 | |
| 2c | HFIP | 77% | Traces | >20/1 | |
| 3 | AcOH (10%) | HFIP | 88% | 10% | 8.8/1 |
| 4 | TFA (10%) | HFIP | 67% | 20% | 3.4/1 |
| 5 | pTSA (10%) | HFIP | Traces | 89% | <1/20 |
| 6 | TfOH (10%) | HFIP | Traces | 86% | <1/20 |
| 7 | pTSA (5%) | HFIP | Traces | 87% | <1/20 |
| 8 | pTSA (2%) | HFIP | Traces | 79% | <1/20 |
| 9 | HFIP/DCE (1/1) | 85% | 8% | 10.6/1 | |
| 10 | HFIP/DCE (3/7) | 85% | 9% | 9.4/1 | |
| 11 | HFIP/DCE (1/9) | 71% | Traces | >20/1 | |
| 12 | DCE | ND | ND | — | |
| 13 | TFE | 29% | 60% | 1/2.1 | |
| 14 | iPrOH | ND | ND | — | |
| 15 | MeNO2 | Traces | Traces | — | |
| 16 | pTSA (5%) | DCE | 11% | 5% | 2.2/1 |
| 17 | pTSA (5%) | TFE | 22% | 12% | 1.8/1 |
| 18 | pTSA (5%) | iPrOH | 13% | 5% | 2.6/1 |
| 19 | pTSA (5%) | MeNO2 | 17% | 20% | 1/1.2 |
On the other hand, introducing a catalytic amount of Brønsted acids into the reaction, as predicted, shifted the selectivity towards the exo-product 3a while maintaining excellent overall yields of both products (Table 1, entries 3–8). Interestingly, weaker acids such as acetic acid (AcOH, entry 2) and trifluoroacetic acid (TFA, entry 4) caused minor shifts in selectivity, whereas stronger acids like p-toluenesulfonic acid (pTSA, entry 5) and triflic acid (TfOH, entry 6) induced a complete alteration in selectivity toward exo-product 3a. pTSA was chosen for further studies of exo-bromolactonization as it is inexpensive and easier to handle. Reducing the amount of pTSA to 5 mol% did not change the reaction efficiency (entry 7) while the yield of exo-product 3a slightly decreased when employing 2 mol% of pTSA (entry 8). We also attempted to reduce the amount of HFIP in endo-bromolactonization by using DCE as a co-solvent, but both yield and selectivity slightly dropped (entries 9–11). To clarify the role of HFIP in this reaction, we carried out the reaction in DCE as a typical solvent for halocyclization chemistry, trifluoroethanol (TFE) as a weaker hydrogen-bonding donor solvent, isopropanol as the respective non-fluorinated alcohol, and nitromethane (MeNO2) as a highly polar solvent for charge stabilization (entries 12–19). Without HFIP as solvent, reactions generally led to unsatisfactory outcomes, regardless of whether or not there was a Brønsted acid catalyst, confirming the crucial role of HFIP in the formation of both endo- and exo-products.
We subsequently explored the versatility of our newly developed method in both endo-bromolactonization (Scheme 2a) and exo-bromolactonization (Scheme 2b). A series of unsaturated carboxylic acid 1 were first subjected to our optimal conditions for endo-bromolactonization. Substrates with diverse electronic and steric effects (1a–j) were well tolerated under endo-bromolactonization conditions, yielding 7-endo-products 2a–j in moderate to excellent yields. Electron-rich thiophene substrate 1k led to poor efficiency, probably due to susceptibility to electrophilic bromination on the electron-rich aromatic ring in HFIP medium.14 Our method also demonstrated good efficiency in 6-endo-bromolactonization, generating 6-endo-product 2l in good yield. However, 8-endo-bromolactonization exhibited unimpressive efficiency (products 2m and 2n), which can presumably be attributed to the challenging formation of eight-membered medium-sized lactones.
 | ||
| Scheme 2 Substrate scope of (a) endo-bromolactonization and (b) exo-bromolactonization. Reaction conditions unless otherwise noted: 1 (0.2 mmol), NBS (1.1 equiv.), with/without pTSA (5 mol%), HFIP (0.1 M). Diastereomeric ratios were all >20/1 without quoted ratios in parentheses. [a] HFIP/DCM (1/4, 0.1 M) was used. [b] HFIP/DCM (1/99, 0.1 M) was used. [c] Reaction was stirred without catalyst for 1 h prior to adding catalyst and further stirring for 1 h. [d] NMR yield. [e] TfOH (10 mol%) was used. | ||
A quite similar trend in reaction yields was also observed when the same set of unsaturated acids 1 was subjected to our optimal conditions of exo-bromolactonization (Scheme 2b). In the case of δ,ε-unsaturated carboxylic acids, except for substrates with an electron-rich aromatic ring (1f and 1k), which are sensitive towards electrophilic aromatic bromination,14,15 other substrates (1a–e and 1g–j) exhibited a complete switch in regioselectivity to 6-exo-bromolactonization with moderate to excellent yields when treated with a Brønsted acid catalyst in HFIP. Absolute exo-regioselectivity and excellent yield were also observed with γ,δ-unsaturated carboxylic acid 1l, while no expected products 3m and 3n were detected for the case of ε,ζ-unsaturated carboxylic acids 1m and 1n. It should be noted that along with controllable regioselectivity, our method also offered excellent to absolute diastereoselectivity for both endo- and exo-cyclization.
