
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSimultaneous detection of 5-methylcytosine and 5-hydroxymethylcytosine at specific genomic loci by engineered deaminase-assisted sequencing†
Neng-Bin
Xie‡
abc,
Min
Wang‡
de,
Tong-Tong
Ji
d,
Xia
Guo
d,
Fang-Yin
Gang
a,
Ying
Hao
d,
Li
Zeng
d,
Ya-Fen
Wang
a,
Yu-Qi
Feng
 a and
Bi-Feng
Yuan
*abcd
a and
Bi-Feng
Yuan
*abcd
aDepartment of Occupational and Environmental Health, School of Public Health, Department of Radiation and Medical Oncology, Zhongnan Hospital of Wuhan University, Wuhan University, Wuhan 430071, China. E-mail: bfyuan@whu.edu.cn
bResearch Center of Public Health, Renmin Hospital of Wuhan University, Wuhan University, Wuhan 430060, China
cCancer Precision Diagnosis and Treatment and Translational Medicine Hubei Engineering Research Center, Zhongnan Hospital of Wuhan University, Wuhan Research Center for Infectious Diseases and Cancer, Chinese Academy of Medical Sciences, Wuhan 430071, China
dCollege of Chemistry and Molecular Sciences, Wuhan University, Wuhan 430072, China
eCollege of Chemical Engineering and Environmental Chemistry, Weifang University, Weifang, 261061, China
First published on 20th May 2024
Abstract
Cytosine modifications, particularly 5-methylcytosine (5mC) and 5-hydroxymethylcytosine (5hmC), play crucial roles in numerous biological processes. Current analytical methods are often constrained to the separate detection of either 5mC or 5hmC, or the combination of both modifications. The ability to simultaneously detect C, 5mC, and 5hmC at the same genomic locations with precise stoichiometry is highly desirable. Herein, we introduce a method termed engineered deaminase-assisted sequencing (EDA-seq) for the simultaneous quantification of C, 5mC, and 5hmC at the same genomic sites. EDA-seq utilizes a specially engineered protein, derived from human APOBEC3A (A3A), known as eA3A-M5. eA3A-M5 exhibits distinct deamination capabilities for C, 5mC, and 5hmC. In EDA-seq, C undergoes complete deamination and is sequenced as T. 5mC is partially deaminated resulting in a mixed readout of T and C, and 5hmC remains undeaminated and is read as C. Consequently, the proportion of T readouts (PT) reflects the collective occurrences of C and 5mC, regulated by the deamination rate of 5mC (R5mC). By determining R5mC and PT values, we can deduce the precise levels of C, 5mC, and 5hmC at particular genomic locations. We successfully used EDA-seq to simultaneously measure C, 5mC, and 5hmC at specific loci within human lung cancer tissue and their normal counterpart. The results from EDA-seq demonstrated a strong concordance with those obtained from the combined application of BS-seq and ACE-seq methods. EDA-seq eliminates the need for bisulfite treatment, DNA oxidation or glycosylation and uniquely enables simultaneous quantification of C, 5mC and 5hmC at the same genomic locations.
Introduction
Apart from the canonical nucleobases, DNA molecules also contain various modifications.1 5-Methylcytosine (5mC) is the most common DNA modification in mammals and is often referred to as the fifth base due to its important roles in various biological processes such as gene expression, embryogenesis, and tumorigenesis.2 Discovered in 2009, 5-hydroxymethylcytosine (5hmC) has garnered significant attention and is now considered the sixth base of mammalian genomes.3–5 In mammalian cells, the ten-eleven translocation (TET) family proteins sequentially oxidize 5mC to generate 5hmC, 5-formylcytosine (5fC), and 5-carboxylcytosine (5caC).6–9 5fC and 5caC can be converted back to unmodified cytosines through the base excision repair pathway or through direct deformylation or decarboxylation.10–13 Despite being the two most prevalent DNA modifications, the biological functions of 5mC and 5hmC in DNA often have opposing effects, and abnormal DNA methylation or hydroxymethylation is closely linked to various diseases.14–17The quantitative and locus-specific analysis of DNA modifications is essential for understanding their dynamic changes and biological significance.18–21 Bisulfite sequencing (BS-seq) is the frequently employed method for detecting 5mC.22 However, it cannot distinguish between 5mC and 5hmC, providing mixed signals of both modifications due to their similar behavior during bisulfite treatment.22 To address this limitation, oxidative bisulfite sequencing (oxBS-seq) and TET-assisted bisulfite sequencing (TAB-seq) have been developed.23,24 Comparing BS-seq with oxBS-seq or TAB-seq can distinguish the sites of 5mC and 5hmC. However, the harsh conditions of bisulfite treatment can cause significant degradation of up to 99% of input DNA.25 Single-molecule, real-time (SMRT) and nanopore sequencing technologies are also capable of detecting DNA modifications.26,27 However, these methods may have a relatively high false-positive rate for mapping modified nucleobases.28
The APOBEC3A (apolipoprotein B mRNA-editing catalytic polypeptide-like 3A, or referred to as wtA3A) protein has been recently characterized to possess efficient deamination activity towards C, 5mC, and 5hmC, but lacks deamination activity towards glycosylated 5hmC (β-glucosyl-5-hydroxymethyl-2′-deoxycytidine, 5gmC).29–31 Leveraging this property of wtA3A, both our team and others developed A3A-mediated deamination sequencing (AMD-seq) and A3A-coupled epigenetic sequencing (ACE-seq) for the detection of 5hmC in DNA.30,32 Additionally, a method termed enzymatic methyl-seq (EM-seq) has been introduced for detecting the combination of 5mC and 5hmC.33 A comparison between EM-seq and ACE-seq also yields information regarding 5mC and 5hmC.33 Furthermore, pyridine borane-mediated sequencing methods enable the detection of different cytosine modifications.34 Comparing TET-assisted pyridine borane sequencing (TAPS) with β-glucosyltransferase blocking TET-assisted pyridine borane sequencing (TAPSβ) also allows for the detection of 5mC and 5hmC.34,35
Until now, most individual analytical methods have been limited to detecting either 5mC or 5hmC, or a combination of both modifications.18,36 Comparing two individual methods for the detection of 5mC and 5hmC can lead to more false positives or negatives and is also inefficient, time-consuming, and complex. Importantly, a method for obtaining the stoichiometric proportions of C, 5mC, and 5hmC at specific genomic loci is still lacking. Therefore, there is a need for an individual analytical method that allows for the simultaneous detection and quantification of C, 5mC, and 5hmC at the same genomic site. In the current study, we engineered the wtA3A protein and identified a particular engineered A3A mutant known as eA3A-M5. This mutant has the ability to efficiently deaminate C and partially deaminate 5mC, but does not exhibit deamination activity towards 5hmC in various DNA sequence contexts. Leveraging the characteristics of eA3A-M5, we proposed a new sequencing technique called engineered deaminase-assisted sequencing (EDA-seq). This method enables the simultaneous detection of C, 5mC, and 5hmC with stoichiometry at the same location.
