
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelectivity descriptors of the catalytic n-hexane cracking process over 10-membered ring zeolites†
Pandong
Ma
a,
Hexun
Zhou
a,
Yubing
Li
b,
Mengheng
Wang
b,
Stefan Adrian F.
Nastase
 c,
Mengsi
Zhu
d,
Jiale
Cui
b,
Luigi
Cavallo
c,
Kang
Cheng
*b and
Abhishek
Dutta Chowdhury
*a
c,
Mengsi
Zhu
d,
Jiale
Cui
b,
Luigi
Cavallo
c,
Kang
Cheng
*b and
Abhishek
Dutta Chowdhury
*a
aCollege of Chemistry and Molecular Sciences, Wuhan University, Wuhan 430072 Hubei, PR China. E-mail: abhishek@whu.edu.cn
bState Key Laboratory of Physical Chemistry of Solid Surfaces, Collaborative Innovation Center of Chemistry for Energy Materials, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen 361005, PR China. E-mail: kangcheng@xmu.edu.cn
cKAUST Catalysis Center (KCC), King Abdullah University of Science and Technology (KAUST), Thuwal 23955, Saudi Arabia
dInnovation Laboratory for Sciences and Technologies of Energy Materials of Fujian Province (IKKEM), Xiamen 361005, PR China
First published on 24th June 2024
Abstract
Zeolite-mediated catalytic cracking of alkanes is pivotal in the petrochemical and refining industry, breaking down heavier hydrocarbon feedstocks into fuels and chemicals. Its relevance also extends to emerging technologies such as biomass and plastic valorization. Zeolite catalysts, with shape selectivity and selective adsorption capabilities, enhance efficiency and sustainability due to their well-defined network of pores, dimensionality, cages/cavities, and channels. This study focuses on the alkane cracking over 10-membered ring (10-MR) zeolites under industrially relevant conditions. Through a series of characterizations, including operando UV-vis spectroscopy and solid-state NMR spectroscopy, we intend to address mechanistic debates about the alkane cracking mechanism, aiming to understand the dependence of product selectivity on zeolite topologies. The findings highlight topology-dependent mechanisms, particularly the role of intersectional void spaces in zeolite ZSM-5, influencing aromatic-based product selectivity. This work provides a unique understanding of zeolite-catalyzed hydrocarbon conversion, linking alkane activation steps to the traditional hydrocarbon pool mechanism, contributing to the fundamental knowledge of this crucial industrial process.
Introduction
Zeolite-mediated catalytic cracking is the core process of the modern petrochemical and refining industry.1–7 Catalytic cracking typically refers to breaking down heavier hydrocarbon feedstocks into valuable lighter products, especially gasoline and propylene.1,7 While fluid catalytic cracking (FCC) and hydrocracking have long been recognized as prevalent reaction classes in the oil refinery industry, catalytic cracking is gaining prominence in various emerging technologies.7 It is now being increasingly utilized in biomass valorization processes,8,9 and depolymerization of plastics10,11 as well as for heat removal and temperature control in hypersonic aircrafts.12,13 The efficacy of zeolite catalysts in catalytic cracking can be attributed to their distinctive properties, including shape selectivity and selective adsorption capabilities.14–19 These characteristics play a vital role in improving the efficiency and sustainability characteristics of the catalytic cracking process.Zeolites are crystalline aluminosilicate minerals with unique properties that make them highly effective refinery catalysts.14–19 The structure of zeolites consists of a well-defined network of pores, cavities/cages, and interconnected channels of molecular dimensions, allowing them to adsorb selectively and catalytically convert incoming hydrocarbon molecules based on their size, shape, and polarity.6 These properties make zeolites particularly suitable for catalytic cracking, where the selective cracking of larger hydrocarbon molecules into smaller, more valuable ones is desired. Hence, it is imperative to understand the operational scope of selectivity descriptors of zeolite-catalyzed cracking processes to improve process efficiency. Deriving appropriate structure–performance relationships between zeolite catalysts and product selectivity proves challenging during the zeolite-catalyzed alkane cracking process due to the utilization of various classes of medium and large pore zeolites, along with a wide range of reaction conditions, as documented in the existing literature.3,16,20–30 To bridge this knowledge gap, this work systematically evaluates the significance of 10-membered ring (MR) zeolites only for the catalytic cracking of n-hexane. With this objective, the following 10-MR zeolites with variable channel (a) topology (i.e., ring shape and size), (b) pathway (e.g., straight vs. sinusoidal), and (c) dimensionality (e.g., 1D vs. 2D vs. 3D) and (d) cages have been considered: ZSM-5 [MFI topology-3D-straight (10-MR)⊥sinusoidal (10-MR) channels], ferrierite [FER topology-2D-straight channels (10-MR⊥8-MR); hereafter also termed FER], MCM-22 [MWW topology-2D-cages⊥sinusoidal (10-MR) channels] and ZSM-22 [TON topology-1D-straight (10-MR) channels] (Fig. 1 and S1†).
 | ||
| Fig. 1 The project design: mechanism of catalytic cracking of alkanes will be investigated over the following 10-membered ring (MR) zeolites: ZSM-5 (3D MFI topology), FER (2D FER topology), MCM-22 (2D MWW topology), and ZSM-22 (1D TON topology). Typically, 10-MR zeolites provide superior molecular traffic control compared to other MR zeolites. Despite their structural and topological diversity, these zeolites are intriguing for mechanistic investigations in zeolite catalysis. In this context, the zeolite's channels, presented in yellow (D < 6 Å), are illustrated in italics, while the cages or intersections are depicted in blue (D ∼ 6–8 Å) and purple (D > 8 Å) spheres.27 | ||
Despite achieving industrial maturity, the fundamental understanding of the catalytic cracking of alkanes over zeolites has remained the Achilles' heel since its inception.5,31,32 Currently, there is a mechanistic consensus regarding zeolite-mediated catalytic cracking, which involves three consecutive steps: initiation, propagation, and termination.32 Within the contemporary literature, the initiation step has emerged as a highly debated topic, with differing perspectives on whether it proceeds through unimolecular protolytic (also known as the Haag–Dessau mechanism31) or classical bimolecular pathways involving carbonium (i.e., ‘‘non-classical’’ carbocations with a five-coordinated positive carbon: ‘‘three-center–two-electron’’, [3c–2e] bonds33), or carbenium (i.e., ‘‘classical’’ carbocations with a three-coordinated positive carbon: ‘‘two-center–two-electron’’ [2c–2e] bonds34,35) reactive intermediates, respectively.5,32,36 In the current study, taking into account the higher reaction temperature used, high reactant coverage, and the spacious characteristics of 10-MR zeolite catalysts, the binuclear carbenium mechanism emerges as the most viable mechanistic candidate.32,36 Yet, the first step of alkane cracking is difficult to understand because paraffinic hydrocarbons are saturated, which makes it challenging to directly form carbenium intermediates necessary for the binuclear mechanism. One possible explanation is that zeolite's Lewis acid sites (LAS) can abstract a hydrogen atom from an alkane molecule, creating the first alkene.2,7,37 This alkene can then be cracked by the zeolite's Brønsted acid sites (BAS) via the carbenium mechanism.38,39 Another possibility is that alkene impurities in the alkane feed or produced by non-catalytic thermal cracking can react with the zeolite in a similar way to produce carbenium intermediates.31,40,41 In any case, the fundamental understanding of catalytic alkane cracking over zeolites has remained a persistent challenge.