Next, we extended the substrate scope of HFIP-mediated bromolactonization to other alkenoic acid scaffolds (Scheme 3). Estrone-derived δ,ε-unsaturated carboxylic acid 1o, obtained by modifying estrone through a three-step procedure as reported in our recent work,13 smoothly underwent 7-endo-bromolactonization, yielding tetracyclic lactone 2o as an equimolar mixture of two diastereomers in 88% yield. Similar to substrates in Scheme 2b, the regioselectivity completely shifted to 6-exo-product 3o, also as an equimolar mixture of two diastereomers in excellent yield, when a catalytic amount of pTSA was used to promote the reaction (Scheme 3a). This example demonstrated a quick and efficient way for the late-stage modification of complex cyclic ketones into lactones with various ring-sizes.
 | ||
| Scheme 3 Expanding the substrate scope of bromolactonization: (a) rapid access to two analogues of estrone-based lactone and (b) bromolactonization on other scaffolds. | ||
As discussed earlier in Scheme 2, we believe that the selective formation of endo-product 2 is kinetically favored due to benzylic stabilization, and the selective formation of exo-product 3 is thermodynamically supported due to generation of a more stable ring size. To support this hypothesis, we carried out some negative testing studies with two other structures often encountered in halolactonization chemistry, terminal alkenoic acid 1p and trans-stilbene-type acid 1q. Gratifyingly, both of these exclusively yielded exo-product 3p and endo-product 2q, respectively in good to excellent yields, regardless of whether catalytic pTSA was used or not (Scheme 3b). These outcomes align well with our hypothesis, as 3p and 2q are both kinetically and thermodynamically favorable, owing to both benzylic stabilization and the formation of a more stable ring size. As a result, this leads to an unswitchable regioselectivity with substrates 1p and 1q.
We also attempted to exclude benzylic stabilization by carrying out reactions on aliphatic unsaturated acids 1r–t, which have nearly identical electronic effects on both reactive sites. Isopropyl-substituted acids 1r and 1s exclusively yielded 6-exo-product 3r and 5-exo-product 3s in excellent yields in the presence or absence of the pTSA catalyst, respectively. Unswitchable regioselectivity and excellent yield were also recorded on a less sterically hindered methyl-substituted acid 1t. These results confirmed the importance of the benzylic effect on the controllable regioselectivity.
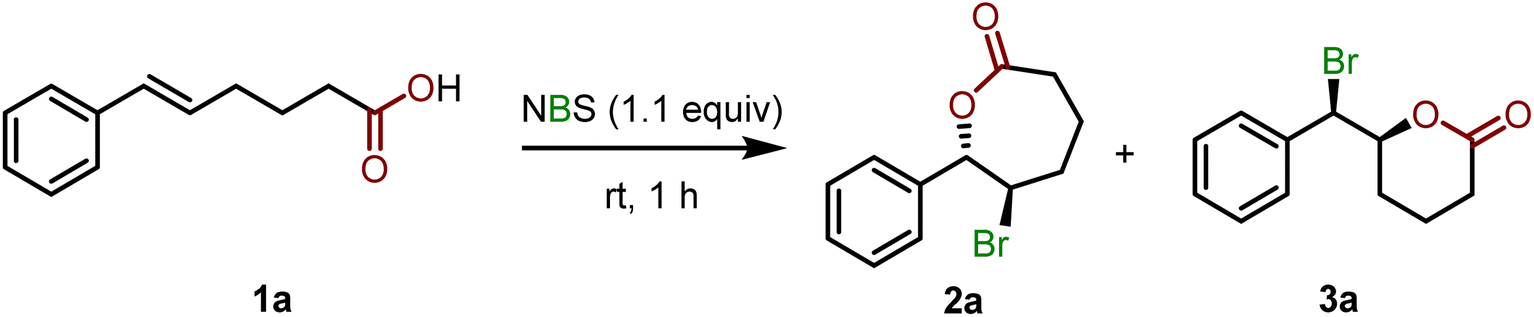
To gain better insights into the reaction mechanism and effect of catalyst/solvent on the regioselectivity for the bromolactonization, we then turn our effort to density functional theory (DFT) calculations at the MN15/6-311+G(2d,2p)/SMD//M06-2X/6-31G(d,p)/SMD level of theory (see page S97 in the ESI† for computational details). The proposed reaction mechanism for the bromolactonization is shown in Scheme 4. Traditionally, this transformation is expected to take place via a stepwise AdE2-type mechanism. The first step of this reaction is the electrophilic addition of the bromine atom to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond of the substrate generating a reactive cyclic bromonium intermediate, which is followed by a nucleophilic addition leading to endo- and exo-cyclic