
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanistic insights into bismuth(III) inhibition of SARS-CoV-2 helicase†
Xueying
Wei
 ab,
Chun-Lung
Chan
a,
Ying
Zhou
a,
Kaiming
Tang
b,
Jingxin
Chen
a,
Suyu
Wang
a,
Jasper Fuk-Woo
Chan
b,
Shuofeng
Yuan
b,
Hongyan
Li
a and
Hongzhe
Sun
*a
ab,
Chun-Lung
Chan
a,
Ying
Zhou
a,
Kaiming
Tang
b,
Jingxin
Chen
a,
Suyu
Wang
a,
Jasper Fuk-Woo
Chan
b,
Shuofeng
Yuan
b,
Hongyan
Li
a and
Hongzhe
Sun
*a
aDepartment of Chemistry and CAS-HKU Joint Laboratory of Metallomics on Heath and Environment, The University of Hong Kong, Pokfulam, Hong Kong Special Administrative Region, China. E-mail: hsun@hku.hk
bDepartment of Microbiology and State Key Laboratory of Emerging Infectious Diseases, The University of Hong Kong, Pokfulam, Hong Kong Special Administrative Region, China
First published on 3rd June 2024
Abstract
The COVID-19 pandemic caused by SARS-CoV-2 resulted in a global public health crisis. In addition to vaccines, the development of effective therapy is highly desirable. Targeting a protein that plays a critical role in virus replication may allow pan-spectrum antiviral drugs to be developed. Among SARS-CoV-2 proteins, helicase (i.e., non-structural protein 13) is considered as a promising antiviral drug target due to its highly conserved sequence, unique structure and function. Herein, we demonstrate SARS-CoV-2 helicase as a target of bismuth-based antivirals in virus-infected mammalian cells by a metal-tagged antibody approach. To search for more potent bismuth-based antivirals, we further screened a panel of bismuth compounds towards inhibition of ATPase and DNA unwinding activity of nsp13 and identified a highly potent bismuth compound Bi(5-aminotropolonate)3, namely Bi(Tro-NH2)3 with an IC50 of 30 nM for ATPase. We show that bismuth-based compounds inhibited nsp13 unwinding activity via disrupting the binding of ATP and the DNA substrate to viral helicase. Binding of Bi(III) to nsp13 also abolished the interaction between nsp12 and nsp13 as evidenced by immunofluorescence and co-immunoprecipitation assays. Finally, we validate our in vitro data in SARS-CoV-2 infected mammalian cells. Notably, Bi(6-TG)3 exhibited an EC50 of 1.18 ± 0.09 μM with a selective index of 847 in VeroE6-TMPRSS2 infected cells. This study highlights the important role of helicase for the development of more effective antiviral drugs to combat SARS-CoV-2 infection.
Introduction
According to World Health Organization statistics, there were 7.7 billion confirmed cases of COVID-19 globally till December 2023, including 6.9 million deaths.1 Although vaccination campaigns are running, vaccines fail to provide full protection. This is partially due to the emergence of more SARS-CoV-2 variants that have enhanced transmissibility, leading to a reduced sensitivity to vaccines.2 Therefore, the development of effective antiviral drugs against SARS-CoV-2 and its variants is highly desirable.SARS-CoV-2 is an enveloped positive-sense single-stranded RNA virus with the largest viral genome of ∼30 kb.3–6 Following entry into host cells, the opening reading frame 1a and 1ab (ORF1a and ORF1ab) of SARS-CoV-2 encodes two large polyproteins, pp1a and pp1ab, respectively,7 which are further cleaved into 16 non-structural proteins (nsps) after processing through encoded viral proteases. All these 16 proteins are mainly involved in viral replication,8 and some enzymes may serve as promising targets for the development of antiviral drugs.9–14 Among these nsps, SARS-CoV-2 helicase (nsp13) has received growing attention as an antiviral drug target due to its unique structure and function.15 It is highly conserved among the coronaviridae family,16 consisting of two canonical RecA ATPase domains and three unique domains, i.e., N-terminal zinc-binding domain (ZBD), stalk domain, and 1B domain.17,18 Nsp13 is an ATPase and possesses DNA or RNA unwinding activity in an NTP-dependent manner with a 5′ > 3′ polarity,17,19–22 and hydrolysis of ATP provides energy for its DNA or RNA unwinding activity. In addition, SARS-CoV-2 nsp13 can form a stable complex with RNA polymerase (RdRp, i.e., nsp12), an RNA scaffold,23 as an important component of the replication–transcription complex (RTC).24,25 It is noteworthy that nsp13 has greater potential druggable prospect than other proteins according to the druggability evaluation of SARS-CoV-2 by SiteMap (Schrodinger, NY).26
Metal compounds are known for their antimicrobial activity and their roles in the fight against antimicrobial resistance have gained momentum.27–30 Recently, metal-based strategies against SARS-CoV-2 infection have also attracted more interest from scientific communities.13,31–34 For example, rhenium(I) picolinic acid complexes were shown to inhibit viral replication by targeting SARS-CoV-2 main protease,35 and certain gold and iron compounds targeted PLpro, also disrupting S protein and ACE2 interaction.33 We have demonstrated previously that bismuth-based compounds and anti-ulcer drugs, e.g. ranitidine bismuth citrate (RBC) and colloidal bismuth subcitrate (CBS), used alone or in combination with N-acetyl cysteine (NAC) showed good inhibition towards SARS-CoV-2 and other coronaviruses in the cells and animal infection models.13,31 Bismuth-based cocktail therapy via oral administration has been in clinical trial in Hong Kong since July 2022 (PR/CT0096/2022). However, the exact molecular mechanism of action of bismuth-based antivirals, in particular at a cellular level, is not fully understood. It is highly desirable to further explore more potent broad-spectrum metal-based antivirals for combating SARS-CoV-2 infection.
Herein, we used a target enrichment approach based on metal-tagged antibody to validate SARS-CoV-2 nsp13 as a target of bismuth drugs in virus-infected mammalian cells. We then further explored more potent inhibitors of SARS-CoV-2 nsp13 via screening more bismuth compounds. We also investigated the mode of inhibition of these inhibitors in detail. We finally validated the efficacy of bismuth compounds in virus-infected mammalian cells. The importance of nsp13 as a target for the development of anti-SARS-CoV-2 drugs is discussed.
Results
Nsp13 is a target of bismuth(III) in SARS-CoV-2 infected mammalian cells
Currently, there appears to be a lack of direct evidence showing that SARS-CoV-2 nsp13 is a target in virus-infected models for almost all nsp13 inhibitors. Previously, we showed that bismuth drugs or compounds inhibited nsp13 activity through displacement of zinc from the ZBD of nsp13;13 however, whether this is the case in SARS-CoV-2 infected cells remains unknown. Here, we used an enrichment method to “fish out” helicase from virus-infected mammalian cells and examined whether bismuth bound to helicase in cellulo (Fig. 1A). In brief, Omicron BA.5 (MOI = 0.1, where MOI represents multiplicity of infection) infected VeroE6-TMPRSS2 cells (cell line was obtained from Japanese Collection of Research Bioresources (JCRB) Cell Bank, JCRB1819) were treated with a complex of bismuth citrate with N-acetyl cysteine in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 molar ratio (i.e. Bi(NAC)3), a drug in clinical trial, and then, cell lysates collected 24 hours post-infection (h.p.i.) were incubated with nsp13 antibody-modified magnetic beads to enrich and immunoprecipitate nsp13 from the cell lysates. The 165Ho-conjugated secondary nsp13 detection antibody was then introduced to recognize the captured nsp13. The intensities of 165Ho, 209Bi, 66Zn and 115In (as the internal standard) were then measured by ICP-MS simultaneously to quantify nsp13 and its corresponding contents of bound Bi and Zn.
