DOI:
10.1039/D4SC00841C
(Edge Article)
Chem. Sci., 2024,
15, 5589-5595
Efficient near-infrared emission benefits from slowing down the internal conversion process†
Received
4th February 2024
, Accepted 12th March 2024
First published on 18th March 2024
Abstract
Organic deep-red (DR) and near-infrared (NIR) emitters with high photoluminescence quantum yield (PLQY) are rare due to the strong non-radiative (knr) decay. Here, we report two DR/NIR emitters with high PLQY, TPANZPyPI and TPANZ3PI. Interestingly, the TPANZPyPI film exhibits 46.5% PLQY at 699 nm. Theoretical calculations indicate that TPANZPyPI can achieve this high PLQY in the near-infrared emission region due to its small S1 to S0 internal conversion (IC) rate. Meanwhile, research has found that, compared to TPANZ3PI, TPANZPyPI with a more rigid structure can effectively suppress the T2 to T1 IC process, which is conducive to higher exciton utilization efficiency (EUE). TPANZPyPI's non-doped OLED shows NIR emission with 4.6% @ 684 nm maximum external quantum efficiency (EQEmax). Its doped OLEDs radiate DR with an EQEmax of 6.9% @ 666 nm. These EQEs are among the highest values for hybridized local charge transfer state materials emitting more than 640 nm. This work demonstrates for the first time, based on a combination of theory and experiment, that increasing the molecular rigidity can inhibit the excited state IC process in addition to the S1 to S0 IC, realizing efficient electroluminescence.
Introduction
Organic light-emitting diodes (OLEDs), a third-generation display technology, have been used in flat panel displays and solid-state lighting devices in recent years. As one of the three primary colors in panchromatic displays, the development of pure organic red emitters lags behind that of blue and green emitters.1,2 The reason for this is that the energy gap (ΔE) from the first singlet excited state (S1) to the ground state (S0) is narrow. According to the law of the energy gap (knr is directly proportional to ae−bΔE, where a and b are constants of proportionality),3,4 it can be seen that the smaller the ΔE, the redder the luminescence and the non-radiative decay rate will exponentially increase with the decrease of ΔE, which means that the non-radiative decay rate (knr) of red fluorescent materials is much larger than that of blue and green materials. This phenomenon is more significant in the deep-red (DR) and near-infrared (NIR) domains. To realize efficient luminescence in the DR and even in the NIR, it is necessary to suppress the non-radiative decay process of fluorescent materials.
Non-radiative decay consists of two main aspects: on the one hand, the internal conversion (IC) process from S1 to S0, and on the other hand, the intersystem crossing (ISC) process from S1 to the neighboring triplet state (T). The ISC process is prolonged because the singlet excited and triplet excited states are usually forbidden transitions. Even though singlet excitons can undergo ISC processes, reverse intersystem crossing (RISC) processes may also occur, allowing for the reuse of this portion of singlet excitons. There is no significant heavy atom effect in pure organic fluorescent materials, the IC process is a spin-allowed process between the same states, and the IC rate is usually much larger than the ISC rate. Thus, IC plays a dominant role in non-radiative decay. Therefore, to reduce the IC rate (kIC) is to minimize the non-radiative decay rate. The following equation can describe the IC rate,5
where
α is a rigidity factor influenced by the molecular structure's rigidity, and
α will increase with increasing molecular rigidity. Therefore, one way to reduce the IC rate is to increase the rigidity of the molecules. For example, Xu
et al. reported that the pTPA-DPPZ DR fluorescent material with a large rigid plane achieved a photoluminescence quantum yield (PLQY) of up to 80.0%.
6 At the same time, the non-doped device prepared with pTPA-DPPZ achieved a maximum external quantum efficiency (EQE) of 12.3% @ 652 nm. However, the PLQY of most non-doped films of DR fluorescent materials with large planes is usually very low, such as the 6.0% and 9.0% PLQY of the non-doped films of ANQDC-MSTA and ANQDC-PSTA reported by Yang
et al., and the 14.0% PLQY of the non-doped thin films of TPA-DCPP reported by Wang
et al.7,8 There are two main reasons for this. The first is that molecules with large rigidity are usually planar and prone to aggregation-caused quenching (ACQ), leading to lower PLQY. The second is that most of these molecules are thermally activated delayed fluorescence (TADF) materials, whose small overlap between their highest occupied molecular orbitals (HOMOs) and lowest unoccupied molecular orbitals (LUMOs) reduces the oscillator strength and radiative decay rates (
kr) leading to a reduction in the efficiency of the emission.
9–11 The solution to the PLQY reduction due to ACQ is usually to dope these fluorescent materials into suitable host materials at a low concentration, but this usually results in a blue shift emission, making it difficult to achieve DR or NIR emission, and thus achieving efficient DR and NIR emission remains a challenge.