products.16 Alternatively, by means of kinetics studies, NMR spectroscopy, and DFT calculations, Jackson and Borhan17 proposed that the halolactonization can take place via a concerted AdE3-type mechanism in which the nucleophilic and electrophilic additions happen simultaneously and no ionic intermediate is generated during the reaction. Moreover, the additions of the nucleophile and electrophile can occur at the same face (i.e., syn-addition) or opposite faces (i.e., anti-addition) of the CC double bond (Scheme 4).17
C double bond of the substrate generating a reactive cyclic bromonium intermediate, which is followed by a nucleophilic addition leading to endo- and exo-cyclic products.16 Alternatively, by means of kinetics studies, NMR spectroscopy, and DFT calculations, Jackson and Borhan17 proposed that the halolactonization can take place via a concerted AdE3-type mechanism in which the nucleophilic and electrophilic additions happen simultaneously and no ionic intermediate is generated during the reaction. Moreover, the additions of the nucleophile and electrophile can occur at the same face (i.e., syn-addition) or opposite faces (i.e., anti-addition) of the CC double bond (Scheme 4).17
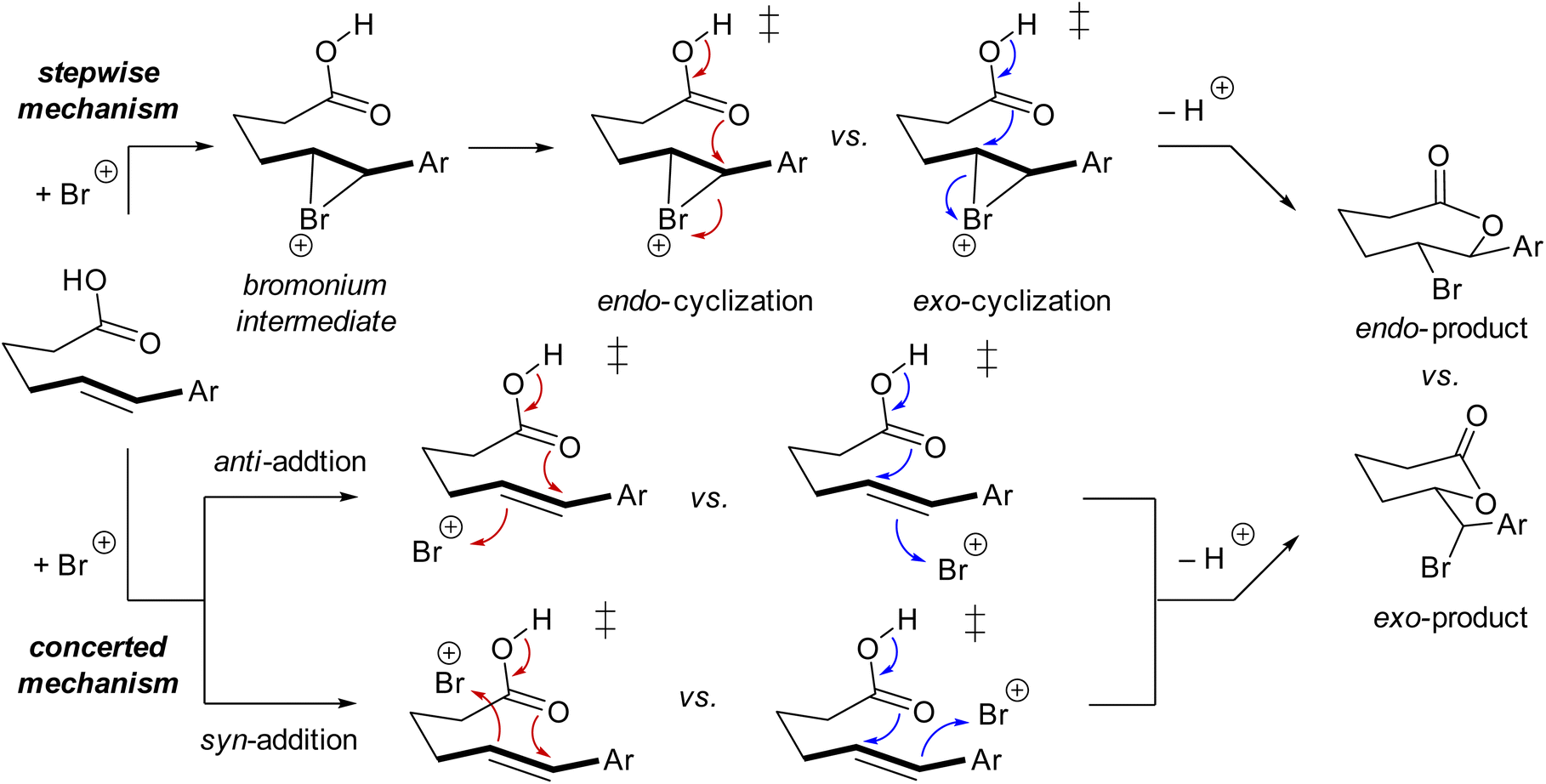 | ||
| Scheme 4 Proposed reaction mechanism for the bromolactonization. | ||
We first performed DFT calculations to elucidate the reaction mechanism for the bromolactonization in the aprotic DCE solvent, using 1c (Ar = p-Br–C6H4–) as the model substrate. Consistent with previous experimental and theoretical studies,17,18 DFT calculations revealed that in DCE, the bromolactonization takes place via a syn-concerted addition pathway (Fig. 1). No transition state for the stepwise mechanism as well as anti-concerted addition pathway could be located. Interestingly, the activation barriers for the syn-additions are calculated to be fairly high, amounting to 36.8 and 39.4 kcal mol−1 for TS-1 and TS-2, respectively. This DFT result is in good agreement with our experimental findings (Table 1) that only a trace amount of the lactonization product can be observed when this reaction is performed in DCE (vide supra).
 | ||
| Fig. 1 Optimized transition states for the concerted bromolactonization in DCE. Transition states TS-1 and TS-2 lead to endo- and exo-cyclic products, respectively. | ||
DFT calculations were subsequently performed to investigate the favorable mechanistic pathway and regioselectivity for the bromolactonization in HFIP. Because of its powerful hydrogen bond (H-bond) donor ability,6a,d it is possible for HFIP to form strong H-bonds with various species along the reaction course. Therefore, a mixed explicit–implicit solvation model is used, in which explicit HFIP molecules were included in DFT calculations.9,12b,19 Based on our kinetic studies suggesting that the reaction order in HFIP is approximately 3 (Fig. 2a and further details in pages S6–S8 in the ESI†), three HFIP molecules were included in our computational investigations. Additionally, we have also performed calculations by involving one and two HFIP molecules (Fig. S1–S4, pages S99–S100 in the ESI†). Although there are some changes in absolute energy values, the conclusion remains similar all through our calculations, which gives a solid validation to the accuracy of the mixed explicit–implicit solvation model.