Experimental methods
Materials and reagents
2′-Deoxynucleoside 5′-triphosphates (dATP, dCTP, dGTP, and TTP) were purchased from Sigma-Aldrich (St. Louis, MO, USA). 5-Hydroxymethyl-2′-deoxycytidine-5′-triphosphate (5hmdCTP) and 5-methyl-2′-deoxycytidine-5′-triphosphate (5mdCTP) were obtained from TriLink BioTechnologies (San Diego, CA, USA). Q5 High-Fidelity DNA polymerase and EpiMark Hot Start Taq DNA polymerase were purchased from New England Biolabs (Ipswich, MA, USA). The human non-small cell lung cancer tissue and the adjacent normal lung tissue were collected from the Zhongnan Hospital of Wuhan University (Wuhan, China). All experiments were conducted in compliance with the guidelines and regulations of the Ethics Committee of Wuhan University.Preparation of DNA with different cytosine modifications
We synthesized a series of double-stranded DNA (dsDNA) substrates that carry different cytosine modifications, with the detailed sequences provided in Tables S1 and S2.† The synthesis of DNA-C1, DNA-5mC1, and DNA-5hmC1 followed the protocols outlined in our previous report.37 For the preparation of DNA-C2, 0.5 ng of synthetic DNA from Takara was used as a template for PCR amplification. The PCR was performed in a 50 μL mixture containing 1 unit of Q5 High-Fidelity DNA polymerase, 4 μL of 2.5 mM dNTPs, 5 μL of 10× reaction buffer, 2 μL of 10 μM forward primer, and 2 μL of 10 μM reverse primer (Table S3†). For DNA-5mC2 and DNA-5hmC2, we conducted PCR amplification with dCTP substituted by 5mdCTP or 5hmdCTP, respectively. The PCR consisted of 95 °C for 5 min, 30 cycles of 95 °C for 1 min, 56 °C for 1 min, and 68 °C for 1 min, followed by an elongation at 68 °C for 10 min. For DNA-5mC2 and DNA-5hmC2, all cytosines were replaced with 5mC or 5hmC, respectively, excluding the cytosines in the PCR primers. For DNA-C/5mC, 0.5 ng of synthetic DNA from Takara was used as a template for PCR amplification. PCR was conducted in a 50 μL solution containing 1 unit of Q5 High-Fidelity DNA polymerase, 4 μL of 2.5 mM dNTPs, 5 μL of 10× reaction buffer, 2 μL of 10 μM forward primer, and 2 μL of 10 μM reverse primer (Table S3†). The PCR consisted of 95 °C for 5 min, 30 cycles of 95 °C for 1 min, 61 °C for 1 min, and 68 °C for 1 min, followed by an elongation at 68 °C for 10 min. We then separated the PCR products using agarose gel electrophoresis and extracted them with a gel extraction kit (Omega Bio-Tek Inc., Norcross, GA, USA). The method for preparing spike-in DNA is described in the ESI.†Expression and purification of wild-type A3A and engineered A3A mutants
To produce wild-type A3A (wtA3A, Gene ID: 200315) and its engineered mutants, we cloned the coding sequences for wtA3A and the engineered A3A (eA3A) mutants into the pET-41a(+) vector using the SpeI and XhoI restriction sites. We also incorporated a human rhinovirus 3C protease (HRV 3C) cleavage site between the glutathione S-transferase (GST) tag and the wtA3A or eA3A sequences (Fig. S1†). These plasmids were then transformed into Escherichia coli (E. coli) BL21(DE3) pLysS cells. The transformed E. coli cells were cultured in LB medium, which included 10 g L−1 tryptone, 5 g L−1 yeast extract, and 10 g L−1 NaCl, and the growth medium was supplemented with 10 μg mL−1 kanamycin and 10 μg mL−1 chloramphenicol. The cultures were maintained at 37 °C with shaking at 180 rpm. Protein expression was induced by adding 0.5 mM isopropyl-β-D-thiogalactoside (IPTG from Sangon) when the optical density at 600 nm (OD600nm) reached between 0.4 and 0.6. The expression of recombinant proteins was carried out for 20 h at 25 °C with shaking at 180 rpm. Following expression, the E. coli cells were harvested by centrifugation at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g for 5 min. The cell pellets were then lysed through sonication in a PBS buffer that included 1 mM dithiothreitol and 50 μg mL−1 phenylmethylsulfonyl fluoride (PMSF). The lysate was centrifuged at 12000g for 30 min, and the supernatant was filtered through a 0.22 μm filter. The filtered supernatant was incubated with Glutathione Sepharose 4B beads (Sangon, Shanghai, China) as per the manufacturer's instructions, allowing the recombinant proteins to bind to the beads. After binding, the proteins were treated with HRV 3C protease (Sangon, Shanghai, China) to release the wtA3A or eA3A mutants from the GST tag. Subsequent purification was performed using a size-exclusion chromatography column (Millipore, Darmstadt, Germany) pre-equilibrated with a buffer containing 50 mM Tris-HCl (pH 7.5), 50 mM NaCl, 0.01 mM EDTA, 0.5 mM dithiothreitol, and 0.01% Tween-20. The purified proteins were subjected to SDS-PAGE analysis (Fig. S2†). Protein concentrations were determined using a BCA protein assay kit (Beyotime, Shanghai, China).
000g for 5 min. The cell pellets were then lysed through sonication in a PBS buffer that included 1 mM dithiothreitol and 50 μg mL−1 phenylmethylsulfonyl fluoride (PMSF). The lysate was centrifuged at 12000g for 30 min, and the supernatant was filtered through a 0.22 μm filter. The filtered supernatant was incubated with Glutathione Sepharose 4B beads (Sangon, Shanghai, China) as per the manufacturer's instructions, allowing the recombinant proteins to bind to the beads. After binding, the proteins were treated with HRV 3C protease (Sangon, Shanghai, China) to release the wtA3A or eA3A mutants from the GST tag. Subsequent purification was performed using a size-exclusion chromatography column (Millipore, Darmstadt, Germany) pre-equilibrated with a buffer containing 50 mM Tris-HCl (pH 7.5), 50 mM NaCl, 0.01 mM EDTA, 0.5 mM dithiothreitol, and 0.01% Tween-20. The purified proteins were subjected to SDS-PAGE analysis (Fig. S2†). Protein concentrations were determined using a BCA protein assay kit (Beyotime, Shanghai, China).