Based on these considerations, this work is premised on the hybridized local charge transfer (HLCT) states with prominent overlapping features between HOMO and LUMO,12–18 and the rigidity of the molecules is appropriately enhanced in anticipation of achieving high PLQY and high device efficiency. To gain a deeper understanding of the effect of enhanced molecular rigidity on the inhibition of the IC process, in this paper, we have designed and synthesized two novel DR and NIR fluorescent molecules with different rigidity, TPANZ3PI and TPANZPyPI. The emitters use triphenylamine donors and naphtho[2,3-c][1,2,5]thiadiazole as acceptors. In addition, TPANZ3PI uses 1,2,4,5-tetraphenyl-1H-imidazole as an auxiliary donor, while TPANZPyPI uses 9,10-diphenyl-9H-pyreno[4,5-d]imidazole as an auxiliary donor (Fig. 1). Among them, the structure of 1,2,4,5-tetraphenyl-1H-imidazole has multiple benzene rings that can rotate freely. In comparison, 9,10-diphenyl-9H-pyreno[4,5-d]imidazole reduces the number of benzene rings that can rotate freely, making its structure more rigid. Through theoretical calculations, it was found that these emitters have similar electron cloud distributions in the S1 states (Fig. 1), and their energy level arrangements are identical (Fig. S5†), indicating that they may have similar photophysical properties. The emission peak of the more planar TPANZPyPI non-doped film was red-shifted by 33 nm compared to TPANZ3PI (666 nm) by testing, located in the NIR region at 699 nm. TPANZPyPI's and TPANZ3PI's PLQYs are 46.5% and 45.5% respectively. To understand this discrepancy more deeply, the IC rate between S1 and S0 was calculated and found to be much smaller for TPANZPyPI (1.62 × 1011 s−1) than for TPANZ3PI (1.73 × 1011 s−1) using the fcclasses and Gaussian software, and the S1 state of TPANZPyPI had more LE state components.19–22 These results indicate that increasing molecular rigidity is beneficial for inhibiting the IC from S1 to S0 while increasing the LE state composition of the molecule and improving PLQY. In addition, the EQEs of the non-doped devices prepared with TPANZPyPI and TPANZ3PI as the emitting layer are 4.6% @ 684 nm and 4.0% @ 676 nm, making them some of the most efficient devices among DR/NIR non-doped emitters with HLCT characteristics. The exciton utilization efficiency of the TPANZPyPI device is 49.5%, higher than that of TPANZ3PI's 43.9%. Based on theoretical calculations, it was found that the IC rate from T2 to T1 (ICT2–T1) of TPANZPyPI with a smaller T2–T1 energy level difference is lower than that of TPANZ3PI's ICT2–T1, with rates of 7.54 × 1010 s−1 and 3.02 × 1011 s−1, respectively. At the same time, their reverse intersystem crossing (RISC) rates (RISCT2–S1) are 1.21 × 106 s−1 for TPANZ3PI and 4.25 × 105 s−1 for TPANZPyPI. The almost merging of T2 and T3 energy levels in TPANZPyPI may result in additional RISC channels, with a corresponding RISC rate of 5.53 × 107 s−1. Therefore, enhancing molecular rigidity improves PLQY and suppresses the IC of triplet excitons, achieving higher exciton utilization efficiency. In addition, PPI33PPPBO was used as the host to achieve the EQE of 6.9% @ 666 nm in the TPANZPyPI doped device, which is one of the highest values of HLCT DR emission.
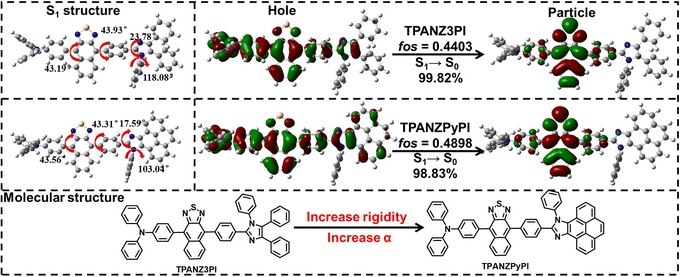 |
| | Fig. 1 Molecular structures, optimized singlet excited state (S1) structure, and electron cloud distributions of TPANZ3PI and TPANZPyPI. fos is the oscillator strength. | |
Results and discussion
Synthesis and characterization
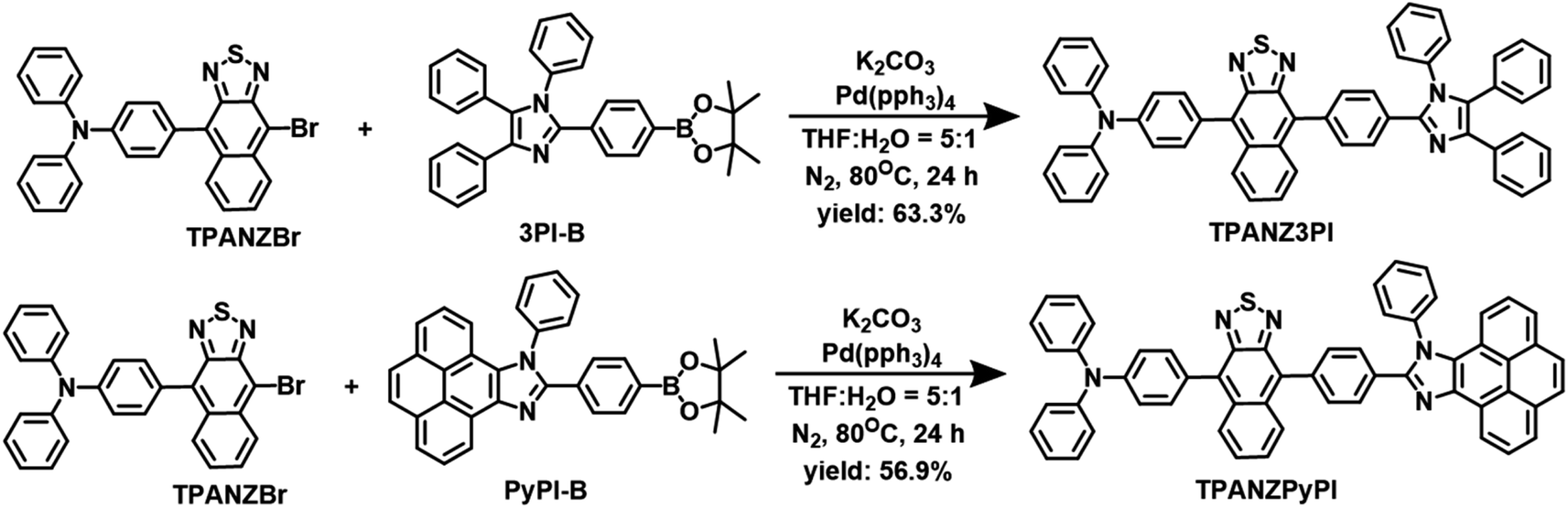
The target molecules TPANZPyPI and TPANZ3PI were efficiently synthesized (see Scheme 1 and S1 in the ESI† for detailed synthesis). Due to the molecule's rigidity and poor solubility, the 13C NMR spectrum of TPANZPyPI could not be obtained. Still, suitable 1H NMR spectra and high-resolution mass spectrometry (HRMS) were available, proving the product's characterization and purity (Fig. S1–S3†). TPANZPyPI and TPANZ3PI are obtained at a sublimation temperature of over 250 °C under a 1.0 × 10−5 Pa pressure, exhibiting high thermal stability. Their decomposition temperatures (Td, equivalent to 5% weight loss, Fig. S4†) are 496 °C for TPANZPyPI and 328 °C for TPANZ3PI, which is beneficial for the preparation of OLED devices. Besides, the glass transition temperatures (Tg) of TPANZPyPI and TPANZ3PI are 184 °C and 158 °C, respectively. The electrochemical behavior of emitters was also tested using cyclic voltammetry (Fig. S4†). The reduction potentials of TPANZPyPI and TPANZ3PI are −1.20 V, and the oxidation potentials are 0.46 V and 0.60 V, respectively. The corresponding LUMO energy levels are estimated to be approximately −3.52 eV, while the HOMO energy levels are −5.05 eV and −5.19 eV, respectively.