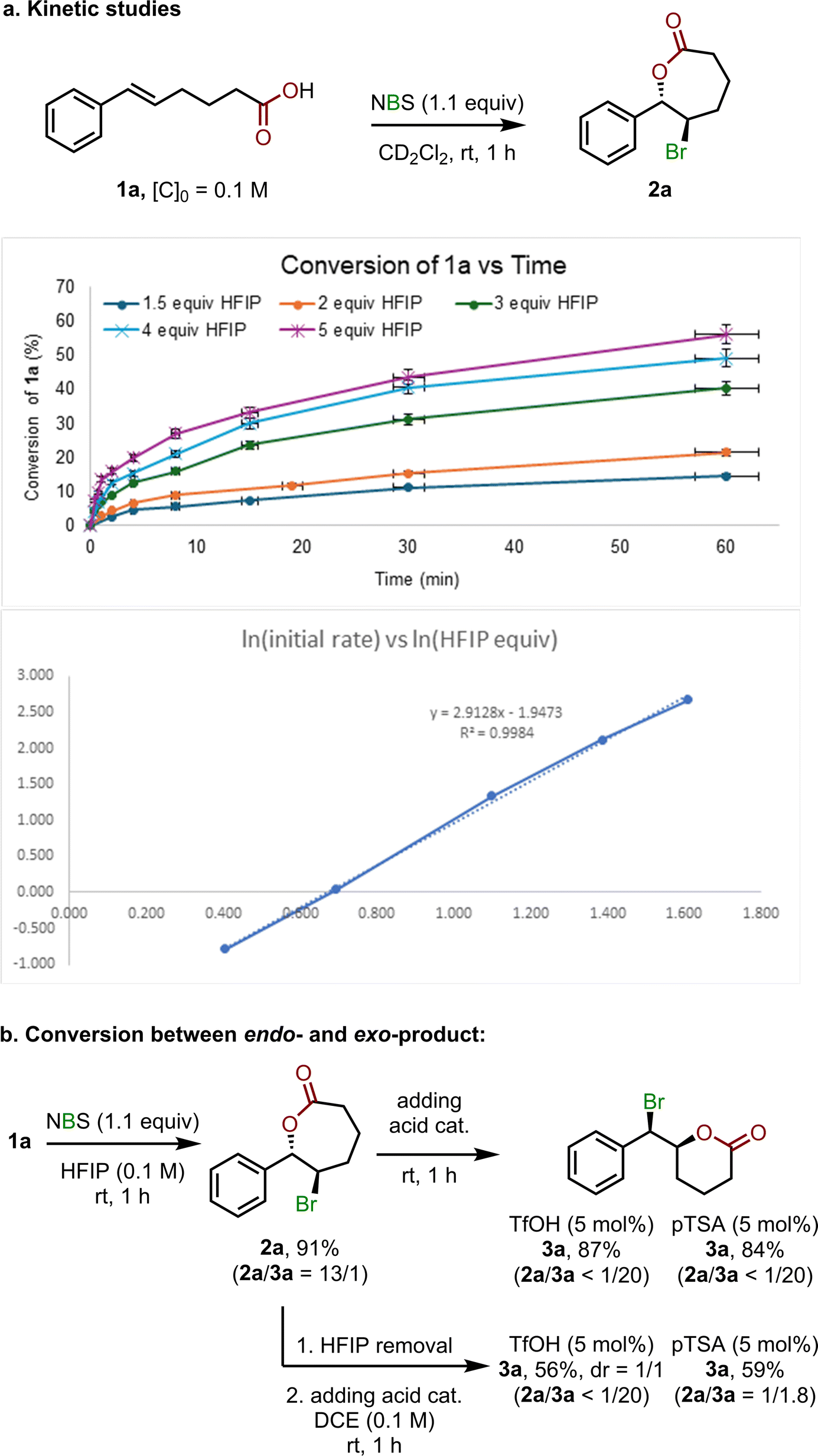 | ||
| Fig. 2 Experimental mechanistic studies: (a) kinetics studies and (b) conversion between endo- and exo-product. See pages S6–S9 in the ESI† for more details. | ||
Our DFT calculations indicated that the favorable mechanism for the bromolactonization in HFIP is the stepwise pathway (Fig. 3), which is consistent with the previous calculation for the halolactonization in protic solvent.18 The barrier height for the formation of the bridged bromonium species 5viaTS-3 is calculated to be 15.7 kcal mol−1. It should be noted that we have also considered the concerted mechanism for this transformation in HFIP. However, we can only locate the transition state for the syn-concerted addition pathway (Fig. S5 in the ESI†), which is calculated to be 9.7 kcal mol−1 higher in energy than TS-3. Therefore, the concerted mechanism is unlikely to occur. Additionally, the stepwise mechanism is also calculated to be the favorable pathway for the reaction of 1l in HFIP (see Fig. S6 in the ESI† for more details). This result is in agreement with the fact that HFIP has an exceptional cation stabilization ability6a,20 due to the low nucleophilicity and high dielectric constant, and, thus, the stepwise mechanism is supposed to be the preferable pathway.16b
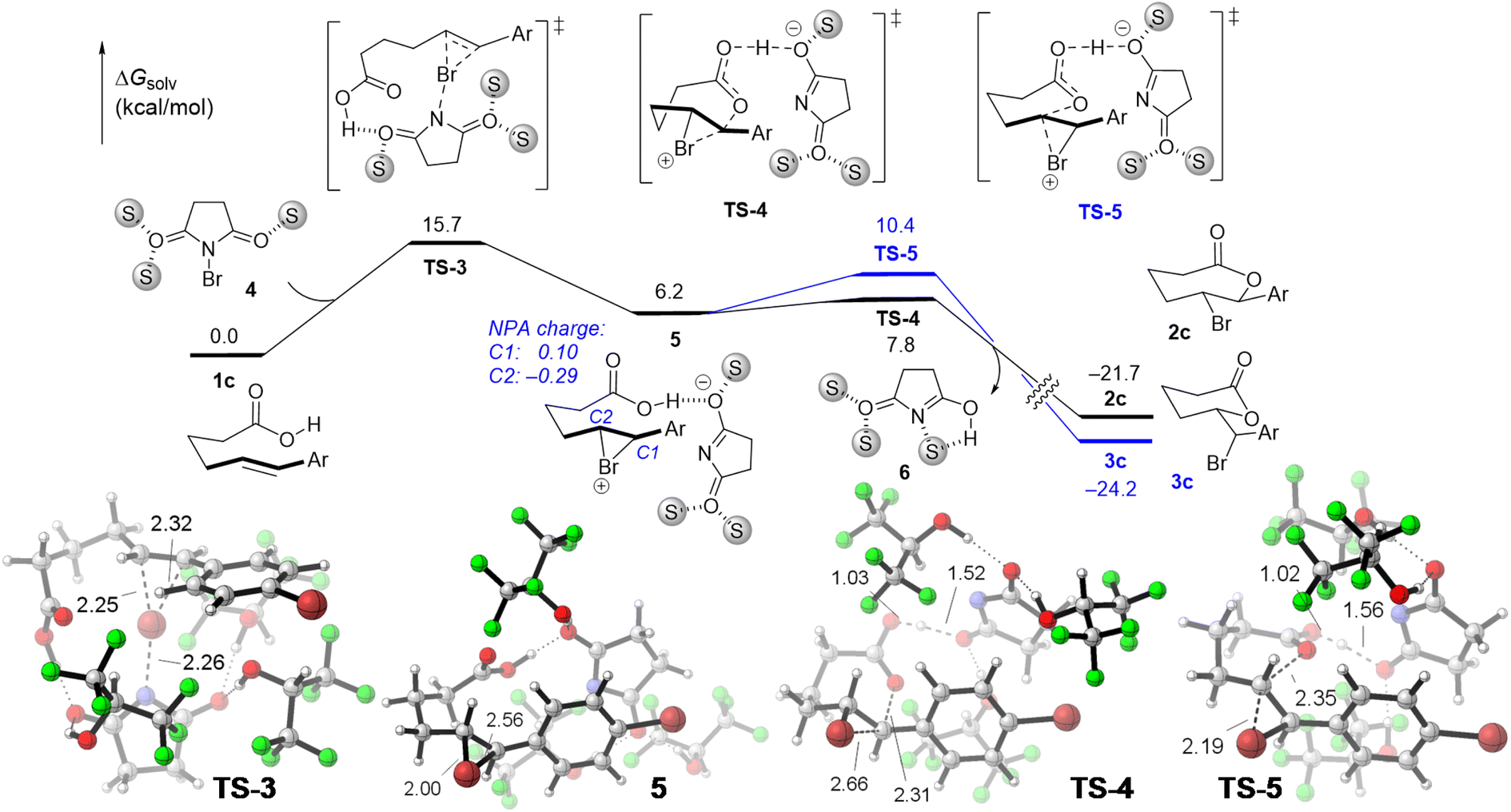 | ||
| Fig. 3 Computed free energy profile for the stepwise bromolactonization in HFIP. S represents the HFIP molecule. | ||
In the bridged bromonium intermediate 5, the natural charge of the C1 atom is higher than that of the C2 atom (Fig. 3). Therefore, the activation barrier for the nucleophilic addition from the oxygen atom of the carboxyl moiety to the C2 atom of the bromonium cation leading to the endo-cyclic product is calculated to be 2.6 kcal mol−1 lower in energy than that for the nucleophilic