DOI:
10.1039/D4SC01040J
(Edge Article)
Chem. Sci., 2024,
15, 8974-8981
A nine-ring fused terrylene diimide exhibits switching between red TADF and near-IR room temperature phosphorescence†
Received
14th February 2024
, Accepted 3rd May 2024
First published on 6th May 2024
Abstract
Herein, we report the first example of a terrylene diimide derivative that switches emission between thermally activated delayed fluorescence (TADF) and room temperature phosphorescence (RTP) in the red region. By design, the molecule TDI-cDBT boasts a symmetrical, consecutively fused nine-ring motif with a kite-like structure. The rigid core formed by the annulated dibenzothiophene moiety favoured efficient intersystem crossing and yielded a narrow-band emission with a full-width half maxima (FWHM) of 0.09 eV, along with high colour purity. A small ΔES1–T1 of 0.04 eV facilitated thermally activated delayed fluorescence, enhancing the quantum yield to 88% in the red region. Additionally, it also prefers a direct triplet emission from the aggregated state. The room temperature phosphorescence observed from the aggregates has a longer emission lifetime of 1.8 ms, which is further prolonged to 8 ms at 77 K in the NIR region. Thus, the current strategy is successful in not only reducing ΔES1–T1 to favour TADF but also serves as a novel platform that can switch emission from TADF to RTP depending upon the concentration.
Introduction
Advancement in technology has driven a thrust for the development of energy-efficient molecules with intense emission in the red to near IR region.1–4 Despite their increased demand, the development of such molecules is quite challenging. According to the energy gap law, this difficulty stems from the two associated contradictory requirements: (i) decreased band gap and (ii) high photoluminescence quantum yields.5 An efficient way to increase the quantum efficiency of these emitters is by harvesting the triplet state, which contributes to 75% of the total exciton populations.6 For this purpose, a handful of small molecules have been developed.7,8 However, this requires multi-step synthesis to incorporate suitable functional groups onto simpler π-conjugated molecules. An alternate approach would be to utilize an inherent red emitter, and minimal functionalization on these molecules can be beneficial for harvesting their triplet excitons. In this direction, terrylene diimide (TDI), a pure red emitter of the rylene diimide (RDI) family with excellent thermal and photochemical stability can be a potential candidate.9 A few TDI derivatives have been shown to emit in the 600–700 nm range.10–14 Ironically, they suffer from reduced quantum yields compared to the parent TDI. It is to be noted that in these cases, the emission presumably emanates only from the singlet excitons, which contribute only 25% of the total excitons.15 Thus, if the triplet excitons can also be harvested, the quantum efficiency would increase, and one can achieve a strongly red-emitting TDI.
In smaller RDIs, either inducing twists to the molecular geometry and/or incorporating hetero atoms into the core has been proven to be useful in efficiently populating the triplet states.16 However, in the case of TDI, a stabilized T1 state often prefers a nonradiative deactivation pathway. It has been observed recently that nitro-TDIs favour intersystem crossing (ISC) but lack any triplet emission due to the stabilized T1 state.17 Thus, this substantiates that only attaining T1 efficiently will not be sufficient for an intense emission. The triplet excitons should be facilitated either to undergo a direct radiative decay or to decay via a thermally mediated reverse intersystem crossing (RISC) to S1. For the former ΔES0–T1 should be larger, and for the latter ΔES1–T1 should be smaller.18 Thus, destabilizing the T1 state of TDI should be beneficial for improving the quantum yield.
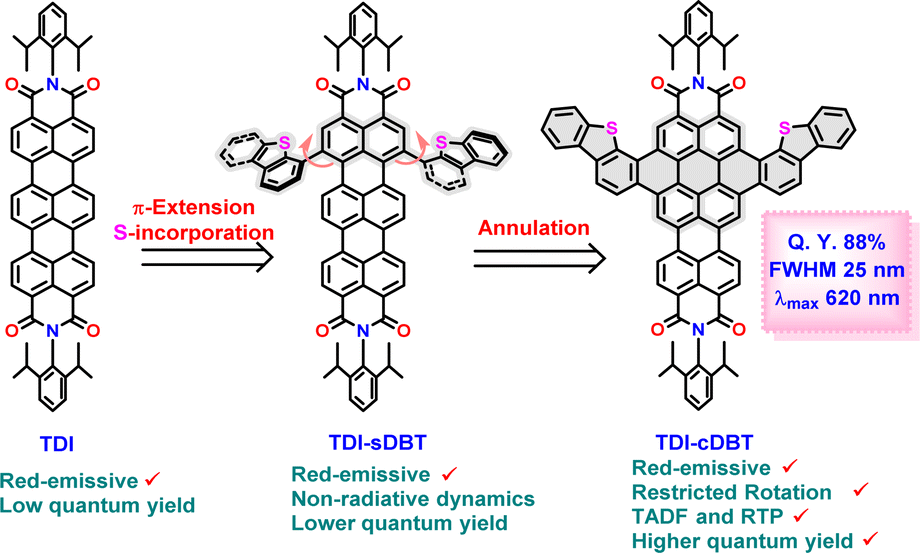
To accomplish this, we propose a novel π-expanded TDI derivative (TDI-cDBT) with a kite-like structure. Our design strategy involves (Scheme 1) (i) an annulation at the bay position of the TDI to increase the HOMO–LUMO gap;19 (ii) introduction of a heteroatom into the annulation frame to facilitate effective intersystem crossing (ISC);20 (iii) the introduced heteroaromatic unit has an electron-rich nature imparting a donor–acceptor character that may further help in reducing ΔES1–T1;21 (iv) a rigid molecular framework to restrict non-radiative decay losses. To probe this strategy, a new TDI derivative with two dibenzothiophene wings fused at the bay positions had been envisaged (Scheme 1). The fusion of a heteroaromatic unit helped to increase the HOMO–LUMO gap and favoured efficient ISC. The rigid molecular framework hindered non-radiative dynamics. These factors combinedly facilitated a narrow-band red emission with an improved intensity. The presence of a smaller S1–T1 energy gap manifested in thermally activated delayed fluorescence (TADF) and a destabilized T1 helped to achieve room temperature phosphorescence (RTP).
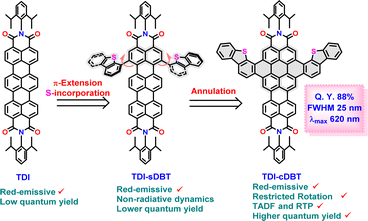 |
| | Scheme 1 Pictorial representation of the design strategy involved in the present work. | |
Results and discussion
Synthesis and characterization of TDI-cDBT
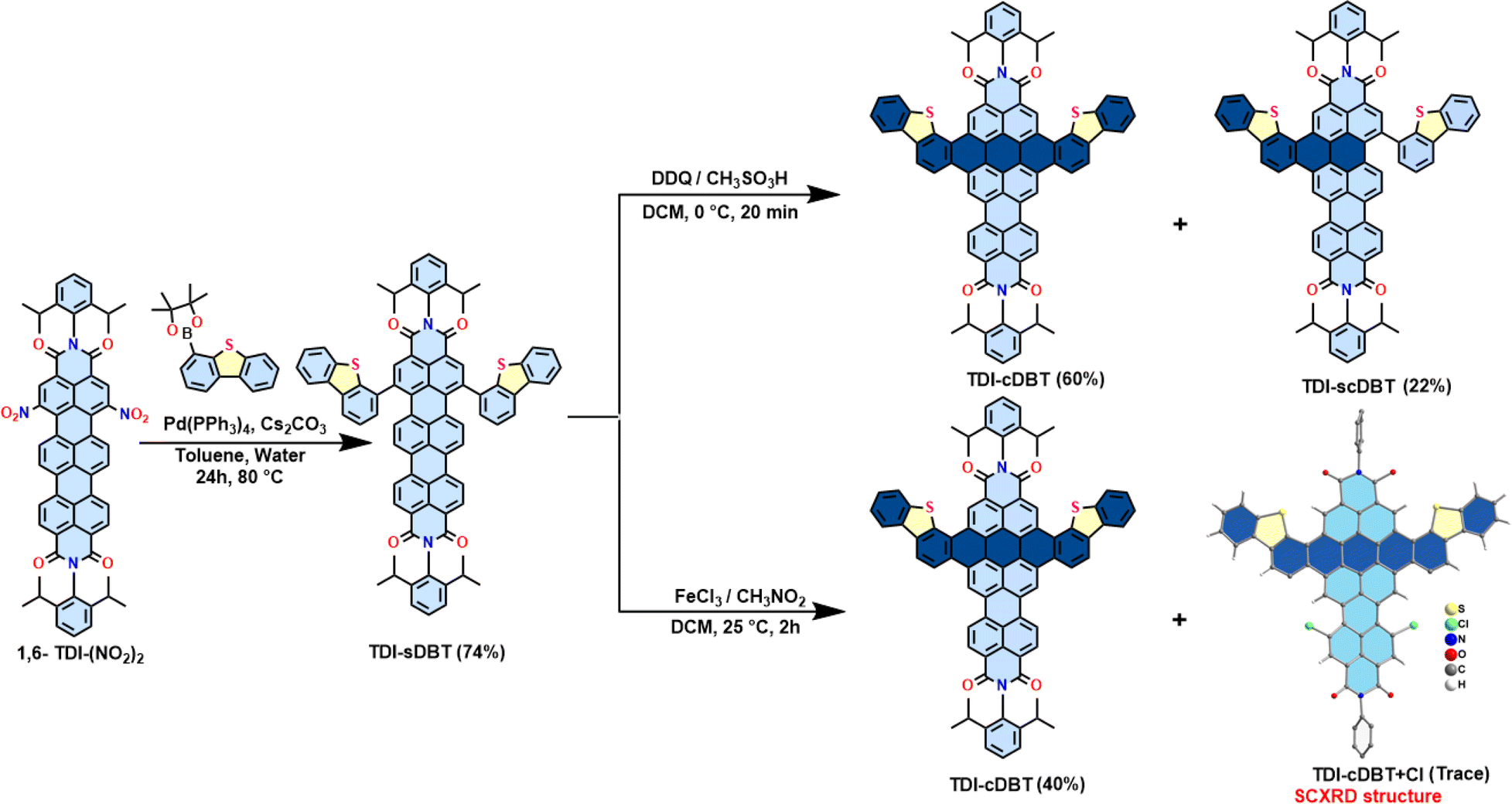
A Suzuki–Miyaura cross-coupling reaction between 1,6-TDI-(NO2)2 and dibenzothiophene-4-boronic acid pinacol ester in the presence of Pd(0) yielded the singly linked product TDI-sDBT as a blue solid (Scheme 2 and Fig. S27, S28, S34†). Subsequently, TDI-sDBT was subjected to a Scholl reaction using ferric chloride in nitromethane to obtain a dark powder. This dark powder on column chromatography using basic alumina with pyridine as the eluent yielded a major pink fraction in 40% yield, followed by another pink fraction in traces. The major pink product was characterized by 1H-NMR (Fig. S30 and S31†) and APCI-HRMS to be TDI-cDBT (Fig. S36†). However, all the efforts to obtain solid-state structural proof for this compound were unsuccessful. Surprisingly, the minor fraction from the above reaction provided single crystals suitable for X-ray diffraction measurements (CCDC 2293830). The structure obtained from the SCXRD study confirms this to be chlorinated TDI-cDBT.22 A modified Scholl reaction using DDQ/methane sulphonic acid was used to avoid undesired chlorination. As shown in Scheme 2 (top right), the desired symmetrical TDI-cDBT was obtained in 60% yield along with single-side fused TDI-scDBT (Fig. S29 and S35†).
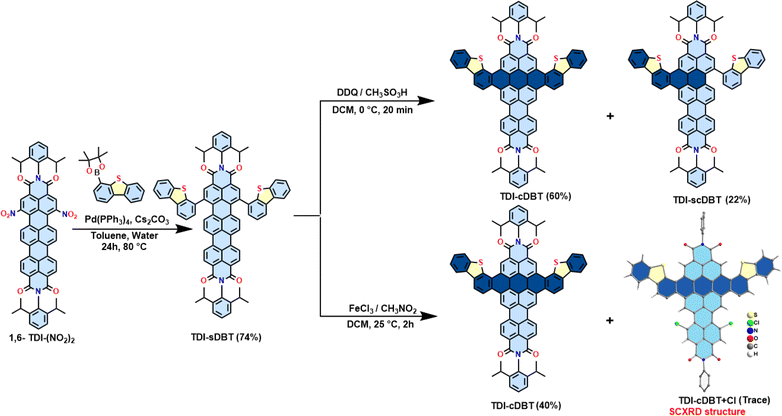 |
| | Scheme 2 Synthesis of bis-dibenzothiophene-fused TDI (TDI-cDBT). | |
Absorption and emission studies
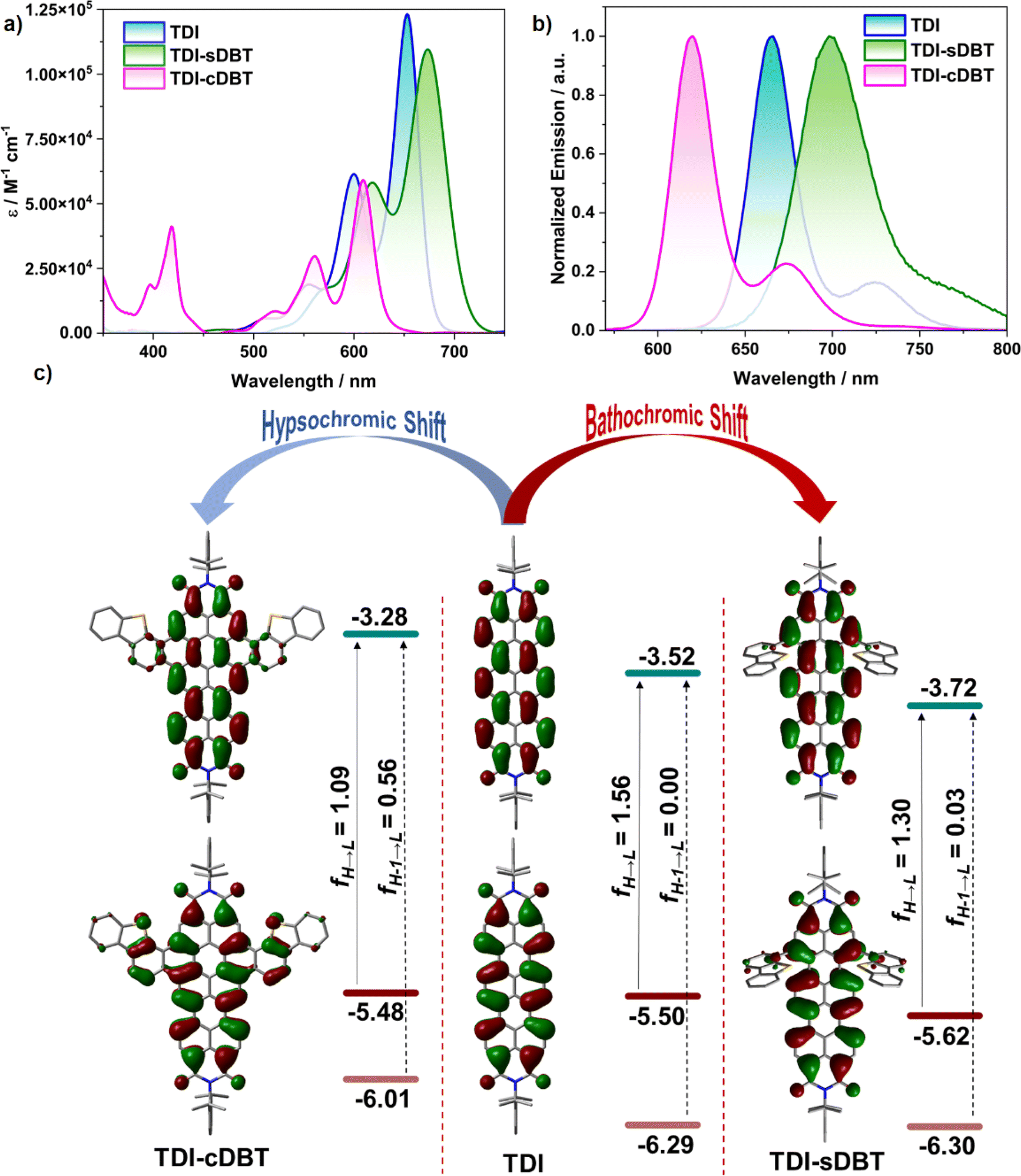
The absorption spectra for both the molecules TDI-sDBT and TDI-cDBT in toluene show an S1 ← S0 transition in the 500–700 nm range with a well-resolved vibronic pattern, similar to TDI (Fig. 1a). The redshift (∼20 nm) in the λmax of TDI-sDBT can be attributed to the extended conjugation. The annulation along the shorter axis in TDI-cDBT has resulted in a huge hypsochromic shift with λmax at 609 nm and a reduced molar extinction coefficient (Fig. 1a and Table 1). This band can be assigned to the HOMO–LUMO transition as supported by spin-flip TDDFT (SF-TDDFT) studies (Fig. 1c and Table S5†).
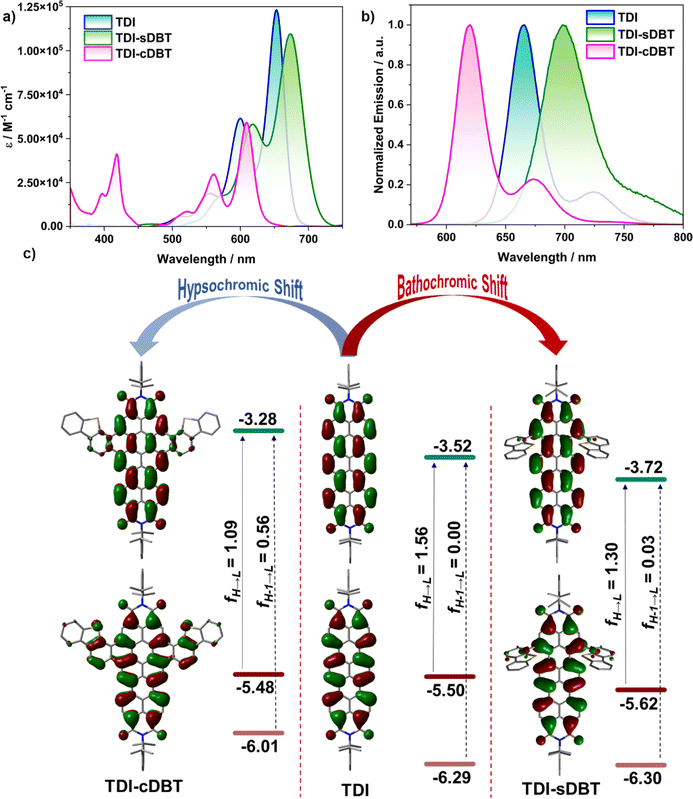 |
| | Fig. 1 (a) Absorption and (b) emission (λex = 550 nm) spectra of the synthesized molecules in 2 × 10−6 M toluene solution and (c) the frontier molecular orbital diagrams representing the oscillator strength. | |
Table 1 Summarized optical data for all the synthesized molecules
| Compound |
λ
max,abs [nm] |
ε [M−1cm−1] |
λ
max,em [nm] |
Stokes shift [cm−1] |
FWHM [nm] |
ϕ
Fl
[%] |
τ
Fl [ns] |
k
r,Fl (s−1) |
|
Absolute fluorescence quantum yield.
|
|
TDI
|
653 |
123![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
666 |
300 |
30 |
27 |
3.2 |
2.2 × 108 |
|
TDI-sDBT
|
673 |
109000 |
698 |
530 |
48 |
13 |
3.6 |
2.4 × 108 |
|
TDI-cDBT
|
609 |
59000 |
620 |
290 |
25 |
41 |
6.0 |
1.6 × 108 |
The π-expansion resulting from annulation offers a coronene-like core, with a slightly destabilized HOMO and a highly destabilized LUMO. Therefore, the main band belonging to the HOMO–LUMO transition gets hypsochromically shifted compared to that of TDI. The detailed cause for this change in HOMO–LUMO energies can be understood from the disruption in conjugation due to the coronene core (Fig. S23†). An additional intense band from the LUMO ← HOMO−1 transition appears at 418 nm, in line with the shorter-axis functionalized RDIs with a highly destabilized HOMO−1.19
As these molecules absorb in the orange-red region, they are expected to fluoresce in the red region. Any emission originating in the longer wavelength region will be hampered by significant nonradiative losses, thus reducing the quantum yields.5 To verify the case with the current set of molecules, the emission spectra for both TDI-sDBT and TDI-cDBT were measured in toluene (Fig. 1b, S1–S4† and Table 1). Both molecules inherited the mirror-image emission pattern from the parent TDI. TDI-sDBT showed a broad emission with a λmax at 698 nm having a large Stokes shift of 530 cm−1 (absolute quantum yield 13%). TDI-cDBT showed a narrow emission with a λmax at 620 nm with a Stokes shift of 290 cm−1 (absolute quantum yield 41%). The reduction in the quantum yield for TDI-sDBT (TDI = 27%) is attributed primarily to the free rotation of the heteroaromatic unit around the single bond connected to the TDI core. However, TDI-cDBT addresses this: the increased band gap and the structural rigidity achieved by annulation together hinder any non-radiative pathways, increasing the emission quantum yields to much higher than that of the parent TDI. The rigidity of the core can be further corroborated through the strikingly narrow emission with an FWHM of 0.09 eV and color purity of CIE 0.69, 0.30 (CIE for pure red 0.70, 0.29).
Thermally activated delayed fluorescence studies
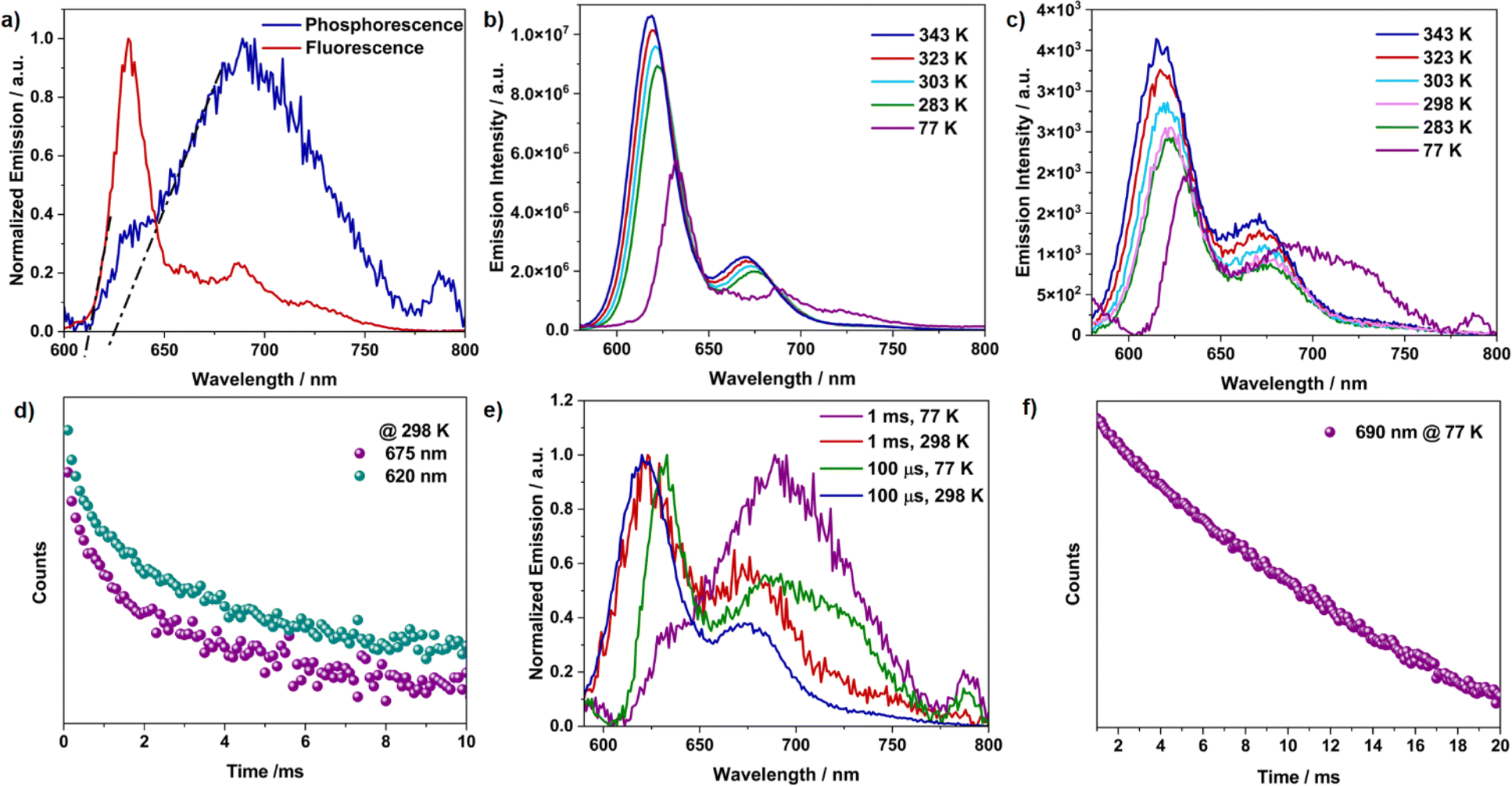
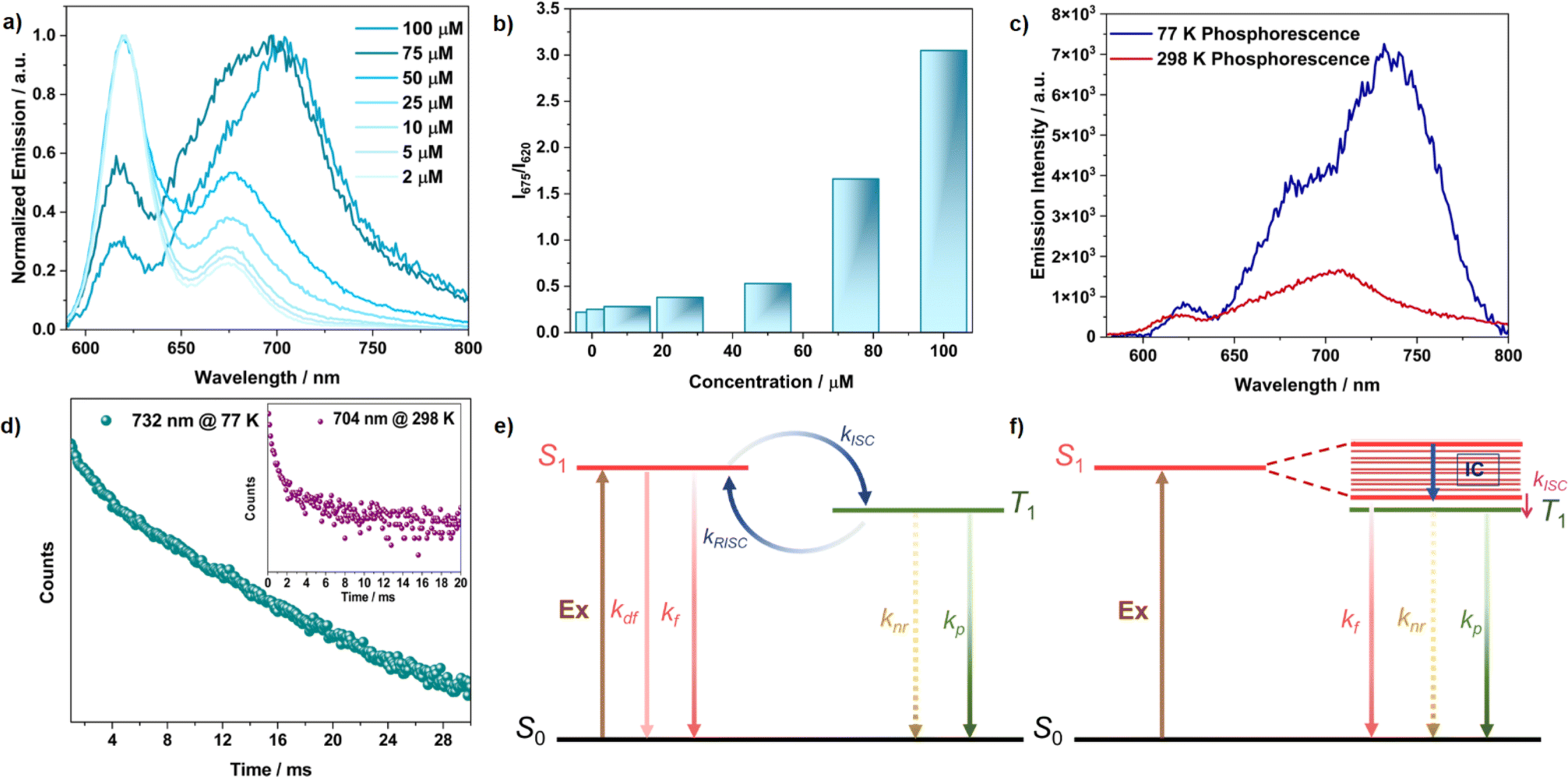
The measured absolute quantum yield of 41% for TDI-cDBT can be enhanced if the proposed triplet contribution is effectively harvested. To evaluate this possibility, steady-state and time-resolved emission measurements at 77 K were performed in toluene. The low-temperature fluorescence spectrum showed a sharp peak (λmax at 630 nm) and a broad emission in the longer wavelength region (Fig. 2a and b). After an initial delay of 1 ms, a broad red-shifted emission was observed at λmax 690 nm with a lifetime of 6 ms (Fig. 2a and f). This red-shifted emission and prolonged lifetime are attributed to phosphorescence.23 The energy gap (ΔES1–T1) calculated from the onsets of low-temperature fluorescence and phosphorescence spectra was found to be 0.04 eV (Fig. 2a). Such a smal energy gap is appropriate for RISC and may result in delayed emission.24 To verify this, variable temperature steady-state and time-resolved emission experiments were carried out (Fig. 2b and c). With an increase in temperature, an increase in emission intensity with a blue shift in both prompt and delayed emission was observed. This temperature dependence of the emission intensity confirms that the delayed emission is TADF.25 Intriguingly, the TADF persisted even at 77 K, suggesting the presence of dual emission; the sharp peak at 630 nm stems from the delayed singlet emission and broad band from 650 to 775 nm from triplet emission (Fig. 2c and e). The lifetime calculated from the transient PL decay at room temperature was 1.1 ms and 1.2 ms for the 620 nm and 675 nm bands, respectively (Fig. 2d). These lifetimes grew shorter at higher temperatures (Fig. S8 and Table S1†), validating the thermal contribution.26 The RISC rate calculated experimentally was of the order of 103 s−1 (Table S2†). Taken together, the overall emission quantum yield sums up to 88% from the S1 to S0 state (41% and 47% for prompt and delayed emission, respectively). Thus, as proposed, the incorporation of sulphur has effectively enhanced the ISC and RISC efficiency. Moreover, the annulation of heteroaromatics rigidified the TDI core and has supressed the nonradiative losses, resulting in the improved emission quantum yields. This annulation has further resulted in destabilization of the T1 state; thus increasing ΔET1–S0, (Table S6†) favouring phosphorescence.18
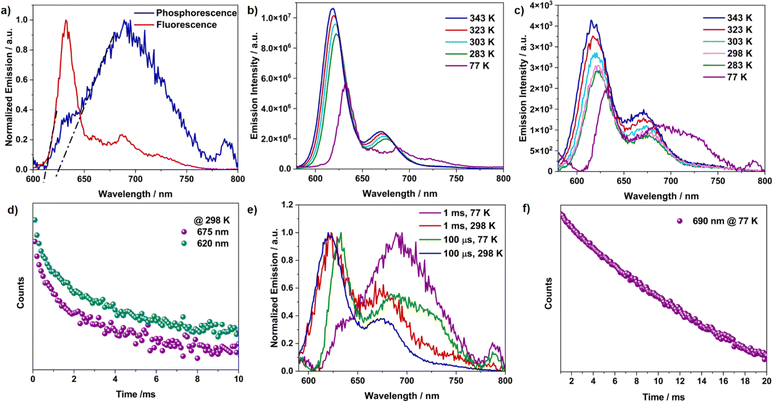 |
| | Fig. 2 (a) Normalized fluorescence and phosphorescence emission at 77 K (initial delay = 1 ms), dotted lines represent the onset for both the bands; variable temperature (b) fluorescence emission, and (c) delayed emission (initial delay = 100 μs); (d) time-resolved decay profile recorded at room temperature; (e) normalized delayed emission with variable temperature and initial delay; (f) time-resolved decay profile recorded at 77 K for TDI-cDBT. All measurements were performed for TDI-cDBT with a 550 nm excitation in 2 × 10−6 M toluene solution. | |
Aggregate studies
The room temperature 1H-NMR spectrum of TDI-cDBT was considerably broader in common organic solvents, suggesting that the molecule has strong π–π interactions and might undergo aggregation (Fig. S31†). Concentration-dependent emission and absorption studies were carried out to gain more insight into the aggregate state. In steady state measurements, on increasing the concentration, the emission intensity at λmax 620 nm decreased while the intensity for the 675 nm band comparatively increased with a slight bathochromic shift (Fig. 3a, b and S11†). This red-shifted emission with a large Stokes shift (∼3000 cm−1) and decreased quantum yield of 15% at 100 μM can be attributed to the emission arising predominantly from aggregates (Fig. S13 and S14†).27 In H-aggregates, the emission intensity of the 0–0 band is supposed to be reduced due to its forbidden nature, and the 0–1 peak should be prominent.28 This results in a perceived larger Stokes shift and reduced quantum yields. Thus, the observed change in emission with respect to concentration suggests the formation of H-aggregates in TDI-cDBT in line with larger planar rylene diimides.28b,c The concentration-dependent absorption studies also supported the formation of aggregates. Under dilute conditions, the molecule showed a well-resolved vibronic pattern in the absorption spectra originating from the S1 ← S0 transition: the 0–0 transition contributes to the λmax (609 nm) while the hump at 560 nm belongs to the 0–1 transition. At higher concentrations, the intensity of the 560 nm (1 ← 0) peak increases in comparison to that of the 609 nm peak (Fig. S11†). This decrease in the absorbance ratio A0–0/A0–1 from 1.8 (μM) to 0.95 (mM) confirms the formation of H-aggregates at higher concentrations (Fig. S12†).29
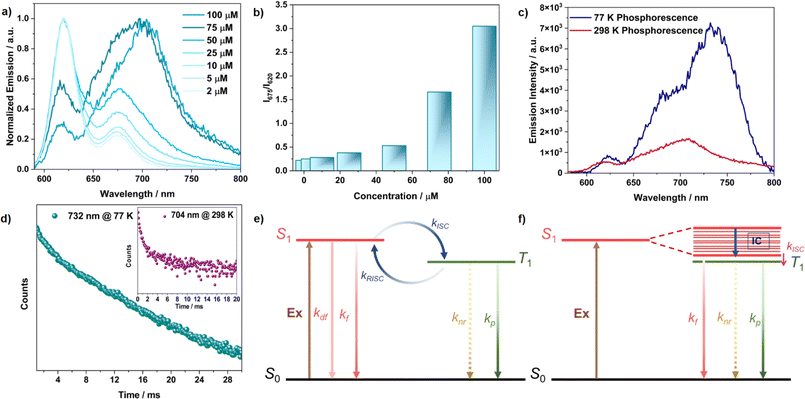 |
| | Fig. 3 (a) Concentration-dependent emission studies; (b) intensity ratio of aggregates upon monomer emission; (c) phosphorescence emission recorded with an initial delay of 100 μs; (d) time-resolved decay profile recorded at 77 K, and the inset shows the one recorded at 298 K; plausible Jablonski diagram for TDI-cDBT (e) at dilute concentration (monomer); (f) at higher concentration (aggregate) for TDI-cDBT; all measurements are performed for TDI-cDBT with 550 nm excitation in 1 × 10−4 M toluene solution. | |
Phosphorescence studies in aggregates
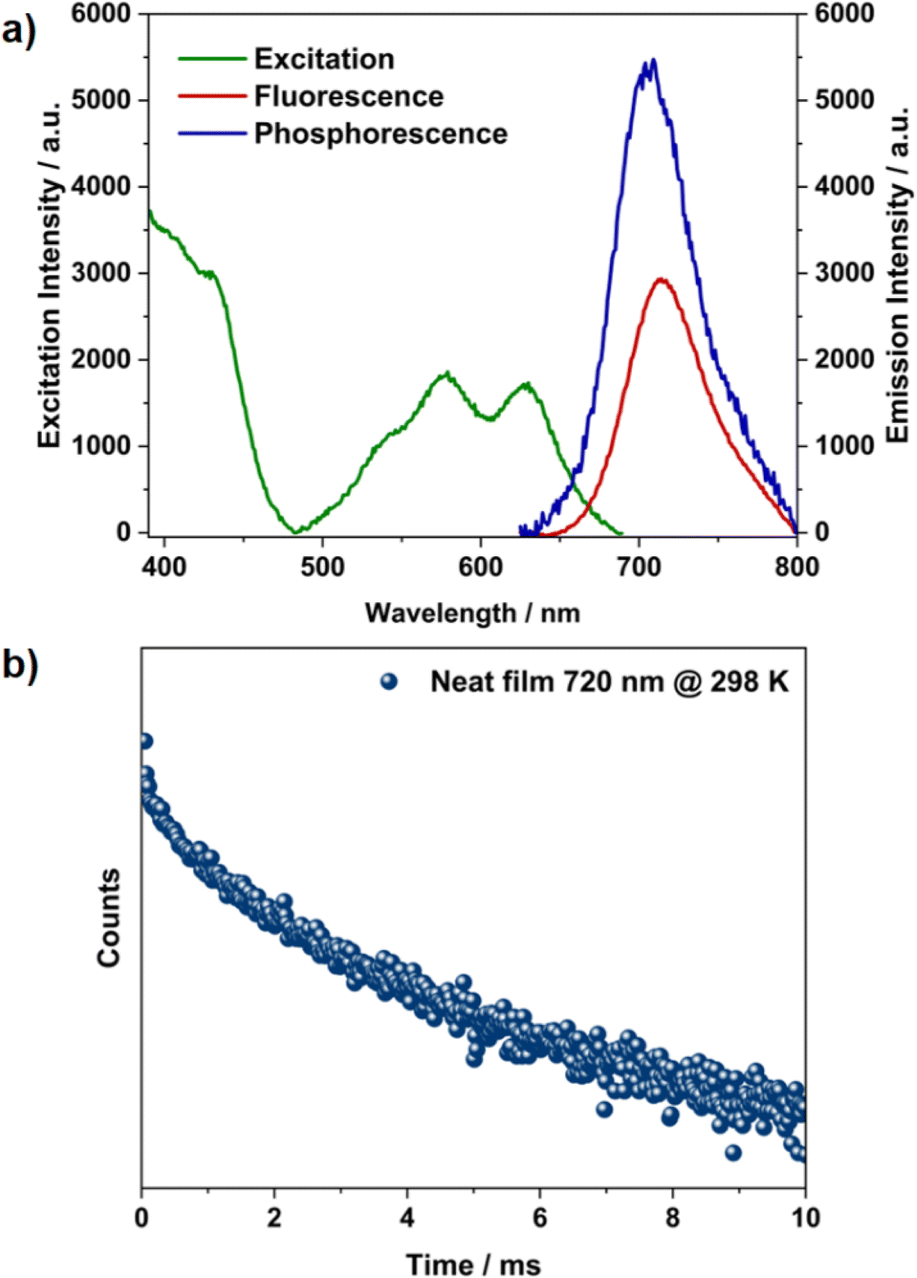
Aggregates are known to facilitate intersystem crossing due to reduced ΔES–T, and this, in a few cases, has led to direct triplet emission.30,31 The time-resolved emission experiment was performed for the aggregates to verify this. After an initial delay of 100 μs, the emission persisted with a longer lifetime of 1.8 ms at room temperature (Fig. 3c, S16 and S17†). This delayed emission overlapped with the phosphorescence emission observed at low concentration at 77 K, revealing the possibility of RTP from the aggregated state (Fig. S9†). The emission intensity for the aggregates increased significantly at 77 K both in prompt and delayed emission, along with a prolonged lifetime of 8 ms (Fig. 3c and d). Unlike the monomer case, this increment in emission intensity at 77 K (Fig. S15, S17 and Table S2†) shows that the origin of delayed emission from the aggregated state is phosphorescence and not TADF (Fig. 3f).32 Aggregation induced splitting of the excitonic bands acts as an energy trap and reduces the ΔES–T.30,31 This energetically favours ISC, leading to direct triplet emission. An identical behaviour is expected from the solid state as well, owing to the close packing of these fluorophores. The excitation and emission spectra obtained from neat films were in good agreement with the absorption and emission spectra obtained at higher concentrations in toluene solution, reiterating the aggregate origin at higher concentrations (Fig. 4a). The prolonged lifetime observed from the neat film (2.4 ms, Fig. 4b) and aggregated state (1.8 ms) are attributed to the room temperature phosphorescence.
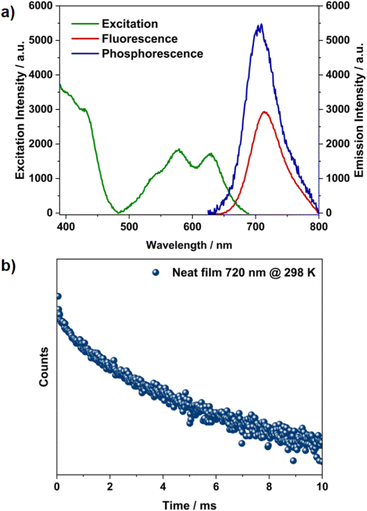 |
| | Fig. 4 (a) Excitation, fluorescence emission, and phosphorescence (initial delay = 30 μs) spectra at 298 K and (b) transient decay profile of TDI-cDBT in a neat film. | |
Density functional theory calculations
Density functional theory (DFT) calculations were carried out to validate our experimental results and hypothesis. As expected, the SF-TDDFT calculations on TDI-cDBT supported the increased HOMO–LUMO gap compared to that of TDI (Fig. 1c, 3e, S22 and Table S6†). This resulted in enhanced radiative rates and increased quantum efficiency. The spin–orbit coupling matrix element calculated for S1 and T1 states were 0.22 cm−1 and 0.02 cm−1 for TDI-cDBT and TDI, respectively (Table S7†). Thus, the incorporation of sulphur has indeed helped in the enhancement of ISC compared to that of parent TDI.
Conclusion
In conclusion, a novel sulphur-embedded terrylene diimide with a wing-like structure having intense red emission has been successfully obtained using a simple synthetic method in high yields. The π-expansion at the bay position helped to widen the bandgap and imparted structural rigidity, thus improving the radiative decay. The introduction of a donor–acceptor character between the heteroaromatic unit and TDI core further helped to reduce the ΔES1–T1, favouring a TADF emission with a quantum yield of 88%. The problem of aggregation in rylene diimides became a boon for this molecule, resulting in RTP. Therefore, it can be highlighted that TDI-cDBT shows excellent TADF emission under dilute conditions, whereas it demonstrates phosphorescence at higher concentrations. Hence, rigidification of the TDI core, along with heteroatom incorporation, can be an efficient strategy to achieve molecules with switching between red TADF and near-IR RTP.
Data availability
Complete characterization, spectral, and crystallographic data along with computational details have been provided in the ESI.†
Author contributions
S. J. performed all the synthesis, characterization, and experimental work. S. J., K. S. M., and J. S. analysed the data and wrote the manuscript. P. K. M. and M. P. contributed to SCXRD experiments and structure refinement. S.S. contributed to SCXRD data collection and refinement at IISER Bhopal. M. D. and D. G. carried out the computational studies. J. S. supervised the project. All authors have given approval to the final version of the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
S. J., S. S. and M. D. thank DST, New Delhi for the INSPIRE fellowship. K. S. M. thanks CSIR, New Delhi, for the SRF. The authors thank SERB, New Delhi (CRG/2020/004632) for the financial support. The authors thank Mr Giorgio Bais for technical support at the XRD1 beamline, Elettra Synchrotron. The work at Elettra, Italy, was funded by a grant from the Italian Ministry of Foreign Affairs and International Cooperation and DST, New Delhi. We thank Dr Ruchika Mishra for fruitful suggestions.
Notes and references
-
(a) U. Mahmood and R. Weissleder, Mol. Cancer Ther., 2003, 2, 489–496 CAS
 ;
(b) A. J. Wilson, D. Devasia and P. K. Jain, Chem. Soc. Rev., 2020, 49, 6087–6112 RSC ;
(c) P. Bon and L. Cognet, ACS Photonics, 2022, 9, 2538–2546 CrossRef CAS PubMed ;
(d) X. Zhen, Y. Tao, Z. An, P. Chen, C. Xu, R. Chen, W. Huang and K. Pu, Adv. Mater., 2017, 29, 1606665 CrossRef PubMed ;
(e) T. Wu, J. Huang and Y. Yan, Cell Rep., 2022, 3, 100771 CAS ;
(f) S. Yoon, S. Y. Cheon, S. Park, D. Lee, Y. Lee, S. Han, M. Kim and H. Koo, Biomater. Res., 2022, 26, 57 CrossRef PubMed .
;
(b) A. J. Wilson, D. Devasia and P. K. Jain, Chem. Soc. Rev., 2020, 49, 6087–6112 RSC ;
(c) P. Bon and L. Cognet, ACS Photonics, 2022, 9, 2538–2546 CrossRef CAS PubMed ;
(d) X. Zhen, Y. Tao, Z. An, P. Chen, C. Xu, R. Chen, W. Huang and K. Pu, Adv. Mater., 2017, 29, 1606665 CrossRef PubMed ;
(e) T. Wu, J. Huang and Y. Yan, Cell Rep., 2022, 3, 100771 CAS ;
(f) S. Yoon, S. Y. Cheon, S. Park, D. Lee, Y. Lee, S. Han, M. Kim and H. Koo, Biomater. Res., 2022, 26, 57 CrossRef PubMed .
-
(a) Y. Shirota and H. Kageyama, Chem. Rev., 2007, 107, 953–1010 CrossRef CAS PubMed ;
(b) G. Hong, X. Gan, C. Leonhardt, Z. Zhang, J. Seibert, J. M. Busch and S. Bräse, Adv. Mater., 2021, 33, 2005630 CrossRef CAS PubMed ;
(c) J. Li, T. Nakagawa, J. MacDonald, Q. Zhang, H. Nomura, H. Miyazaki and C. Adachi, Adv. Mater., 2013, 25, 3319–3323 CrossRef CAS PubMed .
-
(a) Z. Shen, P. E. Burrows, V. Bulovic, S. R. Forrest and M. E. Thompson, Science, 1997, 276, 2009–2011 CrossRef CAS ;
(b) C. D. Müller, A. Falcou, N. Reckefuss, M. Rojahn, V. Wiederhirn, P. Rudati, H. Frohne, O. Nuyken, H. Becker and K. Meerholz, Nature, 2003, 421, 829–833 CrossRef PubMed ;
(c) M. Vasilopoulou, D. G. Georgiadou, A. Soultati, A. M. Douvas, G. Papadimitropoulos, D. Davazoglou, G. Pistolis, N. A. Stathopoulos, T. Kamalakis, D. Alexandropoulos, N. Vainos, C. T. Politi, L. C. Palilis, S. Couris, A. G. Coutsolelos and P. Argitis, Microelectron. Eng., 2015, 145, 21–28 CrossRef CAS .
-
(a) Y. Yuan, Y. Hu, Y.-X. Zhang, J.-D. Lin, Y.-K. Wang, Z.-Q. Jiang, L.-S. Liao and S.-T. Lee, Adv. Funct. Mater., 2017, 27, 1700986 CrossRef ;
(b) J. H. Kim, J. H. Yun and J. Y. Lee, Adv. Opt. Mater., 2018, 6, 1800255 CrossRef .
-
(a) S. D. Cummings and R. Eisenberg, J. Am. Chem. Soc., 1996, 118, 1949 CrossRef CAS ;
(b) J. V. Caspar, E. M. Kober, B. P. Sullivan and T. J. Meyer, J. Am. Chem. Soc., 1982, 104, 630 CrossRef CAS .
-
(a) F. B. Dias, K. N. Bourdakos, V. Jankus, K. C. Moss, K. T. Kamtekar, V. Bhalla, J. Santos, M. R. Bryce and A. P. Monkman, Adv. Mater., 2013, 25, 3707–3714 CrossRef CAS PubMed ;
(b) A. Endo, K. Sato, K. Yoshimura, T. Kai, A. Kawada, H. Miyazaki and C. Adachi, Appl. Phys. Lett., 2011, 98, 083302 CrossRef .
- P. Data, P. Pander, M. Okazaki, Y. Takeda, S. Minakata and A. P. Monkman, Angew. Chem., Int. Ed., 2016, 55, 5739–5744 CrossRef CAS PubMed .
- K. R. Naveen, K. P. CP, R. Braveenth and J. H. Kwon, Chem.–Eur. J., 2022, 28, e202103532 CrossRef PubMed .
-
(a) F. Holtrup, G. Müller, H. Quante, S. de Feyter, F. C. De Schryver and K. Müllen, Chem.–Eur. J., 1997, 3, 219–225 CrossRef CAS PubMed ;
(b) L. Chen, C. Li and K. Müllen, J. Mater. Chem. C, 2014, 2, 1938–1956 RSC ;
(c) K. Peneva, G. Mihov, F. Nolde, S. Rocha, J.-i. Hotta, K. Braeckmans, J. Hofkens, H. Uji-i, A. Herrmann and K. Müllen, Angew. Chem., Int. Ed., 2008, 47, 3372–3375 CrossRef CAS PubMed .
- B. Pigulski, K. Shoyama and F. Würthner, Angew. Chem., Int. Ed., 2020, 59, 15908–15912 CrossRef CAS PubMed .
- N.-H. Xie, C. Li, J.-X. Liu, W.-L. Gong, B. Z. Tang, G. Li and M.-Q. Zhu, Chem. Commun., 2016, 52, 5808–5811 RSC .
- Y. Zhang, H. Zhou, X. Wang, X. Li, J. Wei, Y. Qiao, Y. Song and B. Gao, Chem. Commun., 2021, 57, 651–654 RSC .
-
(a) R. Regar, K. S. Mehra, R. Bhowal and J. Sankar, Eur. J. Org Chem., 2019, 2019, 6278–6284 CrossRef CAS ;
(b) S. Jha, K. S. Mehra, A. Hasija, D. Chopra, R. Regar and J. Sankar, J. Org. Chem., 2022, 87, 3770–3774 CrossRef CAS PubMed .
- K. S. Mehra, S. Jha, S. Bhandary, D. Mandal, R. Mishra and J. Sankar, Angew. Chem., Int. Ed., 2022, 61, e202205600 CrossRef PubMed .
- M. A. Baldo, D. F. O'Brien, Y. You, A. Shoustikov, S. Sibley, M. E. Thompson and S. R. Forrest, Nature, 1998, 395, 151–154 CrossRef CAS .
-
(a) B. Ventura, H. Langhals, B. Böck and L. Flamigni, Chem. Commun., 2012, 48, 4226–4228 RSC ;
(b) A. J. Tilley, R. D. Pensack, T. S. Lee, B. Djukic, G. D. Scholes and D. S. Seferos, J. Phys. Chem. C, 2014, 118, 9996–10004 CrossRef CAS ;
(c) K. Nagarajan, A. R. Mallia, K. Muraleedharan and M. Hariharan, Chem. Sci., 2017, 8, 1776–1782 RSC ;
(d) N. Liang, G. Liu, D. Hu, K. Wang, Y. Li, T. Zhai, X. Zhang, Z. Shuai, H. Yan, J. Hou and Z. Wang, Adv. Sci., 2022, 9, 2103975 CrossRef CAS PubMed .
- K. S. Mehra, S. Jha, A. M. Menon, D. Chopra and J. Sankar, Chem. Sci., 2023, 14, 3147–3153 RSC .
-
(a) J. S. Wilson, N. Chawdhury, M. R. A. Al-Mandhary, M. Younus, M. S. Khan, P. R. Raithby, A. Köhler and R. H. Friend, J. Am. Chem. Soc., 2001, 123, 9412–9417 CrossRef CAS PubMed ;
(b) H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed ;
(c) Q. Zhang, H. Kuwabara, W. J. Potscavage, S. Huang, Y. Hatae, T. Shibata and C. Adachi, J. Am. Chem. Soc., 2014, 136, 18070–18081 CrossRef CAS PubMed .
- M. Adachi and Y. Nagao, Chem. Mater., 2001, 13, 662–669 CrossRef CAS .
-
(a) T. Wang, J. De, S. Wu, A. K. Gupta and E. Z. Colman, Angew. Chem., Int. Ed., 2022, 61, e202206681 CrossRef CAS PubMed ;
(b) R. Ahmed and A. K. Manna, J. Chem. Phys., 2022, 157, 214301–214308 CrossRef CAS PubMed ;
(c) M. A. El-Sayed, J. Chem. Phys., 1963, 38, 2834 CrossRef CAS .
-
(a) M. G. Kim, S. K. Jeon, S. H. Hwang and J. Y. Lee, Adv. Mater., 2015, 27, 2515 CrossRef CAS PubMed ;
(b) J. Lee, K. Shizu, H. Tanaka, H. Nomura, T. Yasuda and C. Adachi, J. Mater. Chem. C, 2013, 1, 4599 RSC .
- J. Luo, X. Xu, R. Mao and Q. Miao, J. Am. Chem. Soc., 2012, 134, 13796–13803 CrossRef CAS PubMed .
-
(a) Y.-L. Zhang, Q. Ran, Q. Wang, Y. Liu, C. Hänisch, S. Reineke, J. Fan and L.-S. Liao, Adv. Mater., 2019, 31, 1902368 CrossRef CAS PubMed ;
(b) C. Si, T. Wang, A. K. Gupta, D. B. Cordes, A. M. Z. Slawin, J. S. Siegel and E. Z. Colman, Angew. Chem., Int. Ed., 2023, 62, e202309718 CrossRef CAS PubMed .
-
(a) J. Fan, Y. Xu, N. Li, J. Miao, C. Zhou, T. Liu, M. Zhu and X. Yin, J. Mater. Chem. C, 2022, 10, 17059–17065 RSC ;
(b) C. Adachi, G. Xie, S. Reineke and E. Z. Colman, Front. Chem., 2020, 8, 625910 CrossRef PubMed .
-
(a) A. Endo, M. Ogasawara, A. Takahashi, D. Yokoyama, Y. Kato and C. Adachi, Adv. Mater., 2009, 21, 4802–4806 CrossRef CAS PubMed ;
(b) C. A. M. Salla, G. Farias, M. RouziHres, P. Dechambenoit, F. Durola, H. Bock, B. de Souza and I. H. Bechtold, Angew. Chem., Int. Ed., 2019, 58, 6982–6986 CrossRef CAS PubMed .
-
(a) B. Xu, Y. Mu, Z. Mao, Z. Xie, H. Wu, Y. Zhang, C. Jin, Z. Chi, S. Liu, J. Xu, Y.-C. Wu, P.-Y. Lu, A. Lienc and M. R. Bryce, Chem. Sci., 2016, 7, 2201–2206 RSC ;
(b) Y. Yang, Y. Liang, Y. Zheng, J.-A. Li, S. Wu, H. Zhang, T. Huang, S. Luo, C. Liu, G. Shi, F. Sun, Z. Chi and B. Xu, Angew. Chem., Int. Ed., 2022, 61, e202201820 CrossRef CAS PubMed .
-
(a) F. Ni, Z. Zhu, X. Tong, M. Xie, Q. Zhao, C. Zhong, Y. Zou and C. Yang, Chem. Sci., 2018, 9, 6150–6155 RSC ;
(b) X.-Y. Wang, J. Gong, H. Zou, S. H. Liu and J. Zhang, Aggregate, 2023, 4, e252 CrossRef CAS ;
(c) Z. Wu, F. Dinkelbach, F. Kerner, A. Friedrich, L. Ji, V. Stepanenko, F. Würthner, C. M. Marian and T. B. Marder, Chem.–Eur. J., 2022, 28, e202200525 CrossRef CAS PubMed ;
(d)
C. Si, D. Sun and E. Z. Colman, ChemRxiv, 2023, preprint, DOI:10.26434/chemrxiv-2023-lxlmf.
-
(a) F. C. Spano, Acc. Chem. Res., 2010, 43, 429–439 CrossRef CAS PubMed ;
(b) C. Kaufmann, W. Kim, A. Nowak-Król, Y. Hong, D. Kim and F. Würthner, J. Am. Chem. Soc., 2018, 140, 4253–4258 CrossRef CAS PubMed ;
(c) Z. Chen, V. Stepanenko, V. Dehm, P. Prins, L. D. A. Siebbeles, J. Seibt, P. Marquetand, V. Engel and F. Würthner, Chem.–Eur. J., 2007, 13, 436–449 CrossRef CAS PubMed .
-
(a) J. M. Lim, P. Kim, M.-C. Yoon, J. Sung, V. Dehm, Z. Chen, F. Würthner and D. Kim, Chem. Sci., 2013, 4, 388–397 RSC ;
(b) R. F. Fink, J. Seibt, V. Engel, M. Renz, M. Kaupp, S. Lochbrunner, H.-M. Zhao, J. Pfister, F. Würthner and B. Engels, J. Am. Chem. Soc., 2008, 130, 12858–12859 CrossRef CAS PubMed .
-
(a) E. G. McRae and M. Kasha, J. Chem. Phys., 1958, 28, 721 CrossRef CAS ;
(b) G. S. Levinson, W. T. Simpson and W. Curtis, J. Am. Chem. Soc., 1957, 79(16), 4314–4320 CrossRef CAS .
-
(a) A. Forni, E. Lucenti, C. Botta and E. Cariati, J. Mater. Chem. C, 2018, 6, 4603–4626 RSC ;
(b) A. D. Nidhankar, Goudappagouda, V. C. Wakchaure and S. S. Babu, Chem. Sci., 2021, 12, 4216–4236 RSC .
- N. Jiang, G.-F. Li, B.-H. Zhang, D.-X. Zhu, Z.-M. Su and M. R. Bryce, Macromolecules, 2018, 51, 4178–4184 CrossRef CAS .
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a
*a