
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Questing for homoleptic mononuclear manganese complexes with monodentate O-donor ligands†
Alberto
Pérez-Bitrián
 *ab,
Julen
Munárriz
*c,
Konstantin B.
Krause
b,
Johanna
Schlögl
a,
Kurt F.
Hoffmann
a,
Johanna S.
Sturm
a,
Amiera N.
Hadi
a,
Christian
Teutloff
d,
Anja
Wiesner
a,
Christian
Limberg
b and
Sebastian
Riedel
*a
*ab,
Julen
Munárriz
*c,
Konstantin B.
Krause
b,
Johanna
Schlögl
a,
Kurt F.
Hoffmann
a,
Johanna S.
Sturm
a,
Amiera N.
Hadi
a,
Christian
Teutloff
d,
Anja
Wiesner
a,
Christian
Limberg
b and
Sebastian
Riedel
*a
aFachbereich Biologie, Chemie, Pharmazie, Institut für Chemie und Biochemie − Anorganische Chemie, Freie Universität Berlin, Fabeckstraße 34/36, Berlin 14195, Germany. E-mail: alberto.perez-bitrian@hu-berlin.de; s.riedel@fu-berlin.de
bInstitut für Chemie, Humboldt-Universität zu Berlin, Brook-Taylor-Straße 2, Berlin 12489, Germany
cDepartamento de Química Física and Instituto de Biocomputación y Física de Sistemas Complejos (BIFI), Universidad de Zaragoza, Pedro Cerbuna 12, Zaragoza 50009, Spain. E-mail: julen@unizar.es
dFachbereich Physik, Freie Universität Berlin, Arnimallee 14, Berlin 14195, Germany
First published on 5th March 2024
Abstract
Compounds containing Mn–O bonds are of utmost importance in biological systems and catalytic processes. Nevertheless, mononuclear manganese complexes containing all O-donor ligands are still rare. Taking advantage of the low tendency of the pentafluoroorthotellurate ligand (teflate, OTeF5) to bridge metal centers, we have synthesized two homoleptic manganese complexes with monomeric structures and an all O-donor coordination sphere. The tetrahedrally distorted MnII anion, [Mn(OTeF5)4]2−, can be described as a high spin d5 complex (S = 5/2), as found experimentally (magnetic susceptibility measurements and EPR spectroscopy) and using theoretical calculations (DFT and CASSCF/NEVPT2). The high spin d4 electronic configuration (S = 2) of the MnIII anion, [Mn(OTeF5)5]2−, was also determined experimentally and theoretically, and a square pyramidal geometry was found to be the most stable one for this complex. Finally, the bonding situation in both complexes was investigated by means of the Interacting Quantum Atoms (IQA) methodology and compared to that of hypothetical mononuclear fluoromanganates. Within each pair of [MnXn]2− (n = 4, 5) species (X = OTeF5, F), the Mn–X interaction is found to be comparable, therefore proving that the similar electronic properties of the teflate and the fluoride are also responsible for the stabilization of these unique species.
Introduction
Manganese is a key element in biological systems,1 being particularly relevant in the photosynthesis,2–6 as well as in a diversity of catalytic processes.7–13 One of the facts that makes it especially interesting is the wide range of oxidation states that it can present, varying from −III to +VII.14 Whereas high oxidation states are stabilized by oxo ligands, as in the [MnO4]− ion or in the binary Mn2O7, fluoride is only able to stabilize medium oxidation states.15–17 In fact, manganese fluorides are only known up to oxidation state +IV in MnF4 and in the related [MnF6]2−, although oxidation state +VII is attained in the oxyfluoride MnO3F.18Compounds containing Mn–O bonds are involved in catalytic and enzymatic reactions.19–22 Notably, the chemistry of manganese complexes with oxygen ligands is mainly dominated by polymetallic species, including oxo ligands in the higher oxidation states, whereas alkoxides or carboxylates are the preferred ligands for lower oxidation states.14,23,24 To prevent aggregation and enable the formation of mononuclear complexes, bulky alkoxide ligands, as well as fluorinated ones, constitute suitable ligand scaffolds.25,26 In this regard, the pentafluoroorthotellurate group (teflate, OTeF5) also offers unique possibilities, as it provides an O-donor ligand system with a low tendency to bridge metal centers.27,28 Its electron-withdrawing properties, similar to those of fluoride, made us envision the possibility of using this monodentate ligand for the synthesis of unprecedented homoleptic mononuclear manganese compounds containing Mn–O bonds, which would be analogues of the well-studied low-valent fluoromanganates.18 Here, we report the synthesis of two different manganese teflate complexes in oxidation states +II and +III, i.e., [Mn(OTeF5)4]2− and [Mn(OTeF5)5]2−, and the investigation of their structural and electronic properties by means of a combined experimental and theoretical approach.
Results and discussion
Synthesis and characterization of [NEt4]2[Mn(OTeF5)4]
The manganese(II) compound [NEt4]2[Mn(OTeF5)4] (1) can be synthesized by reacting [NEt4]2[MnCl4] with AgOTeF5 in CH2Cl2 and after removal of the formed AgCl via filtration it is isolated as an off-white solid (Scheme 1). The product exhibits a similar IR spectrum to the related [NEt4]2[M(OTeF5)4] compounds (M = Ni, Co, Fig. S2†),29,30 indicating the four-coordinate nature of the manganate anion. The Te–O vibration observed at 854 cm−1 signifies the ionic nature of the Mn–OTeF5 bond.31 | ||
| Scheme 1 Synthetic routes to complexes [Mn(OTeF5)4]2− and [Mn(OTeF5)5]2−. The cation is [NEt4]+ in all cases. | ||
Despite numerous attempts under different conditions, only intergrown and highly twinned crystals of compound 1 could be obtained, which were not suitable for single-crystal X-ray diffraction. Gratifyingly, the use of a different cation, namely [PPh4]+, allowed the preparation and growth of single crystals of [PPh4]2[Mn(OTeF5)4] (1*). Compound 1* crystallizes in the tetragonal space group I41/a (see the ESI† for details). The [Mn(OTeF5)4]2− anion (Fig. 1a), which appears well separated from the [PPh4]+ cations, exhibits a distorted tetrahedral geometry, similar to those observed for the related [M(OTeF5)4]2− anions (M = Ni, Co).29,30 Nevertheless, the distortion at the Mn(II) center is much less pronounced in this case, with a geometry index of τ4 = 0.96.32