addition to the C1 atom. This result is consistent with experimental data where the formation of the endo-product is more favorable in HFIP. Our computational study demonstrates that the regioselectivity for the bromolactonization in HFIP is determined by electronic properties and this reaction is under kinetic control.
On the other hand, our experiments demonstrated that when TfOH is used as a catalyst, the exo-cyclic product is more favorable (Table 1). By using DFT calculations, we found that in the presence of TfOH, the NBS reagent can easily be protonated lowering the LUMO energy from −1.05 eV to −2.04 eV for neutral and protonated NBS, respectively leading to the enhancement of NBS reactivity.21 When TfOH is included, the activation barrier for the electrophile addition TS-6 is calculated to be 9.2 kcal mol−1 relative to 1 (Fig. 4), which is 6.5 kcal mol−1 lower than that without TfOH, i.e., TS-3. In addition, TfOH can also have a great stabilization effect on the bridged bromonium species. From bromonium intermediate 8, although the nucleophilic addition generating the endo-product is kinetically more favorable, the reaction is now under thermodynamic control. The endo-product can isomerize to generate a more stable exo-cyclic product. This computational result is consistent with our additional control experiments, in which the endo-cyclic species 2 can transform into the exo-cyclic species 3 in the presence of TfOH or pTSA (Fig. 2b – upper route, also see page S9 in the ESI† for more details). It should be noted that when the isomerization was carried out in DCE instead of HFIP (Fig. 2b – lower route), average yields of exo-product 3a with poor diastereoselectivity (in the case of TfOH) or incomplete conversion (in the case of pTSA) were observed, highlighting the vital role of HFIP in this conversion.