In addition, a definitive explanation for the varying product selectivity patterns observed during the catalytic cracking of alkanes over different zeolites remains elusive.19,25,42–47 These patterns often align with the zeolite framework, particularly the pore sizes.21,47–52 In order to delve deeper into the understanding of selectivity descriptor features, this study comprehensively and systematically assesses the catalytic significance of structural attributes solely within the 10-MR zeolite family.53 The focus on 10-MR zeolites stems from their ability to offer diverse structural characteristics compared to larger (or smaller) pore zeolites, as depicted in Fig. 1. The underlying concept of topology-dependent selectivity descriptors has gained attention in various zeolite-catalyzed hydrocarbon conversion processes such as methanol-to-hydrocarbon (MTH) and CO/CO2-hydrogenations with bifunctional (metal/zeolite) catalytic systems.54–57 These studies have highlighted that only 10-MR zeolites possess the unique ability to confine the reactants and intermediates optimally (i.e., molecular traffic control), governing the ultimate hydrocarbon product selectivity.58–66 Other zeolite pores typically do not offer this advantageous confinement effect in zeolite catalysis.55,56 Furthermore, it is crucial to understand the catalytic cracking process, specifically the involvement of carbenium intermediates, as it represents a significant sub-category of reactions within several zeolite-catalyzed hydrocarbon conversion processes like MTH, biomass/plastic valorization, and others.34,35 Therefore, this study holds broader implications in the field of zeolite catalysis.
To begin, we evaluated the performance of four commercially available 10-MR zeolites (ZSM-5, FER, MCM-22, and ZSM-22, as depicted in Fig. 1) in the catalytic cracking process using n-hexane as a representative alkane reactant. We observed a preferential selectivity towards propylene-dominating short alkenes and hydrogen-transferred products (paraffins and aromatics) over 2D/1D zeolites (FER, MCM-22, and ZSM-22) and 3D zeolites (ZSM-5), respectively. In addition to employing traditional characterization tools relevant to zeolite-based materials, we utilized advanced operando UV-vis spectroscopy coupled with online mass spectrometry, computational analysis, and solid-state NMR spectroscopy to investigate the topology-dependent reaction mechanism. Finally, we used theoretical simulations in the zeolite intersection and sinusoidal channel to determine the degree of interaction between key intermediates and the Brønsted acid sites. This multimodal spectroscopic and theoretical approach contributed to our understanding that the intersectional void spaces in zeolite ZSM-5 are crucial in providing the optimum confinement effect necessary for aromatization. This essential condition cannot be satisfactorily met by 2D/1D zeolites, where their straight/sinusoidal channels preferentially propagate the propylene-generating alkene cycle. As a result, this study offers a unique understanding of zeolite-catalyzed hydrocarbon conversion chemistry and establishes a connection between alkane activation steps and the traditional hydrocarbon pool mechanism.
Results and discussion
To assess the confinement impact of 10-MR zeolites with comparable Si/Al ratios of 8–18 (Table 1) on the catalytic cracking, first, these zeolites were characterized by numerous standard characterization tools, including powder X-ray diffraction (XRD), N2 physical adsorption and desorption measurements, inductively coupled plasma optical emission spectroscopy (ICP-OES), pyridine-Fourier transform infrared (Py-FTIR), scanning electron microscopy (SEM), transmission electron microscopy (TEM), solid-state NMR, and operando UV-vis diffuse reflectance spectroscopy (UV-vis DRS) coupled to online mass spectrometry (MS). We refer to the following items for a comprehensive understanding of the physicochemical properties of zeolites: Table 1 and Fig. S2–S7.† The structural characterization studies suggest that all four zeolites have good crystallinity and reasonable surface areas corresponding to their topologies (Fig. S2–S5†). Although their morphologies are different, we expect the performance of catalytic cracking to be mainly determined by the zeolite and topology. The acidity analysis conducted on all zeolites reveals the presence of both Brønsted acid sites (BAS) and Lewis acid sites (LAS), a typical characteristic of highly acidic zeolites (Fig. S6†). Simultaneously, 27Al magic angle spinning (MAS) solid-state NMR spectra of all zeolites (Fig. S7†) exhibited two peak maxima: one in the range of 50–60 ppm associated with tetra-coordinated Al sites and another at ∼0 ppm related to hexa-coordinated Al sites.67 These findings are presumed to correspond to BAS and LAS, respectively. Hence, the preliminary zeolite characterization suggests the potential for alkane activation mediated by Lewis acid sites in all instances.| Catalyst | Surface area (m2 g−1) | Pore volume (cm3 g−1) | Acid density (μmol g−1) | ICPd | ||||
