
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructure and photochemistry of di-tert-butyldiphosphatetrahedrane†‡
Gabriele
Hierlmeier§
 *a,
Roger Jan
Kutta
b,
Peter
Coburger¶
a,
Hans-Georg
Stammler
c,
Jan
Schwabedissen
c,
Norbert W.
Mitzel
*c,
Maria
Dimitrova
de,
Raphael J. F.
Berger
e,
Patrick
Nuernberger
*b and
Robert
Wolf
*a
*a,
Roger Jan
Kutta
b,
Peter
Coburger¶
a,
Hans-Georg
Stammler
c,
Jan
Schwabedissen
c,
Norbert W.
Mitzel
*c,
Maria
Dimitrova
de,
Raphael J. F.
Berger
e,
Patrick
Nuernberger
*b and
Robert
Wolf
*a
aUniversität Regensburg, Institut für Anorganische Chemie, 93040 Regensburg, Germany. E-mail: robert.wolf@ur.de
bUniversität Regensburg, Institut für Physikalische und Theoretische Chemie, 93040 Regensburg, Germany. E-mail: Patrick.Nuernberger@chemie.uni-regensburg.de
cUniversität Bielefeld, Lehrstuhl für Anorganische Chemie und Strukturchemie, Universitätsstraße 25, 33615 Bielefeld, Germany. E-mail: nmitzel@uni-bielefeld.de
dDepartment of Chemistry, University of Helsinki, Faculty of Science, FI-00014 Helsinki, Finland
eParis Lodron Universität Salzburg, Chemie und Physik der Materialien, 5020 Salzburg, Austria
First published on 8th March 2024
Abstract
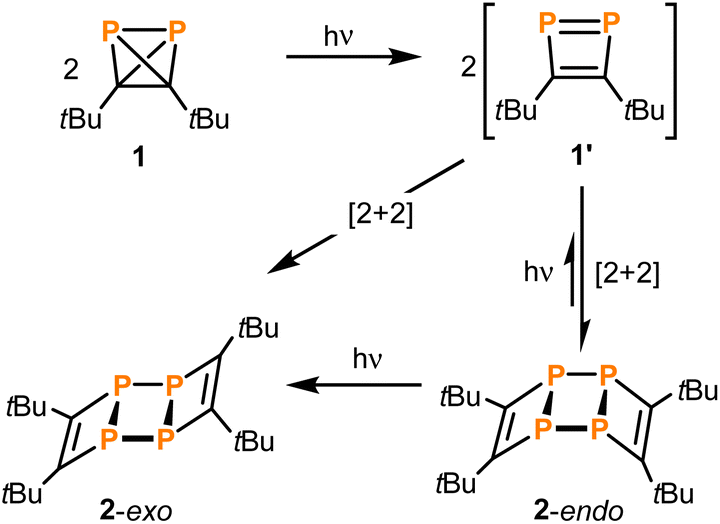
Di-tert-butyldiphosphatetrahedrane (tBuCP)2 (1) is a mixed carbon- and phosphorus-based tetrahedral molecule, isolobal to white phosphorus (P4). However, despite the fundamental significance and well-explored reactivity of the latter molecule, the precise structure of the free (tBuCP)2 molecule (1) and a detailed analysis of its electronic properties have remained elusive. Here, single-crystal X-ray structure determination of 1 at low temperature confirms the tetrahedral structure. Furthermore, quantum chemical calculations confirm that 1 is isolobal to P4 and shows a strong largely isotropic diamagnetic response in the magnetic field and thus pronounced spherical aromaticity. A spectroscopic and computational study on the photochemical reactivity reveals that diphosphatetrahedrane 1 readily dimerises to the ladderane-type phosphaalkyne tetramer (tBuCP)4 (2) under irradiation with UV light. With sufficient thermal activation energy, the dimerisation proceeds also in the dark. In both cases, an isomerisation to a 1,2-diphosphacyclobutadiene 1′ is the first step. This intermediate subsequently undergoes a [2 + 2] cycloaddition with a second 1,2-diphosphacyclobutadiene molecule to form 2. The 1,2-diphosphacyclobutadiene intermediate 1′ can be trapped chemically by N-methylmaleimide as an alternative [2 + 2] cycloaddition partner.
Introduction
The tetrahedron is a fundamental structure motif in chemistry. Tetrahedral molecules can incorporate various elements of the periodic table, including s-, p- and d-block elements. Among the most prominent examples are the first “organic” tetrahedrane, (tBuC)4,1 and the prototype of “inorganic” tetrahedrane, P4.2,3 While the chemical properties and reactivity patterns of P4 and (tBuC)4 have been studied for a long time, the physical properties, in particular the thermal stability and photochemistry, of both molecules have also attracted considerable attention.4The transformation of white phosphorus into its red allotrope by irradiation is well known (Fig. 1A).5 It was proposed that this reaction takes place via photoinduced dissociation of P4 into P2 molecules, which then recombine to form polymeric red phosphorus.6 The same equilibrium P4 ⇌ P2 can also be reached thermally in the gas phase at temperatures above 1000 °C.7 A solvent-dependent radical mechanism has also been proposed for the polymerisation of P4 to red phosphorus under irradiation with ionising γ-rays.8 The photochemistry of P4 can be exploited for the synthesis of new diphosphenes via [2 + 4] cycloadditions of P2 with dienes.9
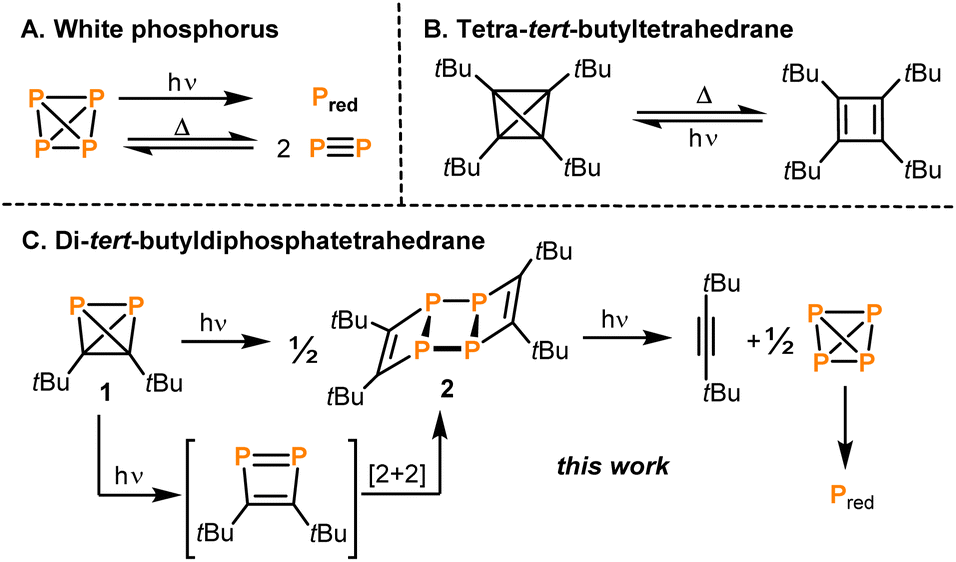 | ||
| Fig. 1 Thermal and photochemical reactions of group 14/group 15 tetrahedranes. | ||
A photochemical pathway was pursued to access the purely carbon-based tetrahedrane tetra-tert-butyltetrahedrane (tBuC)4 which can be generated by irradiation of its cyclobutadiene isomer tetra-tert-butylcyclobutadiene (λ > 300 nm; Fig. 1B).1 Above 130 °C, (tBuC)4 undergoes back-isomerisation to the tetra-tert-butylcyclobutadiene.
Recently, the synthesis and reactivity of di-tert-butyldiphosphatetrahedrane (1), the “hybrid” of P4 and (tBuC)4,3,10,11 as well as the related tri-tert-butylphosphatetrahedrane (tBu3C3P)12 and triphosphatetrahedrane (HCP3)12 have been described. Diphosphatetrahedrane 1 is readily prepared by a nickel-catalysed dimerisation reaction of tert-butylphosphaalkyne tBuCP and is isolated as a metastable liquid with a melting point of −32 °C. Above its melting point, 1 slowly dimerises to give the known ladderane-type phosphaalkyne tetramer (tBuCP)4 (2).13 As a result, attempts to grow single crystals of 1 by standard techniques were unsuccessful previously, while the structure of the silver(I) complex [(η2-1)Ag(2)]2[Al(ORF)4] [1-Ag, RF = C(CF3)3] could be determined. 1-Ag shows an intact P2C2 tetrahedron side-on coordinated to an Ag+ cation.