Deamination assay
The deamination assay was conducted with a slight modification based on a previous report.38 Briefly, 50 ng of the double-stranded (dsDNA) substrate was denatured to single-stranded (ssDNA) by heating at 95 °C for 10 min and then chilling at 0 °C for 5 min in a 10 μL solution containing 20% dimethylsulfoxide (DMSO) (v/v). The denatured DNA was then treated with 20 μM of wtA3A or eA3A mutant. The deamination reaction was carried out at 37 °C for 2 h in a 20 μL solution of 20 mM MES (pH 6.5). The deamination reaction was terminated by heating at 95 °C for 10 min.Evaluation of the deamination activities of eA3A mutants by sequencing
The deamination activities of wtA3A and eA3A mutants towards C, 5mC, and 5hmC were assessed using three dsDNA substrates, DNA-C1, DNA-5mC1, and DNA-5hmC1 (Table S1†). These substrates were denatured and subjected to deamination as per the aforementioned procedure. Subsequently, the deaminase-treated DNA was amplified and subjected to Sanger sequencing. The PCR amplification was carried out in a 50 μL reaction mixture containing 5 ng of the deaminase-treated DNA, 1 unit of EpiMark Hot Start Taq DNA Polymerase, 0.2 mM of each dNTP, 10 μL of 5× reaction buffer, 0.4 μM of the forward primer, and 0.4 μM of the reverse primer (Table S4†). The PCR includes initial denaturation at 95 °C for 5 min, followed by 30 cycles of 95 °C for 30 s, 55 °C for 30 s, and 68 °C for 1 min, with a final extension at 68 °C for 10 min.Simultaneous and quantitative detection of C, 5mC and 5hmC at specific genomic loci using EDA-seq
Genomic DNA was extracted from lung cancer tissue and corresponding normal tissue using a Tissue DNA kit (Omega Bio-Tek Inc., Norcross, GA, USA). The isolated DNA was then fragmented to an average size of approximately 300 bp using a JY92-II N ultrasonic homogenizer (Scientz). To assess the deamination rate of 5mC in different sequence contexts, 0.1% of THRA 5mC spike-in and ALS2CL 5mC spike-in were added to the fragmented DNA. 50 ng of this mixture was denatured and incubated with 20 μM of the eA3A-M5 protein for either 1 or 2 h. The resulting DNA was subsequently PCR-amplified using a set of specific primers (Table S4†). The PCR products were then analyzed by Sanger sequencing. The sequencing data obtained from the spike-in DNA were used to calculate the deamination rate of 5mC, represented as R5mC. The sequencing data from the genomic DNA were used to calculate the proportion of T reads (PT) in the sequence output. Different R5mC and the corresponding PT values were applied to identify the levels of C, 5mC, and 5hmC at the individual cytosine sites in genomic DNA using the eqn (5)–(7).Quantitative detection of C, 5mC and 5hmC at specific genomic loci by combined BS-seq and ACE-seq
Genomic DNA was extracted from lung cancer tissue and corresponding normal tissue and fragmented as aforementioned. for BS-seq, 200 ng of fragmented DNA was subjected to bisulfite treatment using an EpiTect Fast DNA Bisulfite Conversion kit (Qiagen GmbH, Hilden, German) according to the manufacturer's protocol. The deaminated DNA was amplified using site-specific primers (Table S4†) and PCR products were analyzed by Sanger sequencing. The sequencing results of BS-seq provided the total proportion of 5mC and 5hmC (P5mC+5hmC) at the individual cytosine sites in the gene bodies of THRA and ALS2CL. As for ACE-seq, 1 μg of the fragment DNA was first treated with β-glucosyltransferase (New England Biolabs) at 37 °C for 2 h followed by purification using 0.9 × KAPA pure beads (Roche). The resulting DNA (100 ng) was denatured and treated with 20 μM of wtA3A. The deaminated DNA was amplified using site-specific primers (Table S4†) and PCR products were analyzed by Sanger sequencing. The sequencing results of ACE-seq provided the proportion of 5hmC (P5hmC) at the individual cytosine sites in the gene bodies of THRA and ALS2CL. The proportion of 5mC (P5mC) at those individual cytosine sites could be calculated by subtracting P5hmC detected by ACE-seq from P5mC+5hmC detected by BS-seq.Results and discussion
Principle of engineered deaminase-assisted sequencing for quantitative detection of C, 5mC and 5hmC at the same site
Given the comparable performance of 5mC and 5hmC in bisulfite treatment, TET oxidation, and A3A-mediated deamination, a combination of two methods is typically essential for distinguishing between 5mC and 5hmC (Fig. S3†). This can be achieved through the concurrent use of BS-seq and oxBS-seq or TAB-seq. In this study, we aimed to develop a straightforward technique for the simultaneous and quantitative detection of C, 5mC and 5hmC at the same site. It has been proven that wtA3A can deaminate C, 5mC, and 5hmC to yield U, T, and 5-hydroxymethyluracil (5hmU), respectively.29,39–41 Moreover, wtA3A does not affect A, T, and G. Therefore, following wtA3A treatment, C and 5mC are deaminated and interpreted as T during sequencing, while 5hmC is partially deaminated and read as both C and T (Fig. S4†).Building on our previous research, we engineered the wtA3A protein and identified a novel engineered A3A mutant (eA3A-M5), which displayed distinctly different deamination activities towards C, 5mC, and 5hmC (Fig. 1A). Upon treatment with eA3A-M5, C was completely deaminated and read as T during sequencing; 5mC was partially deaminated and read as both C and T; while 5hmC was not deaminated and read as C (Fig. 1A). With the screened eA3A-M5, we proposed an engineered deaminase-assisted sequencing (EDA-seq) method for the simultaneous and quantitative analysis of C, 5mC, and 5hmC at the same location. In this respect, the proportion of T readouts during sequencing at a specific cytosine site is equivalent to the sum of the proportion of C and the proportion of 5mC multiplied by the deamination rate of 5mC (Fig. 1B). The equations are as follows:
| Pc + P5mC + P5hmC = 100% | (1) |
| Pc + P5mC × R5mC = PT | (2) |
| Pc + P5mC × R5mC1 = PT1 | (3) |
| Pc + P5mC × R5mC2 = PT2 | (4) |
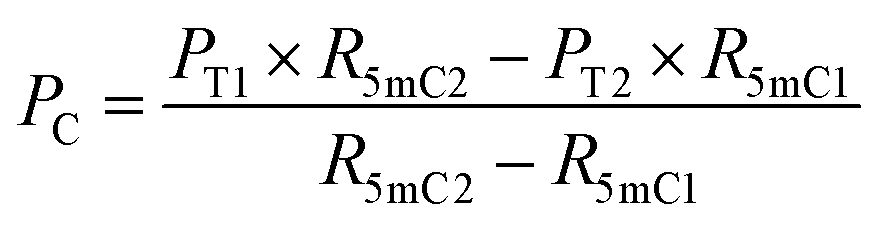 | (5) |
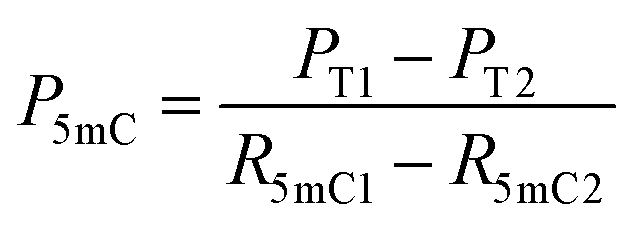 | (6) |
| P5hmC = 100% − Pc − P5mC | (7) |
 | ||
| Fig. 1 Principle of EDA-seq. (A) C can be deaminated by eA3A-M5 to form U, which pairs with A; 5mC is partially deaminated by eA3A-M5, resulting in partial pairing with A and partial pairing with C; 5hmC is not deaminated by eA3A-M5 and still pairs with G. (B) In EDA-seq, upon treatment with eA3A-M5, C was completely deaminated and read as T during sequencing; 5mC was partially deaminated and read as both C and T; while 5hmC was not deaminated and read as C. The proportion of T readouts in sequencing at specific cytosine sites is equivalent to the sum of the proportion of C and the proportion of 5mC multiplied by the deamination rate of 5mC. The PT1 and PT2 values can be determined from the sequencing results of the analyzed DNA treated with eA3A-M5 at different times. The R5mC1 and R5mC2 values can be obtained from the sequencing results of the 5mC spike-in DNA. The proportions of C, 5mC and 5hmC can be obtained according to eqn (5)–(7). | ||
Screening of eA3A proteins
It has been shown that wtA3A exhibits significantly weaker deamination activity towards 5hmC compared to C/5mC, and the deamination selectivity can be enhanced by altering the amino acid compositions around loop 1 (residues 20 to 31) and loop 7 (residues 130 to 135).38,42 Previous study has also indicated that wtA3A displays higher deamination capability towards cytosine in TC and CC sites than that in AC and GC sites.43 Therefore, the deamination of engineered A3A proteins towards C, 5mC, and 5hmC was assessed using three types of dsDNA (DNA-C1, DNA-5mC1, and DNA-5hmC1, Table S1†) with C, 5mC, and 5hmC located in different sequence contexts of AC, GC, TC, and CC sites.We aimed to identify an engineered A3A variant that could fully deaminate C, partially deaminate 5mC, and not deaminate 5hmC. Initially, we created eA3A-M1 by replacing the G25 (G, glycine) and I26 (I, isoleucine) of wtA3A with the oligopeptide “EPWVR” (E, glutamic acid; P, proline; W, tryptophan; V, valine; R, arginine, Fig. S5A†). Sanger sequencing confirmed that eA3A-M1 efficiently deaminated C and 5mC. However, eA3A-M1 also partially deaminated 5hmC (Fig. S5B†). Based on eA3A-M1, we subsequently generated four types of eA3A mutants (eA3A-M2 to eA3A-M5, Fig. 2A). eA3A-M2 was created with H29R (H, histidine; R, arginine) and K30Q (K, lysine; Q, glutamine) mutations in the loop1 of eA3A-M1 (Fig. S6A†). The results showed that eA3A-M2 fully deaminated C and 5mC, while also partially deaminating 5hmC (Fig. S6B†). We obtained eA3A-M3 with P134T (P, proline; T, threonine) and L135D (L, leucine; D, aspartic acid) mutations in the loop 7 of eA3A-M2 (Fig. S7A†). The results demonstrated that eA3A-M3 fully deaminated C and 5mC, without deaminating 5hmC (Fig. S7B†). We generated eA3A-M4 and eA3A-M5 by replacing the V28 of eA3A-M3 with glutamic acid (E) and proline (P), respectively (Fig. S8A† and 2A). eA3A-M4 partially deaminated C and 5mC (Fig. S8B†). Encouragingly, eA3A-M5 fully deaminated C and partially deaminated 5mC but showed no deamination activity towards 5hmC (Fig. 2B). Consequently, eA3A-M5 meets the requirement for the development of EDA-seq.