:3 molar ratio (i.e. Bi(NAC)3), a drug in clinical trial, and then, cell lysates collected 24 hours post-infection (h.p.i.) were incubated with nsp13 antibody-modified magnetic beads to enrich and immunoprecipitate nsp13 from the cell lysates. The 165Ho-conjugated secondary nsp13 detection antibody was then introduced to recognize the captured nsp13. The intensities of 165Ho, 209Bi, 66Zn and 115In (as the internal standard) were then measured by ICP-MS simultaneously to quantify nsp13 and its corresponding contents of bound Bi and Zn.
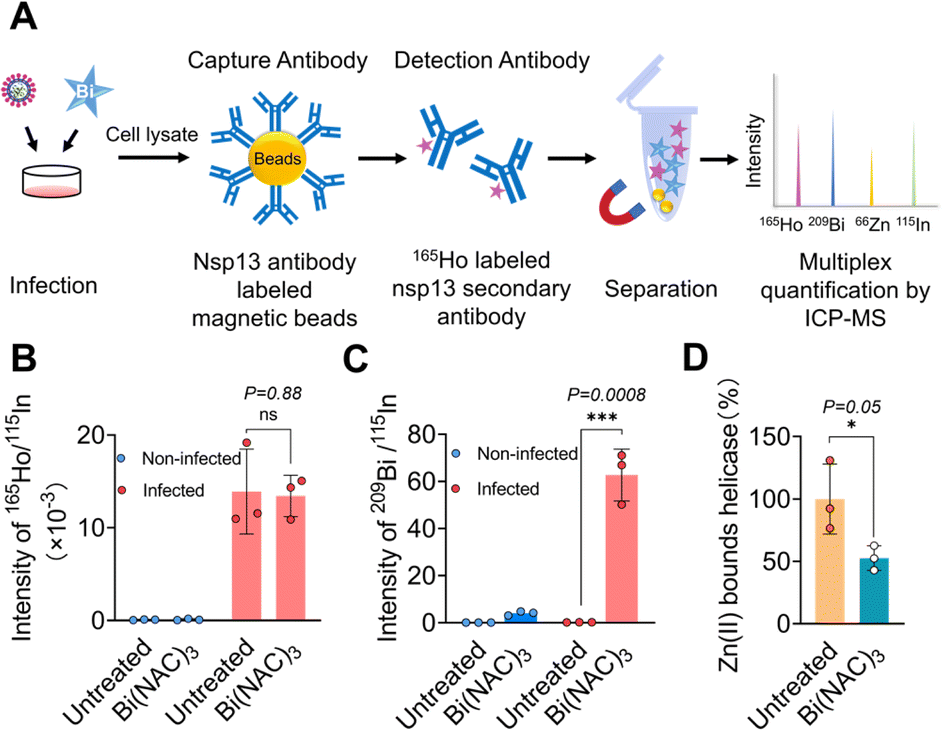 | ||
| Fig. 1 SARS-CoV-2 helicase is the authentic target of bismuth(III) in virus-infected mammalian cells. (A) Schematic chart of the enrichment approach for detecting interactions between targets and metallodrugs in cellulo. (B) Relative intensity of 165Ho/115In for nsp13 content with or without treatment of Bi(NAC)3 in the lysates of non-infected and infected cells by ICP-MS. (C) Cellular nsp13-bound Bi(III) content. (D) Cellular nsp13-bound Zn(II) content. Data are shown as mean ± SD. The statistical significance was calculated by unpaired two-tailed Student's t-test (n = 3), *P < 0.05, **P < 0.01, and ***P < 0.001. | ||
As shown in Fig. 1B, the amounts of nsp13 quantified by the intensity of 165Ho/115In were similar in the lysates of infected cells treated with or without Bi(NAC)3, indicating the treatment of bismuth resulted in no obvious changes in the level of nsp13. While no nsp13 could be detected in the lysates of non-infected cells treated with or without Bi(NAC)3. Importantly, a significantly higher bismuth content (209Bi/115In) could be detected only from the nsp13 extracted from the lysates of virus-infected cells after treatment with Bi(NAC)3. In contrast, almost no observable 209Bi/115In could be found either in non-infected cell lysates or lysates of the cells without treatment of Bi(NAC)3 (Fig. 1C).
Moreover, by measuring the content of Zn(II) and Bi(III) in the extracted nsp13 from the lysate of the cells treated with or without Bi(NAC)3, we found that 47.4 ± 9.8% of zinc was released from the nsp13 after bismuth treatment (Fig. 1D), providing solid evidence that Bi(III) binds to SARS-CoV-2 nsp13 and releases zinc ions from ZBD not only in vitro13 but also in cellulo. Collectively, we demonstrate that nsp13 is a target of Bi(III) in vitro and in SARS-CoV-2 infected mammalian cells. The methodology we used is applicable for the investigation of metallodrug targets in cellulo in general.
Identification of potent bismuth-based inhibitors of nsp13
To further search for more bismuth-based antiviral agents by targeting nsp13 against SARS-CoV-2 infection, we designed and synthesized 10 bismuth complexes. The structures and detailed synthesis information (e.g. NMR and HR-MS) are shown in Fig. S1, S13–15 and Table S1.†We overexpressed and purified SARS-CoV-2 helicase in E. coli expression system as reported previously13 and carried out primary screening by examination of the effects of these compounds on ATPase activity and DNA unwinding activity of nsp13. The ATPase activity was examined by a typical phosphate release assay, where the phosphate release due to ATP hydrolysis was presented as a relative percentage of the ATPase activity in the absence and presence of bismuth compounds. As shown in Fig. S2,† the ATPase activity was inhibited by all the bismuth compounds tested. We selected Bi(GSH)3, Bi(TMPP), Bi(6-TG)3 and Bi(Tro-NH2)3 as showcases to determine their half-maximal inhibitory concentrations (IC50). As shown in Fig. 2B, the ATPase activity was significantly decreased with an increase in the concentrations of selected bismuth compounds. Notably, Bi(Tro-NH2)3 has the lowest IC50 value of 0.03 ± 0.01 μM, which is over 30-fold lower than that of Bi(NAC)3 reported previously,31 followed by 2.99 ± 1.78, 0.85 ± 0.15, and 3.39 ± 0.32 μM for Bi(GSH)3, Bi(6-TG)3 and Bi(TMPP) respectively.