Here, the structure of 1 in the solid state by single-crystal X-ray diffraction is determined and, according to quantum-chemical calculations, its electronic structure reveals a pronounced spherical aromaticity comparable to that in P4 and (tBuC)4. Furthermore, the temperature and light sensitivity of 1 is presented in more detail and the remarkable photochemistry of 1 is reported. Irradiation of 1 with UV light affords 2via [2 + 2] cycloaddition of two intermediate 1,2-diphosphacyclobutadienes (Fig. 1C). The photochemistry of 1 reveals notable differences in comparison with the photochemistry of P4, (tBuCP)4 and tBuCP (the monomer of 1). Moreover, the present study complements recent results on the related tri-tert-butylphosphatetrahedrane (tBu3C3P), which was shown by Cummins and co-workers to isomerise to a planar phosphacyclobutadiene upon addition of the Lewis acid BPh3.14
Results and discussion
X-ray crystallography
Crystallographic structure determination of the title compound 1 by X-ray-diffraction (XRD) was challenging because 1 is a liquid at ambient temperature, which is simultaneously sensitive to air, light and temperature. Numerous attempts to grow a single crystal from the liquid phase were unsuccessful, because the decomposition of 1 started immediately after melting, producing crystal seeds that led to polycrystalline specimens. Moreover, the crystallisation process is difficult to monitor and control, because the observation with polarised light also leads to decomposition. In one of the crystallisation experiments, a crystalline region in the capillary was observed, which turned out to be di-tert-butylacetylene (the structure of which had already been determined earlier).15To overcome these problems, mixing 1 with n-pentane lowered the melting point, allowing crystal growth at a lower temperature, where the sample is sufficiently stable for the time needed to grow a crystalline sample. For this, a freshly prepared mixture of 1 and n-pentane was transferred to a capillary and using the cryo-stream of the diffractometer immediately cooled in the dark from 240 K with 5 K h−1 to 180 K and subsequently to 94 K with a max. cooling rate. A colourless crystal was obtained, which was twinned with ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. The second domain was rotated by −169.1° around [010] (reciprocal). Both domains were taken into account for data integration and only the non- or minor-overlapping reflections of the first domain were used for refinement. 1 crystallised in the monoclinic space group P21 with one molecule per asymmetric unit (Fig. 2). Important bond lengths are summarised in Table 1 and compared with those calculated by coupled cluster theory or found in the molecular structure of the silver(I) complex 1-Ag (Fig. 3 and Table 1).
:1. The second domain was rotated by −169.1° around [010] (reciprocal). Both domains were taken into account for data integration and only the non- or minor-overlapping reflections of the first domain were used for refinement. 1 crystallised in the monoclinic space group P21 with one molecule per asymmetric unit (Fig. 2). Important bond lengths are summarised in Table 1 and compared with those calculated by coupled cluster theory or found in the molecular structure of the silver(I) complex 1-Ag (Fig. 3 and Table 1).
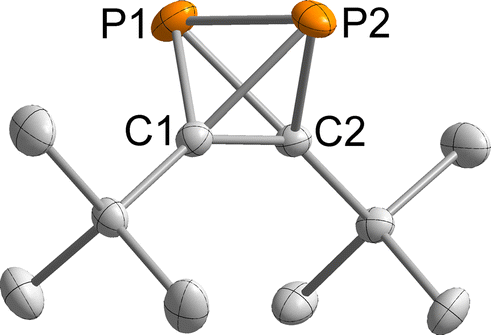 | ||
| Fig. 2 Molecular structure of 1 in the solid state. Thermal ellipsoids are set at the 50% probability level. Hydrogen atoms are omitted for clarity. Selected bond lengths [Å] and angles [°]: P1–P2 2.205(1), P1–C1 1.845(2), P1–C2 1.847(2), P2–C1 1.849(2), P2–C2 1.846(2), C1–C2 1.458(2), C1–P1–P2 53.4(1), C2–P1–P2 53.3(1), C2–P1–C1 46.5(1), C1–P2–P1 53.3(1), C1–P2–C2 46.5(1), C2–P2–P1 53.3(1), P2–C1–P1 73.3(1), C2–C1–P1 66.8(1), C2–C1–P2 66.7(1), P1–C2–P2 73.3(1), C1–C2–P1 66.7(1), C1–C2–P2 66.9(1). | ||
| Bond | Bond length [Å] XRD (1) | Bond length [Å] DLPNO CCSD(T) (1) | Bond length [Å] XRD (1-Ag) |
|---|---|---|---|
| P1–P2 | 2.205(1) | 2.203 | 2.308(3) |
| C1–C2 | 1.458(2) | 1.458 | 1.462(12) |
| P–C | 1.849(2)–1.845(2) | 1.852 | 1.820(8)–1.836(9) |
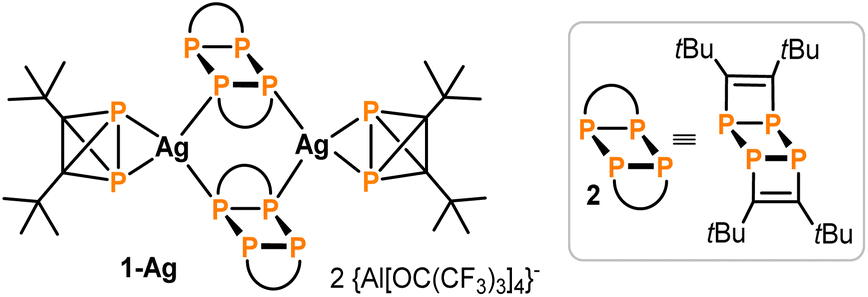 | ||
| Fig. 3 Structure of 1-Ag (see Table 1 for selected structural data). | ||
The P1–P2 bond length of 2.205(1) Å in 1 compares well to the bond length in P4 determined by gas phase electron diffraction (rg = 2.1994(3) Å).2 Moreover, the P–C bond lengths range from 1.845(2) to 1.849(2) Å and the C1–C2 bond length of 1.458(2) Å is very similar to that found in (tBuC)4 (average: 1.485 Å).16
Overall, the bond lengths compare very well to the calculations at the DLPNO-CCSD(T)/def2-QZVPP level of theory (Table 1). Moreover, comparison of the bond metrics with the previously reported Ag(I) complex 1-Ag reveals the expected extension of the P–P bond by ca. 0.1 Å to 2.308(3) Å upon coordination. The differences in C1–C2 and P–C bond lengths were less pronounced.
It should be noted that despite numerous attempts to determine the structure of 1 by gas electron diffraction, suitable conditions could not be found to record consistent diffraction patterns.
Electronic structure
Previously, an analogy in the reactivity of 1 and P4 had been noticed, which may be attributed to the isolobal analogy. In this concept, the frontier orbitals of two molecules are compared with respect to their energies, shape, occupation and symmetry.17DFT calculations on the TPSS-D3BJ/def2-TZVP level of theory have shown that the frontier orbitals of both molecules are related in energy and shape (Fig. 4). As expected, the degeneracy of t- and e-orbitals in P4 is not preserved in 1. The HOMO (−5.3 eV) of 1 has mainly P–C bond character, whereas the HOMO-1 (−5.7 eV) has large contributions of the P–P and C–C bonds (for a comparison of all occupied orbitals see the ESI‡).