 | ||
| Fig. 4 Computed free energy profile for the stepwise bromolactonization in HFIP catalyzed by TfOH. S represents the HFIP molecule. | ||
Conclusion
In summary, we introduce an effective and straightforward method to access two different analogues of bromolactones by precisely manipulating the regioselectivity of the bromolactonization reaction in HFIP solvent. DFT calculations underscored the significance of HFIP in both endo-bromolactonization under kinetic conditions and exo-bromolactonization under thermodynamic conditions. This method, when combined with our recently developed ring-opening olefination of cyclic ketone ketals, offers a novel pathway for late-stage modifications of cyclic ketones into functionalized lactones.Data availability
All data are available in the main text or the ESI.†Author contributions
Conceptualization: TAT and TVN. Experimental work: NTAP and TAT. Computational work: BKM. Supervision: TVN. Writing – original draft: TAT, BKM, and TVN. Writing – review & editing: TVN.Conflicts of interest
The authors declare that they have no competing interests.Acknowledgements
Australian Research Council: FT180100260 (TVN) and DP200100063 (TVN). The authors thank the Australian Research Council and the UNSW Faculty of Science for financial support.References
- (a) G. H. Schmid, in Double-Bonded Functional Groups, 1989, pp. 679–731 Search PubMed; (b) M.-F. Ruasse, in Advances in Physical Organic Chemistry, ed. D. Bethell, Academic Press, 1993, vol. 28, pp. 207–291 Search PubMed; (c) A. Castellanos and S. P. Fletcher, Current Methods for Asymmetric Halogenation of Olefins, Chem.–Eur. J., 2011, 17, 5766–5776 CrossRef CAS PubMed; (d) S. E. Denmark, W. E. Kuester and M. T. Burk, Catalytic, Asymmetric Halofunctionalization of Alkenes—A Critical Perspective, Angew. Chem., Int. Ed., 2012, 51, 10938–10953 CrossRef CAS PubMed; (e) U. Hennecke, New Catalytic Approaches towards the Enantioselective Halogenation of Alkenes, Chem.–Asian J., 2012, 7, 456–465 CrossRef CAS PubMed; (f) J. Chen and L. Zhou, Recent Progress in the Asymmetric Intermolecular Halogenation of Alkenes, Synthesis, 2014, 46, 586–595 CrossRef; (g) Y. A. Cheng, W. Z. Yu and Y.-Y. Yeung, Recent advances in asymmetric intra- and intermolecular halofunctionalizations of alkenes, Org. Biomol. Chem., 2014, 12, 2333–2343 RSC; (h) A. J. Cresswell, S. T.-C. Eey and S. E. Denmark, Catalytic, Stereoselective Dihalogenation of Alkenes: Challenges and Opportunities, Angew. Chem., Int. Ed., 2015, 54, 15642–15682 CrossRef CAS PubMed.
- (a) S. A. Snyder, D. S. Treiler and A. P. Brucks, Halonium-Induced Cyclization Reactions, Aldrichimica Acta, 2011, 44, 27–40 CAS; (b) A. Mendoza, F. J. Fananas and F. Rodríguez, Asymmetric Halocyclizations of Unsaturated Compounds: An Overview and Recent Developments, Curr. Org. Synth., 2013, 10, 384–393 CrossRef CAS; (c) S. Zheng, C. M. Schienebeck, W. Zhang, H.-Y. Wang and W. Tang, Cinchona Alkaloids as Organocatalysts in Enantioselective Halofunctionalization of Alkenes and Alkynes, Asian J. Org. Chem., 2014, 3, 366–376 CrossRef CAS; (d) J. R. Wolstenhulme and V. Gouverneur, Asymmetric Fluorocyclizations of Alkenes, Acc. Chem. Res., 2014, 47, 3560–3570 CrossRef CAS PubMed; (e) M. H. Gieuw, Z. Ke and Y.-Y. Yeung, Lewis Base Catalyzed Stereo- and Regioselective Bromocyclization, Chem. Rec., 2017, 17, 287–311 CrossRef CAS PubMed; (f) B. Maji, Stereoselective Haliranium, Thiiranium and Seleniranium Ion-Triggered Friedel–Crafts-Type Alkylations for Polyene Cyclizations, Adv. Synth. Catal., 2019, 361, 3453–3489 CrossRef CAS; (g) A. Maria Faisca Phillips and A. J. L. Pombeiro, Recent Developments in Enantioselective Organocatalytic Cascade Reactions for the Construction of Halogenated Ring Systems, Eur. J. Org Chem., 2021, 2021, 3938–3969 CrossRef CAS; (h) J. Yan, Z. Zhou, Q. He, G. Chen, H. Wei and W. Xie, The applications of catalytic asymmetric halocyclization in natural product synthesis, Org. Chem. Front., 2022, 9, 499–516 RSC; (i) R. Nishiyori, T. Mori, K. Okuno and S. Shirakawa, Chiral sulfide and selenide catalysts for asymmetric halocyclizations and related reactions, Org. Biomol. Chem., 2023, 21, 3263–3275 RSC.