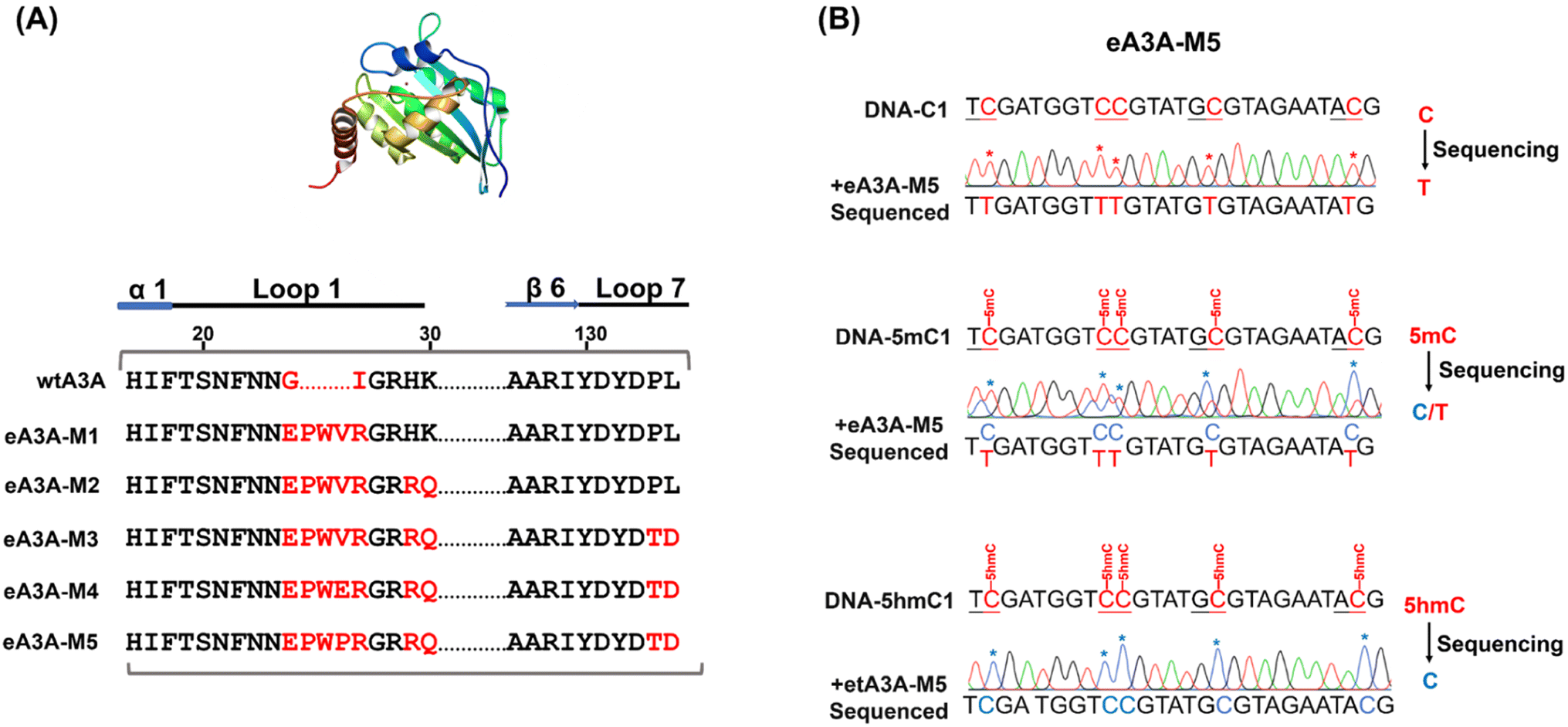 | ||
| Fig. 2 Assessment of eA3A-M5 specificity for C, 5mC, and 5hmC across various sequence contexts. (A) The amino acid compositions of wtA3A and engineered A3A mutants (eA3A-v1 to eA3A-M5). (B) Sanger sequencing results for DNA substrates containing C, 5mC, and 5hmC (DNA-C1, DNA-5mC1, and DNA-5hmC1, respectively) as analyzed by EDA-seq. eA3A-M5 converted all the C sites to U, which were subsequently read as T during sequencing; 5mC sites underwent partial deamination, resulting in a mixed read of C and T; 5hmC sites remained unaltered by eA3A-M5 and continued to be read as C. | ||
When the original sites only contain C and 5mC, after the treatment of eA3A-M5, R5mC and PT can be applied to eqn (1) and (2). Thus, P5mC could be calculated using the following eqn (8):
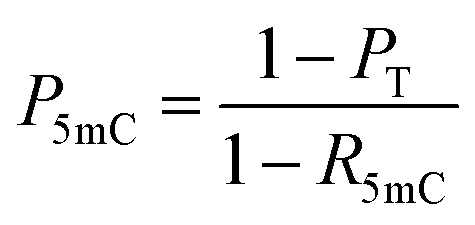 | (8) |
The PT can be determined from the sequencing results of the detected DNA, while the R5mC can be calculated from the sequencing results of the spike-in DNA. To assess the effectiveness of EDA-seq, DNA-C2 and DNA-5mC2 (Table S1†) were mixed at varying ratios, with 5mC in different sequence contexts (TC, CC, GC, and AC sites) ranging from 0 to 100%. Approximately 0.1% of DNA-5mC2 spike-in (Table S2†) was added to determine the deamination rate of 5mC in different sequence contexts (Fig. 3A). The mixtures were then analyzed using EDA-seq with Sanger sequencing (Fig. 3B and S10–S12†). P5mC was calculated according to eqn (8). The results indicated that the measured proportion of 5mC (P5mC) at different sites increased proportionally with the theoretical percentage of 5mC in the mixture of DNA-C2 and DNA-5mC2 (Fig. 3C). This suggests that the EDA-seq method can quantitatively measure 5mC at different stoichiometries in specific sites that contain C and 5mC.