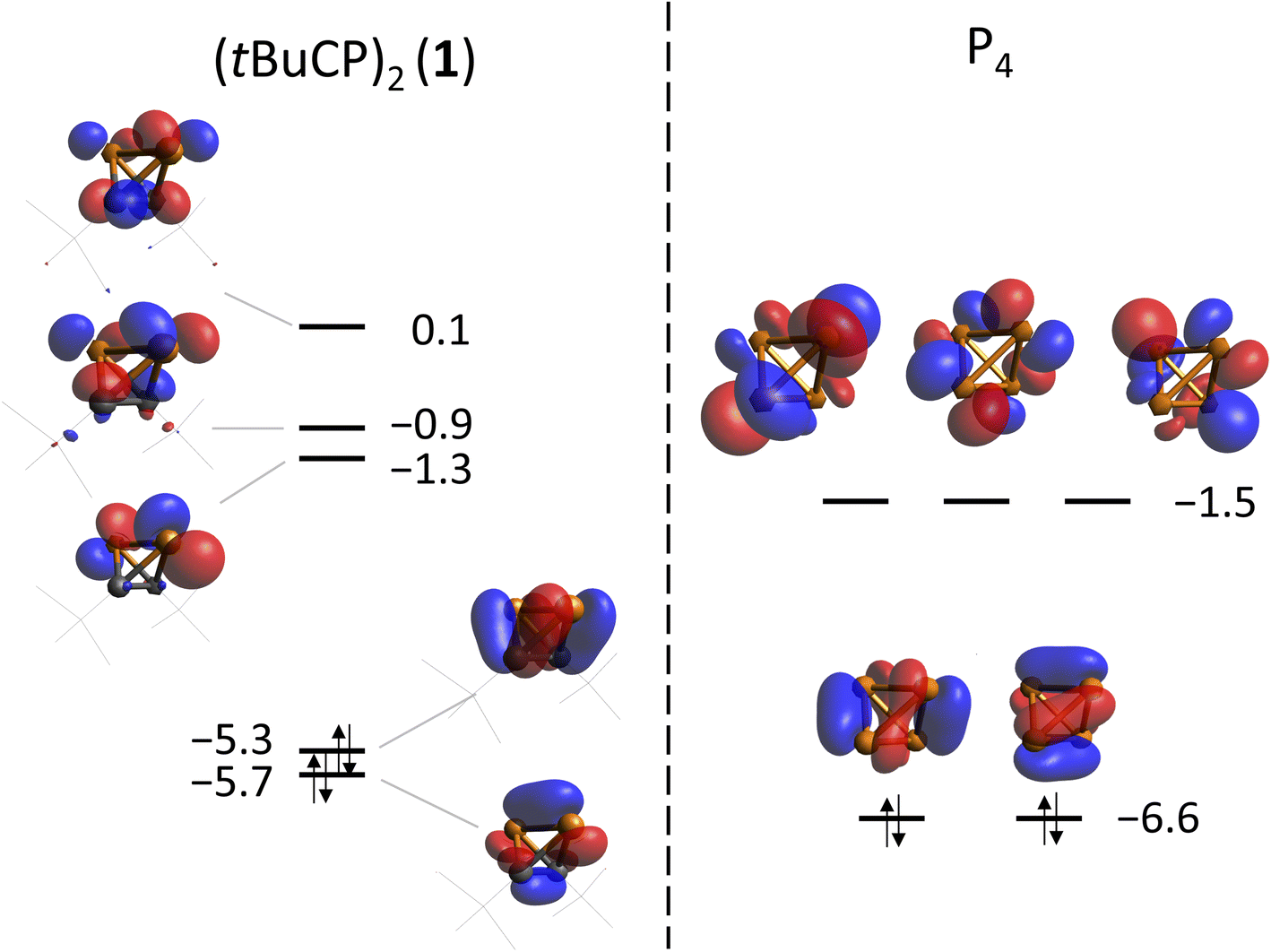 | ||
| Fig. 4 Frontier orbitals of 1 (left) and P4 (right) calculated at the TPSS-D3BJ/def2-TZVP level of theory. Energies are given in eV. | ||
Both P4 and (tBuC)4 have been demonstrated to possess spherical aromaticity.2,18 As a probe for such aromatic ring currents the nucleus independent chemical shifts (NICS) have been an effective tool in previous studies.19 For comparison, the NICS values were calculated on the TPSS/pcSseg-2 level of theory at the centre of the cages of P4, (tBuC)4 and 1, affording strongly negative values of −60.1, −48.2 and −50.3 ppm, respectively. Notably, the value for P4 compares well with a recent calculation (−61.9).20,21 The value for 1 (−50.3) is in between the values for P4 and (tBuC)4, which suggests large induced diatropic ring currents. A calculation of the magnetically induced total ring current flux in P4, 1 and (tBuC)4 yielded 24, 15 (and 16 in one orthogonal direction) and 16 nA T−1, respectively (Fig. 5, B3LYP/def2-TZVP; see the ESI‡ for details).22 The strong diamagnetic response of P4 underlines its aromatic nature, while the directional independence (isotropy) of the flux in 1 suggests the presence of spherical aromaticity (as it is already imposed by symmetry upon P4 and (tBuC)4). Moreover, the high flux values demonstrate a significant amount of aromatic cluster current in 1 and (tBuC)4 (for comparison, benzene shows a total ring current of approx. 12 nA T−1).23
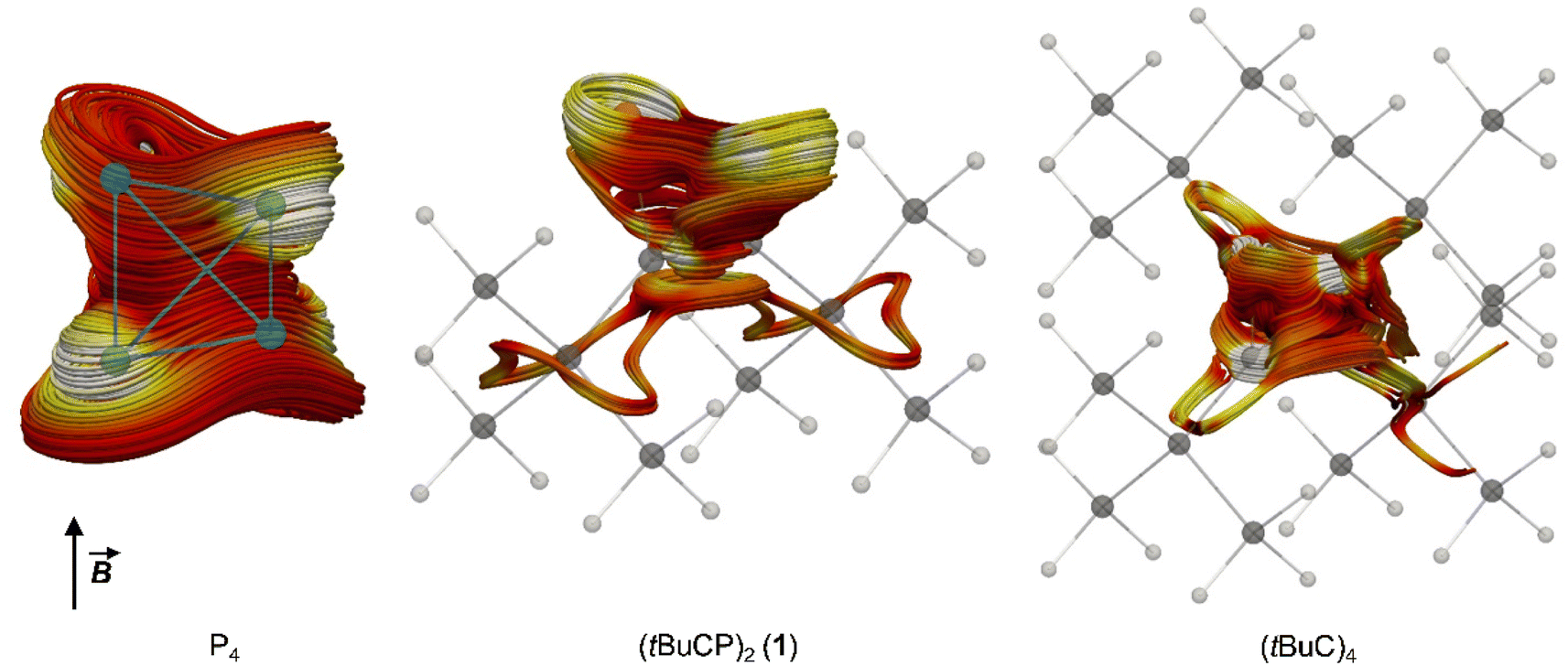 | ||
| Fig. 5 Spaghetti plots of selected current density streamlines induced by an external magnetic field (B) showing the main cluster current contributions for P4, 1 and (tBuC)4, respectively. For details on the calculations, see the ESI.‡ | ||
Photochemistry
As evidenced by monitoring the characteristic 31P NMR signal at −468.2 ppm (see Fig. S21, ESI‡) the concentration of 1 stays constant over 15 hours in the dark at ambient temperature in solution. However, at temperatures above 40 °C, the formation of the ladderane (tBuCP)4 (2) (i.e. a dimer of 1, see Fig. 1) was observed by a singlet resonance at −22.2 ppm (Fig. S19 in the ESI‡).13 Correspondingly, conversion of 1 to 2 increases with higher temperatures. For instance, keeping the solution at 80 °C for ca. 2.5 hours results in ca. 8% conversion (based on integration) of 1 to 2 (Fig. S21‡). This thermal instability of 1 is in agreement with a previous report.10Upon exposure to ambient light, 1 showed a significantly lower stability as demonstrated by preparing two identical samples containing 1 (with a concentration of 0.44 mol L−1) and an internal standard (PPh3) in C6D6 and monitoring the corresponding characteristic 31P NMR signals quantitatively in the dark and in the light, respectively (Fig. S9 and S10‡). While the sample kept in the dark did not degrade notably over time, the sample kept under ambient light was partly converted into 2 after 17 hours. Note that the conversion was not complete with a final concentration of 0.04 mol L−1 (corresponding to 90% conversion) for 1 and 0.15 mol L−1 (corresponding to 68% yield) for 2, which is presumably explained by the formation of a precipitate in the NMR tube (vide infra).