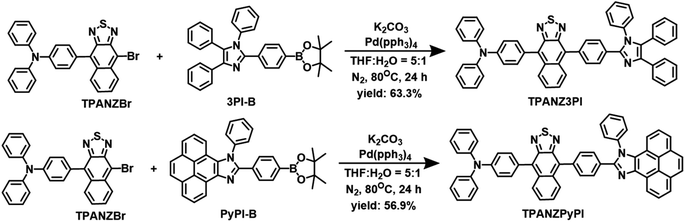 |
| | Scheme 1 The synthesis routes of TPANZPyPI and TPANZ3PI. | |
Theoretical calculations
Fig. 1 shows the molecular geometry and electron cloud distribution of the optimized S1 states of the emitters. The torsion angle of TPANZPyPI is generally smaller than that of TPANZ3PI at the same position, indicating that TPANZPyPI has a planarized structure, which is beneficial for increasing the oscillator strength of molecules. Meanwhile, these emitters' hole and particle distributions are similar, with the holes mainly concentrated on the triphenylamine and naphthothiadiazole portions and only a small portion extending to the imidazole portion. On the other hand, the particle is focused primarily on the naphthothiadiazole acceptor, with only a small portion extending into the adjacent benzene ring. This hole and particle exhibit overlapping and separated HLCT features in the S1 state. In addition, we also calculated the proportion of LE and CT states in the S1 state of these two molecules. The CT/LE components of TPANZPyPI and TPANZ3PI are 65.435%/35.565% and 66.806%/33.194%, respectively. Among them, the S1 states of TPANZPyPI have more LE components that help to achieve higher luminescence efficiency. Their emission energies are similar at 2.08 eV and 2.06 eV, respectively (Table S1†). These similar results imply that their luminescence properties may be similar. Furthermore, the corresponding excited state energy level arrangements (Tables S1 and S2, and Fig. S5†) were calculated with the ground state structures of these emitters. It can be found that the energy level difference between T1 and S1 (ΔET1–S1) of these emitters exceeds 1.0 eV, which implies that the RISC process from T1 to S1 cannot occur and that the ΔET2–T1 for TPANZPyPI and TPANZ3PI is 1.129 eV and 1.206 eV, respectively. Most importantly, these emitters' 2T1 < S1, suggesting the triplet–triplet annihilation mechanism is ruled out.
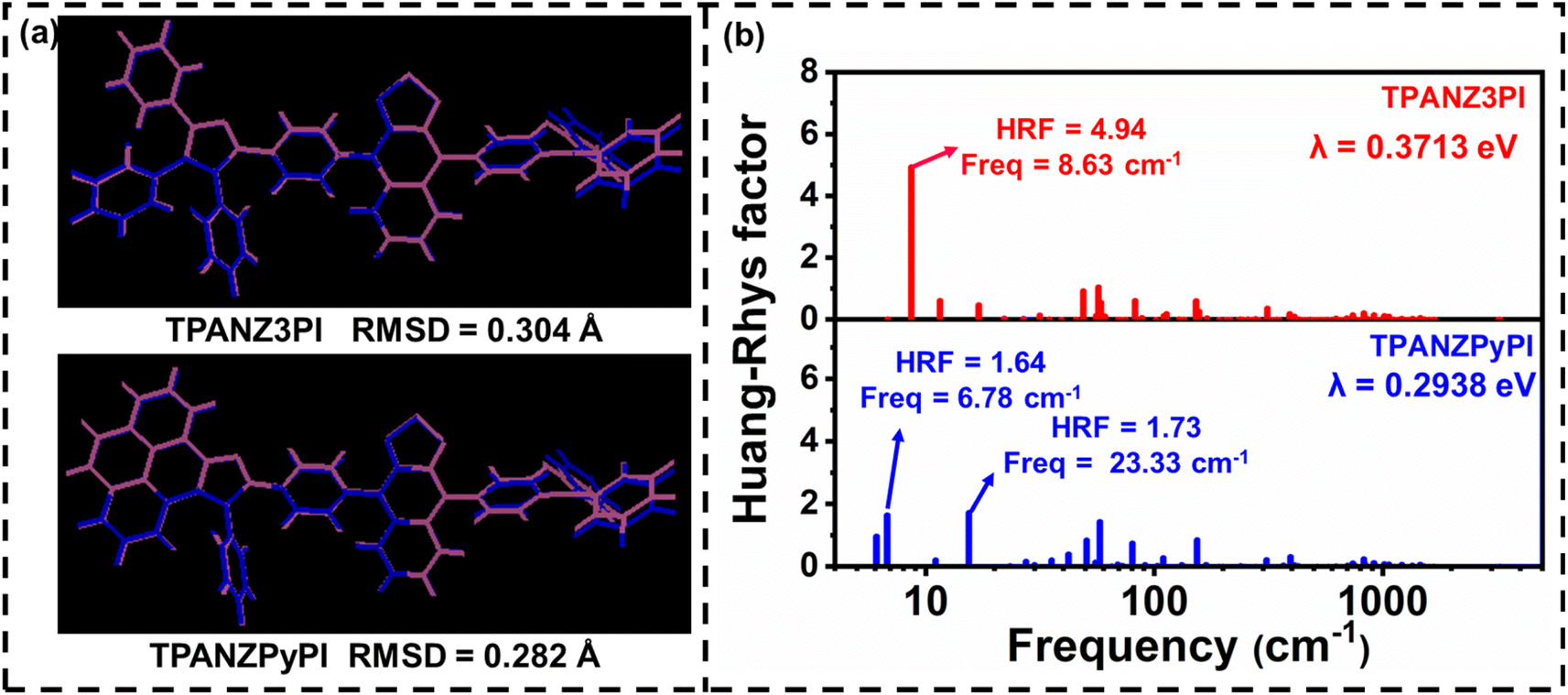
In order to understand the effect of enhanced molecular rigidity on the structural changes between the ground and excited states, the root mean square deviation (RMSD) values and corresponding reorganization energies (λ) between the ground (S0) and excited states (S1) were calculated, as shown in Fig. 2.6 In Fig. 2, it can be observed that TPANZPyPI has a smaller RMSD value and reorganization energy, indicating that increasing molecular rigidity can suppress structural changes, which may reduce energy loss during the excitation process caused by excited state changes. To explain this difference, the Huang–Rhys