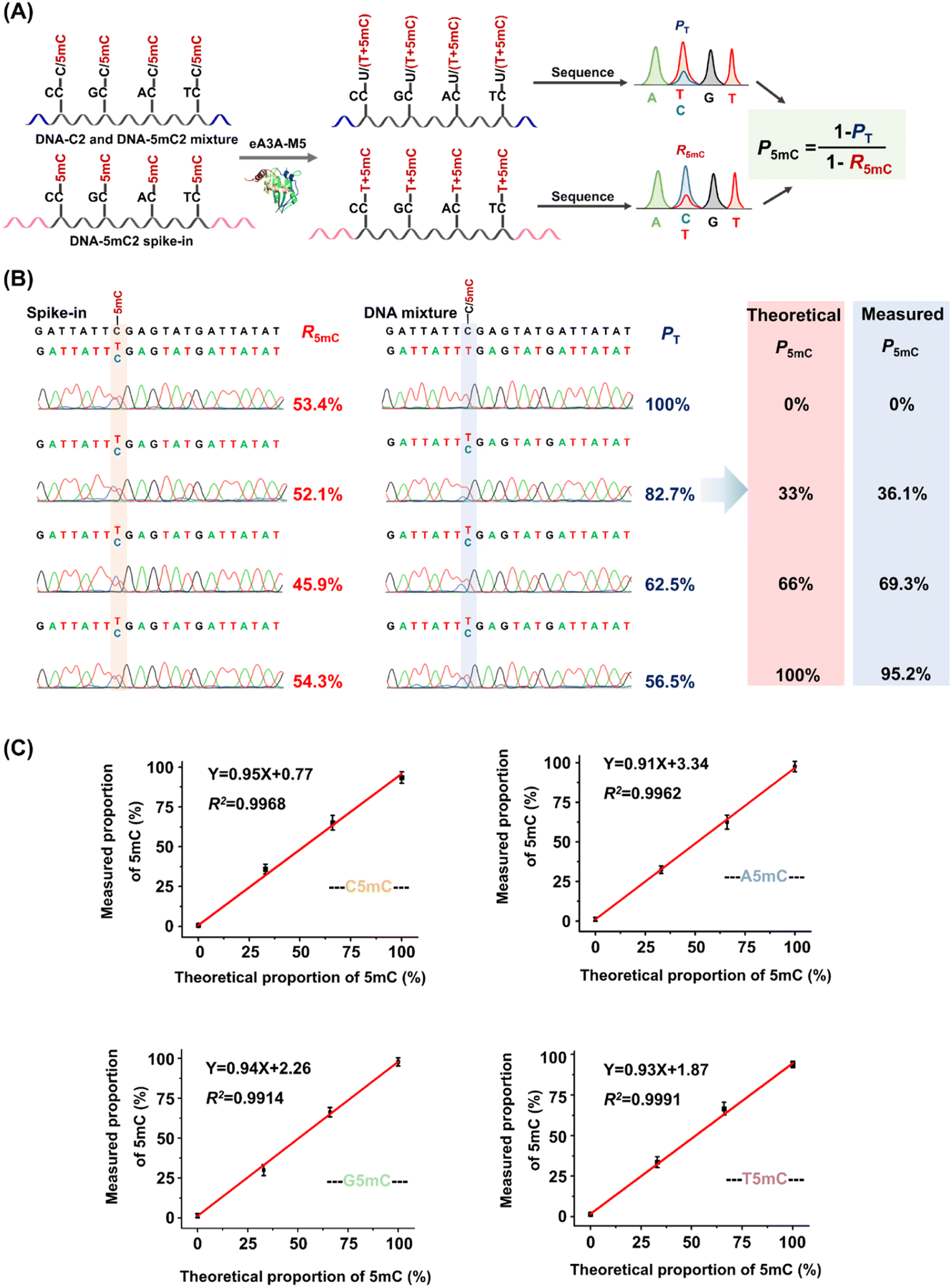 | ||
| Fig. 3 Quantitative assessment of the 5mC level at cytosine sites containing C and 5mC by EDA-seq. (A) DNA-C2 and DNA-5mC2 were mixed at varying ratios, with DNA-5mC2 ranging from 0% to 100%. A 0.1% DNA-5mC2 spike-in was introduced to determine the deamination rate of 5mC. (B) Sanger sequencing results of TC sites in the DNA-C2 and DNA-5mC2 mixture, along with the DNA-5mC2 spike-in. (C) Linear regression analysis of the measured proportion of 5mC at different sequence contexts against the corresponding theoretical proportion of 5mC at those sequence contexts. | ||
When the original sites contain C, 5mC and 5hmC, after the treatment of eA3A-M5 at different times, PT1, PT2, R5mC1 and R5mC2 can be applied to eqn (3) and (4). The PT1 and PT2 values can be determined from the sequencing results of the detected DNA treated with eA3A-M5 at different times. The R5mC1 and R5mC2 values can be obtained from the sequencing results of the DNA-5mC2 spike-in (Fig. 4A). Thus, the proportions of C, 5mC and 5hmC can be obtained according to eqn (5) – (7). In this context, DNA-C2, DNA-5mC2, and DNA-5hmC2 were mixed, with the proportions of C, 5mC, and 5hmC in different sequence contexts setting at 30%, 30%, and 40%, respectively. Approximately 0.1% of DNA-5mC2 spike-in was added to determine the deamination rate of 5mC by treating it with eA3A-M5 at different times (Fig. 4A). The mixtures were then subjected to EDA-seq with Sanger sequencing. The results indicated that the measured proportions of C, 5mC, and 5hmC at different sites were comparable to the theoretical percentages of C, 5mC, and 5hmC in the mixture of DNA-C2, DNA-5mC2, and DNA-5hmC2 (Fig. 4B, C, S13–S15†). This suggests that the EDA-seq method is capable of simultaneously quantifying C, 5mC and 5hmC at specific sites. The EDA-seq method involves sequencing experiments at two different time points to determine the deamination rate for 5mC. This extended process increases the overall duration of the workflow. Nevertheless, the method remains straightforward compared to utilizing two separate techniques to measure 5mC and 5hmC concurrently at identical sites.
 | ||
| Fig. 4 Quantitative assessment of C, 5mC, and 5hmC levels at cytosine sites containing C, 5mC, and 5hmC. (A) DNA-C2, DNA-5mC2, and DNA-5hmC2 were mixed at proportions of 30%, 30%, and 40%, respectively. A 0.1% DNA-5mC2 spike-in was included in the mixture to determine the deamination rate of 5mC. (B) Sanger sequencing results of TC sites in the DNA-C2, DNA-5mC2, and DNA-5hmC2 mixture, as well as the DNA-5mC2 spike-in, after treatment with eA3A-M5 for varying durations. (C) The proportions of C, 5mC, and 5hmC at different sequence contexts measured by EDA-seq. Dashed lines indicate the theoretical proportions of C, 5mC, and 5hmC. | ||
Simultaneous quantification of C, 5mC and 5hmC at specific genomic loci
Aberrant methylation, particularly the genome-wide reduction of 5hmC, is a common epigenetic feature of various cancers.44–46 The dysregulation of tumor-suppressor genes, such as THRA and ALS2CL, is often associated with tumor invasion and metastasis.47–49 A previous study has shown that the level of 5hmC at specific cytosine sites in the gene bodies of THRA and ALS2CL (Table S5†) is lower in lung cancer tissue compared to adjacent normal tissue.50 The EDA-seq method allows for the simultaneous and quantitative detection of 5mC and 5hmC at specific genomic loci, which was then applied in the detection of 5mC and 5hmC in the gene bodies of THRA and ALS2CL from both lung cancer tissue and adjacent normal tissue.The genomic DNA from lung cancer tissue and adjacent normal tissue was isolated and fragmented into an average size of 300 bp. Subsequently, 0.1% THRA 5mC spike-in DNA and ALS2CL 5mC spike-in DNA (Table S2†) were added to determine the deamination rate of 5mC in different sequence contexts. Following this, 100 ng of the DNA mixture was treated with eA3A-M5 for 1 h or 2 h. The Sanger sequencing results of the gene bodies of THRA and ALS2CL provided the proportion of T reads (PT). Additionally, the Sanger sequencing results of 5mC spike-in DNA provided the deamination rates of 5mC (R5mC) in different sequence contexts (Fig. 5A). The different PT and R5mC values were utilized to calculate the proportions of C, 5mC, and 5hmC at individual cytosine sites in the gene bodies of THRA and ALS2CL using eqn (5)–(7).