The photochemical instability of 1 allowed the development of a convenient protocol for the synthesis of 2 from 1. Irradiation of solutions of 1 in THF for three hours using a UV lamp and subsequent extraction with benzene results in pure 2 in 77% isolated yield (Scheme 1). No side products were detected when following the reaction by 31P NMR spectroscopy.
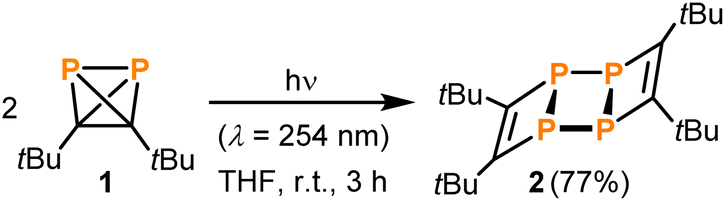 | ||
| Scheme 1 Synthesis of 2 by irradiating 1. | ||
It is important to note that tri-tert-butylphosphatetrahedrane (tBu3C3P) undergoes a very similar reaction with substoichiometric amounts of a Lewis acid such as triphenylborane, affording a planar phosphacyclobutadiene.14 In the absence of other reagents, this species undergoes head-to-head dimerisation to form a ladderane-type dimer similar to 2. In the presence of alkenes or a transition metal complex, however, it could be trapped as cycloaddition product or as a ligand. The photochemistry (when exciting at 254 nm) of (tBu3C3P) is rather unselective, affording a mixture of products including the dicyclopropenyl substituted diphosphene. In contrast, the dimerisation of 1 to 2 is driven photochemically and does not require the addition of further reagents.
Under prolonged irradiation of 2 in THF with either 365 nm or 450 nm a reaction mixture consisting of an orange precipitate and a colourless solution is obtained (see Fig. S5 in the ESI‡). Analysis of the solution by 1H NMR spectroscopy revealed clean conversion to di-tert-butylacetylene as evidenced by a singlet resonance at a chemical shift of 1.34 ppm and the absence of starting material (see Fig. S15 in the ESI;‡ as described above, this formation of di-tert-butylacetylene was already observed in attempts to crystallise pure 1in situ on the diffractometer). The 13C{1H} NMR spectrum also showed the three signals corresponding to the alkyne at chemical shifts of 27.4, 31.7 and 87.5 ppm (Fig. S16‡). The 31P{1H} NMR spectrum showed a small amount of P4 at −520.7 ppm (see Fig. S17 in the ESI‡). Thus, the orange precipitate is assigned to red phosphorus (or a related phosphorus containing polymer), which has been observed to form upon polymerisation of white phosphorus in other reactions induced by ionising γ-radiation (Scheme 2).8 Notably, this is reminiscent of the reactivity of tetrahedral Co2(CO)6(η2-alkyne) complexes, where the Co2(CO)6 can act as protecting group.24
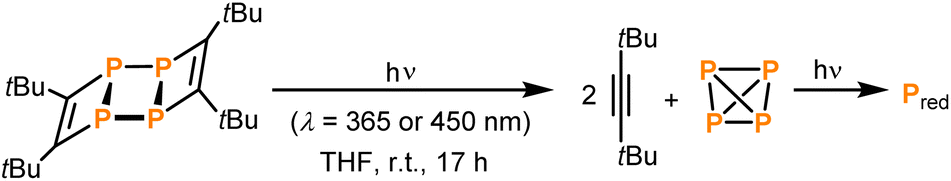 | ||
| Scheme 2 Photochemistry of 2. | ||
At significantly lower concentrations of 1, the photoconversion of 1 to 2 proceeds via a further intermediate, which can be monitored as a complete step prior to further degradation, as evidenced by recording the electronic absorption spectra in the UV/vis after stepwise illumination at 340 nm of a 500 μM solution of 1 in n-hexane (Fig. 6). Within the first 5 s of illumination, the absorption spectrum of 1 with two characteristic bands at ca. 230 and 280 nm decreases while an absorption spectrum with two distinct bands peaking at ca. 350, 300 and 210 nm is formed. On prolonged illumination the formed absorption spectrum becomes particularly more defined in the spectral range from 260 to 380 nm and resembles the absorption spectrum of purified 2 (Fig. 6).