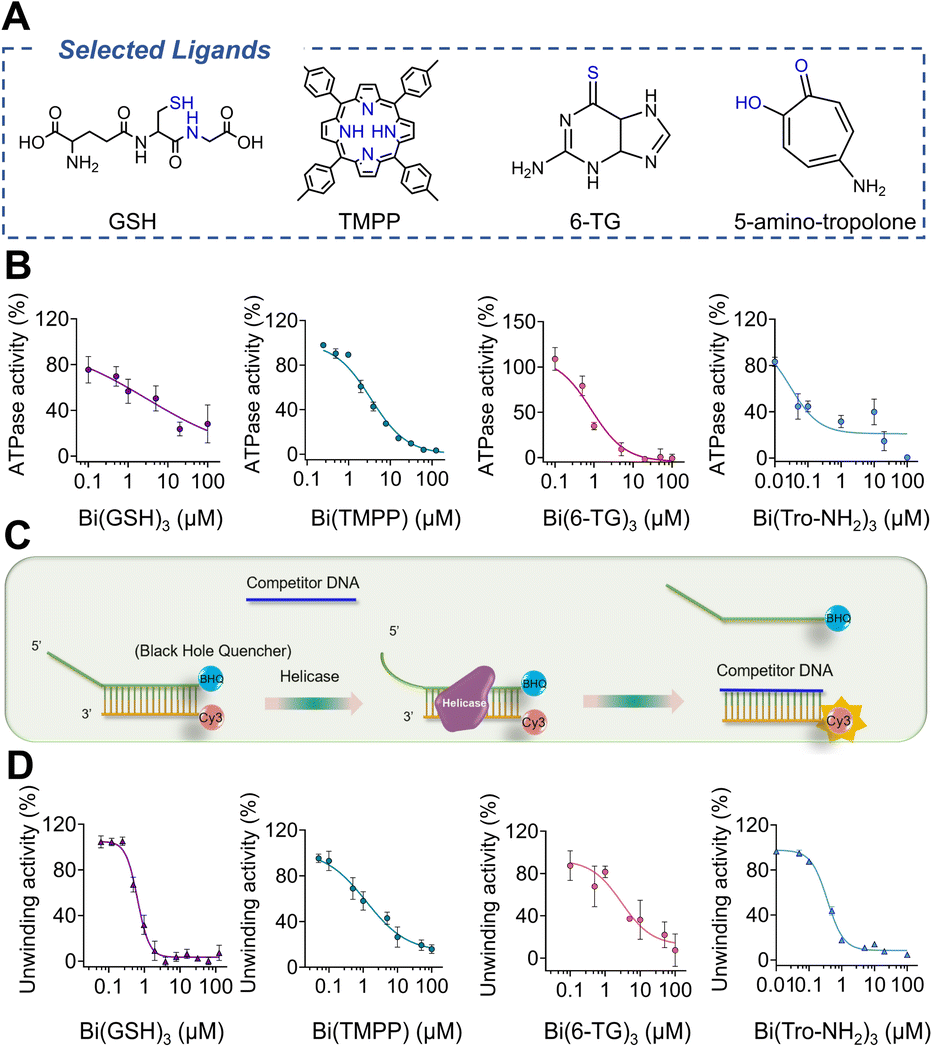 | ||
| Fig. 2 Inhibition of selected bismuth compounds against SARS-CoV-2 helicase. (A) Selected ligands to form complexes with Bi(III). (B) ATPase activity inhibited by Bi(GSH)3, Bi(TMPP), Bi(6-TG)3 and Bi(Tro-NH2)3. (C) Schematic chart of FRET-based DNA duplex unwinding assay. (D) Inhibition of DNA unwinding activity by selected bismuth compounds. IC50 was calculated by GraphPad Prism with nonlinear regression (n = 3). In inhibitory experiments, the final concentration of DMSO was kept at 1%. | ||
We next examined the effects of these metal compounds on the DNA unwinding activity of nsp13 by a fluorescence resonance energy transfer (FRET)-based assay.36 The DNA-duplex substrate was prepared by annealing an oligomer with a Cy3 fluorophore at the 3′ end and an oligomer with a BHQ-2 quencher at the 5′ end (oligomers were purchased from Integrated DNA Technologies). The DNA-duplex and nsp13 were mixed in the presence of different concentrations of bismuth compounds prior to fluorescence titration. The unwinding of the Cy3 strand from the DNA-duplex by helicase resulted in a drastic increase in the signal intensity of the DNA-duplex (Fig. 2C). However, the fluorescence in the primary screening increased much less evidently upon the addition of increasing concentrations of bismuth compounds (Fig. S2†). Similarly, the IC50 values of the these compounds against duplex-unwinding activity of the enzyme were determined to be 0.36 ± 0.02, 0.64 ± 0.09, 1.22 ± 0.69 and 2.96 ± 0.51 μM for Bi(Tro-NH2)3, Bi(GSH)3, Bi(TMPP) and Bi(6-TG)3 respectively, indicative of the inhibition of duplex-unwinding activity of nsp13 in a dose-dependent manner by these bismuth compounds (Fig. 2D). Notably, Bi(Tro-NH2)3 also exhibited slightly better activity compared with Bi(NAC)3 as judged by two-fold lower IC50 value, while being four-fold for CBS. Taken together, we have demonstrated that bismuth-based compounds show good performance on inhibiting SARS-CoV-2 nsp13 activities at an enzyme level.
Bismuth compounds interfere with the binding of nsp13 to ATP
Despite observed phenomena that bismuth-based compounds inhibited SARS-CoV-2 helicase ATPase activity, the underlying mechanism remained unknown. To address this issue, an ATP analogue, adenosine 5′-O-(3-thiotriphosphate) (BODIPY™ FL ATP-γ-S), which is hydrolysis-resistant and thioester fluorophore linked through γ-thiol, was selected for the study. The fluorescence of BODIPY ATP-γ-S thioesters is quenched relative to that of the free dye but is recovered upon binding to ATP-binding proteins. We incubated BODIPY™ FL ATP-γ-S (10 nM) with nsp13 at different concentrations ranging from 20 to 50 to 100 nM and observed a dose-dependent increase in fluorescence intensity (Fig. S4†). We next mixed 100 nM nsp13 with selected bismuth compounds, e.g., CBS, Bi(NAC)3, Bi(GSH)3, Bi(6-TG)3 and Bi(Hino)3 at 1 μM prior to addition of BODIPY™ FL ATP-γ-S, and surprisingly observed decreased fluorescence intensities to different extents, indicating that these compounds interfered with the binding of SARS-CoV-2 nsp13 to ATP. To further examine whether the fluorescence quenching resulted from accompanying ligands, we used N-acetyl cysteine (NAC), glutathione (GSH), 6-thioguanine (6-TG) and hinokitiol (Hino) as controls. As expected, we did not observe any apparent decrease in the fluorescence intensity, confirming that it is the metal ion Bi(III) that plays an essential role (Fig. 3A). We also evaluated fluorescence changes with addition of different concentrations of metal compounds using Bi(NAC)3 as a showcase, observing a dose-dependent decrease in the fluorescence intensities when Bi(NAC)3 at concentrations of 0.1, 0.5, 1 μM was added into the reaction solutions (Fig. 3B). Collectively, we demonstrate that bismuth-based compounds can interfere with the binding of nsp13 to ATP, leading to a reduction in ATPase activity.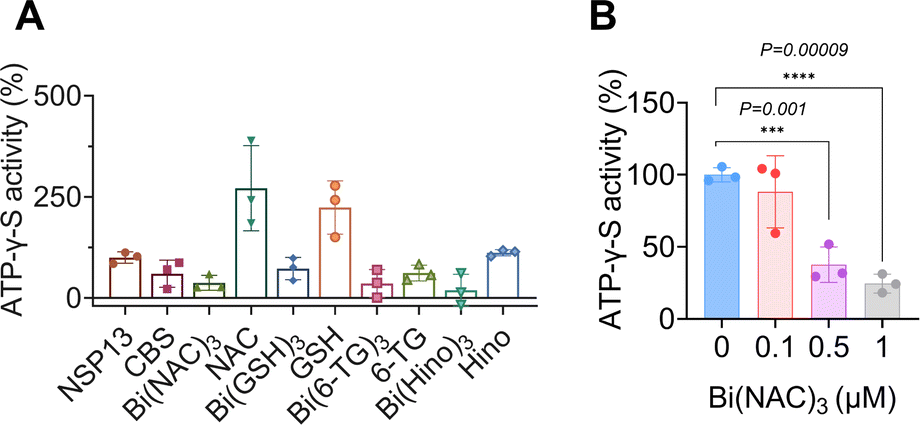 | ||
| Fig. 3 Bismuth compounds interfere with nsp13 binding to ATP. (A) Fluorescence changes of BODIPY ATP-γ-S after treatment with different bismuth compounds, ligands serving as negative controls. (B) Fluorescence intensity of BODIPY ATP-γ-S was decreased by Bi(NAC)3 in a dose-dependent manner. All data are shown as mean ± SD. Statistical significance was calculated by an unpaired two-tailed Student's t-test (n = 3), *P < 0.05, **P < 0.01 and ***P < 0.001. | ||
Bismuth compounds disrupt DNA substrate binding to nsp13
Nsp13 activities could be inhibited through disrupting the bindings of the enzyme to ATP, nucleic acid or blocking helicase translocation.15 We next investigated whether the binding of nsp13 to bismuth(III) affected its substrate binding by electrophoretic mobility shift assay (EMSA).37 We incubated DNA substrate (100 nM) with nsp13 at different concentrations ranging from 0, 0.5, 1, 2 to 5 μM first and observed a dose-dependent increase in the intensity of DNA-bound nsp13 complex accompanied by a decrease in the intensity of free DNA substrate (Fig. 4A).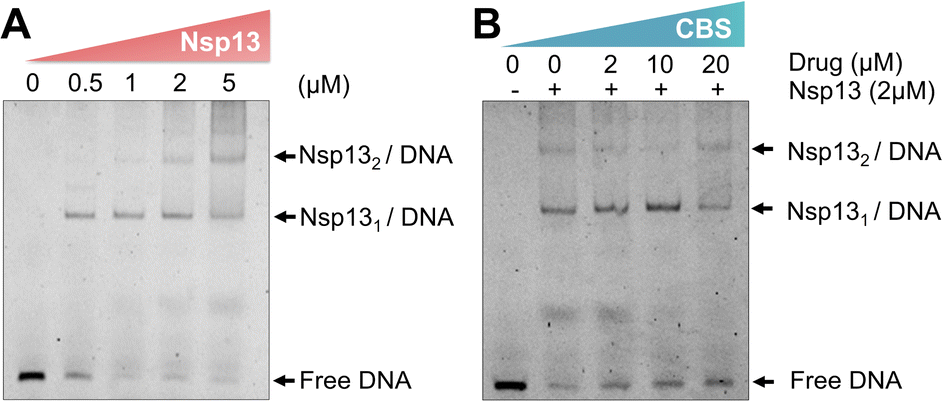 | ||
| Fig. 4 Electrophoretic mobility shift assay showing that bismuth(III) disrupts the DNA substrate binding to nsp13. (A) Dose-dependent binding of native nsp13 (Zn3-nsp13) to its DNA substrate. (B) A dose-dependent inhibition of the substrate binding to nsp13 by a bismuth compound (n = 2). | ||
Subsequently, we pre-incubated nsp13 with different concentrations of a bismuth compound (CBS) for 2 hours prior to the DNA binding assay to ensure that nsp13 is in the Bi(III)-bound form. EMSA images showed a dose-dependent decrease in the DNA-bound nsp13 accompanied by an increase in the intensities of free DNA substrate, clearly indicating that Bi(III) inhibits the binding of nsp13 to its DNA substrate (Fig. 4B). Our combined data demonstrate that Bi(III) inhibits the nsp13 unwinding activity through a dual action, i.e., blocking nsp13 binding to ATP and interfering with nsp13 binding to the DNA substrate.
Bismuth compounds inhibit SARS-CoV-2 variants in infected mammalian cells
We next evaluated the potential antiviral activity of bismuth compounds that exhibited good in vitro efficacy towards inhibition of nsp13 in infected mammalian VeroE6-TMPRSS2 cells. We first examined the cytotoxicity of these metal compounds to ensure that these compounds at selected concentrations would not cause any toxicity to virus-infected mammalian cells. As shown in Fig. S2,† almost all bismuth compounds did not exhibit any cytotoxicity with CC50 (half cytotoxicity concentration) larger than 1000 μM. It is worth mentioning that the cytotoxicity of Bi(6-TG)3 was much lower than that of its original ligand, 6-TG, in the VeroE6 cell line (Fig. S5†). We therefore selected certain compounds to evaluate their antiviral activity against SARS-CoV-2 Omicron BA.5 and the novel XBB variant, which show stronger immune escape ability and observed a dose-dependent inhibition by Bi(TMPP) and Bi(6-TG)3 with EC50 (half-maximal effect concentration) values against the BA.5 variant of 7.28 ± 2.81 μM and 1.18 ± 0.19 μM, respectively, by plaque reduction assay (Fig. 5). Unexpectedly, Bi(Tro-NH2)3 did not show antiviral activity as good as that at the enzyme level, possibly due to the poor cellular uptake of Bi(III), which was much lower than that for CBS, Bi(GSH)3, Bi(6-TG)3 and Bi(TMPP) (Fig. S7†). Further optimization of the structure of the accompanying ligand is needed to improve its uptake.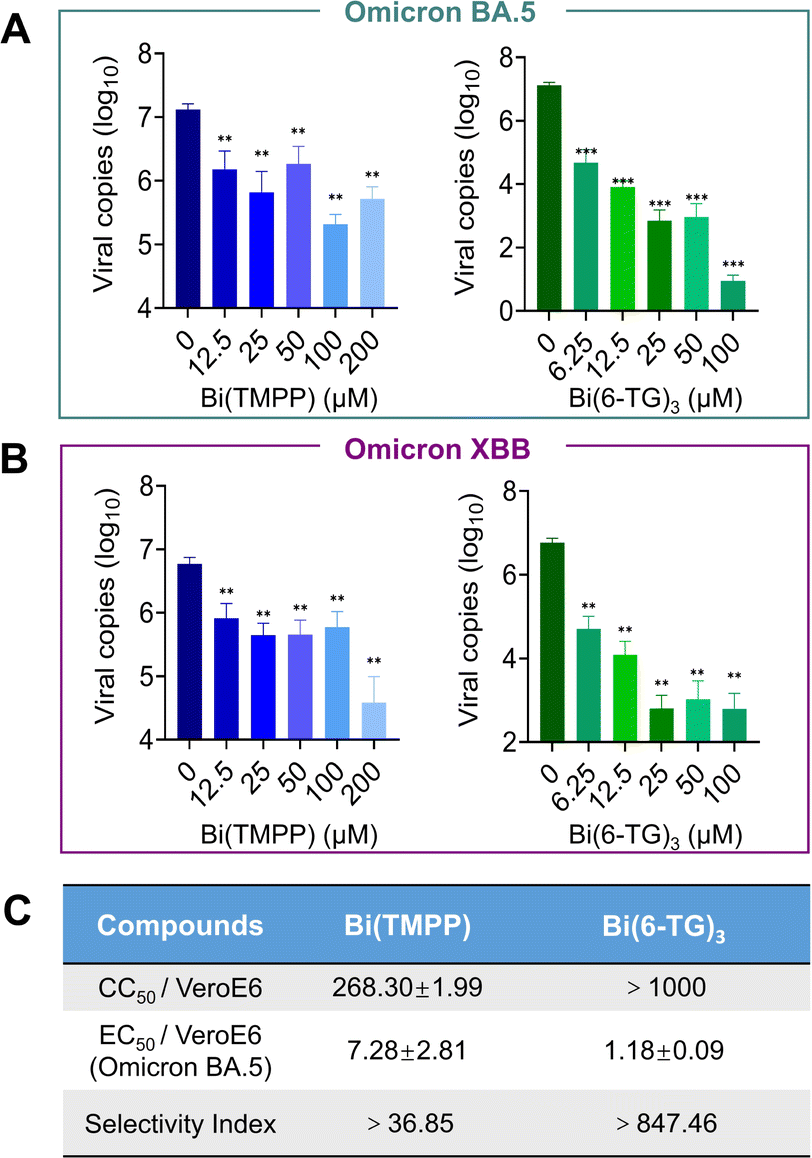 | ||
| Fig. 5 Bismuth compounds inhibit different SARS-CoV-2 Omicron variants in virus-infected mammalian cells. (A and B) Dose-dependent inhibition of SARS-CoV-2 Omicron BA.5 and XBB viral replication in VeroE6-TMPRSS2 cells after treatment with Bi(TMPP) and Bi(6-TG)3 (n = 3). The cell culture supernatant was collected to quantify viral load by reverse transcription (RT-qPCR). (C) Half-maximal effective concentration (EC50) by plaque reduction assay and the selectivity index. For those compounds dissolved in DMSO, the final concentration of DMSO was kept at 1%. Data are shown as mean ± SD. The statistical significance was calculated by unpaired two-tailed Student's t-test (n = 3), *P < 0.05, **P < 0.01, and ***P < 0.001. | ||
Bismuth compounds disrupt nsp13-nsp12 interaction in cellulo
The interaction between RNA-dependent polymerase (RdRp, nsp12) and nsp13 plays an essential role in the process of viral replication. Such an interaction may improve the efficiency of replication; for example, it is reported that nsp12 enhances the catalytic efficiency of nsp13 by 2-fold via increasing the step size of nucleic acid (RNA/RNA or DNA/DNA) unwinding.38 Recently, a new hypothesis was proposed on the mechanism of bismuth inhibition of nsp13 based on a multilevel computational study.39 Replacement of Zn(II) by Bi(III) in the zinc-finger domain could cause slight but appreciable structural modifications in the zinc-binding domain of nsp13, resulting in the decreased extension of a specific hydrophobic portion of nsp13, which is responsible for the interaction with the nsp12 protein. However, there is no experimental data to confirm this hypothesis.To examine whether bismuth binding disrupted the nsp12–nsp13 interaction, we expressed both nsp12 and nsp13 in HEK293T cells (cell line was purchased from ATCC, CRL-3216), then incubated with 50 μM Bi(TMPP) or 50 μM Bi(6-TG)3 for 24 hours. Immunofluorescence images showed nsp13 was closely surrounded by nsp12 in the absence of a bismuth compound. In contrast, upon treatment with Bi(TMPP), nsp12 was no longer of its original shape and moved far away from nsp13 (Fig. 6A and S8†), indicating the presence of a bismuth compound disrupted the complexation of nsp12-nsp13. To further validate this, co-immunoprecipitation (CO-IP) was performed. We first examined whether bismuth compounds affected the expression of nsp12 and nsp13 to exclude the potential effect of protein content in later CO-IP experiments. The cells were transfected with nsp12 and nsp13, and treated with Bi(TMPP) and Bi(6-TG)3 at different doses, and cell lysates were collected 24 hours post-treatment for western blot analysis. As shown in Fig. 6B, there were no obvious changes in the expression levels of nsp12 and nsp13 upon bismuth treatment. In addition, CO-IP results showed that a bismuth compound (i.e., Bi(TMPP)) disrupted the nsp13-nsp12 interaction in a dose-dependent manner (Fig. 6C). Meanwhile, we quantified the content of nsp12 that interacts with nsp13 by lanthanide-tagged antibody CO-IP, a similar approach to that shown in Fig. 1A. In brief, nsp13 antibody-functionalized magnetic beads were used to immunoprecipitate nsp13 or nsp13/nsp12 complex from cell lysates. The nsp12 bound to nsp13 was then recognized by 163Dy-labeled nsp12 detection antibody and quantified by ICP-MS.
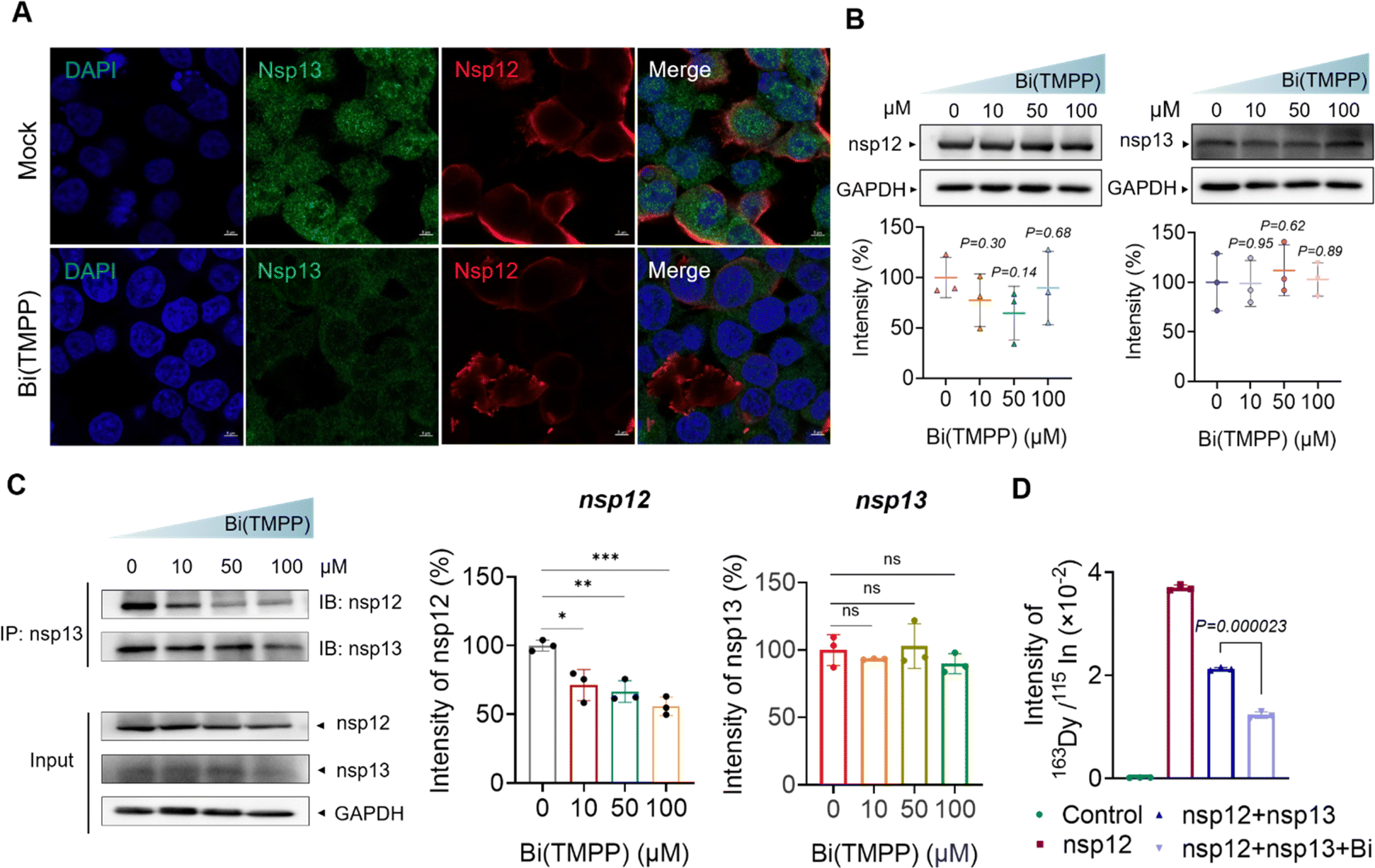 | ||
| Fig. 6 Bismuth disturbs the nsp13−nsp12 interaction. (A) Immunofluorescence staining images show the nsp13–nsp12 interaction. Mock group was the normal condition of the interaction. The other group was treated by 50 μM Bi(TMPP). Transfection, fixation and staining were performed on HEK293T cells. SARS-CoV-2 nsp12 and nsp13 antigens were stained in red and green, respectively, while cell nuclei were stained in blue by DAPI (scale bar = 5 μm). (B) Western blotting images of nsp12 and nsp13 expression after treatment with different concentrations of bismuth compound, Bi(TMPP). (C) Western blotting results for CO-IP. (D) The signal intensity of 163Dy for nsp12 content quantified by ICP-MS. Data are shown as mean ± SD. The statistical significance was calculated by unpaired two-tailed Student's t-test (n = 3). *P < 0.05, **P < 0.01, and ***P < 0.001. | ||
As shown in Fig. 6D, the 163Dy-labeled nsp12 signal was much higher in the absence of bismuth than that with bismuth treatment, confirming that bismuth disrupted the interaction between nsp12 and nsp13. Collectively, we provide the first solid evidence that bismuth can disrupt the nsp13-nsp12 interaction in cellulo.
Discussion and conclusion
The outbreak of the COVID-19 epidemic caused by SARS-CoV-2 highlights the importance of developing pan-spectrum antivirals. Among which, metal-based compounds hold great promise to be developed as pan-spectrum antivirals owing to their ability to target conserved viral enzymes that are essential for virus replication.32 In this study, we further explored metal-based antivirals for combating SARS-CoV-2 infection. Despite nsp13 being considered as a perfect drug target for the development of antivirals against SARS-CoV and SARS-CoV-2 according to previous reports,15,16,36,40–43 it is unknown whether these inhibitors targeted nsp13 in virus-infected mammalian cells. Herein, we validated for the first time that nsp13 is a target of bismuth-based antivirals using an enrichment strategy based on our previous report.34,44 This method enables simultaneous quantification of a protein and corresponding bound metals, providing a general strategy to identify targets of different metalloagents or any drug/drug candidate with a heavy element. We subsequently searched for more potent bismuth-based antivirals using SARS-CoV-2 nsp13 as a target. We synthesized 10 novel bismuth compounds and examined their ability towards inhibition of the ATPase activity and DNA unwinding activity of nsp13 (Fig. S1 and S2†), and indeed, we observed that a certain bismuth compound, Bi(Tro-NH2)3, exhibited potent inhibition of nsp13 enzymatic activities (Fig. 2).Importantly, the broad-spectrum antiviral activity of Bi(III)-based inhibitors of nsp13 was also validated in mammalian cells infected with different Omicron strains of SARS-CoV-2 (Fig. 5 and S6†). In particular, we identified Bi(6-TG)3, which exhibited better antiviral activity (EC50 of 1.2 μM) than previously reported bismuth compounds including RBC (EC50 = 2.3 μM)13 and Bi(NAC)3 (EC50 = 5.8 μM) and also better than that for SARS-CoV-2 Omicron BA.5 (Fig. S6†). However, it is noted that certain compounds, e.g. Bi(Tro-NH2)3, that exhibited excellent activity at an enzyme level did not show potent antiviral activity in virus-infected cells owing to poor cellular uptake (Fig. S7†), indicating that initial screening hits obtained must be validated in the cells and further extending the library of bismuth compounds may allow more potent antivirals to be developed.
Bismuth has a long history in medicine for the treatment of many diseases, especially gastrointestinal disorders caused by Helicobacter pylori28,45 and has also been repurposed for combating NDM-1 carrying superbugs when used together with antibiotics.28 We have demonstrated that RBC and co-therapy of a bismuth drug with NAC exhibited excellent inhibition on SARS-CoV-2 infection in vitro and in vivo. In addition, according to a recent clinical case report, patients of COVID-19, who displayed diarrhea or gastric complaints, showed beneficial effects when taking bismuth subsalicylate (BSS) evidenced by showing improvement in oxygen requirements.46 Other clinical data for BSS also confirmed the feasibility of BSS for COVID-19 co-therapy.47 All these data from an enzyme level to clinical stage supported our conclusion that bismuth-based therapy holds a great potential to be further developed in terms of pan-spectrum anti-coronavirus drugs.
Importantly, we investigated the molecular mechanism of action of bismuth compounds towards inhibition of nsp13. We show that bismuth compounds interfered with the binding of nsp13 to ATP, leading to reduced ATPase activity (Fig. 3). Moreover, the binding of Bi(III) to nsp13 disrupts the DNA substrate binding of nsp13, thus inhibiting nsp13 unwinding activity (Fig. 4). Significantly, we found that bismuth compounds disrupted the nsp13–nsp12 interaction in RTC, which plays an essential role in the process of viral replication (Fig. 6). All these observed phenomena might be attributable to the structural changes of the enzyme caused by the bismuth binding. This may warrant further studies in the future. Using metal-tagged antibody CO-IP combined with ICP-MS, we could quantify the content of a protein in a protein–protein complex easily. Our studies highlight nsp13 as an excellent drug target for the development of anti-SARS-CoV-2 agents.
In summary, we demonstrate that nsp13 serves as a target of bismuth-based antivirals in virus-infected mammalian cells for the first time. We further show that bismuth inhibited nsp13 via interference with ATP and DNA substrate binding. Moreover, the binding of bismuth to nsp13 disrupted the nsp12–nsp13 interaction in cellulo. The metal-tagged antibody approach we developed may provide a new arsenal for precise quantification of the amount of a drug binding to its protein target in vivo as long as the drug contains a heavy element. We identified more potent bismuth-based antivirals through screening more bismuth compounds both at an enzyme level and in infected cells. Our work highlights the importance of nsp13 (i.e. helicase) as a druggable target for the development of anti-SARS-CoV-2 and relevant antiviral drugs.
Data availability
All experimental data and procedures are provided in the ESI.† Other data are available from the corresponding author upon reasonable request.Author contributions
X. W., S. Y., H. L., H. S designed the experiments. C. C. synthesized compounds. X. W., Y. Z., K. T., J. C., S. W performed the experiments. X. W., Y. Z., C. C., J. C., S. W. analyzed the data. X. W. and H. L. wrote the manuscript with conceptual advice provided from other authors. H. S. supervised the study and provided the grant support.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Research Grants Council (17318322, SRFS2122-7S04, T11-709/21N, 17306323) and ITF (ITS/278/20) of Hong Kong SAR and the University of Hong Kong (URC and Norman & Cecilia Yip Foundation) for their support.References
- World-Health-Organization, WHO Coronavirus (COVID-19) Dashboard With Vaccination Data, https://covid19.who.int/.
- R. Wang, Y. Hozumi, C. Yin and G. W. Wei, Genomics, 2020, 112, 5204–5213 CrossRef CAS PubMed.
- P. V'Kovski, A. Kratzel, S. Steiner, H. Stalder and V. Thiel, Nat. Rev. Microbiol., 2021, 19, 155–170 CrossRef.
- D. Kim, J. Y. Lee, J. S. Yang, J. W. Kim, V. N. Kim and H. Chang, Cell, 2020, 181, 914–921.e910 CrossRef CAS PubMed.
- Y. Zhao, Y. Zhu, X. Liu, Z. Jin, Y. Duan, Q. Zhang, C. Wu, L. Feng, X. Du, J. Zhao, M. Shao, B. Zhang, X. Yang, L. Wu, X. Ji, L. W. Guddat, K. Yang, Z. Rao and H. T. Yang, Proc. Natl. Acad. Sci. U.S.A., 2022, 119, e2117142119 CrossRef CAS.
- H. Yang and Z. Rao, Nat. Rev. Microbiol., 2021, 19, 685–700 CrossRef CAS.
- R. Yadav, J. K. Chaudhary, N. Jain, P. K. Chaudhary, S. Khanra, P. Dhamija, A. Sharma, A. Kumar and S. Handu, Cells, 2021, 10, 821 CrossRef CAS PubMed.
- A. Shamsi, T. Mohammad, S. Anwar, S. Amani, M. S. Khan, F. M. Husain, M. T. Rehman, A. Islam and M. I. Hassan, Int. J. Biol. Macromol., 2021, 177, 1–9 CrossRef CAS.
- L. Zhang, D. Lin, X. Sun, U. Curth, C. Drosten, L. Sauerhering, S. Becker, K. Rox and R. Hilgenfeld, Science, 2020, 368, 409–412 CrossRef CAS.
- Z. Jin, X. Du, Y. Xu, Y. Deng, M. Liu, Y. Zhao, Z. Rao and H. T. Yang, Nature, 2020, 582, 289–293 CrossRef CAS PubMed.
- S. Yuan, X. Gao, K. Tang, J. P. Cai, M. Hu, P. Luo, L. Wen, Z. W. Ye, C. Luo, J. O. Tsang, C. C. Chan, Y. Huang, J. Cao, R. Liang, Z. Qin, B. Qin, F. Yin, H. Chu, D. Y. Jin, R. Sun, J. F. Chan, S. Cui and K. Y. Yuen, Protein Cell, 2022, 13, 940–953 CrossRef CAS PubMed.
- Y. Gao, L. Yan, Y. Huang, F. Liu, Y. Zhao, L. Cao, T. Wang, Q. Sun, Z. Ming, L. Zhang, J. Ge, L. Zheng, Y. Zhang, H. Wang, Y. Zhu, C. Zhu, T. Hu, T. Hua, B. Zhang, X. Yang, J. Li, H. Yang, Z. Liu, W. Xu, L. W. Guddat, Q. Wang, Z. Lou and Z. Rao, Science, 2020, 368, 779–782 CrossRef CAS PubMed.
- S. Yuan, R. Wang, J. F. Chan, A. J. Zhang, T. Cheng, K. K. Chik, Z. W. Ye, S. Wang, A. C. Lee, L. Jin, H. Li, D. Y. Jin, K. Y. Yuen and H. Sun, Nat. Microbiol., 2020, 5, 1439–1448 CrossRef CAS PubMed.
- S. Yuan, X. Yin, X. Meng, J. F. Chan, Z. W. Ye, L. Riva, I. F. Hung, R. A. Li, H. Chen, H. Sun, D. Y. Jin, R. Sun, S. K. Chanda and K. Y. Yuen, Nature, 2021, 593, 418–423 CrossRef CAS PubMed.
- G. R. Perez-Lemus, C. A. Menendez, W. Alvarado, F. Bylehn and J. J. de Pablo, Sci. Adv., 2022, 8, eabj4526 CrossRef CAS.
- D. Berta, M. Badaoui, S. A. Martino, P. J. Buigues, A. V. Pisliakov, N. Elghobashi-Meinhardt, G. Wells, S. A. Harris, E. Frezza and E. Rosta, Chem. Sci., 2021, 12, 13492–13505 RSC.
- Z. Jia, L. Yan, Z. Ren, L. Wu, J. Wang, J. Guo, L. Zheng, Z. Ming, L. Zhang, Z. Lou and Z. Rao, Nucleic Acids Res., 2019, 47, 6538–6550 CrossRef CAS.
- J. A. Newman, A. Douangamath, S. Yadzani, Y. Yosaatmadja, A. Aimon, J. Brandao-Neto, L. Dunnett, T. Gorrie-Stone, R. Skyner, D. Fearon, M. Schapira, F. von Delft and O. Gileadi, Nat. Commun., 2021, 12, 4848 CrossRef CAS PubMed.
- K. A. Ivanov and J. Ziebuhr, J. Virol., 2004, 78, 7833–7838 CrossRef CAS PubMed.
- N. R. Lee, H. M. Kwon, K. Park, S. Oh, Y. J. Jeong and D. E. Kim, Nucleic Acids Res., 2010, 38, 7626–7636 CrossRef CAS PubMed.
- A. Seybert, L. C. van Dinten, E. J. Snijder and J. Ziebuhr, J. Virol., 2000, 74, 9586–9593 CrossRef CAS PubMed.
- J. A. Tanner, R. M. Watt, Y. B. Chai, L. Y. Lu, M. C. Lin, J. S. Peiris, L. L. Poon, H. F. Kung and J. D. Huang, J. Biol. Chem., 2003, 278, 39578–39582 CrossRef CAS PubMed.
- J. Chen, B. Malone, E. Llewellyn, M. Grasso, P. M. M. Shelton, P. D. B. Olinares, K. Maruthi, E. T. Eng, H. Vatandaslar, B. T. Chait, T. M. Kapoor, S. A. Darst and E. A. Campbell, Cell, 2020, 182, 1560–1573.e1513 CrossRef CAS PubMed.
- J. Zeng, F. Weissmann, A. P. Bertolin, V. Posse, B. Canal, R. Ulferts, M. Wu, R. Harvey, S. Hussain, J. C. Milligan, C. Roustan, A. Borg, L. McCoy, L. S. Drury, S. Kjaer, J. McCauley, M. Howell, R. Beale and J. F. X. Diffley, Biochem. J., 2021, 478, 2405–2423 CrossRef CAS PubMed.
- J. Chen, Q. Wang, B. Malone, E. Llewellyn, Y. Pechersky, K. Maruthi, E. T. Eng, J. K. Perry, E. A. Campbell, D. E. Shaw and S. A. Darst, Nat. Struct. Mol. Biol., 2022, 29, 250–260 CrossRef CAS.
- S. Yazdani, N. De Maio, Y. Ding, V. Shahani, N. Goldman and M. Schapira, J. Proteome Res., 2021, 20, 4212–4215 CrossRef PubMed.
- H. Sun, Q. Zhang, R. Wang, H. Wang, Y. T. Wong, M. Wang, Q. Hao, A. Yan, R. Y. Kao, P. L. Ho and H. Li, Nat. Commun., 2020, 11, 5263 CrossRef CAS.
- R. Wang, T. P. Lai, P. Gao, H. Zhang, P. L. Ho, P. C. Woo, G. Ma, R. Y. Kao, H. Li and H. Sun, Nat. Commun., 2018, 9, 439 CrossRef.
- Q. Zhang, R. Wang, M. Wang, C. Liu, M. Koohi-Moghadam, H. Wang, P. L. Ho, H. Li and H. Sun, Proc. Natl. Acad. Sci. U.S.A., 2022, 119, e2119417119 CrossRef CAS PubMed.
- H. Wang, M. Wang, X. Xu, P. Gao, Z. Xu, Q. Zhang, H. Li, A. Yan, R. Y. Kao and H. Sun, Nat. Commun., 2021, 12, 3331 CrossRef CAS.
- R. Wang, J. F. Chan, S. Wang, H. Li, J. Zhao, T. K. Ip, Z. Zuo, K. Y. Yuen, S. Yuan and H. Sun, Chem. Sci., 2022, 13, 2238–2248 RSC.
- H. Li, S. Yuan, X. Wei and H. Sun, Chem. Commun., 2022, 58, 7466–7482 RSC.
- M. Gil-Moles, S. Turck, U. Basu, A. Pettenuzzo, S. Bhattacharya, A. Rajan, X. Ma, R. Bussing, J. Wolker, H. Burmeister, H. Hoffmeister, P. Schneeberg, A. Prause, P. Lippmann, J. Kusi-Nimarko, S. Hassell-Hart, A. McGown, D. Guest, Y. Lin, A. Notaro, R. Vinck, J. Karges, K. Cariou, K. Peng, X. Qin, X. Wang, J. Skiba, L. Szczupak, K. Kowalski, U. Schatzschneider, C. Hemmert, H. Gornitzka, E. R. Milaeva, A. A. Nazarov, G. Gasser, J. Spencer, L. Ronconi, U. Kortz, J. Cinatl, D. Bojkova and I. Ott, Chemistry, 2021, 27, 17928–17940 CrossRef CAS.
- J. Chen, Y. Zhou, X. Wei, X. Xu, Z. Qin, C. P. Ong, Z. W. Ye, D. Y. Jin, B. Boitrel, S. Yuan, J. F. Chan, H. Li and H. Sun, ACS Infect. Dis., 2024, 10, 858–869 CrossRef CAS PubMed.
- J. Karges, M. A. Giardini, O. Blacque, B. Woodworth, J. L. Siqueira-Neto and S. M. Cohen, Chem. Sci., 2023, 14, 711–720 RSC.
- N. Yang, J. A. Tanner, B. J. Zheng, R. M. Watt, M. L. He, L. Y. Lu, J. Q. Jiang, K. T. Shum, Y. P. Lin, K. L. Wong, M. C. Lin, H. F. Kung, H. Sun and J. D. Huang, Angew. Chem., Int. Ed. Engl., 2007, 46, 6464–6468 CrossRef CAS PubMed.
- N. Maio, M. K. Raza, Y. Li, D. L. Zhang, J. M. Bollinger, Jr., C. Krebs and T. A. Rouault, Proc. Natl. Acad. Sci. U.S.A., 2023, 120, e2303860120 CrossRef CAS.
- A. O. Adedeji, B. Marchand, A. J. Te Velthuis, E. J. Snijder, S. Weiss, R. L. Eoff, K. Singh and S. G. Sarafianos, PLoS One, 2012, 7, e36521 CrossRef CAS PubMed.
- I. Tolbatov, L. Storchi and A. Marrone, Inorg. Chem., 2022, 61, 15664–15677 CrossRef CAS.
- N. Yang, J. A. Tanner, Z. Wang, J. D. Huang, B. J. Zheng, N. Zhu and H. Sun, Chem. Commun., 2007, 4413–4415 RSC.
- L. Lu, Y. Peng, H. Yao, Y. Wang, J. Li, Y. Yang and Z. Lin, Antiviral Res., 2022, 206, 105389 CrossRef CAS.
- S. Habtemariam, S. F. Nabavi, M. Banach, I. Berindan-Neagoe, K. Sarkar, P. C. Sil and S. M. Nabavi, Arch. Med. Res., 2020, 51, 733–735 CrossRef CAS PubMed.
- N. Mehyar, J. Virus Erad., 2023, 9, 100327 CrossRef CAS.
- Y. Zhou, S. Yuan, K. K. To, X. Xu, H. Li, J. P. Cai, C. Luo, I. F. Hung, K. H. Chan, K. Y. Yuen, Y. F. Li, J. F. Chan and H. Sun, Chem. Sci., 2022, 13, 3216–3226 RSC.
- H. Li and H. Sun, Curr. Opin. Chem. Biol., 2012, 16, 74–83 CrossRef CAS PubMed.
- D. C. Wolf, C. H. Wolf and D. T. Rubin, Am. J. Gastroenterol., 2020, 115, 1298 CrossRef PubMed.
- M. B. Yacyshyn, J. Collins, M. Chua, A. Siegwald, S. Yacyshyn, V. Briones-Pryor and B. Yacyshyn, Curr. Ther. Res. Clin. Exp., 2022, 96, 100667 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc06961c |
| This journal is © The Royal Society of Chemistry 2024 |