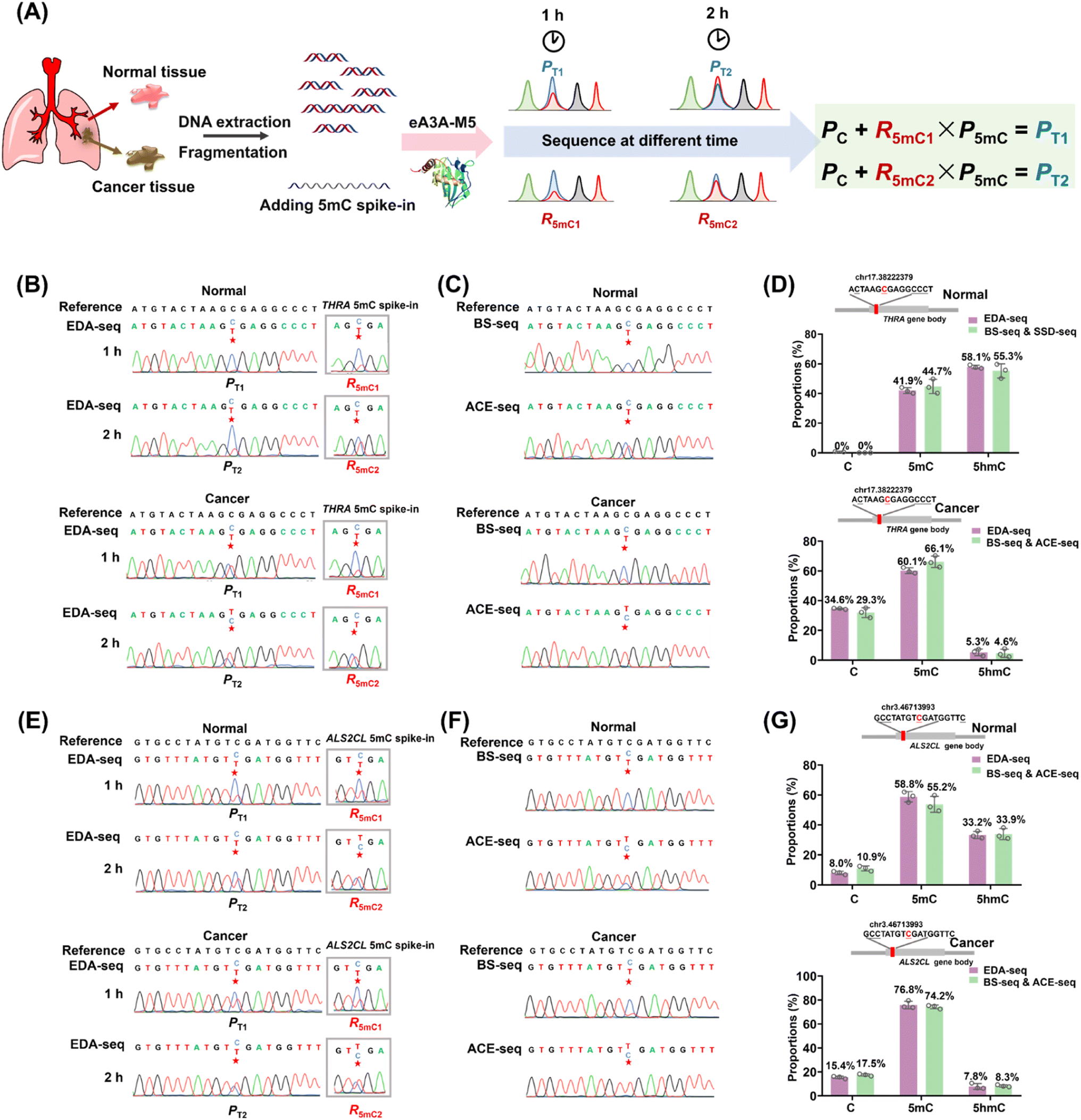 | ||
| Fig. 5 Quantitative assessment of C, 5mC, and 5hmC at specific genomic loci from lung cancer tissue and adjacent normal tissue using EDA-seq and combined BS-seq and ACE-seq. (A) Schematic illustration of the quantitative detection of C, 5mC, and 5hmC at specific genomic loci by EDA-seq. THRA 5mC spike-in and ALS2CL 5mC spike-in were added to determine the deamination rate of 5mC. (B) Sanger sequencing results of the chr17.38222379 site from normal lung tissue and corresponding lung cancer tissue, along with the THRA 5mC spike-in treated with eA3A-M5 for varying durations. (C) Sanger sequencing results of the chr17.38222379 site from normal lung tissue and corresponding lung cancer tissue using BS-seq and ACE-seq. (D) The proportions of C, 5mC, and 5hmC at the chr17.38222379 site detected by EDA-seq and the combined BS-seq and ACE-seq. (E) Sanger sequencing results of the chr3.46713993 site from normal lung tissue and corresponding lung cancer tissue, along with the ALS2CL 5mC spike-in treated with eA3A-M5 for varying durations. (F) Sanger sequencing results of the chr3.46713993 site from normal lung tissue and corresponding lung cancer tissue using BS-seq and ACE-seq. (G) The proportions of C, 5mC, and 5hmC at the chr3.46713993 site detected by EDA-seq and the combined BS-seq and ACE-seq. | ||
To validate the accuracy of the EDA-seq method, we also utilized BS-seq and ACE-seq for the quantitative detection of 5mC and 5hmC at these sites (Fig. S16†). With BS-seq, we obtained the total proportion of 5mC and 5hmC (P5mC+5hmC) at the individual cytosine sites. In ACE-seq, 5hmC in DNA was protected by β-glucosyltransferase and then C and 5mC were deaminated by wtA3A. Following this treatment, C and 5mC were deaminated and read as T during sequencing, while 5hmC was not deaminated and still read as C during sequencing. Therefore, the sequence results provided the proportion of 5hmC (P5hmC) at the individual cytosine sites. The proportion of 5mC at the individual cytosine sites could be calculated by subtracting P5hmC detected by ACE-seq from P5mC+5hmC detected by BS-seq (Fig. S16†).