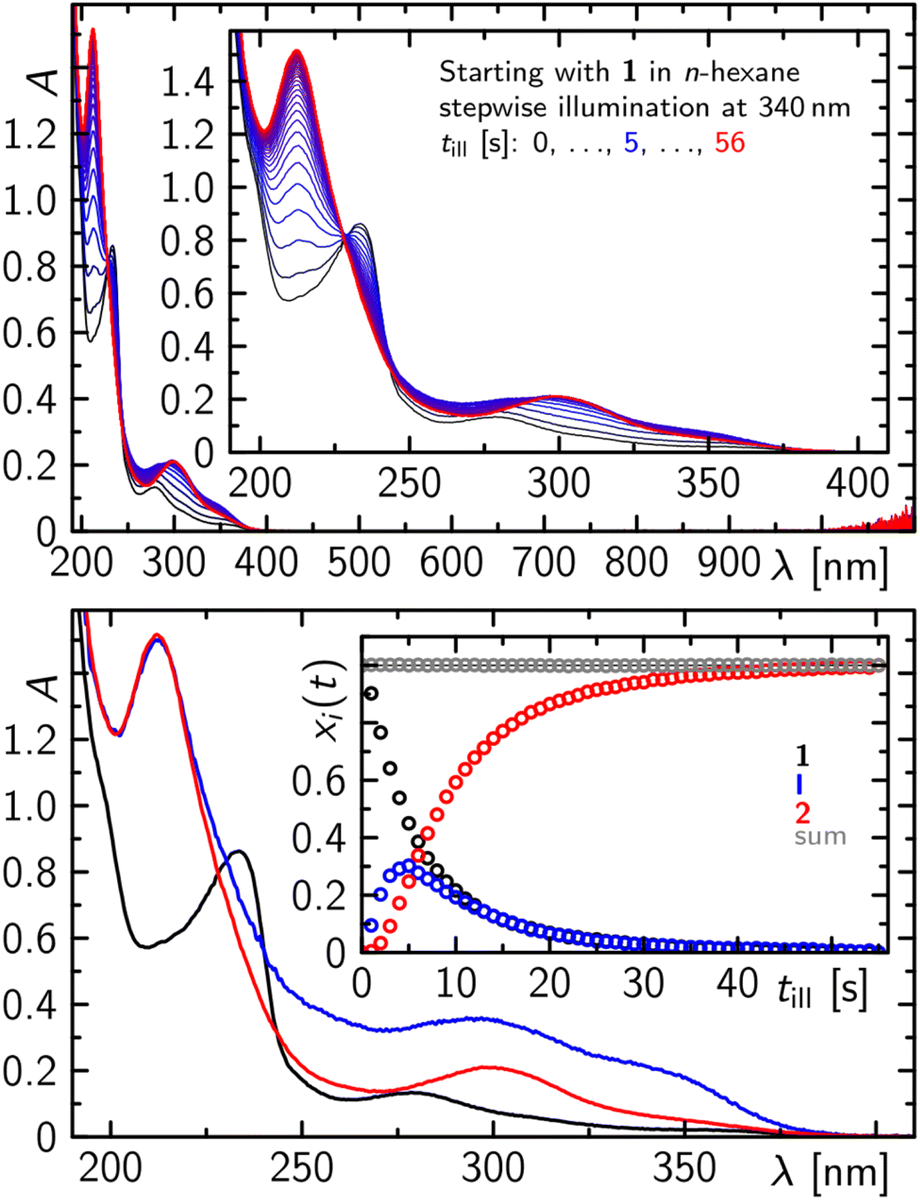 | ||
| Fig. 6 Sequence of absorption spectra of 1 (2 mM) in n-hexane after stepwise illumination at 340 nm with rectangular pulses of 1 s pulse width (top) and the corresponding decomposition into species spectra and concentration time profiles showing clearly an intermediate I prior to the final formation of 2 (bottom). | ||
With even longer illumination, the additional formation of elemental phosphorus and di-tert-butylacetylene is evidenced by scatter contributions in the UV/vis absorption signal (data not shown). Based on the structure of 2, it can be assumed that this ladderane compound is formed by cycloaddition of two P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) P bonds of two planar 1,2-diphosphacyclobutadienes 1′ (Scheme 3).25 To investigate this possibility, molecular dynamics simulations of 1 were performed starting in the electronic ground state and switching into the first excited singlet state after 1 ps of propagation. These simulations provide striking evidence for a formation of planar 1,2-diphosphacyclobutadienes upon illumination (Fig. 7 and S24 and ESI movie‡). Directly after changing to the first excited singlet state the P–P bond length increases instantaneously (Fig. 7a). Within a time-window of 2 ps of propagation, the molecular framework flips into a rather planar trapezoid shape (1′). Notably, the formation of 1′ from 1 in the absence of light has a computed activation barrier of 55.3 kcal mol−1 according to DFT calculations on the ωB97X-D3/def2-SVP level of theory using the NEB method.
P bonds of two planar 1,2-diphosphacyclobutadienes 1′ (Scheme 3).25 To investigate this possibility, molecular dynamics simulations of 1 were performed starting in the electronic ground state and switching into the first excited singlet state after 1 ps of propagation. These simulations provide striking evidence for a formation of planar 1,2-diphosphacyclobutadienes upon illumination (Fig. 7 and S24 and ESI movie‡). Directly after changing to the first excited singlet state the P–P bond length increases instantaneously (Fig. 7a). Within a time-window of 2 ps of propagation, the molecular framework flips into a rather planar trapezoid shape (1′). Notably, the formation of 1′ from 1 in the absence of light has a computed activation barrier of 55.3 kcal mol−1 according to DFT calculations on the ωB97X-D3/def2-SVP level of theory using the NEB method.
 | ||
| Scheme 3 Proposed mechanism for the dimerisation of 1 to 2. | ||
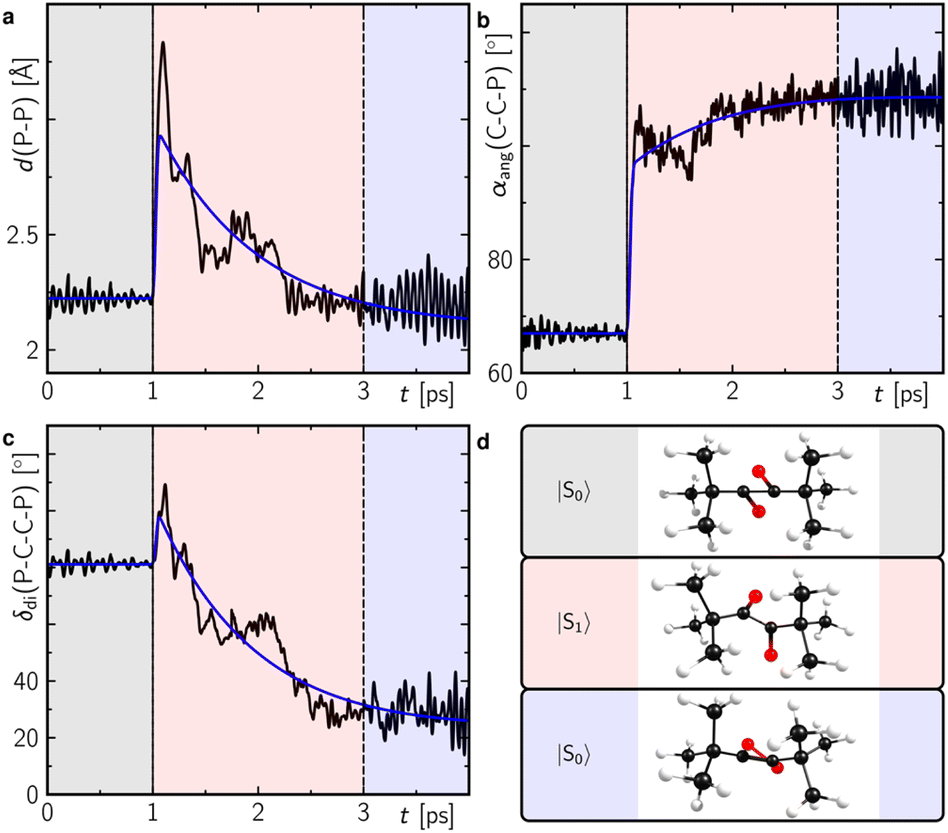 | ||
| Fig. 7 Structural analysis of the molecular dynamics trajectories showing the average of the P–P bond length (a), the carbon–carbon–phosphorus angle (b) and the P–C–C–P dihedral angle (c) as defined in (d) of Fig. S24.‡ The colour shaded temporal ranges indicate propagation in different electronic state as indicated in (d). Corresponding representative structures during this temporal range are also shown in (d). The blue lines in (a–c) correspond to a global monoexponential fit to the data with a lifetime of ca. 960 fs. | ||
Dimerisation of 1′ should generally be allowed to form either the endo- or exo-configured ladderane 2. In both cases no noticeable activation barrier is expected (Fig. 8). Since the observed intermediate shows a spectrum rather similar to the final spectrum of 2 and the fact that the exo-isomer is more stable by 0.5 eV (12 kcal mol−1), it is tempting to speculate that the intermediate represents the endo-form of 2, which can be photochemically converted into the more stable exo-form.26 The calculated absorption spectrum of the exo-configured isomer shows a rather defined absorption band peaking at ca. 300 nm with a shoulder at around 350 nm as observed in the experiment for the final product 2-exo. Importantly, the calculated spectrum of the endo-isomer shows a higher absorptivity red and blue shifted compared to the main absorption band of the exo-isomer peaking at ca. 270 and 350 nm, respectively, which is also observed in the experiment. This provides evidence for the assignment of a photoinduced endo to exo isomerisation of 2. Alternatively, a photoinduced retro-[2 + 2] cycloaddition from 2-endo to 1′ and subsequent [2 + 2] to 2-exo could take place.27
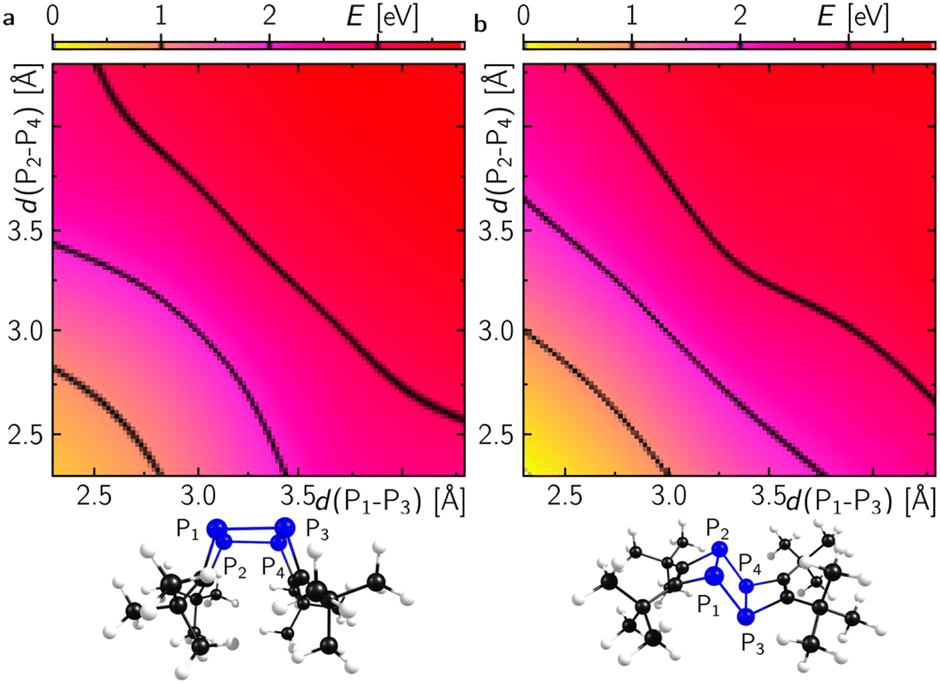 | ||
| Fig. 8 Potential energy surfaces along the dimerisation coordinates of two molecules of 1′ to 2 in either the endo- (a) or the exo-configuration (b). Calculations were performed at the B3LYP-D4/def2-TZVP level of theory. | ||