factor (HRF) was calculated using fcclasses to quantify the coupling strength between structural changes and vibration modes. For TPANZ3PI and TPANZPyPI, the HRF of S1 → S0 is mainly composed of low-frequency rotational and torsional vibration modes (below 50 cm−1), with large HRF values as high as 4.94 and 1.73, respectively. TPANZPyPI has smaller HRF values due to the near absence of low-frequency rotational and torsional vibrational modes in 9,10-diphenyl-9H-pyreno[4,5-d]imidazole compared to 1,2,4,5-tetraphenyl-1H-imidazole in TPANZ3PI (Fig. S6†). These results indicate that TPANZPyPI has a more rigid structure, and according to our previous discussion, a more rigid structure would be favorable for inhibiting the IC process. To this end, we first qualitatively calculated the internal conversion rates (kIC(S1–S0)) between these emitters S1 and S0 at room temperature. We found that the kIC(S1–S0) of TPANZPyPI is 1.62 × 1011 s−1, while the kIC(S1–S0) of TPANZ3PI is 1.73 × 1011 s−1, which is consistent with our expectations. We also believe that enhancing molecular rigidity can help suppress the IC process of triplet-excited states. For this purpose, the IC rate between T1 and T2 of these emitters was also qualitatively calculated at room temperature. Although the energy level difference between T1 and T2 is more minor for TPANZPyPI (Fig. S5†), the kIC(T2–T1) rate is smaller than for TPANZ3PI, where kIC(T2–T1,TPANZPyPI) = 7.54 × 1010 s−1, while kIC(T2–T1,TPANZ3PI) = 3.02 × 1011 s−1. The lower IC rate is more favorable for keeping electrically generated triplet excitons at the T2 energy level. Although the energy level differences in T1 and T2 are so large, there are IC rates up to 1010 to 1011 s−1 orders of magnitude. And the usual RISC rates for high-energy triplet to singlet states of pure organic molecules are 107 to 108 s−1 orders of magnitude.23 Therefore, pure organic molecules usually consider only the RISC process from T2 to S1. Thus, the RISC rate from T2 to S1 was calculated, and the RISC rate for TPANZ3PI was 1.21 × 106 s−1, and the RISC rate for TPANZPyPI was 4.25 × 105 s−1. However, considering that the small T3–T2 gap of TPANZPyPI supports IC and the dynamic equilibrium of reverse IC through vibration coupling, it is reasonable for triplet excitons to be converted into singlet excitons through thermal exciton channels (T2/T3 → S1). Therefore, the RISC rate corresponding to TPANZPyPI is 5.53 × 107 s−1 between T3 and S1. Combining a smaller IC rate of triplets with a larger RISC rate compared to TPANZ3PI, TPANZPyPI is expected to have higher exciton utilization efficiency.
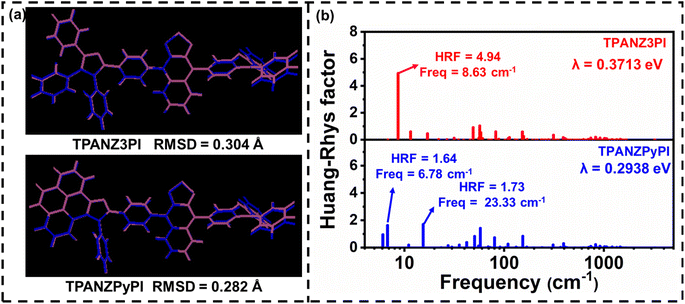 |
| | Fig. 2 (a) The RMSD value between S1 and S0, where the S1 structure is mauve and S0 is blue, and (b) the Huang–Rhys factors versus frequencies. | |
Photophysical properties
Fig. 3a shows the ultraviolet-visible (UV-vis) absorption and photoluminescence (PL) spectra of TPANZPyPI and TPANZ3PI in dilute solutions of THF (10−5 M) and pure films. Detailed data are shown in Table 1. The emitters show similar absorption in the pure films and THF, but the absorption peak in the films is red-shifted compared to that in THF, which may be attributed to the aggregation of molecules in the film state. The absorption bands at about 300–400 nm are mainly due to molecular π–π* transition, whereas the absorption bands after 400 nm originate from the emitter backbone's intramolecular charge transfer (ICT). The emission in THF/film is 698/671 nm and 699/666 nm for TPANZPyPI and TPANZ3PI, respectively. The solvatochromic test is shown in Fig. S7,† and detailed data are recorded in Table S3.†TPANZPyPI and TPANZ3PI showed a redshift of 91 nm and 90 nm from low-polarity hexane to high-polarity acetone, respectively, which indicates they have similar excited state properties. The redshifts in the different solutions indicate that the CT component enhances with increasing polarity, consistent with a gradual decrease in PLQY (Table S3†). However, it is worth noting that TPANZ3PI and TPANZPyPI have PLQYs of 46.5% @ 699 nm and 45.5% @ 666 nm in the pure film, respectively. Especially TPANZPyPI has a very high PLQY, which is rare in the NIR field. This result is consistent with theoretical calculations. In addition, the Lippert–Mataga solvation model was applied to the emitters. In Fig. 3c, the relationship between the Stokes shift of two emitters and solvent polarity shows two fitting lines. The dipole moments at low polarity (f < 0.15) are 11.85 D and 11.28 D, respectively, which are larger than typical pure LE excited states (∼8 D). The dipole moment in the higher polarity environment (f > 0.15) is 18.36 D and 19.24 D, respectively. The transient PL spectra exhibit a single exponential lifetime curve in solvents of different polarities (Fig. S8†) or pure film states (Fig. 3d), indicating only one hybrid state in S1. Considering the electron cloud distribution of S1, these emitters should be considered HLCT emitters.
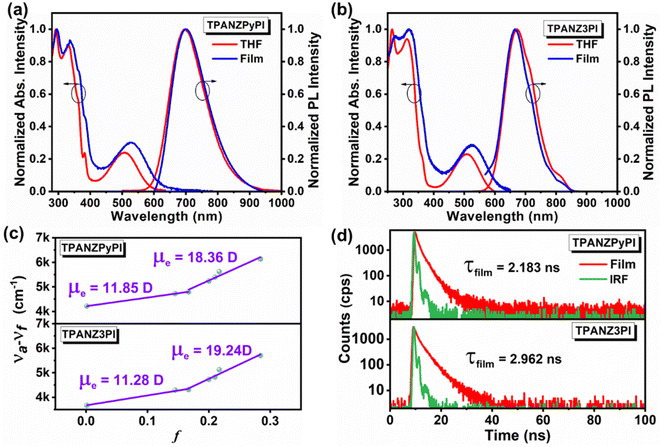 |
| | Fig. 3 The UV-vis absorption spectra and the PL spectra of the films and solutions (10−5 M) of (a) TPANZPyPI and (b) TPANZ3PI; (c) linear fitting of the Lippert–Mataga model, and (d) the transient PL decay spectra of TPANZPyPI and TPANZ3PI in films. | |
Table 1 The photophysical data of TPANZ3PI and TPANZPyPI films
| Compound |
λ
abs
[nm] |
λ
em
[nm] |
Φ
F
[%] |
τ
[ns] |
k
r
[107 s−1] |
k
nr
[107 s−1] |
Φ
F
[%] |
|
Absorption peaks of the neat film.
Maximum emission wavelength of the neat film.
Absolute PLQY of the neat film.
Fluorescence lifetime of the neat film.
k
r and knr for the neat film (kr = ΦF/τ, knr = (1 − ΦF)/τ).
10 wt% emitters in PPI33PPPBO host absolute PLQY.