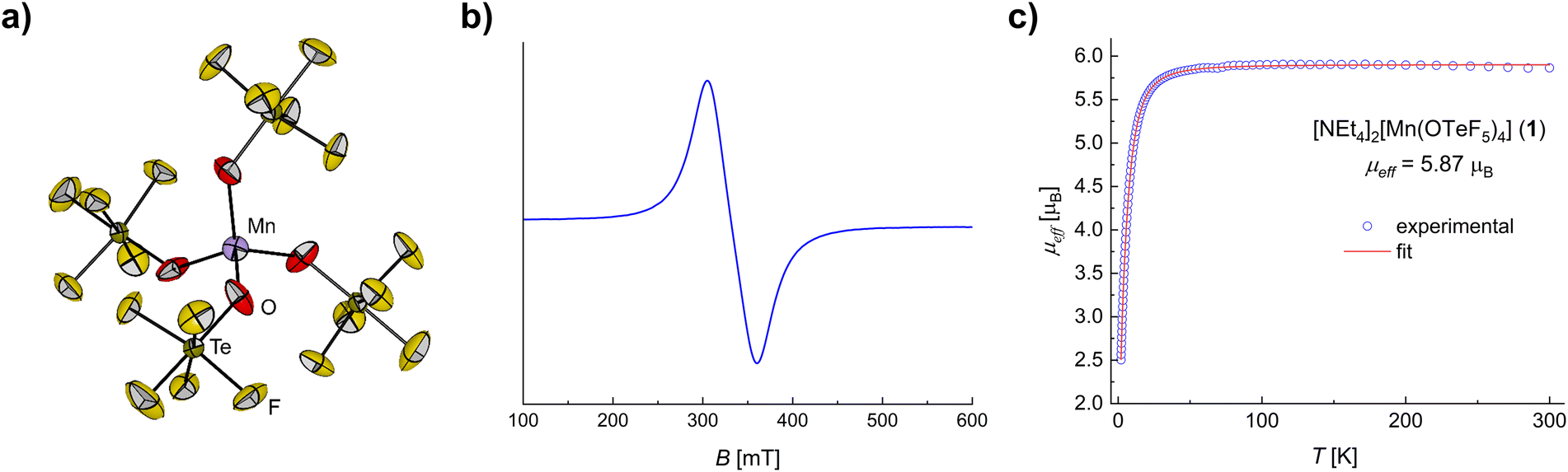 | ||
| Fig. 1 (a) Molecular structure of the [Mn(OTeF5)4]2− anion in the solid state as found in crystals of 1*. The [PPh4]+ cations have been omitted for clarity. Displacement ellipsoids are set at 50% probability. Selected bond lengths [pm] and angles [°]: Mn–O 202.1(3), O–Mn–O 112.23(11)/104.1(2). For crystallographic details see the ESI.† (b) X-band EPR spectrum of 1 in DCM (5.0 mM) at 293 K. The spectrum yields geff = 2.02. The rather high g-value is attributed to the contribution of the zero-field splitting, and the unresolved lines to a yet unknown broadening mechanism. (c) Experimental μeffversus T plot and fit for compound 1. | ||
The combination of manganese and the teflate ligand was already known, yet only with manganese in oxidation state +I in compound [Mn(CO)5(OTeF5)].33–35 The uniqueness of the [Mn(OTeF5)4]2− anion lies in the presence of four single Mn–O bonds, compound 1* actually entailing the first example of a homoleptic mononuclear Mn(II) species with four monodentate O-donor ligands. A compound close to this situation is the coordination polymer [Mn(pyc)4(μ2-SO4)·H2O]∞ (pyc = 4-carboxy-1-methylpyridinium), where each Mn(II) center is coordinated by four pyc ligands in an almost square planar arrangement, but these subunits are connected by η4,μ2-SO4 bridges.36 Noteworthily, in this context, compound [MnII(pinF)2]2− (pinF = perfluoropinacolate) was recently reported, containing two chelating ligands.37 This compound exhibits a pseudotetrahedral geometry around the metal center with a much more prominent distortion (τ4 = 0.43) than the anion in compound 1* (Fig. 1a), probably because of the two chelating ligands. In our case, a perfectly tetrahedral geometry at the metal center should be expected, yet it is slightly distorted probably due to steric reasons. These structures are unusual, as most of the known manganese(II) complexes with O-donors are heteroleptic, ranging from mono- and dinuclear coordination compounds to clusters of different sizes, or even polymeric chain structures.14,23,24 In this regard, also Mn(II) complexes containing two alkoxido or aryloxido ligands and additional solvent molecules are known.38–40 In our case, the use of a non-donor solvent has further helped isolate the homoleptic species, which in conjunction with the low tendency of the teflate to bridge metal centers gives rise to the monomeric nature of the compound.
The existence of a Mn(II) center within the anion [Mn(OTeF5)4]2− (see the ESI† for bond valence sum analyses) was confirmed via electron paramagnetic resonance (EPR) spectroscopy. The X-band EPR spectrum of 1 recorded at room temperature in CH2Cl2 (Fig. 1b) does not show six distinct lines, as would be expected for an I = 5/2 nucleus. Only the corresponding W-band spectrum (Fig. S10†) reveals the expected hyperfine splitting and gives giso = 2.000 (A(55Mn) = 255 MHz), as expected for a high spin (HS), mononuclear Mn(II) complex.41 We attribute the unresolved hyperfine splitting at the X-band to a yet unknown broadening mechanism involving the metal center and the [OTeF5]− ligand.
Due to the stabilization of the half-filled d shell and the low charge of the metal center, typically Mn(II) complexes exhibit a HS configuration (S = 5/2),42 Mn(II) centers in low spin (LS) configurations being very scarce.14,23 Additionally, because of the weak/medium-field character of the teflate ligand, a HS is expected for 1 all the more.29 In line with this, 1 was determined to have an effective magnetic moment of μeff = 5.87 μB (Fig. 1c), being very close to the spin-only value of a d5 HS system, i.e., μs.o. = 5.92 μB. This excellent agreement can be explained by the lack of orbital contribution to the magnetic moment due to the HS electronic configuration leading to a 6S ground term,43 similarly to the [MnX4]2− anions (X = Cl, Br, I).44 The magnetic data were successfully simulated, as shown in Fig. 1c and the fit parameters for 1 are reported in the ESI.† Typically the magnetic anisotropy of a HS Mn(II) center is characterized by a small zero-field splitting (corresponding D values < 1 cm−1)45 and indeed a D = 0.62 cm−1 was inferred for 1.
To further understand the nature of the [Mn(OTeF5)4]2− anion, we investigated its electronic structure by means of a theoretical study. First, we performed a DFT structure optimization of the sextet ground state by using B3LYP-D3BJ, M06, M06-L and TPPSh functionals. All of them provided similar geometries with structural parameters τ4 = 0.99 (B3LYP-D3BJ, M06 and TPPSh) or τ4 = 0.98 (M06-L), which are in good agreement with the experimental one (vide supra). Given the problems that might arise when studying the electronic structure of first–row transition metals by means of DFT,46–49 we also applied multi-reference calculations. Namely, on the B3LYP-D3BJ-optimized structure, we combined the state-average complete active space self-consistent field (SA-CASSCF) that accounts for static electron correlation,50 with n-electron valence state perturbation theory (NEVPT2)51–53 to account for dynamic electron correlation. In the active space, we incorporated the Mn–O bonding orbitals based on the 3d orbitals, along with the primarily 3d orbitals of the metal and the corresponding 4d orbitals, to accurately consider the effects of double-d shell correlation.54–56 As a result, we obtained an active space composed of 11 electrons in 13 molecular orbitals, SA-CASSCF(11,13)/NEVPT2 (Fig. S11†). As anticipated, the ground state corresponds to the sextet based on the 6A1 term, where the five 3d orbitals of the manganese are partially occupied. Note that this configuration has a weight of 98.8%, which allows for the consideration that the sextet ground state has a prominent single-reference character. The first excited state would correspond to a quartet that lies 286 kJ mol−1 above in energy (see Table S6† for the full set of states). Overall, this picture justifies the use of DFT.