Next, reactions of 1 with suitable trapping agents for [2 + 2] cycloadditions, i.e., alkenes or alkynes, were tested as experimental indications for the occurrence of a 1,2-diphosphacyclobutadiene as an intermediate in the formation of 2 from 1. While irradiating 1 at 254 nm in the presence of either bis(trimethylsilyl)acetylene or maleic anhydride resulted only in the formation of 2 as observed for 1 alone,28 the reaction of 1 with the more electron-deficient N-phenylmaleimide gave two singlet resonances (δ = −63.0 and −84.4 ppm) in the 31P{1H} NMR spectrum after irradiation for two hours (see Fig. S18 in the ESI‡). This reaction was more selective (affording less 2) when excess amounts of the alkene were employed. However, from a synthetic point of view, the removal of this excess proved difficult due to similar solubilities and retention factors during chromatographic separation. Therefore, the smaller N-methylmaleimide was chosen for the trapping reaction. The 31P{1H} NMR spectrum of the product showed two distinct signals at chemical shifts of −62.4 and −84.6 ppm. Removal of the excess N-methylmaleimide by sublimation and recrystallisation of the residue from n-hexane afforded single crystals of both of the isomeric [2 + 2] cycloaddition products 3-endo and 3-exo (Scheme 4).
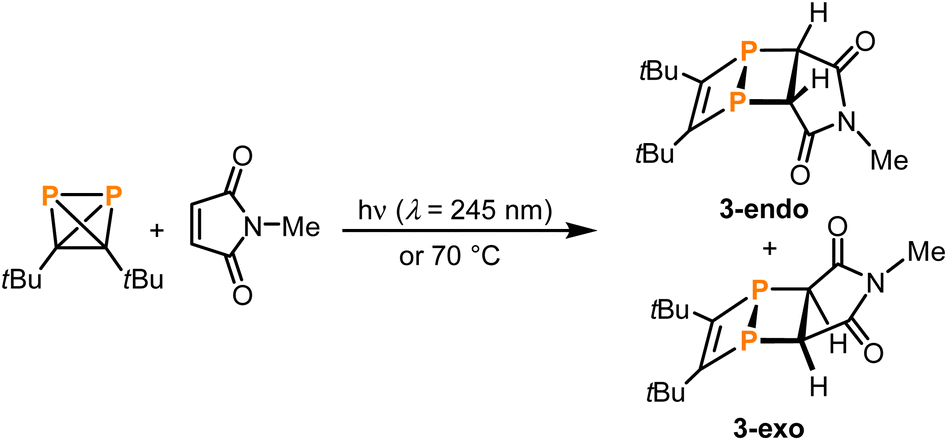 | ||
| Scheme 4 [2 + 2]-Cycloadditions of in situ generated 1,2-diphosphacyclobutadiene 1′ with N-methylmaleimide. | ||
Single-crystal XRD diffraction revealed that 3-exo and 3-endo co-crystallise in the space group P21/c with eight molecules of 3-exo and four molecules 3-endo in the unit cell. The molecular structures of 3-exo and 3-endo in the solid state (Fig. 9) strongly support the formation of diphosphabicyclo[2.2.0]hexene rings formed upon [2 + 2] cycloaddition of an in situ generated 1,2-diphosphacyclobutadiene with N-methylmaleimide. This is consistent with the proposed mechanism for the dimerisation of 1 to 2via a 1,2-diphosphacyclobutadiene. In both 3-exo and 3-endo, the P–P bond lengths [P1–P2 2.242(1) Å, P5–P6 2.233(1) Å] are indicative of a single bond and the C–C bonds originating from 1 have bond lengths typical for double bonds [C1–C2 1.362(3) Å, C32–C31 1.367(3) Å].29 The C–C bonds originating from the maleimide possess typical bond lengths for single bonds [C11–C12 1.524(2) Å, C41–C42 1.523(2) Å]. The C2–P2–C12 and C1–P1–C11 angles of 97.8(1)° and 95.5(1)°, respectively, indicate an almost perpendicular arrangement of the two C2P2 rings in 3-exo. In 3-endo, these angles are larger [C31–P5–C41: 100.8(1)°, C32–P6–C42: 100.7(1)°] possibly resulting from steric hindrance of the tert-butyl groups and the imide moiety.
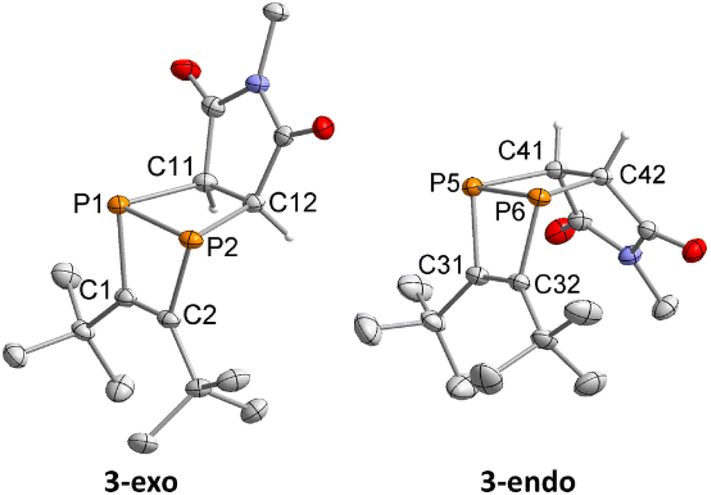 | ||
| Fig. 9 Molecular structures of 3-endo and 3-exo in the solid state. Thermal ellipsoids are set at 50% probability level. H-atoms (except for those binding to C11, C12, C41 and C42) are omitted for clarity. Selected bond lengths [Å] and angles [°]: P1–P2 2.242(1), P1–C1 1.844(2), P1–C11 1.916(2), P2–C2 1.852(2), P2–C12 1.910(2), C1–C2 1.362(3), C11–C12 1.524(2), P5–P6 2.233(1), P5–C31 1.841(2), P5–C41 1.928(2), P6–C32 1.841(2), P6–C42 1.923(2), C32–C31 1.367(3), C41–C42 1.523(2), C1–P1–P2 76.4(1), C2–P2–P1 75.9(1), C1–C2–P2 103.8(1), C2–C1–P1 103.7(1), C11–P1–P2 79.5(1), C12–P2–P1 78.7(1), C12–C11–P1 100.0(1), C11–C12–P2 101.5(1), C2–P2–C12 97.8(1), C1–P1–C11 95.5(1), C31–P5–P6 76.2(1), C32–P6–P5 76.5(1), C32–C31–P5 103.8(1), C31–C32–P6 103.3(1), C41–P5–P6 79.1(1), C42–P6–P5 79.5(1), C41–C42–P6 100.4(1), C42–C41–P5 100.7(1), C31–P5–C41 100.8(1), C32–P6–C42 100.7(1). | ||
According to the 1H and 31P{1H} (single and multinuclear) NMR spectra as well as mass spectra, 3-exo and 3-endo were isolated as a mixture containing ca. 53% of the endo- and 47% of the exo-isomer. The 31P{1H} NMR spectrum shows two singlet signals at chemical shifts of −85.2 and −63.1 ppm. In the proton-coupled 31P NMR spectrum the signal at −85.2 ppm shows coupling to the diphosphacyclobutane protons and has the shape of a pseudo-triplet, whereas the signal at −63.1 ppm shows no coupling. DFT calculations at the TPSS pcSseg-2 level of theory confirm that the high-field resonance, which is assigned to 3-endo, shows higher JPH coupling constants than the low-field species. This is in agreement with the experimental values and also confirmed by a study on the effect of the lone pair conformation on P–H coupling constants.30 Simulation of the 31P NMR signal of 3-endo reveals a 2JPH coupling constant of 13.1 Hz and a 3JPH coupling constant of −9.7 Hz.