- (a) M. D. Dowle and D. I. Davies, Synthesis and synthetic utility of halolactones, Chem. Soc. Rev., 1979, 8, 171–197 RSC; (b) Y. I. Gevaza and V. I. Staninets, Electrophilic heterocyclization of unsaturated carboxylic acids in the synthesis of lactones (review), Chem. Heterocycl. Compd., 1988, 24, 1073–1088 CrossRef; (c) M. Grabarczyk, K. Wińska and W. Mączka, An Overview of Synthetic Methods for the Preparation of Halolactones, Curr. Org. Synth., 2019, 16, 98–111 CrossRef CAS PubMed; (d) W. Liu and N. Winssinger, Synthesis of α-exo-Methylene-γ-butyrolactones: Recent Developments and Applications in Natural Product Synthesis, Synthesis, 2021, 53, 3977–3990 CrossRef CAS.
- (a) C. K. Tan, L. Zhou and Y.-Y. Yeung, Organocatalytic Enantioselective Halolactonizations: Strategies of Halogen Activation, Synlett, 2011, 2011, 1335–1339 CrossRef; (b) J. M. J. Nolsøe and T. V. Hansen, Asymmetric Iodolactonization: An Evolutionary Account, Eur. J. Org Chem., 2014, 2014, 3051–3065 CrossRef; (c) R. Kristianslund, J. E. Tungen and T. V. Hansen, Catalytic enantioselective iodolactonization reactions, Org. Biomol. Chem., 2019, 17, 3079–3092 RSC.
- J. D. Griffin, C. L. Cavanaugh and D. A. Nicewicz, Reversing the Regioselectivity of Halofunctionalization Reactions through Cooperative Photoredox and Copper Catalysis, Angew. Chem., Int. Ed., 2017, 56, 2097–2100 CrossRef CAS PubMed.
- (a) I. Colomer, A. E. R. Chamberlain, M. B. Haughey and T. J. Donohoe, Hexafluoroisopropanol as a highly versatile solvent, Nat. Rev. Chem., 2017, 1, 0088 CrossRef CAS; (b) V. Pozhydaiev, M. Power, V. Gandon, J. Moran and D. Lebœuf, Exploiting hexafluoroisopropanol (HFIP) in Lewis and Brønsted acid-catalyzed reactions, Chem. Commun., 2020, 56, 11548–11564 RSC; (c) T. Bhattacharya, A. Ghosh and D. Maiti, Hexafluoroisopropanol: the magical solvent for Pd-catalyzed C–H activation, Chem. Sci., 2021, 12, 3857–3870 RSC; (d) H. F. Motiwala, A. M. Armaly, J. G. Cacioppo, T. C. Coombs, K. R. K. Koehn, V. M. I. V. Norwood and J. Aubé, HFIP in Organic Synthesis, Chem. Rev., 2022, 122, 12544–12747 CrossRef CAS PubMed.
- M. Piejko, J. Moran and D. Lebœuf, Difunctionalization Processes Enabled by Hexafluoroisopropanol, ACS Org. Inorg. Au, 2024 DOI:10.1021/acsorginorgau.3c00067.
- (a) A. M. Arnold, A. Pöthig, M. Drees and T. Gulder, NXS, Morpholine, and HFIP: The Ideal Combination for Biomimetic Haliranium-Induced Polyene Cyclizations, J. Am. Chem. Soc., 2018, 140, 4344–4353 CrossRef CAS PubMed; (b) J. Binder, A. Biswas and T. Gulder, Biomimetic chlorine-induced polyene cyclizations harnessing hypervalent chloroiodane–HFIP assemblies, Chem. Sci., 2023, 14, 3907–3912 RSC.
- C. Qi, G. Force, V. Gandon and D. Lebœuf, Hexafluoroisopropanol-Promoted Haloamidation and Halolactonization of Unactivated Alkenes, Angew. Chem., Int. Ed., 2021, 60, 946–953 CrossRef CAS PubMed.
- M. A. Hussein, U. P. N. Tran, V. T. Huynh, J. Ho, M. Bhadbhade, H. Mayr and T. V. Nguyen, Halide Anion Triggered Reactions of Michael Acceptors with Tropylium Ion, Angew. Chem., Int. Ed., 2020, 59, 1455–1459 CrossRef CAS PubMed.
- (a) U. P. N. Tran, G. Oss, D. P. Pace, J. Ho and T. V. Nguyen, Tropylium-promoted carbonyl–olefin metathesis reactions, Chem. Sci., 2018, 9, 5145–5151 RSC; (b) G. Oss and T. V. Nguyen, Iodonium-Catalyzed Carbonyl–Olefin Metathesis Reactions, Synlett, 2019, 30, 1966–1970 CrossRef CAS; (c) U. P. N. Tran, G. Oss, M. Breugst, E. Detmar, D. P. Pace, K. Liyanto and T. V. Nguyen, Carbonyl–Olefin Metathesis Catalyzed by Molecular Iodine, ACS Catal., 2019, 9, 912–919 CrossRef CAS; (d) K. Omoregbee, K. N. H. Luc, A. H. Dinh and T. V. Nguyen, Tropylium-promoted prenylation reactions of phenols in continuous flow, J. Flow Chem., 2020, 10, 161–166 CrossRef CAS; (e) D. P. Pace, R. Robidas, U. P. N. Tran, C. Y. Legault and T. V. Nguyen, Iodine-Catalyzed Synthesis of Substituted Furans and Pyrans: Reaction Scope and Mechanistic Insights, J. Org. Chem., 2021, 86, 8154–8171 CrossRef CAS PubMed; (f) J. S. Mann, B. K. Mai and T. V. Nguyen, Carbocation-Catalyzed Intramolecular and Intermolecular Carbonyl-Alkyne Metathesis Reactions, ACS Catal., 2023, 13, 2696–2701 CrossRef CAS; (g) B. Pu and T. V. Nguyen, HFIP-assisted Brønsted acid catalysed synthesis of furan derivatives, Aust. J. Chem., 2023, 76, 58–62 CrossRef CAS.