|---|---|---|---|---|---|---|---|---|
| S BET | S micro | V total | V micro | BASc | LASc | B/L | Si/Al | |
| a Determined by the t-plot method. b Single point pore volume measured at P/P0 of ≈0.99. c Py-FTIR. d ICP-OES. | ||||||||
| ZSM-5 | 347 | 315 | 0.18 | 0.13 | 85 | 37 | 2.3 | 16 |
| MCM-22 | 481 | 361 | 1.18 | 0.15 | 41 | 34 | 1.2 | 12 |
| FER | 359 | 337 | 0.22 | 0.14 | 90 | 50 | 1.8 | 8 |
| ZSM-22 | 219 | 150 | 0.31 | 0.10 | 43 | 25 | 1.7 | 18 |
Next, all four zeolites were subjected to the catalytic performance evaluation with respect to the cracking of n-hexane at 803 K under atmospheric pressure (Fig. 2 and S9†). Due to the distinct physicochemical properties of various zeolites, diverse conversion levels have been attained across different zeolite types. Fig. 2 illustrates the key hydrocarbon product distributions at very early [first GC injection: time-on-stream (TOS) of ∼0.2 h] and steady (reaction for ∼6.2 h) phases of catalysis. This presentation style highlights the influence of zeolite physicochemical properties throughout the catalyst lifetime, with the first GC injection point also serving as a reference point for operando studies. Since the aim of our mechanistic studies is primarily to investigate the very early stages of the alkane cracking reaction, the product distribution data obtained from the first GC injection can be correlated with the time period employed in operando and solid-state NMR investigations (vide infra). Generally, the overall product selectivity did not change much during the reactions over four zeolites. Among the predominant effluent products, only zeolite ZSM-5 produced aromatic hydrocarbons with a selectivity of up to 30% along with the selectivity of C2–C4 short alkanes of 45%. Remarkably, the hydrogen-transferred products (i.e., the sum of aromatics and paraffins) constituted ∼78%, representing the highest reported value compared to the other 10-MR zeolites. Upon lowering the conversion level over the micron-sized ZSM-5 catalyst to make it comparable to other zeolites, aromatics were still produced, though with expectedly lower selectivity (Fig. S10†). A similar conversion-selectivity pattern was also noted over the nano-sized ZSM-5 catalyst (see discussion Section S2 in the ESI and Fig. S12†). In contrast, the corresponding value remained 35–42% for other zeolites, with a very limited contribution from the aromatics fraction. In this study, C2–C4 short olefins emerged as the preferential products, accounting for approximately 50–60% over zeolites ZSM-22, FER, and MCM-22, with the predominant contribution primarily attributed to propylene (35–40%). The subsequent thermogravimetric analysis uncovered the prevailing presence of soft coke species across all zeolites (Fig. S13†). Notably, ZSM-5 exhibited a substantial weight loss attributed to hard coke, distinguishing itself as the sole zeolite-generating aromatics through catalytic alkane cracking.
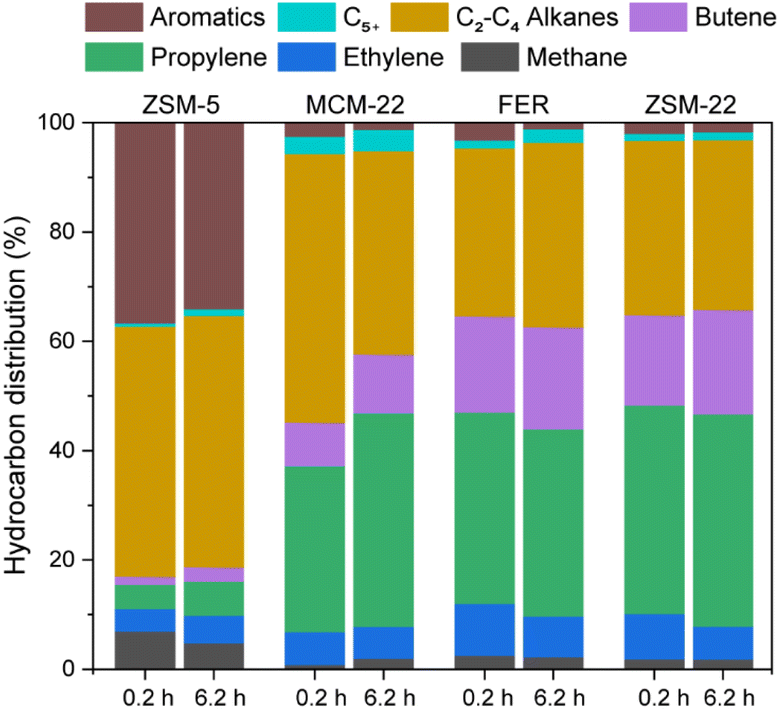 | ||
| Fig. 2 Hydrocarbon distribution in the effluent gas phase over ZSM-5, MCM-22, FER, and ZSM-22 zeolites during early (first GC injection: ∼0.2 h for n-hexane catalytic cracking conversion) and steady states (∼6.2 h) of reaction (reaction conditions: 803 K, 1 atm). Also, see Fig. S9–S12† for additional catalysis data. | ||
The observed pattern of product selectivity provides initial insights into the hydrocarbon pool (HCP) mechanism underlying the reaction. Typically, the steady state of zeolite catalysis is governed by “hybrid” supramolecular reaction centers, which are composed of “organic” HCP species trapped within the “inorganic” zeolites.68,69 The autocatalytic stage of the reaction is regulated by the “dual cycle mechanism,” encompassing catalytic cycles involving arenes and olefins.70–72 In this context, the balance of these interdependent cycles can potentially dictate the favored olefin selectivity in zeolite catalysis. For example, ethylene is postulated to have originated from the arene cycle, while higher olefins (including propylene) are primarily a product of the olefin cycle.73,74 An alternative perspective suggests that propylene is preferentially generated through alkene cycles rather than ethylene, although both olefins could potentially stem from arene cycles.75 Therefore, achieving preferential propylene selectivity may involve selectively promoting the alkene cycle over the arene cycle in zeolite catalysis. Thus, based on the product distribution depicted in Fig. 2, it is reasonable to hypothesize that the arene cycle predominantly governs zeolite ZSM-5, whereas the alkene cycle regulates the alkane cracking mechanism over the other 10-MR zeolites.