The 1H NMR spectrum of the mixture of isomers 3-endo and 3-exo shows singlet resonances for the tert-butyl groups at 1.07 and 1.15 ppm and for the maleimide methyl groups at 2.65 and 2.66 ppm. Moreover, the diphosphacyclobutane proton signals arise as a singlet at 2.75 for 3-exo and as a multiplet between 2.63 and 2.71 ppm for 3-endo. The 13C{1H} NMR spectrum shows two singlet resonances for the maleimide methyl groups and pseudo triplet resonances for the other carbon atoms. Four low-field-shifted resonances were assigned to the diphosphacyclobutene carbon atoms at 167.7 (3-endo) and 168.6 ppm (3-exo) and the carbonyl moieties at 175.2 (3-endo) and 175.9 ppm (3-exo). Analysis of a mixture of 3-endo and 3-exo by GC-MS revealed two peaks at retention times of 14.28 and 14.46 min with mass spectra that show the same, expected molecular ion signal with m/z = 311.0.
It is worth noting that a [2 + 2]-cycloaddition product related to 3-endo/exo was synthesised by Regitz and co-workers by reacting the same maleimide with the isomeric 1,3-diphosphacyclobutadiene (generated by addition of hexachloroethane to a zirconium complex).31 In Regitz's report, the formation of this isomeric diphosphabicyclo[2.2.0]hexene was confirmed by multinuclear (1H, 13C{1H} and 31P{1H}) NMR spectroscopy and mass spectrometry. Only the endo-isomer was observed. Recently, also a different reaction type was reported, in which nickel complexes catalyse the phosphinidene transfer from (tBu3C3P) to styrene, ethylene, neohexene and 1,3-cyclohexadiene, affording phosphiranes.14
Conclusions
The tetrahedral molecular structure of di-tert-butyldiphosphatetrahedrane (1) has been unambiguously determined by single-crystal X-ray diffraction analysis at low temperature. The structure is similar to that of tetra-tert-butyltetrahedrane (tBuC)4 and the P4 molecule. The close analogy between 1 and P4 is also evident from DFT calculations, which reveal their isolobal electronic structure. The cluster-type core of 1 shows spherical aromaticity. The calculated magnetically induced ring currents are between that of P4 and the tetrahedrane (tBuC)4 in magnitude and significantly greater than those in benzene. Diphosphatetrahedrane 1 is fairly stable in solution in the dark at ambient temperature, while it converts to the ladderane (tBuCP)4 (2) upon irradiation or at elevated temperatures. Under UV irradiation, 2 decomposes to P4 (which, in turn, is transformed into red phosphorus) and di-tert-butylacetylene. Based on the NMR and UV/vis spectroscopic monitoring of the photochemical isomerisation of 1 forming 2 together with quantum chemical calculations, a stepwise mechanism for this reaction is proposed: first, photoexcited 1 isomerises to its 1,2-diphosphacyclobutadiene isomer 1′, which forms in the excited state on a ps time scale according to molecular dynamics simulations. Subsequently, [2 + 2] cycloaddition of two 1,2-diphosphacyclobutadienes forms the endo-isomer 2-endo in a purely thermal step without any significant activation barrier. The endo-isomer is then photochemically converted into the exo-isomer, either directely or by a photochemically induced retro-[2 + 2]-reaction via1′. The proposed diphosphacyclobutadiene 1′ was trapped as a [2 + 2] cycloaddition product with N-methylmaleimide. The photochemical isomerisation appears to be an intrinsic feature of the reactivity of diphosphatetrahedranes, resulting in diphosphacyclobutadienes as transient intermediates. It is anticipated that this process can be exploited for the generation of various other bi- and tricyclic organophosphorus compounds by reaction with a broader scope of alkenes, alkynes and other unsaturated organic compounds. Such reactions could give rise to a range of new phosphorus heterocycles with interesting properties.Data availability
The data supporting the findings of this study are available within the paper and its ESI files.‡ Raw data are provided on reasonable request. Crystallographic have been deposited at the CCDC under 2102564 and 2323059.Author contributions
Gabriele Hierlmeier: conceptualisation, investigation – synthesis and characterisation, writing – original draft. Roger Jan Kutta: investigation – spectroscopy and quantum chemical calculations, writing – original draft. Peter Coburger: investigation – quantum chemical calculations. Hans-Georg Stammler: investigation – X-ray structure analysis of 1, writing – original draft. Jan Schwabedissen: investigation – X-ray structure analysis of 1. Norbert W. Mitzel: supervision, writing – review & editing, funding acquisition. Maria Dimitrova: investigation – quantum chemical calculations. Raphael J. F. Berger: investigation – quantum chemical calculations, writing – original draft. Patrick Nuernberger: supervision, writing – review & editing, funding acquisition. Robert Wolf: conceptualization, supervision, writing – review & editing, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Verena Streitferdt for the acquisition of initial kinetic data. Financial support by the Deutsche Forschungsgemeinschaft (grant numbers WO1496/10-1 and Mi477/41-1; project numbers 451444676 and 434445823, as well as TRR 325, projects A4 and A5, 444632635) and the Fonds der Chemischen Industrie (Kekulé fellowship for G. H.) is gratefully acknowledged.Notes and references
- G. Maier, S. Pfriem, U. Schäfer and R. Matusch, Angew Chem. Int. Ed. Engl., 1978, 17, 520–521 CrossRef.
- B. M. Cossairt, C. C. Cummins, A. R. Head, D. L. Lichtenberger, R. J. F. Berger, S. A. Hayes, N. W. Mitzel and G. Wu, J. Am. Chem. Soc., 2010, 132, 8459–8465 CrossRef CAS PubMed.
- R. Jupp and J. C. Slootweg, Angew. Chem., Int. Ed., 2020, 59, 10698–10700 CrossRef PubMed.
- Selected review articles: (a) B. M. Cossairt, N. A. Piro and C. C. Cummins, Chem. Rev., 2010, 110, 4164–4177 CrossRef CAS PubMed; (b) M. Caporali, L. Gonsalvi, A. Rossin and M. Peruzzini, Chem. Rev., 2010, 110, 4178–4235 CrossRef CAS PubMed; (c) M. Scheer, G. Balázs and A. Seitz, Chem. Rev., 2010, 110, 4236–4256 CrossRef CAS PubMed; (d) G. Maier, Angew Chem. Int. Ed. Engl., 1988, 27, 309–332 CrossRef.