- (a) M. A. Hussein, A. H. Dinh, V. T. Huynh and T. V. Nguyen, Synthesis of tertiary amines by direct Brønsted acid catalyzed reductive amination, Chem. Commun., 2020, 56, 8691–8694 RSC; (b) T. A. To, C. Pei, R. M. Koenigs and T. V. Nguyen, Hydrogen Bonding Networks Enable Brønsted Acid-Catalyzed Carbonyl-Olefin Metathesis, Angew. Chem., Int. Ed., 2022, 61, e202117366 CrossRef PubMed; (c) T. A. To, B. K. Mai and T. V. Nguyen, Toward Homogeneous Brønsted-Acid-Catalyzed Intramolecular Carbonyl–Olefin Metathesis Reactions, Org. Lett., 2022, 24, 7237–7241 CrossRef CAS PubMed.
- T. A. To and T. V. Nguyen, Olefination of Aromatic Carbonyls via Site-Specific Activation of Cycloalkanone Ketals, Angew. Chem., Int. Ed., 2024, 63, e202317003 CrossRef CAS PubMed.
- R.-J. Tang, T. Milcent and B. Crousse, Regioselective Halogenation of Arenes and Heterocycles in Hexafluoroisopropanol, J. Org. Chem., 2018, 83, 930–938 CrossRef CAS PubMed.
- W. Wang, X. Yang, R. Dai, Z. Yan, J. Wei, X. Dou, X. Qiu, H. Zhang, C. Wang, Y. Liu, S. Song and N. Jiao, Catalytic Electrophilic Halogenation of Arenes with Electron-Withdrawing Substituents, J. Am. Chem. Soc., 2022, 144, 13415–13425 CrossRef CAS PubMed.
- (a) I. Roberts and G. E. Kimball, The Halogenation of Ethylenes, J. Am. Chem. Soc., 1937, 59, 947–948 CrossRef CAS; (b) G. A. Olah and J. M. Bollinger, Stable carbonium ions. XLVIII. Halonium ion formation via neighboring halogen participation. Tetramethylethylene halonium ions, J. Am. Chem. Soc., 1967, 89, 4744–4752 CrossRef CAS.
- (a) K. D. Ashtekar, M. Vetticatt, R. Yousefi, J. E. Jackson and B. Borhan, Nucleophile-Assisted Alkene Activation: Olefins Alone Are Often Incompetent, J. Am. Chem. Soc., 2016, 138, 8114–8119 CrossRef CAS PubMed; (b) N. Salehi Marzijarani, R. Yousefi, A. Jaganathan, K. D. Ashtekar, J. E. Jackson and B. Borhan, Absolute and relative facial selectivities in organocatalytic asymmetric chlorocyclization reactions, Chem. Sci., 2018, 9, 2898–2908 RSC; (c) R. Yousefi, A. Sarkar, K. D. Ashtekar, D. C. Whitehead, T. Kakeshpour, D. Holmes, P. Reed, J. E. Jackson and B. Borhan, Mechanistic Insights into the Origin of Stereoselectivity in an Asymmetric Chlorolactonization Catalyzed by (DHQD)2PHAL, J. Am. Chem. Soc., 2020, 142, 7179–7189 CrossRef CAS PubMed.
- R. Van Lommel, J. Bock, C. G. Daniliuc, U. Hennecke and F. De Proft, A dynamic picture of the halolactonization reaction through a combination of ab initio metadynamics and experimental investigations, Chem. Sci., 2021, 12, 7746–7757 RSC.
- (a) S. Bag, S. K, A. Mondal, R. Jayarajan, U. Dutta, S. Porey, R. B. Sunoj and D. Maiti, Palladium-Catalyzed meta-C–H Allylation of Arenes: A Unique Combination of a Pyrimidine-Based Template and Hexafluoroisopropanol, J. Am. Chem. Soc., 2020, 142, 12453–12466 CrossRef CAS PubMed; (b) Y. Zhou, R.-C. Xue, Y. Feng and L. Zhang, How Does HOTf/HFIP Cooperative System Catalyze the Ring-Opening Reaction of Cyclopropanes? A DFT Study, Asian J. Org. Chem., 2020, 9, 311–316 CrossRef CAS.
- A. M. Arnold, P. Dullinger, A. Biswas, C. Jandl, D. Horinek and T. Gulder, Enzyme-like polyene cyclizations catalyzed by dynamic, self-assembled, supramolecular fluoro alcohol-amine clusters, Nat. Commun., 2023, 14, 813 CrossRef CAS PubMed.
- D. Barišić, I. Halasz, A. Bjelopetrović, D. Babić and M. Ćurić, Mechanistic Study of the Mechanochemical PdII-Catalyzed Bromination of Aromatic C–H Bonds by Experimental and Computational Methods, Organometallics, 2022, 41, 1284–1294 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures and methods, characterization data, NMR spectra, computational methods and energies and Cartesian coordinates. See DOI: https://doi.org/10.1039/d4sc01503g |
| ‡ Contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |