
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynergistic interface and structural engineering for high initial coulombic efficiency and stable sodium storage in metal sulfides†
Chunrong
Ma
 ab,
Zhengguang
Fu
cd,
Yanchen
Fan
e,
Hui
Li
ab,
Zifeng
Ma
f,
Wei
Jiang
ab,
Guangshuai
Han
*g,
Haoxi
Ben
*ab and
Hui (Claire)
Xiong
*h
ab,
Zhengguang
Fu
cd,
Yanchen
Fan
e,
Hui
Li
ab,
Zifeng
Ma
f,
Wei
Jiang
ab,
Guangshuai
Han
*g,
Haoxi
Ben
*ab and
Hui (Claire)
Xiong
*h
aCollege of Textiles & Clothing, Qingdao University, Qingdao 266071, China
bKey Laboratory of Bio-Fibers and Eco-Textiles, Qingdao University, Qingdao 266071, China
cSchool of Polymer Science and Engineering, Qingdao University of Science and Technology, Qingdao 266110, China
dInstitute of Bioenergy and Bioprocess Technology, Chinese Academy of Sciences, Qingdao 266101, China
ePetroChina Shenzhen New Energy Research Institute, Shenzhen, 518000, China
fSchool of Chemistry and Chemical Engineering, Shanghai Jiao Tong University, Shanghai 200240, China
gInstitute for Advanced Study, Tongji University, Shanghai 200092, China
hMicron School of Materials Science and Engineering, Boise State University, Boise, ID 83725, USA. E-mail: clairexiong@boisestate.edu
First published on 7th May 2024
Abstract
Transition metal sulfides (TMS) have gained significant attention as potential anode materials for sodium ion batteries (SIBs) due to their high theoretical capacity and abundance in nature. Nevertheless, their practical use has been impeded by challenges such as large volume changes, unstable solid electrolyte interphase (SEI), and low initial coulombic efficiency (ICE). To address these issues and achieve both long-term cycling stability and high ICE simultaneously, we present a novel approach involving surface engineering, termed as the “dual-polar confinement” strategy, combined with interface engineering to enhance the electrochemical performance of TMS. In this approach, CoS crystals are meticulously coated with polar TiO2 and embedded within a polar S-doped carbon matrix, forming a composite electrode denoted as CoS/TiO2-SC. Significantly, an ether-based electrolyte with chemical stability and optimized solvation properties synergistically interacts with the Co–S–C bonds to create a stable, ultra-thin SEI. This concerted effect results in a notably high ICE, reaching approximately 96%. Advanced characterization and theoretical simulations confirm that the uniform surface modification effectively facilitates sodium ion transport kinetics, restrains electrode pulverization, and concurrently enhances interaction with the ether-based electrolyte to establish a robust SEI. Consequently, the CoS/TiO2-SC electrode exhibits high reversible capacity, superior rate capability, and outstanding cycling stability.
Introduction
The development of advanced high-energy and high-power electrode materials with long-term durability is of utmost importance for the progress of next-generation sodium-ion batteries (SIBs).1–5 In this pursuit, transition metal sulfides (TMS) have emerged as highly promising candidates for anode materials due to their remarkable attributes, including a high theoretical capacity, abundant raw materials availability, and favorable redox potential.6 Among all TMS materials, CoSx stands out due to its reversible multi-electron redox, offering an impressive theoretical capacity of 872 mA h g−1.7,8 Consequently, CoSx has garnered significant attention and research efforts. However, the practical utilization of CoSx faces substantial challenges, primarily stemming from the volume expansion that occurs during the conversion reaction. This expansion leads to severe mechanical fracture and electronic contact failure within the active materials. Furthermore, akin to lithium sulfur batteries, the polysulfide “shuttling” effect can trigger electrochemical degradation of active CoS materials, resulting in continuous capacity decay.9,10 To address these complex issues, extensive research has been devoted to constructing intricate nanostructures in conjunction with highly conductive carbon frameworks.11–14 Nevertheless, these approaches introduce new problems, such as increased side reactions with the electrolyte due to the higher specific surface area and the rupture of solid electrolyte interphase (SEI) films caused by substantial volume change, resulting in highly irreversible capacity. Additionally, continuous electrolyte decomposition and the formation of undesirable reaction products can exacerbate interfacial instability. The electrochemical reduction of the electrolyte on the electrode is significantly influenced by the surface of electrode materials. Particularly, the initial coulombic efficiency (ICE), a quantifiable indicator of SIB reversibility, closely relates to the structure and quality of SEI, which, in turn, influences the lifespan and rate capability of SIBs.15–18 Therefore, it becomes imperative to consider the interfacial stability at the electrode/electrolyte interface when designing CoS-based electrode materials.Beyond electrode structure engineering, the electrochemical behavior of materials also hinges on the electrolyte. Recent research has shed light on the pivotal role of electrolytes in shaping the structural evolution and electrochemical behavior of SIB anodes.19–22 For instance, Huo et al. demonstrated noticeable differences in SEI films formation on CoS2 electrode when employing electrolytes like NaClO4/propylene carbonate (PC) and NaPF6/diethylene glycol dimethyl ether (DEGDME).23 They achieved superior cycling performance using the NaPF6/DEGDME electrolyte, suggesting that electrolytes containing NaPF6 in glymes are beneficial for forming a stable SEI layer that has good passivating properties for reversible operation. However, this strategy faces challenges from recent research indicating that the presence of NaPF6 during SEI formation may inevitably generate harmful hydrofluoric acid with trace water, which could attack electrolytes and degrade their performance if it is not controlled.24,25 Furthermore, most research efforts have concentrated on comparing the performance of electrode materials in different electrolyte systems, often overlooking the profound influence of the choice of electrolyte on the electrode itself. It is crucial to recognize that understanding the intricate interplay between the electrode and electrolyte is of paramount importance as it directly affects electrode kinetics and SEI chemistry. Therefore, in the pursuit of exceptional electrochemical properties for CoS-based anode materials, it is imperative not only to design composite assembly structures but also to comprehend the effects of various electrolytes.
Here, a new “dual-polar confinement” strategy is proposed, wherein the inner polar TiO2 coated CoS crystals, encapsulated within an outer polar S-doped carbon matrix, are fabricated through a sequence of co-precipitation and self-assembly process, followed by high-temperature sulfuration (denoted as CoS/TiO2-SC). The TiO2 layer plays a crucial role in anchoring dissolved polysulfide intermediates, thereby preserving the structural stability of the CoS particles during charge and discharge cycles. Importantly, we have identified that a DEGDME-based electrolyte, with exceptional electrochemical stability and optimized solvation properties, synergistically interacts with the Co–S–C bonds to form a stable, ultra-thin SEI. This results in an exceptionally high ICE in SIBs. The SEI composition and the composite structural stability have been comprehensively validated through a range of ex situ characterizations and theoretical simulations. As a result of this synergistic approach involving surface engineering with a dual-polar layer and interphase engineering via electrolyte modulation, CoS-based anode materials exhibit impressive high capacity and rate performance, even after 3000 cycles. This study provides a theoretical and technological foundation for understanding the interfacial and structural stability of various metal sulfide anode materials for long-term operation, offering a significant contribution to the field of sodium-ion battery research.
Results and discussion
The synthesis of the superhierarchical porous CoS/TiO2-SC composite is depicted in Fig. 1a. Initially, uniform cubic ZIF-67 nanocrystals (Fig. S1†) were meticulously generated through an organometallic reaction between Co2+ ions and 2-methylimidazole ligands in methanol. Subsequently, these ZIF-67 crystals were enveloped with a layer of TiO2via a solvothermal process. Ultimately, the TiO2-coated ZIF-67 structures self-assembled with a solution containing carboxymethyl cellulose (CMC) and graphene oxide (GO) to yield CoS@TiO2@GO 2D nanosheets. This assembly occurred through the formation of hydrogen bonds and chemical coordination. As the reaction duration increased, a leaf-like protective layer developed around these nanosheets. Following vulcanization, the CoS@TiO2@GO precursors underwent transformation into hierarchical S-doped porous carbon nanosheets containing dispersed CoS/TiO2 nanocubes. The morphology of the resulting CoS/TiO2-SC composite was characterized using scanning electron microscopy (SEM) and transmission electron microscopy (TEM). In Fig. 1b, the CoS/TiO2-SC composite demonstrates a hierarchical structure, with graphene sheets intricately woven into a macroporous framework. This architecture is expected to facilitate ion diffusion and electron transport, ultimately enhancing in the magnified SEM image (Fig. 1c), numerous cubes are evenly distributed within the graphene nanosheets. Upon closer examination (Fig. 1d), it becomes evident that CoS/TiO2 nanocubes, with an average diameter of approximately 150 nm, are fully enveloped by the graphene nanosheets. This configuration provides an efficient electrically conductive network for the encapsulated CoS nanoparticles. TEM (Fig. 1e) further confirms the cubic morphology of the CoS/TiO2 composite, which exhibits a characteristic porous feature attributed to volume contraction during calcination. A high-resolution TEM image (Fig. 1f) reveals that CoS nanocrystallites, approximately 5 nm in size, are uniformly dispersed within the carbon matrix. Additionally, an amorphous shell with a thickness of approximately 3 nm is visible along the edge of particle, attributed to the conformal TiO2 coating. Within the inner region of the CoS/TiO2-SC composite nanocomposite, a zoomed-in image (Fig. 1g) shows lattice fringes with an interplanar distance of 0.254 nm. This interplanar spacing corresponds to the (101) plane of CoS. The elemental composition of the CoS/TiO2-SC composite was further elucidated through energy-dispersive X-ray spectroscopy (EDS). As displayed in Fig. S2,† the detected elements, including C, S, Ti, O, and Co, closely align with the expected composition of the composite material, affirming the successful construction of the composite.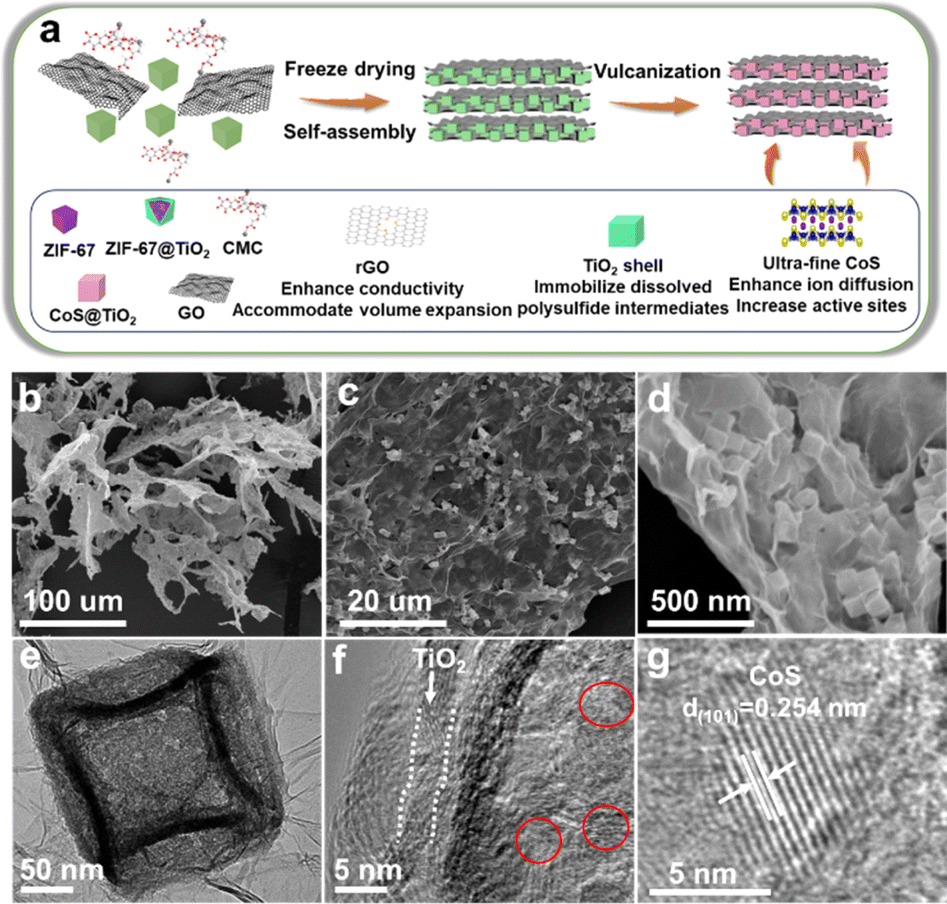 | ||
| Fig. 1 (a) Scheme illustration of the synthesis process of CoS/TiO2-SC composite, (b–d) SEM images, (e–g) TEM images of CoS/TiO2-SC composite. | ||
The crystal structure of the as-prepared CoS/TiO2-SC composite is presented in Fig. 2a. The X-ray diffraction (XRD) pattern of the sample reveals distinct diffraction peaks at various angles, including 30.5°, 35.2°, 46.8°, 54.3°, 62.5°, 66.3°, and 74.5°. These peaks correspond to the (100), (101), (102), (110), (103), (201), and (202) planes of hexagonal CoS (PDF. 75-0605). Additionally, a broad peak at 24.5° corresponds to the diffraction peak of carbon. No distinct TiO2 diffraction peaks are detected, likely due to the amorphous nature of TiO2. Raman spectroscopy was employed to examine the surface properties of the CoS/TiO2-SC composite (Fig. 2b). Two characteristic peaks are observed at 1357 and 1580 cm−1, corresponding to the D and G bands, respectively. The ratio of ID/IG provides insight into the degree of disorder in the carbon structure.26,27 A calculated ID/IG value of 1.35 for CoS/TiO2-SC indicates a high level of disorder and defect, and this disorder may be attributed to S-doping. Moreover, five peaks within the black box in Fig. 2b are consistent with TiO2,28 and the peak at 687 cm−1 is characteristic of CoS.29 These results confirm the successful synthesis of CoS/TiO2 embedded within the carbon structure. The chemical state and composition of CoS/TiO2-SC were further analyzed through X-ray photoelectron spectroscopy (XPS). The XPS survey (Fig. S3a†) reveals the presence of Co, S, Ti, O, and C elements in the CoS/TiO2-SC composite. In the high-resolution Co 2p spectrum (Fig. 2c), peaks at binding energies of 778.3 and 797.2 eV correspond to Co3+, while peaks at 780.6 and 802.2 eV are attributed to Co2+.30,31 A pair of broad satellite peaks accompanies these Co peaks. The peak-fitting analysis of the Ti 2p spectra (Fig. 2d) reveals two peaks at binding energies of 458.5 and 464.2 eV, corresponding to Ti 2p3/2 and Ti 2p1/2, respectively.32 The S 2p spectrum (Fig. 2e) exhibits two peaks at 161.8 and 163.6 eV, corresponding to S 2p3/2 and S 2p1/2, respectively. Additionally, peaks at 165.7 and 169.0 eV can be ascribed to C–S and SOx bonds, with a small peak at 160.3 eV indicating chemical interaction between CoS and carbon through the C–S–Co combination.33 The high-resolution O 1s spectrum (Fig. S3b†) features three peaks at 529.6, 531.5, and 532.2 eV, corresponding to Ti–O, C–O, and H–O bonds, respectively.34 The C 1s spectrum (Fig. 1f) displays two prominent peaks at 284.4 and 288.1 eV, attributed to C–C and C–S bonds, suggesting the presence of S groups within the carbon structure.35 Doped sulfur is known to modify the electronic properties of carbon, facilitating ion transport.36 These findings provide detailed information about the chemical composition and state of the elements within the CoS/TiO2-SC composite. The carbon mass in the CoS/TiO2-C composite was determined via Thermogravimetric Analysis (TGA). As depicted in Fig. S4,† a marginal weight increase was observed between ∼200 and 420 °C, attributed to the oxidation of CoS. Subsequent weight loss occurring within the range of 400–900 °C was attributed to the combustion of carbon and further oxidation of CoS to metal oxides.37 The calculated carbon content in the CoS/TiO2-C composite samples was approximately 38%.
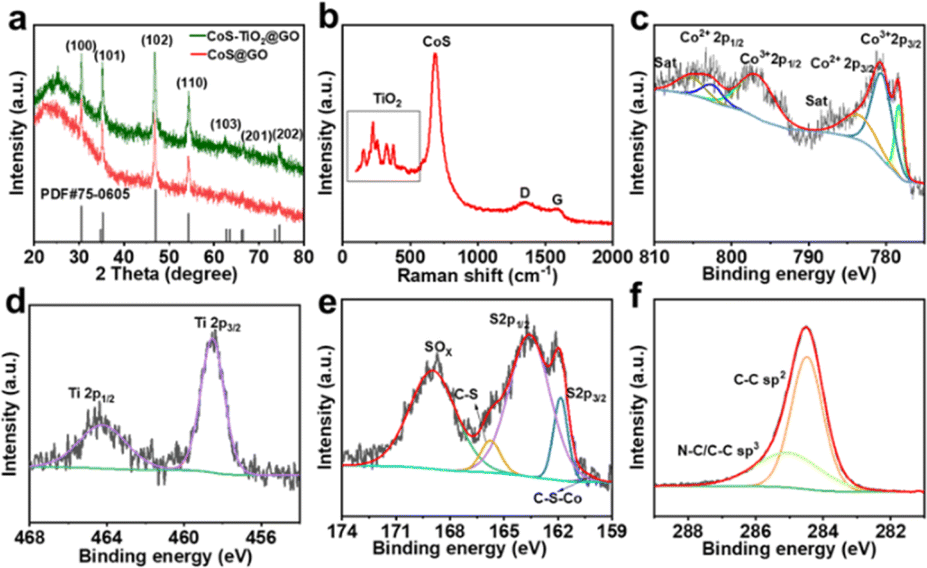 | ||
| Fig. 2 (a) XRD patterns of CoS/TiO2-SC and CoS-SC composites, (b) Raman spectrum of CoS/TiO2-SC composite, and (c–f) High-resolution XPS profiles of the Co 2p, Ti 2p, S 2p, and C 1s for CoS/TiO2-SC composite. | ||
The electrochemical performance of the synthesized CoS/TiO2-SC composite in DEGDME-based electrolyte is thoroughly examined by assembling CR2032 half-cells. Fig. 3a depicts the charge–discharge profiles of the CoS/TiO2-SC electrode at a current density of 0.2 A g−1 within the voltage range of 0.01–3 V. In the initial cycle, the CoS/TiO2-SC electrode exhibits a discharge and charge specific capacity of 813 and 769 mA h g−1, respectively, resulting in a high ICE of 94.5%. This ICE is notably higher than the 79% ICE observed for the CoS-SC electrode (Fig. S5†). In the initial cycle, the CoS/TiO2-SC electrode exhibits a discharge and charge specific capacity of 813 and 769 mA h g−1, respectively, resulting in a high ICE of 94.5%. The improved ICE could be attributed to the incorporation of TiO2, which has the ability to adsorb polysulfides, thereby mitigating the escape of polysulfide intermediates. Importantly, the CoS/TiO2-SC electrode shows a CE that rapidly approaches ∼100% within the first 3 cycles, indicating the formation of a stable SEI. In contrast, the CoS-SC electrode maintains an average CE of ∼95% over the initial 3 cycles. Furthermore, the CoS/TiO2-SC electrode displays a superior specific capacity and better capacity reversibility in subsequent cycles. To gain deeper insights into the Na+ storage behavior of the CoS/TiO2-SC electrode, cyclic voltammetry (CV) are conducted within the voltage window of 0.01–3 V (Fig. 3b). For the CoS/TiO2-SC electrode, a broad peak appears at ∼0.88 V during the initial cathodic scan and disappears in subsequent scans, indicating the formation of the SEI.6,38–40 The peak at ∼0.75 V corresponds to the conversion reaction of CoS to Co and Na2S.7 After the initial cathodic scan, the reduction peak divides into four peaks at 0.63, 0.96, 1.21, and 1.65 eV. In the anodic process, the 1.45 and 1.77 V peaks can be attributed to the reverse reaction, forming metal sulfides. A pair of small peaks near 0 V reflects reversible Na storage in the carbon.41,42 The cycling performances of the CoS/TiO2-SC and CoS-SC electrodes are compared (Fig. 3c). The CoS/TiO2-SC electrode delivers a high reversible capacity of 780 mA h g−1 at a current density of 0.2 A g−1, whereas the CoS-SC electrode achieves only 387 mA h g−1 under the identical conditions. After 100 cycles, the CoS/TiO2-SC electrode maintains a stable capacity, while capacity fading is observed in the CoS-SC electrode. Furthermore, the CE of the CoS/TiO2-SC electrode rapidly stabilizes at 100% after the initial cycles. In contrast, the CE of the CoS-SC electrode continuously fluctuates around an average value of 95%, likely due to the continuous formation of the SEI (Fig. S6†). The rate capability of both electrodes is evaluated (Fig. 3d). The CoS/TiO2-SC electrode consistently outperforms the CoS-SC electrode at all tested current densities, with the capacity gap increasing as the current density rises. Specific capacities of 592, 578, 554, and 512 mA h g−1 are achieved for the CoS/TiO2-SC electrode at current densities of 1, 2, 5, and 8 A g−1, respectively. Even at a high current density of 10 A g−1, the CoS/TiO2-SC electrode still delivers 478 mA h g−1. Upon returning to a current density of 0.2 A g−1, the specific capacity promptly returns to 764 mA h g−1. In contrast, the CoS-SC electrode exhibits a low capacity of 236 mA h g−1 at a high current density of 10 A g−1, with significant capacity fading, primarily due to slow reaction kinetics and poor reversibility. The long-term cycling stability is crucial for evaluating sodium storage performance. The CoS/TiO2-SC electrode is subjected to cycling at a high current density of 5 A g−1. As shown in Fig. 3e, the CoS/TiO2-SC electrode maintains a reversible capacity of 510 mA h g−1 after 3000 cycles, demonstrating excellent electrochemical reversibility. The effect of surface engineering involving the introduction of TiO2 coating on the sodium storage is investigated by density functional theory (DFT) calculations. As shown in Fig. S7,† the introduction of TiO2, providing it as polysulfide adsorbent, exhibits a considerably higher adsorption capacity for polysulfides in comparison to carbon-based materials. Consequently, the designed CoS/TiO2-SC composite, featuring the polar TiO2 and S–C bond, could effectively inhibits the escape of the polysulfide intermediates through a polar–polar interaction mechanism,43 thus ensuring the cycling stability. To better understand the Na+ storage mechanism, CV experiments were conducted at varying scan rates, ranging from 0.1 to 2 mV s−1. Notably, the CV curves maintain their characteristic shape at the scan rate increases (Fig. 3f). Typically, the charge total charge storage can be described by the following formula:44
| i = avb | (1) |
| i = k1v + k2v1/2 | (2) |
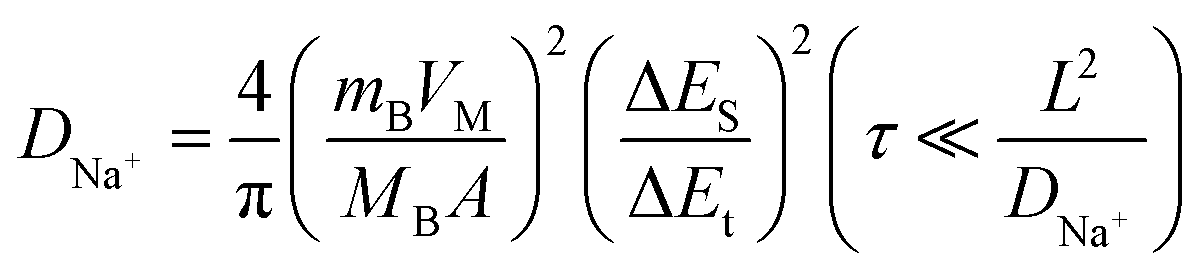 | (3) |
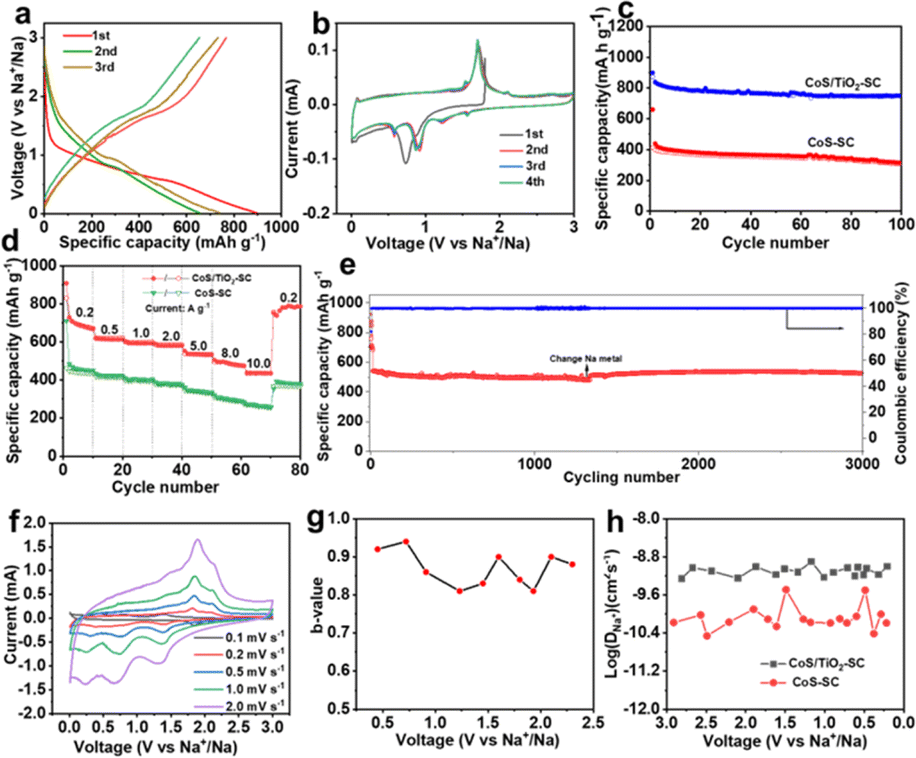 | ||
| Fig. 3 (a) Charge–discharge profiles of CoS/TiO2-SC, (b) CV profiles of CoS/TiO2-SC at 0.1 mV s−1, (c) cycling performance of CoS/TiO2-SC and CoS-SC at 0.2 A g−1, (d) rate capabilities of CoS/TiO2-SC and CoS-SC, (e) long-term cycling performance of CoS/TiO2-SC at 10 A g−1, (f) CV profiles of CoS/TiO2-SC at different scanning rate, (g) b-value of CoS/TiO2-SC electrode, and (h) the Na+ diffusion coefficient derived from the GITT curves of CoS/TiO2-SC electrode. | ||
Notably, the sodium ion diffusion kinetics in CoS/TiO2-SC electrode demonstrate a faster rate compared to those in CoS-SC electrode (Fig. 3h). This distinction stands as a primary contributing factor to the superior rate capability observed in the CoS/TiO2-SC electrode. The average value of corresponding DNa+ in CoS/TiO2-SC electrode was 6 × 10−10 during discharge process. For a deeper insight into the impact of surface engineering through TiO2 coating on sodium storage, DFT calculations are performed to assess the polysulfide adsorption capability of the introduced TiO2 in comparison to conventional carbon materials. As depicted in Fig. S7,† it is observed that the carbon, TiO2, and TiO2@C structures exhibit negative adsorption energies, indicating their suitability for interacting with polysulfides. Remarkably, the TiO2@C structure displays a more robust polysulfide capture capability compared to both carbon and TiO2. This dual-polar effect effectively hinders the escape of polysulfide intermediates, thereby ensuring enhanced cycling stability.
The structural transformation occurring in the CoS/TiO2-SC electrode during charge and discharge processes were further investigated through in situ XRD analysis. As shown in Fig. 4a, the characteristic peaks corresponding to CoS gradually decrease as the discharge process proceeds, indicating the involvement of CoS in the sodiation reaction. Subsequently, new peaks (24.3°) emerge and persist as the discharge progresses to lower potentials, indicating the formation of Na2S during the sodiation reaction. As the discharge process proceeds, the CoS peaks continue to weaken, while the Na2S peaks concurrently intensify until the CoS peaks completely vanish, indicating the full conversion of CoS into Co and Na2S. Throughout the charging process, the Na2S diffraction peaks gradually weaken and eventually disappear, accompanied by the reemergence of CoS peaks, indicating the desodiation process. Simultaneously, the CoS peaks gradually reappear during the desodiation until the end of charging, suggesting the regrowth of the CoS phase. Importantly, no Na2S peaks are detected at the fully charged state, indicating the reversibility of the process. Co peaks are not observed during the charge/discharge process, which could be attributed to the low crystallinity of the generated Co.48 The conversion reaction mechanism of the CoS/TiO2-SC electrode is presented in Fig. 4b. To gain more insight into the kinetic process, the sodium adsorption energy of CoS, TiO2, and CoS/TiO2 was computed. As shown in Fig. 4c–e, CoS/TiO2 exhibits the smallest Na adsorption energy of −2.88 eV, indicating an exceptional sodium affinity of the surface-engineered composite. These interactions effectively inhibit the dissolution of soluble active cobalt species and enhance long-term cycling stability during the charge/discharge process. Furthermore, the charge difference maps (DOS) of the structures involved in this work are detailed in Fig. 4f–h. The adsorbed Na atom loses electrons, facilitating charge transfer. Meanwhile, electrons accumulate on the surfaces of CoS and TiO2, indicating that both materials can capture sodium ions, thus providing a synergistic effect on sodium ion storage performance. When compared to single CoS or TiO2, the CoS/TiO2 composite structure demonstrate a greater propensity for enhancing both Na adsorption and diffusion.
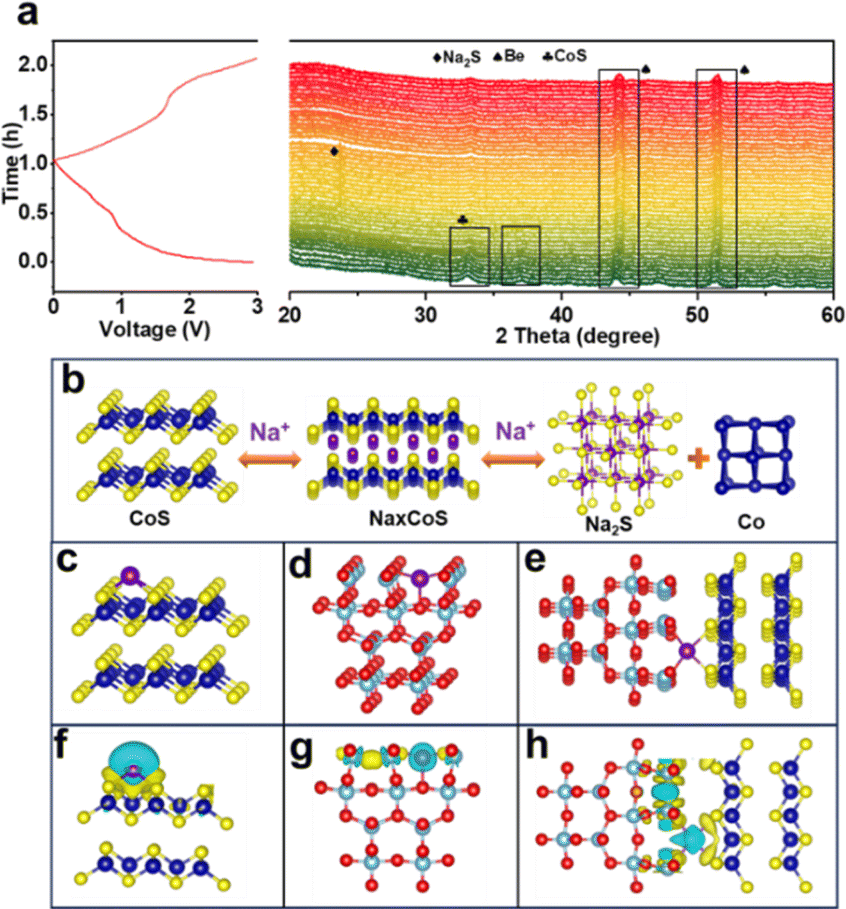 | ||
| Fig. 4 (a) In situ XRD patterns, (b) schematic illustration of sodium storage mechanism of CoS, (c–e) Na atom adsorption at CoS, TiO2, and CoS/TiO2 interface, and (f–h) charge density difference plot for CoS, TiO2, and CoS/TiO2 interface. | ||
To further elucidate the factors contributing to the exceptional electrochemical performance, the morphological evolution of the CoS/TiO2-SC electrode was analyzed using TEM. After 50 cycles, the surface of CoS displayed a porous-like structure (Fig. 5a). With extended cycling, this porous structure became even more pronounced after 100 cycles (Fig. 5b), ultimately resulting in the establishment of a stable porous structure. Notably, this porous nanostructure maintained its interconnected integrity without significant cracks, and the outer TiO2 shell remained intact. The morphological evolution of CoS is schematically illustrated in Fig. 5e. The porous structure observed in the active materials is primarily the outcome of the strong chemical adsorption of DME molecule on the surface of CoS. This interaction induces the movement of CoS surfaces atoms and aids in the formation of a porous structure during cycling, effectively reducing interfacial strain energy.49 This porous structure is advantageous for facilitating electrolyte penetration into the active materials and relieving stress during the charge/discharge process. Consequently, this structure feature significantly contributes to the exceptional rate capability and long-term cycling stability of the electrode. In the high-resolution TEM image of the CoS electrode after 100 cycles, presented in Fig. 5(c1), the observed lattice fringes corresponded to the (101) planes of CoS. Furthermore, selected area electron diffraction (SAED) patterns (Fig. 5d) confirmed the presence of CoS. These results indicate that the crystal structure of CoS remains unchanged during the solid–liquid conversion behavior between solid-state CoS and soluble sodium polysulfide. The surface of the CoS/TiO2-SC electrode was also analyzed for the formation of the SEI film. As a comparison, an electrolyte based on EC/DEC was used. As shown in Fig. 4f, in the DEGDME-based electrolyte, the SEI film appeared uniform and thinner than that in the EC/DEC-based electrolyte (Fig. S10†). Such a uniform and thin SEI film can promote cycling stability. The composition of the SEI film was further analyzed using ex situ XPS. In the DEGDME electrolyte, the main components of the SEI film were sodium alkoxides (RCH2ONa) and polyethers, as indicated by the C 1s and O 1s spectra (Fig. 5g–h). In contrast, the SEI in the EC/DEC-based electrolyte mainly consisted of sodium alkylcarbonates (ROCO2Na) and polyester. ROCO2Na is known to be unstable and further decomposes into RCH2ONa and Na2CO3, leading to an unstable SEI in EC/DEC electrolyte.50 The F 1s high-resolution spectra (Fig. 5i) revealed two distinct peaks corresponding to organic C–F and inorganic NaF components. Notably, the proportion of these two components in the SEI layer was significantly higher (48%) in the DEGDME-based electrolyte. These fluorinated species contribute to the formation of a dense and robust SEI layer on the surface of the active electrode, protecting it from electrolyte corrosion and contributing to the superior electrochemical performance of the CoS/TiO2-SC electrode in DEGDME electrolyte.50 Furthermore, DFT calculations were employed to gain fundamental insights into the effect of the two different electrolytes on the electrode. Specifically, solvation energy (Es) and desolvation energy (Edes,x) calculations were conducted for various [Na–solventx]+ complexes. The corresponding Es values for [Na–DEGDME]+, [Na–DEGDME2]+, [Na–EC]+, [Na–EC2]+, [Na–DEC]+, [Na–DEC2]+, and [Na–EC/DEC]+ complexes were calculated and compared, as shown in Fig. 5j. Relative to the DEGDME-based electrolyte, the EC/DEC-based electrolytes exhibited weaker solvation energy, making the carbonate solvation sheath more prone to accepting electrons and decomposing. In contrast, DEGDME exhibited a higher Es value of −1.38 eV, indicating strong solvation with Na+ ions. This solvation isolates PF6− anions and enables them to preferentially decompose, forming a fluorine-rich inorganic SEI inner layer. Additionally, the desolvation behavior at the interface between the electrodes and electrolyte can significantly impact the electrochemical reaction. The calculated desolvation energy, as shown in Fig. 5k, revealed that the [Na–DEGDME]+ complexes exhibit the highest desolvation energy among the [Na–solventx]+ complexes. This suggests that [Na–DEGDME]+ complexes are more likely to combine with the electrode, thereby promoting rate capability and cycling stability.
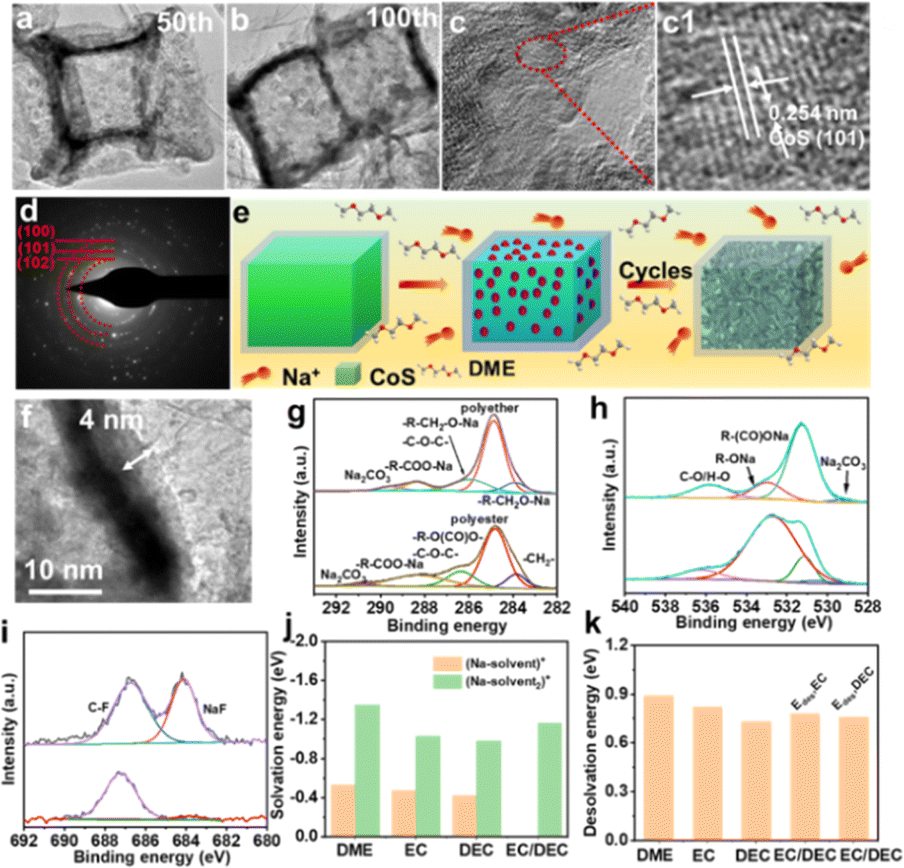 | ||
| Fig. 5 (a–c) TEM images, (d) SAED patterns of CoS/TiO2-SC after 100 cycles, (e) schematic illustration of morphological evolution of CoS/TiO2-SC during cycling, (f) enlarge TEM image of CoS/TiO2-SC after 100 cycles (g–i) high-resolution XPS spectrum of C 1s, O 1s, and F 1s in the electrolyte of DME and EC/DEC, (j and k) solvation and desolvation energy of Na+ solvent complexes. | ||
Conclusions
In conclusion, we successfully employed a “dual-polar confinement” strategy to design a CoS/TiO2-SC composite electrode for efficient sodium storage in sodium-ion batteries (SIBs). Through meticulous surface engineering and the modulation of the electrode–electrolyte interface, the CoS/TiO2-SC electrode has demonstrated exceptional high ICE, high capacity, superior rate capability, and long-term cycling stability. Our investigation, facilitated by an ensemble of physicochemical characterization techniques and DFT calculations, has unveiled critical insights into the underlying mechanisms driving this remarkable performance. Specifically, it has elucidated the importance of reversible electrochemical phase transitions with low reaction energy barriers, which facilitate rapid electrochemical reactions and contribute to the observed excellence of the electrode. Crucially, the incorporation of polar TiO2, synergistic with sulfur-doped carbon, has proven to be highly effective in anchoring and immobilizing dissolved polysulfide intermediates. This synergy enhances reaction reversibility, ensuring long-term cycling stability throughout the charge/discharge cycles. Additionally, the engineered surface facilitates the formation of a ultra-thin fluorine-rich SEI with facile ion transport. In summary, our research deepens our understanding of the structural and component evolution during electrochemical reactions, shedding light on the intricate processes that underlie high-performance sodium-ion battery anodes.Author contributions
Chunrong Ma: writing-original draft, investigation. Zhengguang Fu: reviewing. Yanchen Fan and Hui Li: investigation. Zifeng Ma: resources. Wei Jiang: resources. Guangshuai Han: resources. Haoxi Ben: resources. Claire (Hui) Xiong: resources, supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Shandong Taishan Scholars Project (No. ts20190932), State Key Laboratory of Bio-Fibers and Eco-Textiles, Qingdao University (No. G2RC202021), and Youth Innovation Team Project of Shandong Provincial Education Department (No. 2022KJ143).Notes and references
- B. Sun, P. Li, J. Zhang, D. Wang, P. Munroe, C. Wang, P. H. L. Notten and G. Wang, Dendrite-Free Sodium-Metal Anodes for High-Energy Sodium-Metal Batteries, Adv. Mater., 2018, 29, 1801334 CrossRef PubMed.
- L. Fang, N. Bahlawane, W. Sun, H. Pan, B. B. Xu, M. Yan and Y. Jiang, Conversion-Alloying Anode Materials for Sodium Ion Batteries, Small, 2021, 37, 2101137 CrossRef PubMed.
- Z. Ali, T. Zhang, M. Asif, L. Zhao, Y. Yu and Y. Hou, Transition metal chalcogenide anodes for sodium storage, Mater. Today, 2020, 35, 131–167 CrossRef CAS.
- M. Xuanao, L. Yang, Z. Yunhuai and G. Yun, Layered sodium titanate with a matched lattice: a single ion conductor in a solid-state sodium metal battery, Chem. Sci., 2023, 14, 13812 RSC.
- E. J. Kim, P. R. Kumar, Z. T. Gossage, K. Kubota, T. Hosaka, R. Tatara and S. Komaba, Active material and interphase structures governing performance in sodium and potassium ion batteries, Chem. Sci., 2022, 13, 6121 RSC.
- Y. Yu, M. Yang, W. Xuejie, G. Zicheng, Y. Jiaguo and L. Tao, In-situ evolution of CoS/C hollow nanocubes from metal-organic frameworks for sodium-ion hybrid capacitors, Chem. Eng. J., 2023, 455, 140610 CrossRef.
- Y. Chen, X. Li, K. Park, L. Zhou, H. Huang, Y.-W. Mai and J. B. Goodenough, Hollow Nanotubes of N-Doped Carbon on CoS, Angew. Chem., Int. Ed., 2016, 51, 15831–15834 CrossRef PubMed.
- Z. Limin, Z. Kai, S. Jinzhi, A. Qinyou, T. Zhanliang, K. Yong-Mook, C. Jun and M. Liqiang, Structural and Chemical Synergistic Effect of CoS Nanoparticles and Porous Carbon Nanorods for High-Performance Sodium Storage, Nano Energy, 2017, 35, 281 CrossRef.
- Y. Li and F. Chen, Li-S batteries: Firing for compactness, Nat. Energy, 2017, 7, 17096 CrossRef.
- Y. Gao, Q. Guo, Q. Zhang, Y. Cui and Z. Zheng, Li-S Batteries: Fibrous Materials for Flexible Li-S Battery, Adv. Energy Mater., 2021, 15, 2170058 CrossRef.
- Y. Xiao, D. Su, X. Wang, S. Wu, L. Zhou, Y. Shi, S. Fang, H.-M. Cheng and F. Li, CuS Microspheres with Tunable Interlayer Space and Micropore as a High-Rate and Long-Life Anode for Sodium-Ion Batteries, Adv. Energy Mater., 2018, 22, 1800930 CrossRef.
- Y. Hu, L. Zhang, J. Bai, F. Liu, Z. Wang, W. Wu, R. Bradley, L. Li, H. Ruan and S. Guo, Boosting High-Rate Sodium Storage of CuS via a Hollow Spherical Nanostructure and Surface Pseudocapacitive Behavior, ACS Appl. Energy Mater., 2021, 9, 8901–8909 CrossRef.
- Y. Fang, B. Y. Guan, D. Luan and X. W. Lou, Synthesis of CuS@CoS2 Double-Shelled Nanoboxes with Enhanced Sodium Storage Properties, Angew. Chem., Int. Ed., 2019, 23, 7739–7743 CrossRef PubMed.
- Q. Guo, Y. Ma, T. Chen, Q. Xia, M. Yang, H. Xia and Y. Yu, Cobalt Sulfide Quantum Dot Embedded N/S-Doped Carbon Nanosheets with Superior Reversibility and Rate Capability for Sodium-Ion Batteries, ACS Nano, 2017, 12, 12658–12667 CrossRef PubMed.
- D. Sun, B. Luo, H. Wang, Y. Tang, X. Ji and L. Wang, Engineering the trap effect of residual oxygen atoms and defects in hard carbon anode towards high initial Coulombic efficiency, Nano Energy, 2019, 64, 103937 CrossRef CAS.
- M. Liu, Z. Yang, Y. Shen, S. Guo, J. Zhang, X. Ai, H. Yang and J. Qian, Chemically presodiated Sb with a fluoride-rich interphase as a cycle-stable anode for high-energy sodium ion batteries, J. Mater. Chem. A, 2021, 9, 5639–5647 RSC.
- O. Jin An Sam, D. Grayson, R. Phillip, C. Yu-Ting, C. Diyi, C. Ashley, H. So-Yeon, H. S. T. Darren, J. Jihyun, N. Long Hoang Bao and M. Ying Shirley, High-Performing All-Solid-State Sodium-Ion Batteries Enabled by the Presodiation of Hard Carbon, Adv. Energy Mater., 2023, 26, 2300776 Search PubMed.
- Z. Jiahua, H. Xiang-Xi, L. Wei-Hong, Y. Zhuo, L. Xiao-Hao, L. Lin, Q. Yun, X. Yao, L. Li, W. Xingqiao and C. Shu-Lei, Catalytic Defect-Repairing Using Manganese Ions for Hard Carbon Anode with High-Capacity and High-Initial-Coulombic-Efficiency in Sodium-Ion Batteries, Adv. Energy Mater., 2023, 18, 2300444 Search PubMed.
- Y. Wan, K. Song, W. Chen, C. Qin, X. Zhang, J. Zhang, H. Dai, Z. Hu, P. Yan, C. Liu, S. Sun, S. L. Chou and C. Shen, Ultra-High Initial Coulombic Efficiency Induced by Interface Engineering Enables Rapid, Stable Sodium Storage, Angew. Chem., Int. Ed., 2021, 20, 11481–11486 CrossRef PubMed.
- Y. Jian, G. Xin, G. Hong, W. Tianyi, L. Zhigang, Y. Qing, Y. Hang, L. Jiabao, W. Chengyin and W. Guoxiu, A High-Performance Alloy-Based Anode Enabled by Surface and Interface Engineering for Wide-Temperature Sodium-Ion Batteries, Adv. Energy Mater., 2023, 18, 2300444 Search PubMed.
- K. Li, J. Zhang, D. Lin, D.-W. Wang, B. Li, W. Lv, S. Sun, Y.-B. He, F. Kang, Q.-H. Yang, L. Zhou and T.-Y. Zhang, Evolution of the electrochemical interface in sodium ion batteries with ether electrolytes, Nat. Commun., 2019, 10, 725 CrossRef CAS PubMed.
- L. Gao, J. Chen, Q. Chen and X. Kong, The chemical evolution of solid electrolyte interface in sodium metal batteries, Sci. Adv., 2022, 6, 4606 CrossRef PubMed.
- X. Yin, Y. Ren, S. Guo, B. Sun, L. Wu, C. Du, J. Wang, G. Yin and H. Huo, Investigating the Origin of the Enhanced Sodium Storage Capacity of Transition Metal Sulfide Anodes in Ether-Based Electrolytes, Adv. Funct. Mater., 2022, 22, 2110017 CrossRef.
- D. M. C. Ould, S. Menkin, C. A. O'Keefe, F. Coowar, J. Barker, C. P. Grey and D. S. Wright, New Route to Battery Grade NaPF6 for Na-Ion Batteries: Expanding the Accessible Concentration, Angew. Chem., Int. Ed., 2020, 447, 227363 Search PubMed.
- P. Barnes, K. Smith, R. Parrish, C. Jones, P. Skinner, E. Storch, Q. White, C. Deng, D. Karsann, M. L. Lau, J. J. Dumais, E. J. Dufek and H. Xiong, A non-aqueous sodium hexafluorophosphate-based electrolyte degradation study: Formation and mitigation of hydrofluoric acid, J. Power Sources, 2021, 47, 24882–24887 Search PubMed.
- Y.-S. Sun, C.-F. Lin and S.-T. Luo, Two-Dimensional Nitrogen-Enriched Carbon Nanosheets with Surface-Enhanced Raman Scattering, J. Phys. Chem. C, 2017, 27, 14795–14802 CrossRef.
- V. I. Korepanov, H.-o. Hamaguchi, E. Osawa, V. Ermolenkov, I. K. Lednev, B. J. M. Etzold, O. Levinson, B. Zousman, C. P. Epperla and H.-C. Chang, Carbon structure in nanodiamonds elucidated from Raman spectroscopy, Carbon, 2017, 121, 322–329 CrossRef CAS.
- L. Liu, F. Pan, C. Liu, L. Huang, W. Li and X. Lu, TiO2 Nanofoam-Nanotube Array for Surface-Enhanced Raman Scattering, ACS Appl. Energy Mater., 2018, 12, 6563–6566 Search PubMed.
- W. Ying, X. Yiyuan, Z. Xiaodan, Z. Zuoming and J. Guohua, Mechanistic insights into rapid sulfite activation with cobalt sulfide towards iohexol abatement: contribution of sulfur conversion, Chem. Eng. J., 2021, 429, 132404 Search PubMed.
- X. Song, P. Ning, C. Wang, K. Li, L. Tang, X. Sun and H. Ruan, Research on the low temperature catalytic hydrolysis of CoS and CS2 over walnut shell biochar modified by Fe-Cu mixed metal oxides and basic functional groups, Chem. Eng. J., 2016, 314, 418–433 CrossRef.
- M. Guanyu, Z. Yan, Z. Yong, C. Yanning, L. Fujian, L. Shijing, Z. Yingying and J. Lilong, Oxygen vacancy defects engineering on Cu-doped Co3O4 for promoting effective COS hydrolysis, Green Energy Environ., 2021, 3, 831–841 Search PubMed.
- H. Xu, Y. Liu, T. Qiang, L. Qin, J. Chen, P. Zhang, Y. Zhang, W. Zhang, W. Tian and Z. Sun, Boosting sodium storage properties of titanium dioxide by a multiscale design based on MOF-derived strategy, Energy Storage Mater., 2018, 17, 126–135 CrossRef.
- F. Wang, Z. Liu, H. Feng, Y. Wang, C. Zhang, Z. Quan, L. Xue, Z. Wang, S. Feng, C. Ye, J. Tan and J. Liu, Engineering C-S-Fe Bond Confinement Effect to Stabilize Metallic-Phase Sulfide for High Power Density Sodium-Ion Batteries, Small, 2023, 37, 2302200 CrossRef PubMed.
- Q. Gan, H. He, Y. Zhu, Z. Wang, N. Qin, S. Gu, Z. Li, W. Luo and Z. Lu, Defect-Assisted Selective Surface Phosphorus Doping to Enhance Rate Capability of Titanium Dioxide for Sodium Ion Batteries, ACS Nano, 2019, 8, 9247–9258 CrossRef PubMed.
- X. Chang, Y. Ma, M. Yang, T. Xing, L. Tang, T. Chen, Q. Guo, X. Zhu, J. Liu and H. Xia, In-situ solid-state growth of N, S codoped carbon nanotubes encapsulating metal sulfides for high-efficient-stable sodium ion storage, Energy Storage Mater., 2019, 23, 358–366 CrossRef.
- Z. Hong, Y. Zhen, Y. Ruan, M. Kang, K. Zhou, J.-M. Zhang, Z. Huang and M. Wei, Rational Design and General Synthesis of S-Doped Hard Carbon with Tunable Doping Sites toward Excellent Na-Ion Storage Performance, Adv. Mater., 2018, 29, 1802035 CrossRef PubMed.
- S. Zhang, H. Zhao, W. Ma, J. Mi, J. Zhu, J. Liu, S. Ding and L. Dang, Insight to Se-doping effects on Fe7S8/carbon nanotubes composite as anode for sodium-ion batteries, J. Power Sources, 2022, 536, 231458 CrossRef CAS.
- Y. Zhang, R. Zhan, Q. Xu, H. Liu, M. Tao, Y. Luo, S. Bao, C. Li and M. Xu, Circuit board-like CoS/MXene composite with superior performance for sodium storage, Chem. Eng. J., 2018, 357, 220–225 CrossRef.
- W. Yingxiao, C. Jinqian, L. Zibin, Q. Tianjie, T. Yanqun, S. Jinming, G. Song, Z. Ruiqin and Z. Ruqiang, Construction of CoS-encapsulated in ultrahigh nitrogen doped carbon nanofibers from energetic metal-organic frameworks for superior sodium storage, Carbon, 2022, 198, 353–363 CrossRef.
- H. Gao, T. Zhou, Y. Zheng, Q. Zhang, Y. Liu, J. Chen, H. Liu and Z. Guo, CoS Quantum Dot Nanoclusters for High-Energy Potassium-Ion Batteries, Adv. Funct. Mater., 2017, 43, 1702634 CrossRef.
- X. Zhong, Y. Wu, S. Zeng and Y. Yu, Carbon and Carbon Hybrid Materials as Anodes for Sodium-Ion Batteries, Chem. – Asian J., 2018, 10, 1248–1265 CrossRef PubMed.
- H. Hou, X. Qiu, W. Wei, Y. Zhang and X. Ji, Carbon Anode Materials for Advanced Sodium-Ion Batteries, Adv. Energy Mater., 2017, 24, 1602898 CrossRef.
- L. Feng, P. Yu, X. Fu, Z.-M. Zhang, K. Davey, Y. Wang, Z. Guo and W. Yang, Regulating Polysulfide Diffusion and Deposition via Rational Design of Core-Shell Active Materials in Li-S, Batteries, 2022, 5, 7982–7992 Search PubMed.
- J. Wang, J. Polleux, J. Lim and B. Dunn, Pseudocapacitive Contributions to Electrochemical Energy Storage in TiO2 (Anatase) Nanoparticles, J. Phys. Chem. C, 2007, 40, 14925–14931 CrossRef.
- T. C. Liu, W. G. Pell, B. E. Conway and S. L. Roberson, Behavior of Molybdenum Nitrides as Materials for Electrochemical Capacitors, J. Electrochem. Soc., 1998, 6, 1882 CrossRef.
- Y. Ge, Y. Li, F. Wang, X. Tan, P. Liu, D. Wang, W. Zhou, Q. Yao, M. S. Balogun, D. Huang and J. Deng, Superior high-rate and cycle performances of a single-phase ferrous orthophosphate Na1.2Fe4(PO4)3 anode material for lithium-ion batteries, J. Power Sources, 2022, 535, 231447 CrossRef CAS.
- W. Weppner and R. A. Huggins, Electrochemical investigation of the chemical diffusion, partial ionic conductivities, and other kinetic parameters in Li3Sb and Li3Bi, J. Solid State Chem., 2018, 15, 98–107 Search PubMed.
- Y. Von Lim, S. Huang, Y. Zhang, D. Kong, Y. Wang, L. Guo, J. Zhang, Y. Shi, T. P. Chen, L. K. Ang and H. Y. Yang, Bifunctional porous iron phosphide/carbon nanostructure enabled high-performance sodium-ion battery and hydrogen evolution reaction, Energy Storage Mater., 2018, 15, 98–107 CrossRef.
- L. Zhao, H. Yang, F. He, Y. Yao, R. Xu, L. Wang, L. He, H. Zhang, S. Li and F. Huang, Biomimetic N-doped sea-urchin-structured porous carbon for the anode material of high-energy-density potassium-ion batteries, Electrochim. Acta, 2021, 388, 138565 CrossRef CAS.
- R. C. Cui, H. Y. Zhou, J. C. Li, C. C. Yang and Q. Jiang, Ball-Cactus-Like Bi Embedded in N-Riched Carbon Nanonetworks Enables the Best Potassium Storage Performance, Adv. Funct. Mater., 2021, 33, 2103067 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc02587c |
| This journal is © The Royal Society of Chemistry 2024 |