Synthesis and characterization of [NEt4]2[Mn(OTeF5)5]
The reaction of [NEt4]2[MnCl4] with ClOTeF5 takes place with oxidation of the Mn(II) center to Mn(III) and coordination of five teflate ligands to the metal center (Scheme 1). Compound [NEt4]2[Mn(OTeF5)5] (2) is selectively formed as a deep blue solid, whereas Cl2 is released as a yellow gas. The role of ClOTeF5 as an oxidizer towards various metals has been previously reported, e.g. in the preparation of [Mo(OTeF5)6]57 or [ReO(OTeF5)4].58 This behavior is in contrast with the synthesis of the related [NEt4]2[MII(OTeF5)4] salts (M = Ni, Co), in which the oxidation state and coordination number of the metals remain unaltered upon reaction of the corresponding [NEt4]2[MIICl4] salt with neat ClOTeF5.29,30 Interestingly, compound 2 can also be obtained by oxidation of 1 with ClOTeF5, which is advantageous, as a much lower amount of hypochlorite is needed. In this case, the IR spectrum (see Fig. S4†) exhibits a broad band at 827 cm−1 for the Te–O vibrations, denoting the ionic nature of the Mn–OTeF5 bond also in the Mn(III) species.31All attempts to prepare crystals of 2 proved to be of an overall low quality, resulting in an aggregation of different components that made it difficult to treat the structure as a non-merohedral twin, additionally characterized by severe disorder. Unfortunately, although the use of the [PPh4]+ cation seemed promising after the successful crystallization of 1*, this cation proved to be unstable in the presence of ClOTeF5. Therefore, only the connectivity in compound 2 could be established from our crystallographic analysis. Compound 2 consists of two [NEt4]+ cations and the [Mn(OTeF5)5]2− anion, without any significant interaction among them. Interestingly, the [Mn(OTeF5)5]2− anion shows a monomeric structure with a {MnO5} core displaying an overall square pyramidal geometry (Fig. 2). Such an isolated core in a homoleptic complex with monodentate ligands is without precedence in the literature: the vast majority of species containing {MnO5} cores are clusters and polynuclear species, including both homo- and heterometallic compounds. Mononuclear representatives contain either chelating ligands37,59–65 or solvent molecules66,67 completing the coordination sphere, yet none of them has the same O-donor ligand occupying the five coordination sites around the manganese center.
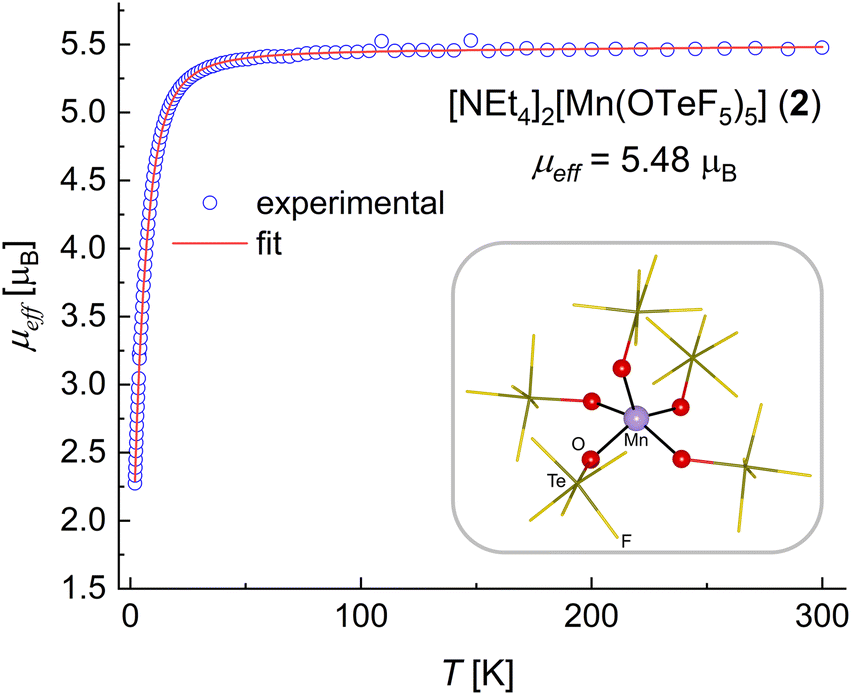 | ||
| Fig. 2 Experimental μeffversus T plot and fit for compound 2. A schematic representation of the [Mn(OTeF5)5]2− anion in the solid state is shown inside the frame, with the first coordination sphere of the Mn(III) center highlighted. | ||
An effective magnetic moment μeff = 5.48 μB was determined for compound 2 (Fig. 2). This is higher than the spin-only value of μs.o. = 4.90 μB expected for four unpaired electrons at a Mn(III) center (S = 2) and also than the value found in the structurally related68–70 [MnCl5]2− anion.71–73 Nevertheless, it is comparable with the magnetic moments determined for other square pyramidal dianionic {MnIIIO5} complexes,37 and it clearly demonstrates a high-spin configuration that is in line with the weak/medium-field character of the teflate ligand.29 The fact that 2 contains a Mn center in the oxidation state +III was further confirmed by the results of a bond valence sum analysis (see the ESI†).
To gain further insights into the geometry of the [Mn(OTeF5)5]2− anion beyond the limitations of our crystallographic data, we undertook a theoretical analysis. As five-coordinate transition-metal complexes can exhibit two different geometries: square pyramidal (SPY-5) and trigonal bipyramidal (TBPY-5),74 we considered both structural possibilities for our calculations. All the optimizations converged to the experimentally observed square pyramid, regardless of the starting structure. Interestingly, we found two intimately related, although slightly different, SPY-5 structures, whose energy difference is lower than 1.0 kJ mol−1 for the B3LYP-based methodology. In order to obtain a structure with the TBPY-5 geometry, geometrical constraints had to be imposed (see the ESI† for additional details). Nevertheless, the obtained TBPY-5 structure (for B3LYP-D3BJ) is almost isoenergetic, being only 6.4 kJ mol−1 higher in energy. In general, the same trend is observed when using M06-L, M06 and TPSSh (Table S9†). Both optimized structures at B3LYP-D3BJ, with indicated bond lengths, calculated geometry indices τ5,75 and relative energies are shown in Fig. 3.
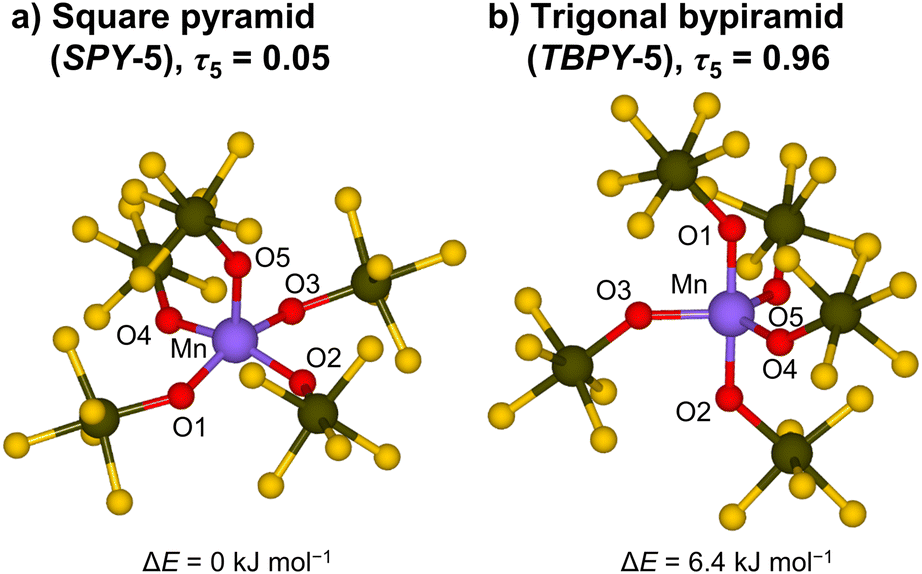 | ||
| Fig. 3 DFT structures of the [Mn(OTeF5)5]2− anion optimized at the B3LYP-D3BJ level of theory and calculated relative energies. (a) Square pyramidal structure. Selected bond lengths [pm]: Mn–O1 191.07, Mn–O2 190.79, Mn–O3 191.18, Mn–O4 190.01, and Mn–O5 201.41. (b) Trigonal bipyramidal structure, obtained after imposition of geometrical constraints. Selected bond lengths [pm]: Mn–O1 188.60, Mn–O2 188.15, Mn–O3 196.00, Mn–O4 196.38, and Mn–O5 197.36. | ||
Furthermore, although the HS d4 electronic configuration of our system is the most common one for the electronic ground state of a Mn(III) center,42 the existence of complexes exhibiting spin crossover (SCO) that involve an intermediate spin (IS) configuration (S = 1, two unpaired electrons) or even with the LS configuration (S = 0) has been reported for some systems.76–84 Therefore, despite SCO not being experimentally observed for compound 2, we optimized the structure at the DFT level (by using the same functionals as for the Mn(II) species), considering not only the HS state as described above but also the IS and LS states. While there was significant dispersion in the relative energies of the various spin states, all the functionals revealed a HS quintet ground state. The energy difference to the IS state is in the range 94.7–188.5 kJ mol−1 (depending on the functional), being 94.7 kJ mol−1 for TPSSh, which has generally been ranked as a suitable choice for the study of the electronic structure of Mn complexes.47,80,85 Note that this value is close to the B3LYP one, which is 112.8 kJ mol−1.
As a further check of consistency we applied multi-reference SA-CASSCF(12,14)/NEVPT2 calculations on the global minimum structure. Consistent with the calculations performed for the [Mn(OTeF5)4]2− anion, our active space comprised Mn–O bonding orbitals that exhibit substantial involvement of Mn 3d orbitals. Additionally, we considered the five 3d-based orbitals that prominently incorporate Mn and the corresponding 4d orbitals in order to account for the double-shell effect (Fig. S12†). According to the NEVPT2 calculations, a quintet ground state was determined, where the Mn 3dx2−y2 orbital is unoccupied (Fig. 4). This specific configuration holds a weight of 96.6%, indicating that the state can be described as a single-reference state in broad terms. All other states are provided in Table S7,† in which it can be seen that the lowest triplet state is 217.8 kJ mol−1 higher in energy, and this difference increases to 333.6 kJ mol−1 for the lowest singlet. The HS quintet state is also in agreement with the weak/medium-field character of the teflate ligand,29 as well as with our magnetic measurements (vide supra).
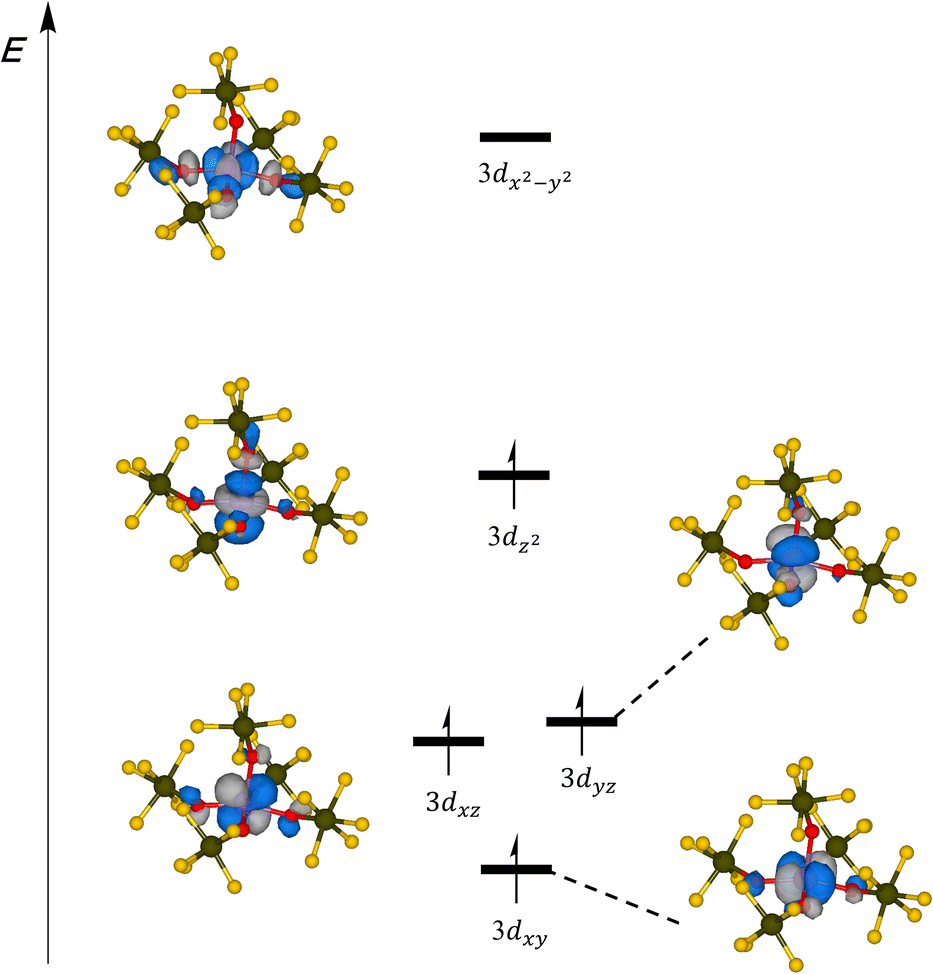 | ||
| Fig. 4 Schematic representation of the energy diagram of the SA-CASSCF(12,14) MOs mainly composed of Mn 3d orbitals for the [Mn(OTeF5)5]2− anion. | ||
Chemical bonding analyses
Compounds 1 and 2 represent unique examples of homoleptic mononuclear low-valent manganese compounds with all monodentate O-donor ligands. These features are enabled by the electronic similarities of the fluoride and teflate ligands, together with the limited tendency of the teflate to bridge metal centers.27,28 In fact, when compared to the corresponding fluoride analogues, it is the latter reason that hinders the formation of extended structures in the solid state, contrary to [MnF4]2−, which exists as layers, or [MnF5]2−, which forms chains.18With the aim of comparing the bonding mode of such electronically similar compounds, namely [Mn(OTeF5)n]2− (n = 4, 5) and the corresponding hypothetical monomeric [MnFn]2− (n = 4, 5), we undertook a bonding analysis by means of the Interacting Quantum Atoms (IQA) energy decomposition scheme,86 which we have previously applied to related [CoX4]2− complexes (X = OTeF5, F, Cl).30 IQA is an orbital-invariant and parameter-free approach that applies a scalar topological partition to divide the space into regions associated with chemically meaningful entities. As it is customarily performed, we coupled IQA with the partition of space provided by the Quantum Theory of Atoms in Molecules (QTAIM).87 This way, the space is divided into different atoms. Within this framework, the total energy is divided into intra-atomic and inter-atomic contributions between pairs of atoms or groups of atoms (say A and B). The latter term (EABinter) can be further decomposed into a classical electrostatic (VABcl) and an exchange-correlation contribution (VABxc), which is directly related to bond covalency.88–90 It should be noted that VABxc has also been proposed as a direct measure of bond strength, as the classical (Coulomb) interaction is significantly affected by long–range interactions between highly charged groups.91 Herein, we considered the interaction between the manganese center and a given group (X) that might be a single atom, as in the case of F in [MnFn]2−, or the combination of various atoms, as the teflate ligand in [Mn(OTeF5)n]2−. For the latter, its interaction with the metal is obtained by adding all the pairwise interactions between it and each of the atoms belonging to the group.
The classical (ionic) and exchange-correlation (covalent) interaction terms for [Mn(OTeF5)4]2− and [MnF4]2− are provided in Table 1. They are referred to as VMXcl and VMXxc, as they account for the interaction between the metal center (M) and the ligand group (X). For comparison purposes, the Co analogues are also provided.30 The VMXxc term for [Mn(OTeF4)4]2− (−251.0 kJ mol−1) is comparable to that of [MnF4]2− (−233.1 kJ mol−1), which is in line with the similar covalent character of both ligands. In this regard, it is noticeable that, albeit they are still quite similar, the difference between these terms (17.9 kJ mol−1) is smaller than that for the Co-based compounds (28.6 kJ mol−1), and that the covalent interaction in [Mn(OTeF4)4]2− is significantly weaker than that for [Co(OTeF4)4]2− (−251.0 vs. −293.3 kJ mol−1).
Electrostatic interactions also deserve a special comment. Given the lower electronegativity of Mn with respect to Co, it is somehow evident that the charge of the Mn center should be higher than that of Co, which is indeed an observed fact (see Table 1). In this line, the classical electrostatic M–OTeF5 interaction is more favorable for [Mn(OTeF5)4]2− than for [Co(OTeF5)4]2− (−705.4 vs. −617.4 kJ mol−1, respectively), which compensates for the decrease in the covalent interaction term.
We now consider the Mn(III) anion. First, we checked if the structure of the hypothetical monomeric [MnF5]2− anion is the same as for [Mn(OTeF5)5]2−. Surprisingly, the global minimum for [MnF5]2− is represented by a TBPY-5 structure instead of the SPY-5 geometry of the [Mn(OTeF5)5]2− anion. Nonetheless, the energy difference between both structures is, as in the case of the teflate compound, very small. Namely, the square pyramidal structure is, at the B3LYP-D3BJ level, 1.4 kJ mol−1 higher in electronic energy and exhibits an imaginary frequency of 9i cm−1 that corresponds to the transition to the TBPY-5 structure via Berry pseudorotation. Note that comparable results were obtained for the other functionals (see Table S13† and additional explanations provided in the ESI†).
With this in hand, we proceed to analyze the energetics of the Mn–OTeF5 interaction in the [Mn(OTeF5)5]2− anion. The VMnXxc term of the IQA framework for the SPY-5 structure is significantly larger (in absolute value) for the bond with the basal teflates than for the apical one (−330.3 and −248.8 kJ mol−1, respectively). This fact is somehow expected, as the Mn–O bond length with the apical oxygen in the SPY-5 structure (201.41 pm) is much longer than that with the basal oxygen atoms (190.76 pm av.), as can be seen in Fig. 3. In the same line, the interaction energy for the axial Mn–O bonds in the TBPY-5 structure is more favorable than that for the equatorial bonds (−352.6 and −287.4 kJ mol−1, respectively), as also anticipated from the shorter Mn–O bonds (Fig. 3) in the axial positions (188.38 pm av.) than in the equatorial ones (196.58 pm av.). When comparing the VMnXxc term for both ligands (X = OTeF5, F), a similar covalent character of the interaction is observed in both cases, yet slightly more favorable for the teflate (about 15 kJ mol−1 at the maximum, Table 2).
| [MnX5]2− | V MnXxc | q (Mn)a | q (X)a | ||
|---|---|---|---|---|---|
| Geometry | Position | X = OTeF5 | X = F | ||
| a Charges correspond to [Mn(OTeF5)5]2−, while values in parentheses are those of [MnF5]2−. | |||||
| SPY-5 | Basal | −330.3 | −315.5 | 1.90 (1.96) | −0.76 (−0.78) |
| Apical | −248.8 | −240.4 | −0.86 (−0.85) | ||
| TBPY-5 | Axial | −352.6 | −339.5 | 1.90 (1.97) | −0.74 (−0.76) |
| Equatorial | −287.4 | −271.1 | −0.81 (−0.82) | ||
Finally, the charge of the Mn center is almost the same for the teflate and the fluoride species within each set of compounds, namely the Mn(II) and the Mn(III) complex anions, which is in agreement with the similar electronegative character of both groups.27,28,30 This way, for the [MnX4]2− anions, the charge of the Mn is 1.58 |e−| when X = OTeF5 and 1.54 |e−| when X = F, similarly to what happens in the Co-based systems (Table 1). In this line, when comparing both Mn(III) species (Table 2), the Mn has a charge of 1.90 |e−| in [Mn(OTeF5)5]2− (for both geometries) and ca. 1.96 |e−| in [MnF5]2−. All in all, the teflate ligand causes similar effects on the manganese center as the fluoride, therefore allowing a similar stabilization of the oxidation states +II and +III.
Conclusions
In this work, two unprecedented motifs in the coordination chemistry of manganese with all identical monodentate O-donor ligands are reported and their characterization and properties are investigated by means of theory and experiment. The reaction of [MnCl4]2− with AgOTeF5 results in the MnII anion [Mn(OTeF5)4]2−, which displays a distorted tetrahedral structure in the solid state. The nature of the Mn(II) center was investigated by EPR spectroscopy and magnetic susceptibility measurements, indicating a high spin d5 electronic configuration (S = 5/2). Additionally, DFT and SA-CASSCF/NEVPT2 calculations show a pseudo-6A1 sextet as the ground state, which is single-reference. On the other hand, when [MnCl4]2− or [Mn(OTeF5)4]2− is reacted with ClOTeF5, oxidation of the manganese center takes place to yield the MnIII anion [Mn(OTeF5)5]2−. This species exhibits preferentially a square pyramidal geometry instead of a trigonal bipyramidal one and contains a Mn(III) center with a high spin d4 electronic configuration (S = 2), as determined experimentally and backed by theoretical calculations. A bond analysis through the IQA energy decomposition scheme in these [Mn(OTeF5)n]2− anions (n = 4, 5) was performed and shows that, in comparison with the hypothetical mononuclear fluoromanganates, the Mn–OTeF5 interactions are slightly stronger than the Mn–F ones. Additionally, the charge in the Mn center is virtually the same in both [MnIIX4]2− analogues (X = OTeF5, F), as well as in the [MnIIIX5]2− pair of complexes (X = OTeF5, F).The teflate ligand is known to exhibit similar electronic properties to fluoride, but normally leads to mononuclear species, i.e., it is much less prone to bridge metal centers.27–30 In fact, it is this combination of properties that has enabled the formation of the unique compounds reported in this work. In this regard, fluoride is only able to stabilize medium oxidation states of manganese,15–17 something also shown now to be possible for the teflate, yet incorporating a novel homoleptic coordination environment of all O-donor monodentate ligands.
Data availability
Data supporting this manuscript are available in the ESI† and also upon request.Author contributions
A. P.-B., J. S. S. and A. N. H. performed synthetic work and collected vibrational spectroscopy data. A. P.-B. interpreted the spectroscopic data. J. M. performed and interpreted theoretical calculations. K. B. K. and C. L. performed and analysed magnetic susceptibility measurements. J. S. and C. T. performed and analysed EPR measurements. K. F. H. and A. W. collected the XRD data and solved and refined the crystal structures. A. P.-B. and S. R. conceptualized and coordinated the project. A. P.-B. and J. M. wrote the manuscript. All authors discussed and commented on the manuscript. A. P.-B., J. M. and S. R. revised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) − Project-ID 387284271 − SFB 1349, as well as through the cluster of excellence “UniSysCat” funded by the DFG under Germany's Excellence Strategy-EXC2008/1-390540038. The assistance provided by the Core Facility BioSupraMol, supported by the DFG, is also acknowledged. A. P.-B. thanks the Alexander von Humboldt Foundation for a postdoctoral research fellowship, as well as the Department of Biology, Chemistry, and Pharmacy of the Freie Universität Berlin for a Rising Star Junior Fellowship. J. M. acknowledges financial support from the Spanish MICINN (grant PID2021-122763NBI00). J. S. acknowledges the Verband der Chemischen Industrie (VCI) for providing PhD funding (Kekulé Fellowship).Notes and references
- E. Salomon, N. Keren, M. Kanteev and N. Adir, in The Chemistry of Organomanganese Compounds, Belonging to Patai's Chemistry of Functional Groups, ed. I. Marek and Z. Rappoport, Wiley, Chichester, 2011, pp. 289–304 Search PubMed.
- T. J. Meyer, M. H. V. Huynh and H. H. Thorp, The possible role of proton-coupled electron transfer (PCET) in water oxidation by photosystem II, Angew. Chem., Int. Ed., 2007, 46, 5284–5304 CrossRef CAS PubMed.
- J. P. McEvoy and G. W. Brudvig, Water-splitting chemistry of photosystem II, Chem. Rev., 2006, 106, 4455–4483 CrossRef CAS PubMed.
- R. D. Britt, K. A. Campbell, J. M. Peloquin, M. L. Gilchrist, C. P. Aznar, M. M. Dicus, J. Robblee and J. Messinger, Recent pulsed EPR studies of the photosystem II oxygen-evolving complex: implications as to water oxidation mechanisms, Biochim. Biophys. Acta, Bioenerg., 2004, 1655, 158–171 CrossRef CAS PubMed.
- S. Mukhopadhyay, S. K. Mandal, S. Bhaduri and W. H. Armstrong, Manganese clusters with relevance to photosystem II, Chem. Rev., 2004, 104, 3981–4026 CrossRef CAS PubMed.
- V. K. Yachandra, K. Sauer and M. P. Klein, Manganese cluster in photosynthesis: where plants oxidize water to dioxygen, Chem. Rev., 1996, 96, 2927–2950 CrossRef CAS PubMed.
- K. Das, S. Waiba, A. Jana and B. Maji, Manganese-catalyzed hydrogenation, dehydrogenation, and hydroelementation reactions, Chem. Soc. Rev., 2022, 51, 4386–4464 RSC.
- Y. Wang, M. Wang, Y. Li and Q. Liu, Homogeneous manganese-catalyzed hydrogenation and dehydrogenation reactions, Chem, 2021, 7, 1180–1223 CAS.
- S. Waiba and B. Maji, Manganese catalyzed acceptorless dehydrogenative coupling reactions, ChemCatChem, 2020, 12, 1891–1902 CrossRef CAS.
- J. R. Carney, B. R. Dillon and S. P. Thomas, Recent Advances of manganese catalysis for organic synthesis, Eur. J. Org Chem., 2016, 3912–3929 CrossRef CAS.
- W. Liu and L. Ackermann, Manganese-catalyzed C–H activation, ACS Catal., 2016, 6, 3743–3752 CrossRef CAS.
- D. A. Valyaev, G. Lavigne and N. Lugan, Manganese organometallic compounds in homogeneous catalysis: past, present, and prospects, Coord. Chem. Rev., 2016, 308, 191–235 CrossRef CAS.
- W. Liu and J. T. Groves, Manganese catalyzed C–H halogenation, Acc. Chem. Res., 2015, 48, 1727–1735 CrossRef CAS PubMed.
- F. A. Cotton, G. Wilkinson, C. A. Murillo and M. Bochmann, in Advanced Inorganic Chemistry, Wiley, New York, 6th edn, 1999, pp. 757–775 Search PubMed.
- A. Higelin and S. Riedel, in Modern Synthesis Processes and Reactivity of Fluorinated Compounds, ed. H. Groult, F. R. Leroux and A. Tressaud, Elsevier, Amsterdam, 2nd edn, 2017, pp. 561–586 Search PubMed.
- S. Riedel, in Comprehensive Inorganic Chemistry II, ed. J. Reedijk and K. Poeppelmeier, Elsevier, Amsterdam, 2nd edn, 2013, pp. 187–221 Search PubMed.
- S. Riedel and M. Kaupp, The highest oxidation states of the transition metal elements, Coord. Chem. Rev., 2009, 253, 606–624 CrossRef CAS.
- A. Haupt, in Organic and Inorganic Fluorine Chemistry, de Gruyter, Berlin/Boston, 2021, pp. 220–235 Search PubMed.
- D. B. Rice, A. A. Massie and T. A. Jackson, Manganese–Oxygen intermediates in O–O bond activation and hydrogen-atom transfer reactions, Acc. Chem. Res., 2017, 50, 2706–2717 CrossRef CAS.
- H. M. Neu, R. A. Baglia and D. P. Goldberg, A balancing act: stability versus reactivity of Mn(O) complexes, Acc. Chem. Res., 2015, 48, 2754–2764 CrossRef CAS.
- K. Ray, F. F. Pfaff, B. Wang and W. Nam, Status of reactive non-heme metal–oxygen intermediates in chemical and enzymatic reactions, J. Am. Chem. Soc., 2014, 136, 13942–13958 CrossRef CAS PubMed.
- V. L. Pecoraro, M. J. Baldwin and A. Gelasco, Interaction of manganese with dioxygen and its reduced derivatives, Chem. Rev., 1994, 94, 807–826 CrossRef CAS.
- M.-N. Collomb and A. Deronzier, Manganese: inorganic & coordination chemistry, in Encyclopedia of Inorganic Chemistry, ed. R. B. King, R. H. Crabtree, C. M. Lukehart, D. A. Atwood and R. A. Scott, Wiley, Chichester, 2006 Search PubMed.
- J. C. Vites and M. M. Lynam, 3. Manganese, Coord. Chem. Rev., 1994, 131, 95–126 CrossRef CAS.
- A. Grass, D. Wannipurage, R. L. Lord and S. Groysman, Group-transfer chemistry at transition metal centers in bulky alkoxide ligand environments, Coord. Chem. Rev., 2019, 400, 213044 CrossRef CAS.
- J. K. Elinburg and L. H. Doerrer, Synthesis, structure, and electronic properties of late first-row transition metal complexes of fluorinated alkoxides and aryloxides, Polyhedron, 2020, 190, 114765 CrossRef CAS.
- M. Gerken, H. P. A. Mercier and G. J. Schrobilgen, in Advanced Inorganic Fluorides: Synthesis, Characterization and Applications, ed. T. Nakajima, B. Žemva and A. Tressaud, Elsevier, Switzerland, 2000, pp. 117–174 Search PubMed.
- K. Seppelt, Stabilization of Unusual oxidation and coordination states by the ligands OSF5, OSeF5, and OTeF5, Angew Chem., Int. Ed. Engl., 1982, 21, 877–888 CrossRef.
- A. Pérez-Bitrián, K. F. Hoffmann, K. B. Krause, G. Thiele, C. Limberg and S. Riedel, Unravelling the role of the pentafluoroorthotellurate group as a ligand in nickel chemistry, Chem.–Eur. J., 2022, 28, e202202016 CrossRef PubMed.
- A. Pérez-Bitrián, J. Munárriz, J. S. Sturm, D. Wegener, K. B. Krause, A. Wiesner, C. Limberg and S. Riedel, Further perspectives on the teflate versus fluoride analogy: the case of a Co(II) pentafluoroorthotellurate complex, Inorg. Chem., 2023, 62, 12947–12953 CrossRef PubMed.
- S. H. Strauss, M. D. Noirot and O. P. Anderson, Preparation and characterization of silver(I) teflate complexes: bridging OTeF5 groups in the solid state and in solution, Inorg. Chem., 1985, 24, 4307–4311 CrossRef CAS.
- The four-coordinate geometry index is defined as τ4 = [360°−(α+β)]/141°, where α and β are the two largest angles in the four-coordinate species: L. Yang, D. R. Powell and R. P. Houser, Structural variation in copper(I) complexes with pyridylmethylamide ligands: structural analysis with a new four-coordinate geometry index, τ4, Dalton Trans., 2007, 955–964 RSC.
- M. C. Crossman, E. G. Hope and L. J. Wootton, Reactions of [M2(CO)10] (M = Mn or Re) with xenon bis[pentafluorooxo-tellurate(VI) and -selenate(VI)], J. Chem. Soc., Dalton Trans., 1998, 1813–1817 RSC.
- K. D. Abney, K. M. Long, O. P. Anderson and S. H. Strauss, Preparation and properties of metal carbonyl teflates, including the structure and reactivity of Mn(CO)5(OTeF5), Inorg. Chem., 1987, 26, 2638–2643 CrossRef CAS.
- S. H. Strauss, K. D. Abney, K. M. Long and O. P. Anderson, Low-valent metal teflates: preparation and characterization of Mn(CO)5(OTeF5), Inorg. Chem., 1984, 23, 1994–1995 CrossRef CAS.
- L. Song, A unique complex based on pyridinium carboxylate ligand involving manganese(II)-catalyzed in situ oxidative cleavage, Monatsh. Chem., 2013, 144, 1617–1620 CrossRef CAS.
- S. M. Moore, C. Sun, J. L. Steele, E. M. Laaker, A. L. Rheingold and L. H. Doerrer, HAA by the first {Mn(III)OH} complex with all O-donor ligands, Chem. Sci., 2023, 14, 8187–8195 RSC.
- J. A. Bellow, M. Yousif, D. Fang, E. G. Kratz, G. A. Cisneros and S. Groysman, Synthesis and reactions of 3d metal complexes with the bulky alkoxide ligand [OCtBu2Ph], Inorg. Chem., 2015, 54, 5624–5633 CrossRef CAS PubMed.
- S. K. Kondaveeti, S. Vaddypally, C. Lam, D. Hirai, N. Ni, R. J. Cava and M. J. Zdilla, Synthesis, structure, and magnetic studies of manganese–oxygen clusters of reduced coordination number, featuring an unchelated, 5-coordinate octanuclear manganese cluster with water-derived oxo ligands, Inorg. Chem., 2012, 51, 10095–10104 CrossRef CAS PubMed.
- G. B. Deacon, L. Y. Goh, W. R. Jackson, B. W. Skelton, W. Chen and A. H. White, A new method of synthesis of monomeric aryloxide complexes of manganese(II): structures of manganese phenoxides, Z. Anorg. Allg. Chem., 2010, 636, 1478–1483 CrossRef CAS.
- C. Duboc, M.-N. Collomb and F. Neese, Understanding the zero-field splitting of mononuclear manganese(II) complexes from combined EPR spectroscopy and quantum chemistry, Appl. Magn. Reson., 2010, 37, 229–245 CrossRef.
- S. Alvarez and J. Cirera, How high the spin? Allowed and forbidden spin states in transition-metal chemistry, Angew. Chem., Int. Ed., 2006, 45, 3012–3020 CrossRef CAS.
- B. N. Figgis and J. Lewis, The magnetic properties of transition metal complexes, Prog. Inorg. Chem., 1964, 6, 37–239 CAS.
- N. S. Gill and R. S. Nyholm, 802. Complex Halides of the transition metals. Part I. Tetrahedral nickel complexes, J. Chem. Soc., 1959, 3997–4007 RSC.
- C. Duboc, Determination and prediction of the magnetic anisotropy of Mn ions, Chem. Soc. Rev., 2016, 45, 5834–5847 RSC.
- D. Mejía-Rodríguez, A. Albavera-Mata, E. Fonseca, D.-T. Chen, H. P. Cheng, R. G. Hennig and S. B. Trickey, Barriers to predictive high-throughput screening for spin-crossover, Comput. Mater. Sci., 2022, 206, 111161 CrossRef.
- J. Cirera, M. Via-Nadal and E. Ruiz, Benchmarking density functional methods for calculation of state energies of first row spin-crossover molecules, Inorg. Chem., 2018, 57, 14097–14105 CrossRef CAS PubMed.
- K. P. Kepp, Theoretical study of spin crossover in 30 iron complexes, Inorg. Chem., 2016, 55, 2717–2727 CrossRef CAS PubMed.
- M. Swart and M. Gruden, Spinning around in transition-metal chemistry, Acc. Chem. Res., 2016, 49, 2690–2697 CrossRef CAS PubMed.
- B. O. Roos, P. R. Taylor and P. E. M. Sigbahn, A complete active space SCF method (CASSCF) using a density matrix formulated super-CI approach, Chem. Phys., 1980, 48, 157–173 CrossRef CAS.
- C. Angeli, R. Cimiraglia and J.-P. Malrieu, n-electron valence state perturbation theory: a spinless formulation and an efficient implementation of the strongly contracted and of the partially contracted variants, J. Chem. Phys., 2002, 117, 9138–9153 CrossRef CAS.
- C. Angeli, R. Cimiraglia, S. Evangelisti, T. Leininger and J.-P. Malrieu, Introduction of n-electron valence states for multireference perturbation theory, J. Chem. Phys., 2001, 114, 10252–10264 CrossRef CAS.
- C. Angeli, R. Cimiraglia and J.-P. Malrieu, n-electron valence state perturbation theory: a fast implementation of the strongly contracted variant, Chem. Phys. Lett., 2001, 350, 297–305 CrossRef CAS.
- K. Pierloot, Q. M. Phung and A. Domingo, Spin state energetics in first-row transition metal complexes: contribution of (3s3p) correlation and its description by second-order perturbation theory, J. Chem. Theory Comput., 2017, 13, 537–553 CrossRef CAS PubMed.
- K. Boguslawski, K. H. Marti, Ö. Legeza and M. Reiher, Accurate ab initio spin densities, J. Chem. Theory Comput., 2012, 8, 1970–1982 CrossRef CAS PubMed.
- K. Pierloot, Transition metals compounds: outstanding challenges for multiconfigurational methods, Int. J. Quantum Chem., 2011, 111, 3291–3301 CrossRef CAS.
- L. Turowsky and K. Seppelt, Molybdän- und Wolframverbindungen mit dem Liganden -OTeF5, Z. Anorg. Allg. Chem., 1990, 590, 23–36 CrossRef CAS.
- L. Turowsky and K. Seppelt, Rheniumverbindungen mit dem Liganden -OTeF5, Z. Anorg. Allg. Chem., 1990, 590, 37–47 CrossRef CAS.
- Y. Zhang, A.-Q. Jia, J.-J. Zhang, Z. Xin and Q.-F. Zhang, Syntheses, structures and catalytic properties of new mononuclear terpyridine-manganese(II) complexes with tetraphenylimido-diphosphinate and diphenylphosphinate ligands, J. Coord. Chem., 2019, 72, 1036–1048 CrossRef CAS.
- A. Panja, N. C. Jana, A. Bauzá, S. Adak, T. M. Mwania, D. M. Eichhorn and A. Frontera, Introducing supramolecular interactions into robust bis(tetrabromocatecholate) chelated manganese(III) systems and biomimetic catalytic activity, ChemistrySelect, 2017, 2, 2094–2105 CrossRef CAS.
- C. J. Rolle III, K. I. Hardcastle and J. D. Soper, Reactions of tetrabromocatecholatomanganese(III) complexes with dioxygen, Inorg. Chem., 2008, 47, 1892–1894 CrossRef.
- N. Shaikh, S. Goswami, A. Panja, X.-Y. Wang, S. Gao, R. J. Butcher and P. Banerjee, New route to the mixed valence semiquinone-catecholate based mononuclear FeIII and catecholate based dinuclear MnIII complexes: first experimental evidence of valence tautomerism in an iron complex, Inorg. Chem., 2004, 43, 5908–5918 CrossRef CAS.
- J. Münchenberg, H. Thönnessen, P. G. Jones and R. Schmutzler, N′,N′,N′′,N′′-Tetramethylguanidine-substituted phosphoryl compounds as ligands in transition metal chemistry – unusual modes of coordination, Chem. Ber., 1997, 130, 1825–1832 CrossRef.
- A. R. Schake, E. A. Schmitt, A. J. Conti, W. E. Streib, J. C. Huffman, D. N. Hendrickson and G. Christou, Preparation and properties of mononuclear and ferromagnetically coupled dinuclear manganese complexes with 2,2'-biphenoxide, Inorg. Chem., 1991, 30, 3192–3199 CrossRef CAS.
- S. K. Larsen, C. G. Pierpont, G. DeMunno and G. Dolcetti, Manganese(III)-catecholate coordination in the bis(tetrabromocatecholato)(triphenylphosphineoxide)manganese(III) anionic dimer, Inorg. Chem., 1986, 25, 4828–4831 CrossRef CAS.
- G.-D. Ge, J.-B. Shen and G.-L. Zhao, Triaquabis{2-methoxy-6-[(phenyliminiumyl)methyl]phenolate-κO1}manganese(II) dinitrate, Acta Crystallogr., E, 2011, 67, m813–m814 CrossRef CAS PubMed.
- H. W. Roesky, M. Scholz and M. Noltemeyer, Über Reaktionen des 2,4,6-Tris(trifluormethyl)phenols mit Verbindungen von Hauptgruppen- und Nebengruppen-Elementen (Li, Na, Mg, Ca, Ba, Ge, Sn und Ti, W, Mn, Cd), Chem. Ber., 1990, 123, 2303–2309 CrossRef CAS.
- Y. Rojas, M. Tadrosse, C. Assaf, I. Bernal and R. A. Lalancette, Preparation and structural studies of manganese polychloride anions in three metal charge states [(II), (III), (IV)]—their stereochemistries, Struct. Chem., 2020, 31, 1309–1316 CrossRef CAS.
- M. Matsui, S. Koda, S. Ooi, H. Kuroya and I. Bernal, The crystal structure of phenanthrolinium pentachloromanganate(III), (phenH2)(MnCl5), Chem. Lett., 1972, 1, 51–53 CrossRef.
- I. Bernal, N. Elliott and R. Lalancette, Molecular configuration of the anion MnCl52–—A square pyramidal pentahalide of the 3d transition series, J. Chem. Soc. D, 1971, 803–804 RSC.
- E. H. Witten and W. M. Reiff, A low temperature magnetic susceptibility study of phenanthrolinium pentachloro manganate(III): [PhenH2][MnCl5], Inorg. Chim. Acta, 1980, 41, 227–228 CrossRef CAS.
- H. A. Goodwin and R. N. Sylva, The magnetic properties of some complexes of higher-valent manganese, Aust. J. Chem., 1967, 20, 629–637 CrossRef CAS.
- H. A. Goodwin and R. N. Sylva, 1,10-Phenanthroline and 2,2'-bipyridine chelates of higher-valent manganese, Aust. J. Chem., 1965, 18, 1743–1749 CrossRef CAS.
- A. R. Rossi and R. Hoffmann, Transition metal pentacoordination, Inorg. Chem., 1975, 14, 365–374 CrossRef CAS.
- The five-coordinate geometry index is defined as τ5 = (β−α)/60°, where α and β are the two largest angles in the five-coordinate species: A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, Synthesis, structure, and spectroscopic properties of copper(II) compounds containing nitrogen–sulphur donor ligands; the crystal and molecular structure of aqua[1,7-bis(N-methylbenzimidazol-2′-yl)-2,6-dithiaheptane]copper(II) perchlorate, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
- C.-Y. Qin, S.-Z. Zhao, H.-Y. Wu, Y.-H. Li, Z.-K. Wang, Z. Wang and S. Wang, The dynamic interplay between intramolecular and intermolecular interactions in mononuclear manganese(III) SCO complexes, Dalton Trans., 2021, 50, 5899–5910 RSC.
- I. A. Kühne, L. C. Gavin, M. Harris, B. Gildea, H. Müller-Bunz, M. Stein and G. G. Morgan, Mn(III) complexes with nitro-substituted ligands—spin states with a twist, J. Appl. Phys., 2021, 129, 213903 CrossRef.
- D. Villaman, C. J. McMonagle, M. R. Probert, O. Peña, Y. Moreno and M. Fuentealba, Structural studies of a manganese(III) complex with spin-crossover and thermochromic properties, CrystEngComm, 2020, 22, 3221–3233 RSC.
- J. Olguín, Unusual metal centres/coordination spheres in spin crossover compounds, Coord. Chem. Rev., 2020, 407, 213148 CrossRef.
- S. Amabilino and R. J. Deeth, DFT analysis of spin crossover in Mn(III) complexes: is a two-electron S = 2 to S = 0 spin transition feasible?, Inorg. Chem., 2017, 56, 2602–2613 CrossRef CAS PubMed.
- Y. Garcia and P. Gütlich, Thermal spin crossover in Mn(II), Mn(III), Cr(II) and Co(III) coordination compounds, Top. Curr. Chem., 2004, 234, 49–62 CrossRef CAS.
- F. Franceschi, J. Hesschenbrouck, E. Solari, C. Floriani, N. Re, C. Rizzoli and A. Chiesi-Villa, The reactivity of dibenzotetramethyltetraaza[14]annulene-Mn(II): functionalisation of manganese in a macrocyclic environment, J. Chem. Soc., Dalton Trans., 2000, 593–604 RSC.
- P. G. Sim and E. Sinn, First manganese(III) spin crossover, first d4 crossover. Comment on cytochrome oxidase, J. Am. Chem. Soc., 1981, 103, 241–243 CrossRef CAS.
- I. D. Chawla and M. J. Frank, Electronic spectra of hexacyanomanganate(III) and hydroxopentacyanomanganate(III) ions in acidic media, J. Inorg. Nucl. Chem., 1970, 32, 555–563 CrossRef CAS.
- M. Orio, D. A. Pantazis, T. Petrenko and F. Neese, Magnetic and spectroscopic properties of mixed valence manganese(III,IV) dimers: a systematic study using broken symmetry density functional theory, Inorg. Chem., 2009, 48, 7251–7260 CrossRef CAS PubMed.
- M. A. Blanco, A. Martín Pendás and E. Francisco, Interacting quantum atoms: a correlated energy decomposition scheme based on the quantum theory of atoms in molecules, J. Chem. Theory Comput., 2005, 1, 1096–1109 CrossRef CAS.
- R. F. W. Bader, Atoms in Molecules – A Quantum Theory, Clarendon Press, Oxford, 1994 Search PubMed.
- Á. Martín Pendás, E. Francisco, D. Suárez, A. Costales, N. Díaz, J. Munárriz, T. Rocha-Rinza and J. M. Guevara-Vela, Atoms in molecules in real space: a fertile field for chemical bonding, Phys. Chem. Chem. Phys., 2023, 25, 10231–10262 RSC.
- J. M. Guevara-Vela, E. Francisco, T. Rocha-Rinza and Á. Matín Pendás, Interacting quantum atoms—a review, Molecules, 2020, 25, 4028 CrossRef CAS PubMed.
- E. Francisco, D. Menéndez Crespo, A. Costales and Á. Martín Pendás, A multipolar approach to the interatomic covalent interaction energy, J. Comput. Chem., 2017, 38, 816–829 CrossRef CAS PubMed.
- A. Martín Pendás, J. L. Casals-Sainz and E. Francisco, On electrostatics, covalency, and chemical dashes: physical interactions versus chemical bonds, Chem.–Eur. J., 2019, 25, 309–314 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2287496. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc00543k |
| This journal is © The Royal Society of Chemistry 2024 |