|
|
TPANZPyPI
|
295/336/527 |
699 |
46.5 |
2.18 |
21.30 |
24.51 |
66.1 |
|
TPANZ3PI
|
273/316/525 |
666 |
45.5 |
2.96 |
15.36 |
18.40 |
74.5 |
Electroluminescent devices
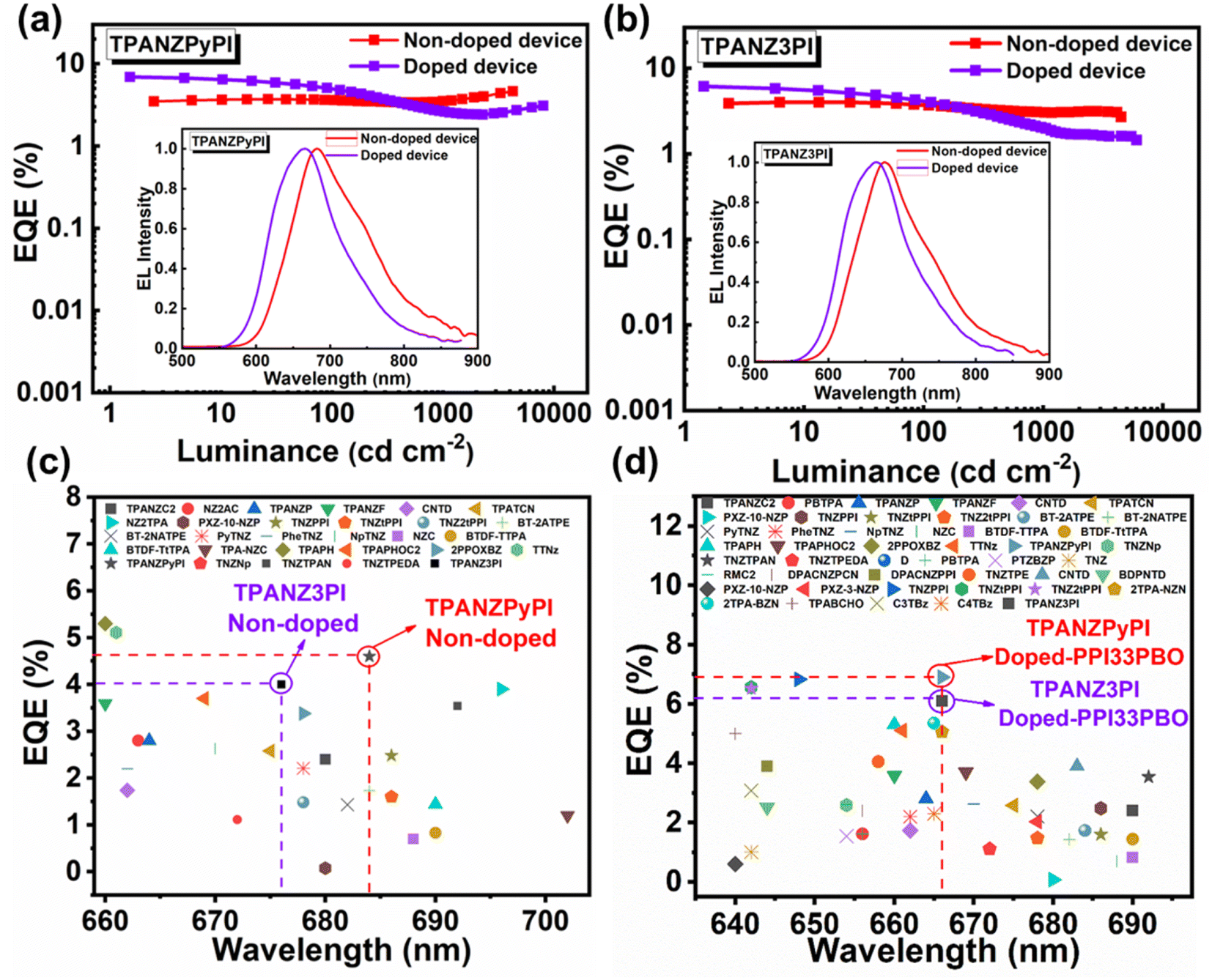
Given the excellent photophysical properties of emitters, their electroluminescence properties were further investigated. Non-doped devices were first prepared with the structure of ITO/HATCN (20 nm)/TAPC (35 nm)/TCTA (10 nm)/EML (20 nm)/TPBi (60 nm)/LiF (1 nm)/Al (100 nm), in which HATCN (1,4,5,8,9,11-hexaazatriphenylenehexacarbonitrile) is the hole-injection layer, TAPC (1,1-bis[(di-4-tolylamino)phenyl]cyclohexane) is the hole-transport layer, TCTA (tris(4-carbazoyl-9-ylphenyl)amine) is the hole-transport layer and electron-blocking layer, and TPBi (2,2′,2′′-(1,3,5-benzinetriyl)-tris(1-phenyl-1-H-benzimidazole)) is the electron-transport layer and hole-blocking layer (Fig. S9†). The corresponding data is recorded in Table 2. The non-doped TPANZPyPI OLED shows NIR emission at 684 nm with a color coordinate of (0.69, 0.30) and provides a maximum EQE of 4.6%, which is comparable to the DR to NIR emission of state-of-the-art non-doped OLEDs with HLCT characteristics. However, the device with TPANZ3PI as the emission layer has an emission peak of 676nm and a maximum EQE of 4.0%. Due to the similar molecular structures of TPANZ3PI and TPANZPyPI, to qualitatively compare the exciton utilization efficiency of their non-doped devices, the optical output coupling efficiency is assumed to be 0.2, and thus the exciton utilization efficiency of TPANZPyPI and TPANZ3PI is 49.5% and 43.9%, respectively. This result matches the theoretical calculations and proves that the appropriate enhancement of molecular rigidity is beneficial to reduce the internal conversion process of the excited state and realize higher exciton utilization efficiency. However, it is worth noting that the EQE curves of the devices of TPANZPyPI show an upward trend at high voltages. This may be attributed to the fact that at high voltages, the internal conversion from T2 to T1 is limited, and the density of the T2 and T3 excitons is elevated, further facilitating the RISC process and improving the efficiency of the devices. There's another possibility that the suppression of T1 exciton density growth at high current density, as stated in the literature, reduces the likelihood of singlet–triplet annihilate (STA) occurrence, thereby enabling the device to achieve higher EQE at high current densities.24 Because the energy level arrangement of these emitters (2T1 < S1, Fig. S5†) and the current density-luminance curve of the device have a linear relationship (Fig. S11†), which excludes TTA behavior, the increase of EQE at high current density is also independent of TTA. The curves of the spectra at different voltages and the corresponding color coordinates did not change significantly (Fig. S12 and Table S4†), ruling out that the device efficiency improvement is due to the change of the composite region.
Table 2 EL performances of the OLEDs based on TPANZPyPI and TPANZ3PI
| EML |
V
turn-on
[V] |
LEmaxb [cd A−1] |
PEmaxc [lm W−1] |
EQEmaxd [%] |
L
max
[cd m−2] |
λ
EL,max
[nm] |
CIE [x,y]g |
|
V
turn-on: turn-on voltage at 1 cd m−2.
LEmax: maximum current efficiency.
PEmax: maximum power efficiency.
EQEmax: maximum external quantum efficiency.
L
max: maximum luminance.
λ
EL,max: electroluminescence peak.
CIE: Commission Internationale de I'Eclairage coordinates.
|
|
TPANZPyPI
|
3.0 |
0.93 |
0.60 |
4.6 |
4756 |
684 |
(0.693, 0.302) |
|
TPANZ3PI
|
3.0 |
0.96 |
0.93 |
4.0 |
4495 |
676 |
(0.692, 0.305) |
| PPI33PPPBO: TPANZPyPI |
3.0 |
3.30 |
3.46 |
6.9 |
8072 |
666 |
(0.675, 0.323) |
| PPI33PPPBO: TPANZ3PI |
2.9 |
3.03 |
3.28 |
6.1 |
6467 |
666 |
(0.678, 0.323) |
Further investigations were conducted on the electroluminescence performance of emitter doped organic light-emitting devices (ITO/HATCN (20 nm)/TAPC (40 nm)/TCTA (20 nm)/host:TPANZPyPI (10 wt%, 20 nm)/TPBI (50 nm)/LiF (1 nm)/Al (100 nm), where the host is our previously reported PPI33PPPBO).25 The device structure is shown in Fig. S9,† and the emission spectra of the PPI33PPPBO film and absorption spectra of the TPANZPyPI and TPANZ3PI films are shown in Fig. S10.† Compared with non-doped organic electroluminescent devices, these doped devices emit a blue shift but significantly improve the electroluminescence efficiency, consistent with the doped films' high PLQY (Table 1). Doped in PPI33PPPBO, the TPANZPyPI and TPANZ3PI devices all show DR emission with peaks at 666 nm and excellent EL performance, with the maximum luminance of 8072 cd m−2 and 6467 cd m−2, and the maximum EQE of 6.90% and 6.1%, respectively, which is one of the highest values reported so far for DR HLCT OLEDs (Fig. 4).
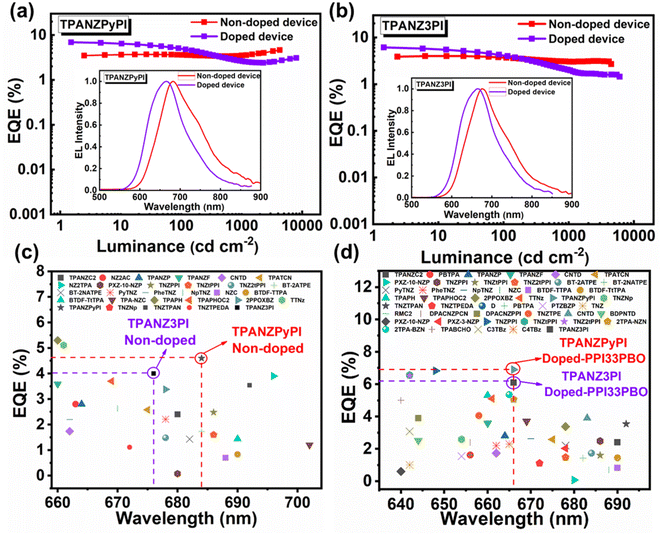 |
| | Fig. 4 (a) and (b) Luminance–EQE plots with EL spectra inserted, (c) reported maximum EQE of red non-doped HLCT OLEDs with electroluminescence peaks ranging from 660 to 710 nm, (d) reported maximum EQE of red doped HLCT OLEDs with electroluminescence peak ranging from 640 to 700 nm. | |
Conclusions
In summary, we synthesized and characterized two DR and NIR molecules, TPANZ3PI and TPANZPyPI, with different rigid structures. Theoretical calculations reveal that increasing the molecular rigidity can inhibit the IC process from S1 to S0, which improves the photoluminescence efficiency and shows NIR emission in neat films. Moreover, increasing the molecular rigidity also slows the internal conversion process between excited states, thus enabling this HLCT material to achieve higher exciton utilization efficiency and superior electroluminescence performance. TPANZPyPI non-doped devices with greater rigidity have achieved higher EQE and exciton utilization efficiency than TPANZ3PI with values of 4.6%/49.5% @ 684 nm and 4.0%/43.9% @ 676 nm, respectively. The doping devices of these two emitters achieved 666 nm DR emission, with an EQE of 6.9% for TPANZPyPI and 6.1% for TPANZ3PI, respectively. To our knowledge, these results are among the highest EL efficiencies reported for HLCT NIR/DR emission. These results suggest the moderate increase in molecular rigidity is a practical approach to designing high-performance DR/NIR fluorescent materials.
Data availability
All data supporting this study are available from article and ESI.†
Author contributions
Mingliang Xie conducted the synthesis and the photophysical characterizations of the materials. Yannan Zhou and Chengling Ma fabricated the OLED for the materials and estimated the OLED performances. Huayi Zhou is responsible for the basic testing and analysis of thermal and electrochemical properties. Shi-Tong Zhang and Qikun Sun did the theoretical calculations. Mingliang Xie wrote the paper, and all the authors revised it. Shanfeng Xue, Yujian Zhang and Wenjun Yang supervised the whole work.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work is supported by the National Natural Science Foundation of China (No. 51873095 and 52273183), Taishan Scholar Constructive Engineering Foundation of Shandong Province of China (No. tsqn202211164), the Natural Science Foundation of Shandong Province (No. ZR2020QE083), and the Natural Science Foundation of Qingdao City of China (No. 23-2-1-239-zyyd-jch). We also thank the Open Project of the State Key Laboratory of Supramolecular Structure and Materials of Jilin University (sklssm2023029) and the Qingdao University of Science and Technology Postgraduate Independent Research and Innovation Program (S2022KY007).
Notes and references
- Y. Xu, P. Xu, D. Hu and Y. Ma, Chem. Soc. Rev., 2021, 50, 1030–1069 RSC
 .
.
- Y. Liu, C. Li, Z. Ren, S. Yan and M. R. Bryce, Nat. Rev. Mater., 2018, 3, 18020 CrossRef CAS .
- S. Sharma and A. K. Pal, J. Mater. Chem. C, 2022, 10, 15681–15707 RSC .
- F. Liu, Y. Tan, H. Liu, X. Tang, L. Gao, C. Du, J. Min, H. Jina and P. Lu, J. Mater. Chem. C, 2020, 8, 6883–6890 RSC .
- J.-X. Chen, K. Wang, C.-J. Zheng, M. Zhang, Y.-Z. Shi, S.-L. Tao, H. Lin, W. Liu, W.-W. Tao, X.-M. Ou and X.-H. Zhang, Adv. Sci., 2018, 1800436 CrossRef PubMed .
- B. Zhao, H. Wang, C. Han, P. Ma, Z. Li, P. Chang and H. Xu, Angew. Chem., Int. Ed., 2020, 132, 19204–19209 CrossRef .
- X. Gong, C.-H. Lu, W.-K. Lee, P. Li, Y.-H. Huang, Z. Chen, L. Zhan, C.-C. Wu, S. Gong and C. Yang, Chem. Eng. J., 2021, 405, 126663 CrossRef CAS .
- S. Wang, X. Yan, Z. Cheng, H. Zhang, Y. Liu and Y. Wang, Angew. Chem., Int. Ed., 2015, 54, 13068–13072 CrossRef CAS PubMed .
- J.-X. Chen, Y.-F. Xiao, K. Wang, D. Sun, X.-C. Fan, X. Zhang, M. Zhang, Y.-Z. Shi, J. Yu, F.-X. Geng, C.-S. Lee and X.-H. Zhang, Angew. Chem., Int. Ed., 2021, 60, 2478–2484 CrossRef CAS PubMed .
- T. Yang, Z. Cheng, Z. Li, J. Liang, Y. Xu, C. Li and Y. Wang, Adv. Funct. Mater., 2020, 2002681 CrossRef CAS .
- Z. Cai, X. Wu, H. Liu, J. Guo, D. Yang, D. Ma, Z. Zhao and B. Z. Tang, Angew. Chem., Int. Ed., 2021, 60, 23635–23640 CrossRef CAS PubMed .
- H. Zhang, G. Li, X. Guo, K. Zhang, B. Zhang, X. Guo, Y. Li, J. Fan, Z. Wang, D. Ma and B. Z. Tang, Angew. Chem., Int. Ed., 2021, 60, 1–8 CrossRef .
- S. Xiao, Y. Gao, R. Wang, H. Liu, W. Li, C. Zhou, S. Xue, S.-T. Zhang, B. Yang and Y. Ma, Chem. Eng. J., 2022, 440, 135911 CrossRef CAS .
- M. Xie, T. Li, C. Liu, C. Ma, S. Zhang, G. Liu, Q. Sun, S.-T. Zhang, W. Yang and S. Xue, Chem. Eng. J., 2023, 472, 144950 CrossRef CAS .
- T. Liu, X. Chen, J. Zhao, W. Wei, Z. Mao, W. Wu, S. Jiao, Y. Liu, Z. Yang and Z. Chi, Chem. Sci., 2021, 12, 5171–5176 RSC .
- J. Zhu, G. Liu, X. Lian, J.-H. Pang, M.-D. Li, Y. Wang and Q.-X. Tong, J. Mater. Chem. C, 2022, 10, 8684–8693 RSC .
- Z. Zhong, X. Zhu, X. Wang, Y. Zheng, S. Geng, Z. Zhou, X. J. Feng, Z. Zhao and H. Lu, Adv. Funct. Mater., 2022, 32, 2112969 CrossRef CAS .
- Q. Wan, J. Tong, B. Zhang, Y. Li, Z. Wang and B. Z. Tang, Adv. Opt. Mater., 2019, 1901520 Search PubMed .
- J. Cerezo and F. Santoro, J. Comput. Chem., 2023, 44, 626–643 CrossRef CAS PubMed .
- F. Neese, Wiley Interdiscip. Rev. Comput. Mol. Sci., 2018, 8, e1327 CrossRef .
- F. Neese, F. Wennmohs, U. Becker and C. Riplinger, J. Chem. Phys., 2020, 152, 224108 CrossRef CAS PubMed .
- A. Humeniuk, M. Bužančić, J. Hoche, J. Cerezo, R. Mitrić, F. Santoro and V. Bonačić-Kouteck, J. Chem. Phys., 2020, 152, 054107 CrossRef CAS PubMed .
- J. Li, M. Zhang, T. Li, D. Guo, T. Tian and H. Zhang, J. Mater. Chem. C, 2022, 10, 13124–13136 RSC .
- Y. Zhang, M. Whited, M. E. Thompson and S. R. Forrest, Singlet–triplet quenching in high intensity fluorescent organic light emitting diodes, Chem. Phys. Lett., 2010, 495, 161–165 CrossRef CAS .
- C. Liu, T. Li, M. Sun, M. Xie, Y. Zhou, W. Feng, Q. Sun, S.-T. Zhang, S. Xue and W. Yang, Adv. Funct. Mater., 2023, 33, 2215066 CrossRef CAS .
Footnote |
| † Electronic supplementary information (ESI) available: Compound syntheses and characterization, other theoretical calculations, spectra, morphology of films and other device performances. See DOI: https://doi.org/10.1039/d4sc00841c |
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *b,
Wenjun
Yang
*b,
Wenjun
Yang