In the case of the THRA gene, analysis of the cytosine site at chr17.38222379 in normal lung tissue using EDA-seq revealed the proportions of C, 5mC, and 5hmC being 0%, 41.9%, and 58.1%, respectively. A combined analysis using BS-seq and ACE-seq at the same site showed similar proportions, with 0%, 44.7%, and 55.3% for C, 5mC, and 5hmC, respectively (Fig. 5B–D). In lung cancer tissue, EDA-seq revealed the proportions of C, 5mC, and 5hmC being 34.6%, 60.1%, and 5.3%, respectively; the combined BS-seq and ACE-seq methods showed 29.3% C, 66.1% 5mC, and 4.6% 5hmC (Fig. 5B–D). For the ALS2CL gene, at the cytosine site chr3.46713993 in normal lung tissue, EDA-seq identified 8.0% C, 58.8% 5mC, and 33.2% 5hmC; BS-seq and ACE-seq showed 10.9% C, 55.2% 5mC, and 33.9% 5hmC (Fig. 5E–G). In lung cancer tissue, EDA-seq identified 15.4% C, 76.8% 5mC, and 7.8% 5hmC; BS-seq and ACE-seq showed 17.5% C, 74.2% 5mC, and 8.3% 5hmC (Fig. 5E–G). Overall, the quantification of C, 5mC, and 5hmC at specific genomic locations by EDA-seq was found to be in close agreement with the results from the combined BS-seq and ACE-seq approach. This confirms the high accuracy of EDA-seq for quantitative detection of 5mC and 5hmC at individual cytosine sites. Furthermore, the data revealed that levels of 5hmC at these specific sites were lower in lung cancer tissue compared to normal lung tissue, while the levels of 5mC showed the opposite trend, consistent with previous studies.50
Compared to previous methods used for quantitative detection of 5mC and 5hmC, EDA-seq offers several notable advantages. Firstly, EDA-seq enables the simultaneous detection of C, 5mC, and 5hmC at individual cytosine sites in DNA without the need for combining two separate methods. Secondly, EDA-seq simplifies the analytical process compared to methods involving glycosylation of 5hmC or TET-mediated oxidation. Thirdly, EDA-seq utilizes a mild deamination reaction, eliminating the need for harsh chemical reactions such as bisulfite treatment. This gentle approach ensures that DNA is not susceptible to degradation, making EDA-seq suitable for quantitative detection of 5mC and 5hmC at individual cytosine sites with limited amounts of input DNA. It is important to note that in addition to 5hmC, 5fC and 5caC are also present in DNA. However, the levels of 5fC and 5caC are significantly lower than that of 5hmC, typically by 2 to 3 orders of magnitude.36,51,52 As a result, their impact on the quantitative detection of 5mC and 5hmC using EDA-seq is minimal and can be disregarded. Overall, EDA-seq overcomes several limitations present in previous 5mC and 5hmC detection methods, making it a cost-effective and versatile approach for simultaneous and quantitative detection of 5mC and 5hmC at the same site in DNA. Based on the calibration curves in Fig. 3, with intercepts ranging from 0.77 to 3.34, the EDA-seq method may not be suitable for accurately mapping low stoichiometric levels of 5mC and 5hmC. Previous study showed that approximately 8.5 to 10.5 million 5mC sites and 0.23 to 0.36 million 5hmC sites were mapped in lung cancer tissues.50 Among these sites, over 90% exhibited stoichiometric levels exceeding 2%. Therefore, the EDA-seq method could effectively analyze the majority of 5mC and 5hmC sites in this context. The genome-wide mapping of 5mC and 5hmC by EDA-seq has not yet been achieved due to the varying deamination activity of eA3A-M5 towards 5mC in different sequence contexts. This limitation may be addressed in future studies by identifying an engineered A3A mutant with consistent deamination ability towards 5mC in various sequence contexts.
Conclusion
In summary, we developed the EDA-seq method, which enables the simultaneous and quantitative detection of C, 5mC, and 5hmC in DNA at single-base resolution. Through the engineering of five A3A mutants (eA3A-M1 to eA3A-M5), we identified eA3A-M5 as the mutant possessing distinct deamination activities for these cytosine modifications. eA3A-M5 is capable of fully deaminating C and partially deaminating 5mC, yet exhibits no deamination activity toward 5hmC, forming the basis for the development of EDA-seq. Employing EDA-seq, we effectively performed simultaneous and quantitative analyses of C, 5mC, and 5hmC at specific genomic loci in lung cancer and adjacent normal tissues. EDA-seq represents an advancement over previous methods for mapping 5mC and 5hmC, as it does not require bisulfite treatment, chemical labeling, or DNA glycosylation. In conclusion, EDA-seq offers a straightforward and precise approach for the simultaneous and quantitative detection of C, 5mC, and 5hmC at specific genomic loci without the need for combining with other methods. EDA-seq's sensitivity and precision in detecting cytosine modifications with minimal DNA input make it a valuable tool for epigenetic studies in scenarios with scarce samples, such as single-cell and cell-free DNA methylation and hydroxymethylation profiling. This technique holds promise for advancing research in areas such as early disease detection and understanding cellular heterogeneity at the epigenetic level.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the National Key R&D Program of China (2022YFA0806600 and 2022YFC3400700), the Fundamental Research Funds for the Central Universities (2042024kf0022), the National Natural Science Foundation of China (22277093 and 22074110), and the Wuhan Knowledge Innovation Project (2022020801010111).References
- M. K. Bilyard, S. Becker and S. Balasubramanian, Curr. Opin. Chem. Biol., 2020, 57, 1–7 CrossRef CAS PubMed.
- C. Luo, P. Hajkova and J. R. Ecker, Science, 2018, 361, 1336–1340 CrossRef CAS PubMed.
- S. Kriaucionis and N. Heintz, Science, 2009, 324, 929–930 Search PubMed.
- M. Tahiliani, K. P. Koh, Y. Shen, W. A. Pastor, H. Bandukwala, Y. Brudno, S. Agarwal, L. M. Iyer, D. R. Liu, L. Aravind and A. Rao, Science, 2009, 324, 930–935 CrossRef CAS PubMed.
- M. Munzel, D. Globisch and T. Carell, Angew Chem. Int. Ed. Engl., 2011, 50, 6460–6468 CrossRef PubMed.
- X. Wu and Y. Zhang, Nat. Rev. Genet., 2017, 18, 517–534 CrossRef CAS PubMed.
- Y. F. He, B. Z. Li, Z. Li, P. Liu, Y. Wang, Q. Tang, J. Ding, Y. Jia, Z. Chen, L. Li, Y. Sun, X. Li, Q. Dai, C. X. Song, K. Zhang, C. He and G. L. Xu, Science, 2011, 333, 1303–1307 CrossRef CAS PubMed.
- S. Ito, L. Shen, Q. Dai, S. C. Wu, L. B. Collins, J. A. Swenberg, C. He and Y. Zhang, Science, 2011, 333, 1300–1303 CrossRef CAS PubMed.
- J. H. Ding, G. Li, J. Xiong, F. L. Liu, N. B. Xie, T. T. Ji, M. Wang, X. Guo, Y. Q. Feng, W. Ci and B. F. Yuan, Anal. Chem., 2024, 96, 4726–4735 CrossRef CAS PubMed.
- Y. Feng, J. J. Chen, N. B. Xie, J. H. Ding, X. J. You, W. B. Tao, X. Zhang, C. Yi, X. Zhou, B. F. Yuan and Y. Q. Feng, Chem. Sci., 2021, 12, 11322–11329 RSC.
- Y. Feng, N. B. Xie, W. B. Tao, J. H. Ding, X. J. You, C. J. Ma, X. Zhang, C. Yi, X. Zhou, B. F. Yuan and Y. Q. Feng, CCS Chem., 2021, 3, 994–1008 CrossRef CAS.
- Y. Feng, Y. Q. Tian, Y. Q. Zhao, S. J. Chen and B. F. Yuan, Chin. Chem. Lett., 2024, 35, 109656 CrossRef.
- Y. Feng, S. J. Chen and B. F. Yuan, Chin. J. Chem., 2024, 42, 645–651 CrossRef CAS.
- A. P. Feinberg and A. Levchenko, Science, 2023, 379, eaaw3835 CrossRef CAS PubMed.
- S. Zhao, C. D. Allis and G. G. Wang, Nat. Rev. Cancer, 2021, 21, 413–430 CrossRef CAS PubMed.
- T. Feng, Y. L. Gao, D. Hu, K. Y. Yuan, S. Y. Gu, Y. H. Gu, S. Y. Yu, J. Xiong, Y. Q. Feng, J. Wang and B. F. Yuan, Chin. Chem. Lett., 2024, 35, 109259 CrossRef CAS.
- M. Y. Chen, Z. Gui, K. K. Chen, J. H. Ding, J. G. He, J. Xiong, J. L. Li, J. Wang, B. F. Yuan and Y. Q. Feng, Chin. Chem. Lett., 2022, 33, 2086–2090 CrossRef CAS.
- L. Y. Zhao, J. Song, Y. Liu, C. X. Song and C. Yi, Protein Cell, 2020, 11, 792–808 CrossRef CAS PubMed.
- E. A. Raiber, R. Hardisty, P. van Delft and S. Balasubramanian, Nat. Rev. Chem, 2017, 1, 1–13 CrossRef.