To probe into the zeolitic topological parameters at the very early stages of alkane cracking, the operando UV-vis diffuse reflectance spectroscopy (DRS) coupled with online mass spectrometry (MS) has been employed (Fig. 3, S14 and S15†). This method proves advantageous for distinguishing between neutral and carbocation zeolite-trapped organic compounds.76–80 These distinctions can be correlated with effluent gas stream analysis to extract valuable mechanistic insights. Typically, all zeolites displayed a time-monitored increase of absorption bands, although at different extent, at around <275, 335–350, ∼420, and >590 nm regions, which typically could be attributed to π–π* transitions associated with neutral alkylbenzene molecules, dienylic carbocations or less-methylated benzenium ions (up to 3–4 alkyl groups), highly alkylated arenium molecules (e.g., hexamethylbenzenium ions, HMB+), and poly-arenium species, respectively.76–79 Curiously, during the initial stages of the reaction, all zeolites exhibited the formation of arenium-based HCP species. However, it was observed that only zeolite ZSM-5 produced a substantial amount of aromatic-based hydrocarbons in the effluent gas feed (Fig. S15†). Except MCM-22, the coke-precursor appearance has quite been evidenced over other zeolites (i.e., >590 nm bands). Nevertheless, the key descriptor role of HMB+ species could be acknowledged over all zeolites due to its consistent rise in intensity as a function of time (Fig. S14†). While diverse UV-vis profiles were acquired across various zeolites, their MS profiles exhibited significant similarities, highlighting an identical lagging period of ∼2 minutes (i.e., the time needed to identify the first hydrocarbon species under operando conditions) and a hydrocarbon product profile dominated by C2–C4 species. While the product distribution profile, as illustrated either by GC in Fig. 2 or by MS in Fig. 3, exhibited a notable degree of similarity among zeolites MCM-22, FER, and ZSM-22, their distinct UV-vis DRS profiles suggest varied reaction mechanisms. To complement our operando studies, we conducted a temperature-resolved investigation monitored by UV-vis DRS coupled with online MS (Fig. S16†). In this approach, we chemisorbed the zeolite with liquid alkane, subjected it to gradual heating, and monitored the process using UV-vis DRS and MS. The instantaneous formation of carbenium species was observed across all zeolites, aligning well with the generation of cracking products. This observation suggests that alkane cracking occurs either simultaneously or following carbenium ion formation, confirming their intra-dependent relationship in zeolite-catalyzed cracking processes. While this observation may be applicable across various zeolites, the comprehensive operando study discussed in this subsection underscores the concept that identical catalytic outcomes, including product selectivity, can be achieved through diverse mechanistic pathways across different zeolites.
 | ||
| Fig. 3 Operando mechanistic study: (a–d) time-monitored UV-vis DRS profile and (e–h) time-resolved mass-spectral profiles of key hydrocarbon effluent species during the catalytic n-hexane conversion over zeolites ZSM-5 (a and e), MCM-22 (b and f), FER (c and g) and ZSM-22 (d and h) for 20 min at 803 K. | ||
Next, 13C-based MAS solid-state NMR spectroscopy has been employed to complement the operando studies (Fig. 4 and S17†). The solid-state NMR spectroscopy was performed over post-reacted zeolite materials after the reaction for 10 minutes at 803 K using hexane-1–13C, where only the primary 1° position is selectively isotope labeled. Herein, solid-state NMR studies were performed over the zeolites ZSM-5, MCM-22, and ZSM-22 to represent 3D, 2D, and 1D zeolites, respectively. The following features were observed primarily in the respective 1H–13C cross-polarization (CP) MAS spectra: (i) 10–35 ppm aliphatic/methyl groups, and (ii) weak intensity of the 120–140 ppm region due to olefinic/aromatic groups.76,77,79 The peaks around 13 ppm, 23 ppm, and 32 ppm were attributed to primary (i.e., alkyl carbons: –![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) H3), secondary (i.e., methylene carbons: –H2–), and tertiary (i.e., methine carbons: –H
H3), secondary (i.e., methylene carbons: –H2–), and tertiary (i.e., methine carbons: –H![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) ) carbons in an aliphatic moiety, respectively (Fig. 4). Since we used selectively isotope-enriched hexane-1–13C feed in making NMR samples, the primary carbons were more intense than others. However, the appearance of signals due to secondary and tertiary carbons indicates the existence of skeletal rearrangement during the process. The aromatic region, when zoomed in [see Fig. 4(a)], displayed a minor peak around ∼130 ppm, suggesting a lower likelihood of isotope scrambling. Furthermore, under our experimental conditions, the primary isotope-enriched carbon did not contribute to the formation of Csp2 carbon. To dig deeper into the zeolite-trapped molecular scaffolds, 2D 1H–13C heteronuclear correlation (HETCOR) spectra were acquired to track the 1H–13C correlations of zeolite-trapped organics (Fig. S17†). The 13C peak at approximately 13 ppm corresponds to at least three 1H signals: (I) around 1–2 ppm, (II) about 3–3.4 ppm, and (III) approximately 6.2 ppm. These signals are indicative of saturated/unsaturated hydrocarbon analogs based on C3/C4. The existence of such a C3/C4-based hydrocarbon backbone was further supported by additional correlations: ∼32 ppm (13C)/∼32–32.5 ppm (1H) and 22–23 ppm (13C)/3.2–3.4 ppm (1H).76,77,79 Among them, the downfield correlations based on ∼13 ppm (13C)/∼6.2 ppm (1H) especially indicate unsaturated olefinic fragments, which strangely could not be observed over the zeolite MCM-22. Although we identify aliphatic-based moieties by 2D 1H–13C HETCOR correlations, we cannot rule out their attachment to aromatics/olefinic fragments. Nevertheless, we can infer a lower likelihood of the presence of aromatic/olefinic fragments over zeolite MCM-22 when compared to the other zeolites.
) carbons in an aliphatic moiety, respectively (Fig. 4). Since we used selectively isotope-enriched hexane-1–13C feed in making NMR samples, the primary carbons were more intense than others. However, the appearance of signals due to secondary and tertiary carbons indicates the existence of skeletal rearrangement during the process. The aromatic region, when zoomed in [see Fig. 4(a)], displayed a minor peak around ∼130 ppm, suggesting a lower likelihood of isotope scrambling. Furthermore, under our experimental conditions, the primary isotope-enriched carbon did not contribute to the formation of Csp2 carbon. To dig deeper into the zeolite-trapped molecular scaffolds, 2D 1H–13C heteronuclear correlation (HETCOR) spectra were acquired to track the 1H–13C correlations of zeolite-trapped organics (Fig. S17†). The 13C peak at approximately 13 ppm corresponds to at least three 1H signals: (I) around 1–2 ppm, (II) about 3–3.4 ppm, and (III) approximately 6.2 ppm. These signals are indicative of saturated/unsaturated hydrocarbon analogs based on C3/C4. The existence of such a C3/C4-based hydrocarbon backbone was further supported by additional correlations: ∼32 ppm (13C)/∼32–32.5 ppm (1H) and 22–23 ppm (13C)/3.2–3.4 ppm (1H).76,77,79 Among them, the downfield correlations based on ∼13 ppm (13C)/∼6.2 ppm (1H) especially indicate unsaturated olefinic fragments, which strangely could not be observed over the zeolite MCM-22. Although we identify aliphatic-based moieties by 2D 1H–13C HETCOR correlations, we cannot rule out their attachment to aromatics/olefinic fragments. Nevertheless, we can infer a lower likelihood of the presence of aromatic/olefinic fragments over zeolite MCM-22 when compared to the other zeolites.
 | ||
| Fig. 4 The 13C NMR solid-state spectra of zeolite-trapped organics after the hexane-1–13C conversion over zeolites ZSM-5 (black), MCM-22 (green) and ZSM-22 (blue) (MAS = 16 kHz): (a) 1D 1H–13C cross-polarization (CP) MAS spectra of all three zeolites and (b–d) 2D 1H–13C CP HETCOR MAS spectra of post-reacted zeolites (b) ZSM-5, (c) MCM-22, and (d) ZSM-22. (e) Identified molecular scaffolds (also see Fig. S17†). | ||
In order to further identify the differences between intersections and sinusoidal channels, a series of theoretical simulations were applied to quantify the interaction between the zeolite acid sites of ZSM-5 and the n-hexane reactant, together with the main representative products of interest: benzene, butane, and butene. As shown by the results presented in Table 2, the topological features of the intersection in ZSM-5 (i) enhance the interaction with their respective adsorbates to a greater extent than in the sinusoidal channel ZSM-5 (s) by 10–20 kJ mol−1. The strong benzene adsorption, in particular, further validates the increased contribution of the intersection acid sites in the aromatic cycle. We conducted these studies only on zeolite ZSM-5 due to its unique product selectivity patterns compared to other 10-MR zeolites used in this work.
| Hexane | Butene | Butane | Benzene | |
|---|---|---|---|---|
| a (i): intersection channel. (s): sinusoidal channel. | ||||
| ZSM-5 (i) | −114 | −119 | −81 | −91 |
| ZSM-5 (s) | −98 | −102 | −74 | −69 |
From the experimental findings presented in this study, the following mechanistic insights dependent on topology can be deduced: (i) zeolite ZSM-5 exclusively demonstrated the capability to produce aromatics through the catalytic cracking of n-hexane, which was unattainable by other 10-MR zeolites (Fig. 2). Consequently, it is reasonable to posit that the zeolite channels, whether straight or sinusoidal, might have had a lesser impact on the aromatization process. Instead, the intersection of straight and sinusoidal channels appears to be crucial for the generation of aromatics.81 (ii) Since the zeolitic vicinal BAS is essential to yielding aromatics, criteria could be delivered by all highly acidic zeolites used in this study; their location matters more than the zeolite topology in facilitating aromatics production. Hence, the presence of vicinal BAS sites at the intersection of zeolite ZSM-5 proves essential for promoting the arene cycle over the alkene cycle in zeolite catalysis.81,82 Although it may be premature to directly correlate the Al-pair concept to vicinal BAS, such a highly acidic commercial zeolite ZSM-5 usually poses a reasonable percentage of the Al-pair, as already verified in the literature.83–87 (iii) The uniform product profile observed across zeolites MCM-22, FER, and ZSM-22 suggests that the alkene cycle could be influential in catalysis, operating from both straight and sinusoidal channels. This preference leads to the production of propylene and higher olefins over these zeolites.88 (iv) Both UV-vis analysis (Fig. 3) and solid-state NMR studies (Fig. 4) suggest that zeolite MCM-22 exhibits the lowest probability of generating aromatic species. This implies that the efficiency of sinusoidal channels in promoting the arene cycle is limited during zeolite catalysis. (v) Our solid-state NMR studies did not reveal the presence of dimerized species, suggesting that surface alkoxide-driven oligomerization is not operative in the current case.76,89 We have summarized our topology-dependent mechanistic information in Scheme 1.
 | ||
| Scheme 1 The simplified illustration of the catalytic cracking mechanism over 10-MR zeolites employed in this study. The interconnectedness of chain initiation, propagation, and termination events during alkane activation within the framework of the dual-cycle-led hydrocarbon pool mechanism is pivotal in governing catalysis. The spacious intersectional voids of 3D zeolite ZSM-5 promote the arene cycle, whereas straight/sinusoidal channels, irrespective of zeolites, propagate the alkene cycle. The distinct preference for either the alkene or arene cycle, based on the zeolite's topological properties, dictates the ultimate product selectivity. | ||
The operando UV-vis DRS (Fig. 3) revealed the instantaneous formation of carbenium ions, indicating LAS-mediated activation of incoming alkanes by the zeolite in the initiation step.7,90 This process leads to the production of activated carbocationic complexes. The co-liberation of hydrogen, as evidenced by online MS, supports this observation (Fig. S15†).1,34 Typically, the product distribution is influenced by subsequent propagation events, with hydride transfer from the alkane reactant and alkoxide formation playing a pivotal role. While surface alkoxide species were not detected in our solid-state NMR studies due to the use of selectively isotope-labeled hexane feed during sample preparation, the identification of numerous isomeric homologs implies the occurrence of skeletal isomerization under our experimental conditions,1 indirectly confirming surface alkoxide-driven chain propagation events in the current study. However, it is worth noting that surface alkoxide-driven oligomerization is not active across all zeolites, as evidenced by the absence of signals for dimerized products in our solid-state NMR studies (Fig. 4) and the lower C5+ selectivity observed (Fig. 2).76,89 In the present study, the desorption and β-scission of the carbenium ion to generate propylene can be regarded as the chain termination step.90
In the case of 10-MR zeolites other than zeolite ZSM-5, steady-state catalysis was primarily governed by the alkene cycle. In contrast, zeolite ZSM-5 facilitated aromatization at the intersection, primarily consuming post-cracking alkene feed. These olefins were typically produced by β-scission from the alkane-derived activated carbocationic complex.1 After cracking larger alkanes into shorter olefins, oligomerization/aromatization of true-oligomers was prevented, except in the case of zeolite ZSM-5. For instance, this phenomenon could be anticipated in 1D zeolite ZSM-22, where cylindrical channels do not allow true oligomers to grow due to diffusion limitations. Similar diffusional limitation arguments could be applied to zeolites MCM-22 and FER due to the presence of sinusoidal channels and the absence of cage-like empty spaces (supercages), respectively.
Therefore, we conclude that alkene cycles were predominant over 2D and 1D 10-MR zeolites, leading to C3-based propylene as a preferential product in the catalytic cracking of C6-based hexanes. In contrast, 3D zeolite ZSM-5 contains optimum void spaces at their inter-channel connecting paths, an inherent consequence of its channel-cage-window structure. This 3D zeolite structure allows true-oligomers to diffuse better, grow, and cyclize, providing an optimum confinement effect to facilitate aromatics production. This observation indirectly justifies the superior efficiency of β-scission over 2D and 1D zeolites, restricting diffusion while simultaneously reducing the size of incoming alkane chains during the zeolite-catalyzed cracking process. The mechanistic insights derived from this study have the potential to contribute to advancements in the current state of industrial processes. For instance, in the industrial FCC process, where propylene is a minor product,7 enhancements could be achieved by introducing more diffusional limitations in the catalyst. Although the zeolite ZSM-5 is currently used as a propylene-selective FCC additive, our studies indicate that 2D/1D zeolites might be superior alternative candidates for this role due to their ability to promote the propylene-producing alkene cycle (cf.Scheme 1). This would impede the easy accessibility of true-oligomers to grow, promoting β-scission and potentially improving propylene yields by promoting the alkene cycle.
Conclusions
To unveil the topology-dependent reaction mechanism of alkane cracking over 10-MR zeolites, namely ZSM-5, MCM-22, FER, and ZSM-22, this project was conceived to gain insights into preferential post-cracking selectivity. Alkane cracking is a crucial subclass in zeolite catalysis, not only playing a significant role in FCC-like industrial processes but also constituting key reaction steps in various zeolite-catalyzed processes (such as methanol conversion) and holding immense potential for emerging catalytic technologies like biomass or plastic valorization. Upon subjecting n-hexane to cracking over 10-MR zeolites, we achieved a preferential selectivity toward propylene-dominating short alkenes and hydrogen-transferred products (paraffins and aromatics) over 2D/1D zeolites (FER, MCM-22, and ZSM-22) and 3D zeolites (ZSM-5), respectively. Subsequently, we employed a comprehensive and complementary approach to investigate topology-dependent mechanisms using traditional zeolite characterization tools, operando characterization, solid-state NMR spectroscopy, and computational analysis. This multimodal spectroscopic and theoretical approach led to the following topology-dependent insights into the reaction mechanism: the intersectional void spaces and straight/sinusoidal channels are crucial in propagating the aromatics-generating arene cycle and propylene-generating alkene cycle, respectively. Consequently, 3D zeolite ZSM-5 positions itself uniquely as a suitable candidate for yielding aromatics, a feat not achievable by 2D/1D zeolites. Conversely, this observation could be applied to enhance the FCC process, where less spacious 2D/1D zeolites play a critical role in boosting propylene selectivity. In summary, this research provides a distinctive understanding of the host–guest chemistry between inorganic zeolites and organic HCP species in the context of the catalytic alkane cracking process.Data availability
The data supporting this article have been included in the manuscript and ESI.†Author contributions
Pandong Ma: investigation, conceptualization, methodology, validation, formal analysis, data curation, writing – original draft, writing – review & editing. Hexun Zhou: investigation, conceptualization, validation. Yubing Li: investigation, validation. Mengheng Wang: investigation. Stefan Adrian F. Nastase and Mengsi Zhu: investigation, formal analysis, methodology. Jiale Cui: investigation. Luigi Cavallo: investigation, formal analysis. Kang Cheng and Abhishek Dutta Chowdhury: conceptualization, supervision, project administration, formal analysis, writing – original draft, writing – review & editing, funding acquisition.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
We are grateful for the financial support provided by the National Key Research and Development Program of the Ministry of Science and Technology (2022YFA1504500), the National Natural Science Foundation of China (NSFC) (Grant No. 22350610243), and the Fundamental Research Funds for the Central Universities (Grant No.: 2042023kf0126) (China).Notes and references
- C. Chizallet, C. Bouchy, K. Larmier and G. Pirngruber, Chem. Rev., 2023, 123, 6107–6196 CrossRef CAS PubMed
.
- A. Corma and A. V. Orchillés, Microporous Mesoporous Mater., 2000, 35, 21–30 CrossRef
- J. Van der Mynsbrugge, A. Janda, L. C. Lin, V. Van Speybroeck, M. Head-Gordon and A. T. Bell, ChemPhysChem, 2018, 19, 341–358 CrossRef CAS PubMed
- P. Del Campo, C. Martínez and A. Corma, Chem. Soc. Rev., 2021, 50, 8511–8595 RSC
- V. Blay, B. Louis, R. Miravalles, T. Yokoi, K. A. Peccatiello, M. Clough and B. Yilmaz, ACS Catal., 2017, 7, 6542–6566 CrossRef CAS
- A. Corma, Chem. Rev., 1995, 95, 559–614 CrossRef CAS
- E. T. C. Vogt and B. M. Weckhuysen, Chem. Soc. Rev., 2015, 44, 7342–7370 RSC
- G. W. Huber and A. Corma, Angew. Chem., Int. Ed., 2007, 46, 7184–7201 CrossRef CAS PubMed
- M. Stöcker, Angew. Chem., Int. Ed., 2008, 47, 9200–9211 CrossRef PubMed
- I. Vollmer, M. J. F. Jenks, M. C. P. Roelands, R. J. White, T. van Harmelen, P. de Wild, G. P. van der Laan, F. Meirer, J. T. F. Keurentjes and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2020, 59, 15402–15423 CrossRef CAS PubMed
- W. Zhang, S. Kim, L. Wahl, R. Khare, L. Hale, J. Hu, D. M. Camaioni, O. Y. Gutiérrez, Y. Liu and J. A. Lercher, Science, 2023, 379, 807–811 CrossRef CAS PubMed
- M. Jacoby, Cool fuel for hypersonic aircraft, 2018, vol. 96 Search PubMed.
- A. C. Nixon and H. T. Henderson, Ind. Eng. Chem. Prod. Res. Dev., 1966, 5, 87–92 CrossRef CAS
- J. Liang, Z. Liang, R. Zou and Y. Zhao, Adv. Mater., 2017, 29, 1701139 CrossRef PubMed
- P. D. Hopkins, J. Catal., 1968, 12, 325–334 CrossRef CAS
- A. Corma, J. Planelles, J. Sánchez-Marín and F. Tomás, J. Catal., 1985, 93, 30–37 CrossRef CAS
- Y. Ono and K. Kanae, J. Chem. Soc., Faraday Trans., 1991, 87, 663–667 RSC
- J. Dwyer and D. J. Rawlence, Catal. Today, 1993, 18, 487–507 CrossRef
- A. Corma, V. Gonzàlez-Alfaro and A. V. Orchillès, Appl. Catal., A, 1995, 129, 203–215 CrossRef CAS
- J. S. Jung, J. W. Park and G. Seo, Appl. Catal., A, 2005, 288, 149–157 CrossRef CAS
- S. Altwasser, C. Welker, Y. Traa and J. Weitkamp, Microporous Mesoporous Mater., 2005, 83, 345–356 CrossRef CAS
- A. M. Ávila, C. M. Bidabehere and U. Sedran, Chem. Eng. J., 2007, 132, 67–75 CrossRef
- J. A. Swisher, N. Hansen, T. Maesen, F. J. Keil, B. Smit and A. T. Bell, J. Phys. Chem. C, 2010, 114, 10229–10239 CrossRef CAS
- P. M. Zimmerman, D. C. Tranca, J. Gomes, D. S. Lambrecht, M. Head-Gordon and A. T. Bell, J. Am. Chem. Soc., 2012, 134, 19468–19476 CrossRef CAS PubMed
- Y. Nakasaka, T. Okamura, H. Konno, T. Tago and T. Masuda, Microporous Mesoporous Mater., 2013, 182, 244–249 CrossRef CAS
- S. M. Sadrameli, Fuel, 2016, 173, 285–297 CrossRef CAS
- A. Janda, B. Vlaisavljevich, L. C. Lin, B. Smit and A. T. Bell, J. Am. Chem. Soc., 2016, 138, 4739–4756 CrossRef CAS PubMed
- J. Van der Mynsbrugge and A. T. Bell, J. Catal., 2021, 404, 832–849 CrossRef CAS
- F. Chen, J. Hao, Y. Yu, D. guo Cheng and X. Zhan, Microporous Mesoporous Mater., 2022, 330, 111575 CrossRef CAS
- F. Bleken, W. Skistad, K. Barbera, M. Kustova, S. Bordiga, P. Beato, K. P. Lillerud, S. Svelle and U. Olsbye, Phys. Chem. Chem. Phys., 2011, 13, 2539–2549 RSC
- S. Kotrel, H. Knö and B. C. Gates, Microporous Mesoporous Mater., 2000, 35–36, 11–20 CrossRef CAS
- X. Dupain, M. Makkee and J. A. Moulijn, Appl. Catal., A, 2006, 297, 198–219 CrossRef CAS
- A. D. Chowdhury and J. Gascon, Chem Catal., 2023, 3, 100525 CrossRef CAS
- X. Gong, M. Çaǧlayan, Y. Ye, K. Liu, J. Gascon and A. Dutta Chowdhury, Chem. Rev., 2022, 122, 14275–14345 CrossRef CAS PubMed
- W. Chen, X. Yi, Z. Liu, X. Tang and A. Zheng, Chem. Soc. Rev., 2022, 51, 4337–4385 RSC
- F. C. Jentoft and B. C. Gates, Top. Catal., 1997, 4, 1–13 CrossRef CAS
- A. Brait, A. Koopmans, H. Weinstabl, A. Ecker, K. Seshan and J. A. Lercher, Ind. Eng. Chem. Res., 1998, 37, 873–881 CrossRef CAS
- P. Cnudde, K. De Wispelaere, J. Van der Mynsbrugge, M. Waroquier and V. Van Speybroeck, J. Catal., 2017, 345, 53–69 CrossRef CAS
- J. S. Buchanan, J. G. Santiesteban and W. O. Haag, J. Catal., 1996, 158, 279–287 CrossRef CAS
- B. S. Greensfelder, H. H. Voge and G. M. Good, Ind. Eng. Chem., 1949, 41, 2572–2584 Search PubMed
- W. F. Pansing, J. Phys. Chem., 1965, 69, 392–399 CrossRef CAS
- S. M. Babitz, B. A. Williams, J. T. Miller, R. Q. Snurr, W. O. Haag and H. H. Kung, Appl. Catal., A, 1999, 179, 71–86 CrossRef CAS
- A. Janda and A. T. Bell, J. Am. Chem. Soc., 2013, 135, 19193–19207 CrossRef CAS PubMed
- P. Matias, J. M. Lopes, S. Laforge, P. Magnoux, M. Guisnet and F. Ramôa Ribeiro, Appl. Catal., A, 2008, 351, 174–183 CrossRef CAS
- S. Inagaki, K. Takechi and Y. Kubota, Chem. Commun., 2010, 46, 2662–2664 RSC
- Y. Kubota, S. Inagaki and K. Takechi, Catal. Today, 2014, 226, 109–116 CrossRef CAS
- N. Afroukhteh-Langaroudi, S. Tarighi and H. A. Khonakdara, Pet. Chem., 2018, 58, 457–463 CrossRef CAS
- A. Miyaji, Y. Iwase, T. Nishitoba, N. Q. Long, K. Motokura and T. Baba, Phys. Chem. Chem. Phys., 2015, 17, 5014–5032 RSC
- C. E. Ramachandran, B. A. Williams, J. A. Van Bokhoven and J. T. Miller, J. Catal., 2005, 233, 100–108 CrossRef CAS
- L. Guan, C. Huang, D. Han, L. Zhu, Y. Mei, D. He and Y. Zu, Microporous Mesoporous Mater., 2022, 330, 111605 CrossRef CAS
- Y. Sang, Q. Jiao, H. Li, Q. Wu, Y. Zhao and K. Sun, J. Nanopart. Res., 2014, 16, 2755 CrossRef
- A. T. Townsend and J. Abbot, Energy Fuels, 1994, 8, 690–699 CrossRef CAS
- S. Teketel, W. Skistad, S. Benard, U. Olsbye, K. P. Lillerud, P. Beato and S. Svelle, ACS Catal., 2012, 2, 26–37 CrossRef CAS
- A. Ramirez, X. Gong, M. Caglayan, S. A. F. Nastase, E. Abou-Hamad, L. Gevers, L. Cavallo, A. Dutta Chowdhury and J. Gascon, Nat. Commun., 2021, 12, 5914 CrossRef CAS PubMed
- X. Gong, A. Ramirez, E. Abou-Hamad, T. B. Shoinkhorova, M. Çağlayan, Y. Ye, W. Wang, N. Wehbe, R. Khairova, A. D. Chowdhury and J. Gascon, Chem Catal., 2022, 2, 2328–2345 CrossRef CAS
-
S. Etemadi, Thesis for the Master's degree in chemistry, University of Oslo, 2014 Search PubMed
- B. Smit and T. L. M. Maesen, Nature, 2008, 451, 671–678 CrossRef CAS PubMed
- A. J. Jones, S. I. Zones and E. Iglesia, J. Phys. Chem. C, 2014, 118, 17787–17800 CrossRef CAS
- S. M. Csicsery, Zeolites, 1984, 4, 202–213 CrossRef CAS
- J. Weitkamp and S. Ernst, Stud. Surf. Sci. Catal., 1988, 38, 367–382 CrossRef
- A. Brenner and P. H. Emmett, J. Catal., 1982, 75, 410–415 CrossRef CAS
- G. B. McVicker, G. M. Kramer and J. J. Ziemiak, J. Catal., 1983, 83, 286–300 CrossRef CAS
- H. Krannila, W. O. Haag and B. C. Gates, J. Catal., 1992, 135, 115–124 CrossRef CAS
- W. O. Haag, R. M. Lago and P. B. Weisz, Faraday Discuss. Chem. Soc., 1981, 72, 317–330 RSC
- Y. W. Bizreh and B. C. Gates, J. Catal., 1984, 88, 240–243 CrossRef CAS
- G. T. Whiting, S. H. Chung, D. Stosic, A. D. Chowdhury, L. I. Van Der Wal, D. Fu, J. Zecevic, A. Travert, K. Houben, M. Baldus and B. M. Weckhuysen, ACS Catal., 2019, 9, 4792–4803 CrossRef CAS
- D. M. McCann, D. Lesthaeghe, P. W. Kletnieks, D. R. Guenther, M. J. Hayman, V. Van Speybroeck, M. Waroquier and J. F. Haw, Angew. Chem., Int. Ed., 2008, 47, 5179–5182 CrossRef CAS PubMed
- C. Wang, Q. Wang, J. Xu, G. Qi, P. Gao, W. Wang, Y. Zou, N. Feng, X. Liu and F. Deng, Angew. Chem., Int. Ed., 2016, 55, 2507–2511 CrossRef CAS PubMed
- X. Sun, S. Mueller, H. Shi, G. L. Haller, M. Sanchez-Sanchez, A. C. Van Veen and J. A. Lercher, J. Catal., 2014, 314, 21–31 CrossRef CAS
- M. Bjørgen, S. Svelle, F. Joensen, J. Nerlov, S. Kolboe, F. Bonino, L. Palumbo, S. Bordiga and U. Olsbye, J. Catal., 2007, 249, 195–207 CrossRef
- I. Yarulina, A. D. Chowdhury, F. Meirer, B. M. Weckhuysen and J. Gascon, Nat. Catal., 2018, 1, 398–411 CrossRef CAS
- S. Svelle, F. Joensen, J. Nerlov, U. Olsbye, K. P. Lillerud, S. Kolboe and M. Bjørgen, J. Am. Chem. Soc., 2006, 128, 14770–14771 CrossRef CAS PubMed
- M. Bjørgen, F. Joensen, K. P. Lillerud, U. Olsbye and S. Svelle, Catal. Today, 2009, 142, 90–97 CrossRef
- X. Sun, S. Mueller, Y. Liu, H. Shi, G. L. Haller, M. Sanchez-Sanchez, A. C. Van Veen and J. A. Lercher, J. Catal., 2014, 317, 185–197 CrossRef CAS
- Z. Ristanović, A. D. Chowdhury, R. Y. Brogaard, K. Houben, M. Baldus, J. Hofkens, M. B. J. Roeffaers and B. M. Weckhuysen, J. Am. Chem. Soc., 2018, 140, 14195–14205 CrossRef PubMed
- A. D. Chowdhury, A. L. Paioni, K. Houben, G. T. Whiting, M. Baldus and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2018, 57, 8095–8099 CrossRef CAS PubMed
- S. P. Verkleij, G. T. Whiting, S. P. Esclapez, M. M. Mertens, A. J. Bons, M. Burgers and B. M. Weckhuysen, Catal. Sci. Technol., 2018, 8, 2175–2185 RSC
- Y. Ye, E. Abou-Hamad, X. Gong, T. B. Shoinkhorova, A. Dokania, J. Gascon and A. D. Chowdhury, Angew. Chem. Int. Ed., 2023, 62, e202303124 CrossRef CAS PubMed
- A. D. Chowdhury, K. Houben, G. T. Whiting, S. H. Chung, M. Baldus and B. M. Weckhuysen, Nat. Catal., 2018, 1, 23–31 CrossRef CAS
- T. Liang, J. Chen, Z. Qin, J. Li, P. Wang, S. Wang, G. Wang, M. Dong, W. Fan and J. Wang, ACS Catal., 2016, 6, 7311–7325 CrossRef CAS
- T. Li, S. H. Chung, S. Nastase, A. Galilea, Y. Wang, I. Mukhambetov, M. Zaarour, J. C. Navarro de Miguel, J. Cazemier, A. Dokania, L. Panarone, J. Gascon, L. Cavallo and J. Ruiz-Martínez, Chem Catal., 2023, 3, 100540 CrossRef CAS
- J. Dedecek, V. Balgová, V. Pashkova, P. Klein and B. Wichterlová, Chem. Mater., 2012, 24, 3231–3239 CrossRef CAS
- S. Wang, P. Wang, Z. Qin, Y. Chen, M. Dong, J. Li, K. Zhang, P. Liu, J. Wang and W. Fan, ACS Catal., 2018, 8, 5485–5505 CrossRef CAS
- J. Dědeček, D. Kaucký, B. Wichterlová and O. Gonsiorová, Phys. Chem. Chem. Phys., 2002, 4, 5406–5413 RSC
- S. Kim, G. Park, M. H. Woo, G. Kwak and S. K. Kim, ACS Catal., 2019, 9, 2880–2892 CrossRef CAS
- J. Dědeček, Z. Sobalík and B. Wichterlová, Catal. Rev.: Sci. Eng., 2012, 54, 135–223 CrossRef
- S. Wang, Z. Li, Z. Qin, M. Dong, J. Li, W. Fan and J. Wang, Chin. J. Catal., 2021, 42, 1126–1136 CrossRef CAS
- M. L. Sarazen, E. Doskocil and E. Iglesia, ACS Catal., 2016, 6, 7059–7070 CrossRef CAS
- W. Chen, M. Huang, X. Yi, Y. Hui, P. Gao, G. Hou, A. G. Stepanov, Y. Qin, L. Song, S. Bin Liu, Z. Chen and A. Zheng, Chem Catal., 2023, 3, 100503 CrossRef CAS
- M. N. Mazar, S. Al-Hashimi, M. Cococcioni and A. Bhan, J. Phys. Chem. C, 2013, 117(45), 23609–23620 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc00603h |
| This journal is © The Royal Society of Chemistry 2024 |