- (a) A. Pedler, J. Chem. Soc., Trans., 1890, 57, 599–613 RSC; (b) M. Serrano-Ruiz, A. Romerosa and P. Lorenzo-Luis, Eur. J. Inorg. Chem., 2014, 1587–1598 CrossRef CAS.
- G. Rathenau, Physica, 1937, 4, 503–514 CrossRef CAS.
- (a) O. J. Scherer, Angew. Chem., Int. Ed., 2000, 39, 1029–1030 CrossRef CAS; (b) H. Bock and H. Mueller, Inorg. Chem., 1984, 23, 4365–4368 CrossRef CAS; (c) N. A. Piro, J. S. Figueroa, J. T. McKellar and C. C. Cummins, Science, 2006, 313, 1276–1279 CrossRef CAS PubMed.
- N. P. Tarasova, Y. V. Smetannikov, I. M. Artemkina, I. A. Lavrov, M. A. Sinaiskii and V. I. Ermakov, Dokl. Chem., 2006, 410, 189–191 CrossRef CAS.
- (a) D. Tofan and C. C. Cummins, Angew. Chem., Int. Ed., 2010, 49, 7516–7518 CrossRef CAS PubMed; (b) L.-P. Wang, D. Tofan, J. Chen, T. van Voorhis and C. C. Cummins, RSC Adv., 2013, 3, 23166 RSC.
- G. Hierlmeier, P. Coburger, M. Bodensteiner and R. Wolf, Angew. Chem., Int. Ed., 2019, 58, 16918–16922 CrossRef CAS PubMed.
- For reactivity studies on di-tert-butyldiphosphatetrahedrane see: (a) G. Hierlmeier, M. K. Uttendorfer and R. Wolf, Chem. Commun., 2021, 57, 2356–2359 RSC; (b) G. Hierlmeier and R. Wolf, Angew. Chem., Int. Ed., 2021, 60, 6435–6440 CrossRef CAS PubMed; (c) G. Hierlmeier, P. Coburger, D. J. Scott, T. M. Maier, S. Pelties, R. Wolf, D. M. Pividori, K. Meyer, N. P. van Leest and B. de Bruin, Chem.–Eur. J., 2021, 27, 14936–14946 CrossRef CAS PubMed; (d) M. K. Uttendorfer, G. Hierlmeier and R. Wolf, Z. Anorg. Allg. Chem., 2022, e202200124 CrossRef CAS.
- (a) M.-L. Y. Riu, R. L. Jones, W. J. Transue, P. Müller and C. C. Cummins, Sci. Adv., 2020, 6, eaaz3168 CrossRef CAS PubMed; (b) M.-L. Y. Riu, M. Ye and C. C. Cummins, J. Am. Chem. Soc., 2021, 143, 16354–16357 CrossRef CAS PubMed.
- S. Geissler, U. Barth, U. Bergsträsser, M. Slany, J. Durkin, P. B. Hitchcock, M. Hofmann, P. Binger, J. F. Nixon, P. von Ragué Schleyer and M. Regitz, Angew Chem. Int. Ed. Engl., 1995, 34, 484–487 CrossRef.
- M.-L. Y. Riu, A. K. Eckhardt and C. C. Cummins, J. Am. Chem. Soc., 2021, 143, 13005–13009 CrossRef CAS PubMed.
- R. Boese, D. Bläser, R. Latz and A. Bäumen, Acta Crystallogr., 1999, C55, IUC9900016 Search PubMed.
- H. Irngartinger, A. Goldmann, R. Jahn, M. Nixdorf, H. Rodewald, G. Maier, K.-D. Malsch and R. Emrich, Angew Chem. Int. Ed. Engl., 1984, 23, 993–994 CrossRef.
- M. Elian, M. M. L. Chen, D. M. P. Mingos and R. Hoffmann, Inorg. Chem., 1976, 15, 1148–1155 CrossRef CAS.
- (a) A. Hirsch, Z. Chen and H. Jiao, Angew. Chem., Int. Ed., 2001, 40, 2834–2838 CrossRef CAS; (b) N. K. V. Monteiro, J. F. de Oliveira and C. L. Firme, New J. Chem., 2014, 38, 5892–5904 RSC; (c) D. Moran, M. Manoharan, T. Heine and P. von Ragué Schleyer, Org. Lett., 2003, 5, 23–26 CrossRef CAS PubMed.
- P. von Ragué Schleyer, C. Maerker, A. Dransfeld, H. Jiao and N. J. R. van Eikema Hommes, J. Am. Chem. Soc., 1996, 118, 6317–6318 CrossRef PubMed.
- P. Weis, D. C. Röhner, R. Prediger, B. Butschke, H. Scherer, S. Weber and I. Krossing, Chem. Sci., 2019, 48, 295 Search PubMed.
- The NICS value of P4 was also calculated by Cummins (−59.4), see: B. M. Cossairt and C. C. Cummins, J. Am. Chem. Soc., 2009, 131, 15501–15511 CrossRef CAS PubMed.
- D. Sundholm, H. Fliegl and R. J. F. Berger, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2016, 6, 639–678 CAS.
- It is noteworthy that a significant fraction of the total current density in (tBuC)4 (about 50% of the diamagnetic contribution) extends over the tBu groups. However, at this stage no further attempts have been made to analyse this phenomenon in more detail.
- K. M. Nicholas and R. Pettit, Tetrahedron Lett., 1971, 12, 3475–3478 CrossRef.
- Alternatively, it was hypothesised that the intermediate observed at dilute concentration conditions may result from the reaction between 1′ and 1 (Fig. S25‡). According to quantum-chemical calculations, the resulting ‘twisted dimer’ would represent a stable structure (see the ESI‡ for details). However, its electronic absorption spectrum is expected to show transitions in the visible range from 400 to 500 nm (Fig. S25‡), which are not observed in any of the experimental data, thus, excluding its formation.
- Intermediate 2-endo shows considerable stabilisation by dispersion interactions; see: J. P. Wagner and P. R. Schreiner, Angew. Chem., Int. Ed., 2015, 54, 12274–12296 CrossRef CAS PubMed.
- A stepwise bond formation via a biradical intermediate is conceivable as an alternative mechanistic pathway for the formation of the endo- and exo-configured ladderane 2 from 1′. We did not observe such an intermediate, but related biradicals were detected for silicon analogues: J. R. Gee, W. A. Howard, G. L. McPherson and M. J. Fink, J. Am. Chem. Soc., 1991, 113, 5461–5462 CrossRef CAS.
- We believe that photodimerisation of maleic anhydride is a major competitive pathway, see T. Horie, M. Sumino, T. Tanaka, Y. Matsushita, T. Ichimura and J. Yoshida, Org. Process Res. Dev., 2010, 14, 405–410 CrossRef CAS.
- (a) B. Cordero, V. Gómez, A. E. Platero-Prats, M. Revés, J. Echeverría, E. Cremades, F. Barragán and S. Alvarez, Dalton Trans., 2008, 2832–2838 RSC; (b) P. Pyykkö and M. Atsumi, Chem.–Eur. J., 2009, 15, 186–197 CrossRef PubMed; (c) P. Pyykkö, J. Phys. Chem. A, 2015, 119, 2326–2337 CrossRef PubMed.
- W. H. Hersh, S. T. Lam, D. J. Moskovic and A. J. Panagiotakis, J. Org. Chem., 2012, 77, 4968–4979 CrossRef CAS PubMed.
- S. Mack, S. Danner, U. Bergsträßer, H. Heydt and M. Regitz, J. Organomet. Chem., 2002, 643–644, 409–415 CrossRef.
Footnotes |
| † We dedicate this manuscript to the memory of Manfred Regitz. |
| ‡ Electronic supplementary information (ESI) available. CCDC 2102564–2323059. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc00936c |
| § Present address: Julius-Maximilians-Universität Würzburg, Institut für Anorganische Chemie, Am Hubland, 97074 Würzburg. |
| ¶ Present address: Technische Universität München, Lehrstuhl für Anorganische Chemie mit Schwerpunkt neue Materialien, Lichtenbergstraße 4, 85748 Garching bei München, Germany. |
| This journal is © The Royal Society of Chemistry 2024 |