- F. Tang, J. Yuan, B. F. Yuan and Y. Wang, J. Am. Chem. Soc., 2022, 144, 454–462 CrossRef CAS PubMed.
- C. J. Ma, G. Li, W. X. Shao, Y. H. Min, P. Wang, J. H. Ding, N. B. Xie, M. Wang, F. Tang, Y. Q. Feng, W. Ci, Y. Wang and B. F. Yuan, ACS Cent. Sci., 2023, 9, 1799–1809 CrossRef CAS PubMed.
- B. Mark and J. F. McGouran, Nat. Rev. Chem, 2018, 2, 332–348 CrossRef.
- M. Yu, G. C. Hon, K. E. Szulwach, C. X. Song, L. Zhang, A. Kim, X. Li, Q. Dai, Y. Shen, B. Park, J. H. Min, P. Jin, B. Ren and C. He, Cell, 2012, 149, 1368–1380 CrossRef CAS PubMed.
- M. J. Booth, M. R. Branco, G. Ficz, D. Oxley, F. Krueger, W. Reik and S. Balasubramanian, Science, 2012, 336, 934–937 CrossRef CAS PubMed.
- K. Tanaka and A. Okamoto, Bioorg. Med. Chem. Lett., 2007, 17, 1912–1915 CrossRef CAS PubMed.
- Y. Wang, Y. Zhao, A. Bollas, Y. Wang and K. F. Au, Nat. Biotechnol., 2021, 39, 1348–1365 Search PubMed.
- S. Ardui, A. Ameur, J. R. Vermeesch and M. S. Hestand, Nucleic Acids Res., 2018, 46, 2159–2168 CrossRef CAS PubMed.
- Y. Kong, E. A. Mead and G. Fang, Nat. Rev. Genet., 2023, 24, 363–381 CrossRef CAS PubMed.
- E. K. Schutsky, C. S. Nabel, A. K. F. Davis, J. E. DeNizio and R. M. Kohli, Nucleic Acids Res., 2017, 45, 7655–7665 CrossRef CAS PubMed.
- Q. Y. Li, N. B. Xie, J. Xiong, B. F. Yuan and Y. Q. Feng, Anal. Chem., 2018, 90, 14622–14628 CrossRef CAS PubMed.
- J. Xiong, K. K. Chen, N. B. Xie, T. T. Ji, S. Y. Yu, F. Tang, C. Xie, Y. Q. Feng and B. F. Yuan, Anal. Chem., 2022, 94, 15489–15498 CrossRef CAS PubMed.
- E. K. Schutsky, J. E. DeNizio, P. Hu, M. Y. Liu, C. S. Nabel, E. B. Fabyanic, Y. Hwang, F. D. Bushman, H. Wu and R. M. Kohli, Nat. Biotechnol., 2018, 36, 1083–1090 CrossRef CAS PubMed.
- R. Vaisvila, V. K. C. Ponnaluri, Z. Sun, B. W. Langhorst, L. Saleh, S. Guan, N. Dai, M. A. Campbell, B. S. Sexton, K. Marks, M. Samaranayake, J. C. Samuelson, H. E. Church, E. Tamanaha, I. R. Corrêa Jr, S. Pradhan, E. T. Dimalanta, T. C. Evans Jr, L. Williams and T. B. Davis, Genome Res., 2021, 31, 1280–1289 CrossRef PubMed.
- Y. Liu, P. Siejka-Zielinska, G. Velikova, Y. Bi, F. Yuan, M. Tomkova, C. Bai, L. Chen, B. Schuster-Bockler and C. X. Song, Nat. Biotechnol., 2019, 37, 424–429 CrossRef CAS PubMed.
- Y. Liu, J. Cheng, P. Siejka-Zielinska, C. Weldon, H. Roberts, M. Lopopolo, A. Magri, V. D'Arienzo, J. M. Harris, J. A. McKeating and C. X. Song, Genome Biol., 2020, 21, 54 CrossRef CAS PubMed.
- Y. Dai, B. F. Yuan and Y. Q. Feng, RSC Chem. Biol., 2021, 2, 1096–1114 RSC.
- M. Wang, N. B. Xie, K. K. Chen, T. T. Ji, J. Xiong, X. Guo, S. Y. Yu, F. Tang, C. Xie, Y. Q. Feng and B. F. Yuan, Anal. Chem., 2023, 95, 1556–1565 CAS.
- N. B. Xie, M. Wang, T. T. Ji, X. Guo, J. H. Ding, B. F. Yuan and Y. Q. Feng, Chem. Sci., 2022, 13, 7046–7056 RSC.
- S. J. Ziegler, Y. Hu, S. C. Devarkar and Y. Xiong, Biochemistry, 2019, 58, 3838–3847 CrossRef CAS PubMed.
- F. Ito, Y. Fu, S. A. Kao, H. Yang and X. S. Chen, J. Mol. Biol., 2017, 429, 1787–1799 CrossRef CAS PubMed.
- J. Xiong, P. Wang, W. X. Shao, G. J. Li, J. H. Ding, N. B. Xie, M. Wang, Q. Y. Cheng, C. H. Xie, Y. Q. Feng, W. M. Ci and B. F. Yuan, Chem. Sci., 2022, 13, 9960–9972 RSC.
- N. B. Xie, M. Wang, W. Chen, T. T. Ji, X. Guo, F. Y. Gang, Y. F. Wang, Y. Q. Feng, Y. Liang, W. Ci and B. F. Yuan, ACS Cent. Sci., 2023, 9, 2315–2325 CrossRef CAS PubMed.
- K. Shi, M. A. Carpenter, S. Banerjee, N. M. Shaban, K. Kurahashi, D. J. Salamango, J. L. McCann, G. J. Starrett, J. V. Duffy, O. Demir, R. E. Amaro, D. A. Harki, R. S. Harris and H. Aihara, Nat. Struct. Mol. Biol., 2017, 24, 131–139 CrossRef CAS PubMed.
- S. M. Janssen and M. C. Lorincz, Nat. Rev. Genet., 2022, 23, 137–153 CrossRef CAS PubMed.
- G. Cavalli and E. Heard, Nature, 2019, 571, 489–499 CrossRef CAS PubMed.
- M. L. Chen, F. Shen, W. Huang, J. H. Qi, Y. Wang, Y. Q. Feng, S. M. Liu and B. F. Yuan, Clin. Chem., 2013, 59, 824–832 CrossRef CAS PubMed.
- E. Hwang, W. K. L. Doolittle, Y. J. Zhu, X. Zhu, L. Zhao, Y. Yu and S. Y. Cheng, Oncogene, 2023, 42, 3075–3086 CrossRef CAS PubMed.
- D. J. Lee, F. Schonleben, V. E. Banuchi, W. Qiu, L. G. Close, A. M. Assaad and G. H. Su, Cancer Biol. Ther., 2010, 10, 689–693 CrossRef CAS PubMed.
- Z. Mou, J. Spencer, J. S. McGrath and L. W. Harries, Hum. Genomics, 2023, 17, 97 CrossRef CAS PubMed.
- X. Li, Y. Liu, T. Salz, K. D. Hansen and A. Feinberg, Genome Res., 2016, 26, 1730–1741 CrossRef CAS PubMed.
- B. F. Yuan and Y. Q. Feng, TrAC, Trends Anal. Chem., 2014, 54, 24–35 CrossRef CAS.
- S. Liu, J. Wang, Y. Su, C. Guerrero, Y. Zeng, D. Mitra, P. J. Brooks, D. E. Fisher, H. Song and Y. Wang, Nucleic Acids Res., 2013, 41, 6421–6429 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Preparation of 5mC spike-in DNA; Tables S1–S5; Fig. S1–S16. See DOI: https://doi.org/10.1039/d4sc00930d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |