
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Palladium/XuPhos-catalyzed enantioselective cascade Heck/intermolecular C(sp2)–H alkylation reaction†
Chao
Fang
a,
Quan-Pu
Wang
a,
Bing
Xu
ac,
Zhan-Ming
Zhang
 *ab and
Junliang
Zhang
*acd
*ab and
Junliang
Zhang
*acd
aDepartment of Chemistry, Fudan University, Shanghai, 200438, P. R. China. E-mail: Zhanmingzhang@fudan.edu.cn; junliangzhang@fudan.edu.cn
bFudan Zhangjiang Institute, Shanghai, 201203, P. R. China
cZhuhai Fudan Innovation Institute, Zhuhai, Guangdong 519000, P. R. China
dSchool of Chemistry and Chemical Engineering, Henan Normal University, Xinxiang, Henan 453007, P. R. China
First published on 9th March 2024
Abstract
Palladium-catalyzed enantioselective domino Heck/intramolecular C–H functionalization reaction, as a valuable strategy for creating molecular diversity, has remained a prominent challenge. Here, we describe a Pd/XuPhos catalyst for asymmetric domino Heck/intermolecular C–H alkylation of unactivated alkenes with diverse polyfluoro- and heteroarenes in a highly chemo- and enantioselective manner. This process enables efficient synthesis of various dihydrobenzofurans, indolines and indanes, which are of interest in pharmaceutical research and other areas. Late-stage modifications of the core structures of natural products are also well showcased. Moreover, synthetic transformations create a valuable platform for preparing a series of functionalized molecules. Several control experiments for mechanistic study are conducted to pursue a further understanding of the reaction.
Introduction
Palladium-catalyzed C–H bond functionalization, as a synthetically significant yet challenging bond-forming process, has been tremendously exploited to realize precision control of site-selectivity for fabricating densely functionalized molecules.1,2 Among others, palladium-catalyzed domino Heck/C–H functionalization reaction involving an σ-alkylpalladium intermediate represents one of the most powerful, step- and atom-economic tools to construct highly functionalized heterocyclics bearing quaternary carbon centers.3–9 Compared with Heck/intramolecular C–H functionalization,3–9 the intermolecular reactions are more challenging owing to the direct C–H functionalization side reactions. In 2009, the group of Fagnou reported a pioneering study on palladium-catalyzed domino Heck/intermolecular C–H alkylation reactions between aryl bromides with sulfur-containing heterocycles.10 Utilizing a similar strategy, Sharma and Van der Eycken demonstrated that acrylamides could react with 1,3,4-oxadiazoles to construct bis-heteroaryl frameworks under microwave irradiation.11,12 Later, the domino process was applied to the synthesis of alkylated polyfluoroarene derivatives employing electron-deficient polyfluoroarenes as the direct arylation coupling partner, which was accomplish by Liang and Xu.13 Recently, Kuram et al. disclosed that 1,2,3-triazoles were also suitable coupling partners to obtain bisheterocycles bearing all-carbon quaternary centers.14 Despite continuous development in the Heck/C–H alkylation reaction (Scheme 1a), the exploration of its asymmetric variants is still dramatically limited. To the best of our knowledge, enantioselective domino Heck/intermolecular C–H bond functionalization was only established by Zhu and co-workers, efficiently creating various 3,3-disubstituted oxindoles and bisoxindoles (Scheme 1b).15 Thus, the identification of new catalysts for this interesting reaction is still highly in demand.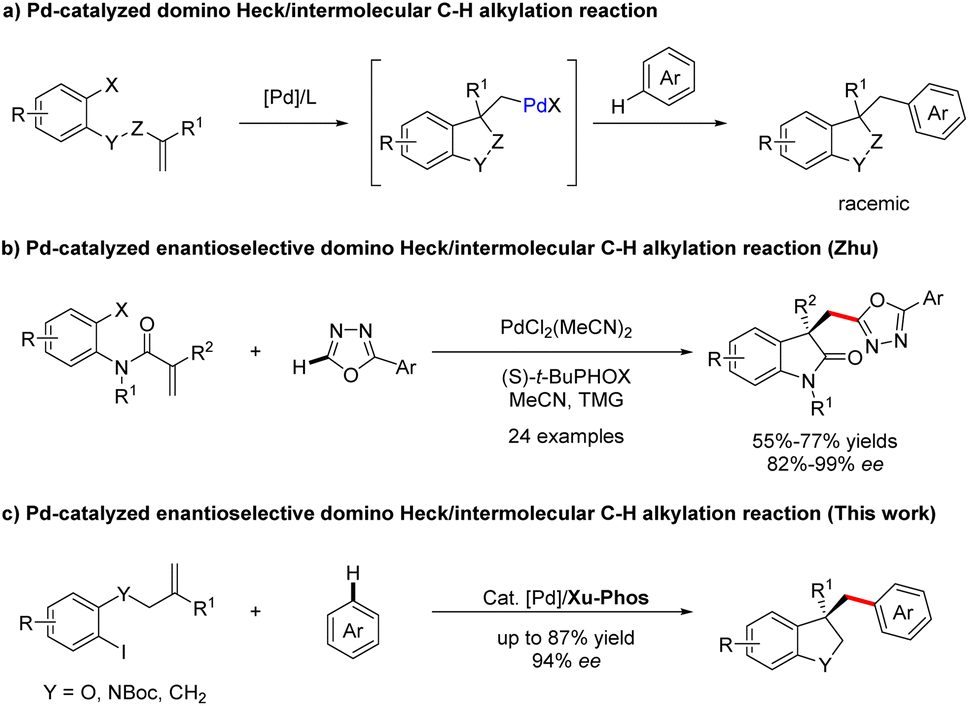 | ||
| Scheme 1 Previous work and this work on Pd-catalyzed domino Heck/intermolecular C–H alkylation reaction. | ||
In 2022, a remarkable example of highly enantioselective domino Heck/intramolecular C–H alkylation for the selective synthesis of chiral strained 5,4- and 5,5-spirocycles was accomplished by our group, employing our own developed Sadphos as the chiral ligand.8 Based on the success of the intramolecular variant and our ongoing interest on domino Heck reactions,8,16–19 we were intrigued to develop newly efficient catalyst systems to realize domino Heck/intermolecular C–H alkylation, which, if successful, would offer a highly efficient route for the construction of various privileged heterocycle skeletons existing in a number of natural products and drugs.20–23
Herein, we establish a Pd/XuPhos system as an effective catalyst for the enantioselective cascade Heck/intermolecular C(sp2)–H alkylation reaction of unactivated alkenes with various polyfluoroarenes, providing expedient access to a wide spectrum of structurally diverse dihydrobenzofuran-, indoline- and indane-containing polyfluoroarene compounds (Scheme 1c). Moreover, the significance of this methodology is also underscored by easily converting products to other classes of functionalized molecules.
Results and discussion
We began our investigation using o-iodophenol-derived allyl ether 1a and pentafluorobenzene 2a as model substrates (Table 1 and Scheme 2). An exhaustive screening of various types of monodentate and bidentate commercial ligands showed that ligands L3 and L5–7 failed to deliver the desired product 3a (Scheme 2). Although ligand L1–2 showed better enantioselectivity and ligand L4 favored this transformation, both of them didn't obtain 3a with satisfactory results. Then, we turned attention to our developed ligands, which have demonstrated potential performances in palladium-catalyzed asymmetric cascade Heck reactions. The examination of the Sadphos ligand kit indicated that only N-Me masked ligands could deliver the desired product, in which Xu4 was the optimal choice, allowing the formation of 3a in 60% yield with 52% ee. Further screening of different solvents indicated that Et2O, iPr2O and MTBE resulted in higher ee (Table 1, entries 1–3). The use of DCM, DMF, CH3CN and DCE as solvent increased neither yield nor ee (entries 4–7). Subsequently, we focused on the optimization of the metal salt and base (entries 8–16). When the metal salt and base were changed to Pd2dba3·CHCl3 and Cs2CO3, respectively, the desired product 3a was obtained in 83% yield with 90% ee (entry 16). To our delight, lowering the temperature to 80 °C provided 3a with a slightly higher ee of 92% (entry 17). Finally, it was found that Ag2CO3 had also a considerable effect on the reactivity (entry 18).|
|
|||||
|---|---|---|---|---|---|
| Entry | [Pd] | Solvent | Base | Yieldb (%) | Eec (%) |
| a Unless otherwise noted, all reactions were performed with 1a (0.1 mmol), 2a (0.3 mmol), Ag2CO3 (0.075 mmol), base (0.2 mmol), 10 mol% [Pd] and 20 mol% ligand in 1.0 mL solvent at 90 °C for 15–48 h. b NMR yield with CH2Br2 as an internal standard. c Enantioselectivity was determined by chiral-phase HPLC. d 80 °C. e No Ag2CO3 added. | |||||
| 1 | Pd2dba3 | Et2O | K2CO3 | 66 | 76 |
| 2 | Pd2dba3 | iPr2O | K2CO3 | 80 | 84 |
| 3 | Pd2dba3 | MTBE | K2CO3 | 74 | 80 |
| 4 | Pd2dba3 | DCM | K2CO3 | 44 | 28 |
| 5 | Pd2dba3 | DMF | K2CO3 | 10 | 5 |
| 6 | Pd2dba3 | CH3CN | K2CO3 | 45 | 49 |
| 7 | Pd2dba3 | DCE | K2CO3 | 48 | 35 |
| 8 | Pd2dba3·CHCl3 | iPr2O | K2CO3 | 82 | 88 |
| 9 | Pd(η-allyl)Cl2 | iPr2O | K2CO3 | 78 | 75 |
| 10 | Pd(OAc)2 | iPr2O | K2CO3 | 24 | 25 |
| 11 | Pd(TFA)2 | iPr2O | K2CO3 | 60 | 32 |
| 12 | Pd(acac)2 | iPr2O | K2CO3 | 15 | 3 |
| 13 | Pd2dba3·CHCl3 | iPr2O | KOH | 70 | 80 |
| 14 | Pd2dba3·CHCl3 | iPr2O | KOtBu | 74 | 63 |
| 15 | Pd2dba3·CHCl3 | iPr2O | CsOPiv | 46 | 7 |
| 16 | Pd2dba3·CHCl3 | iPr2O | Cs2CO3 | 83 | 90 |
| 17d | Pd2dba3·CHCl3 | iPr2O | Cs2CO3 | 84 | 92 |
| 18d,e | Pd2dba3·CHCl3 | iPr2O | Cs2CO3 | Trace | — |
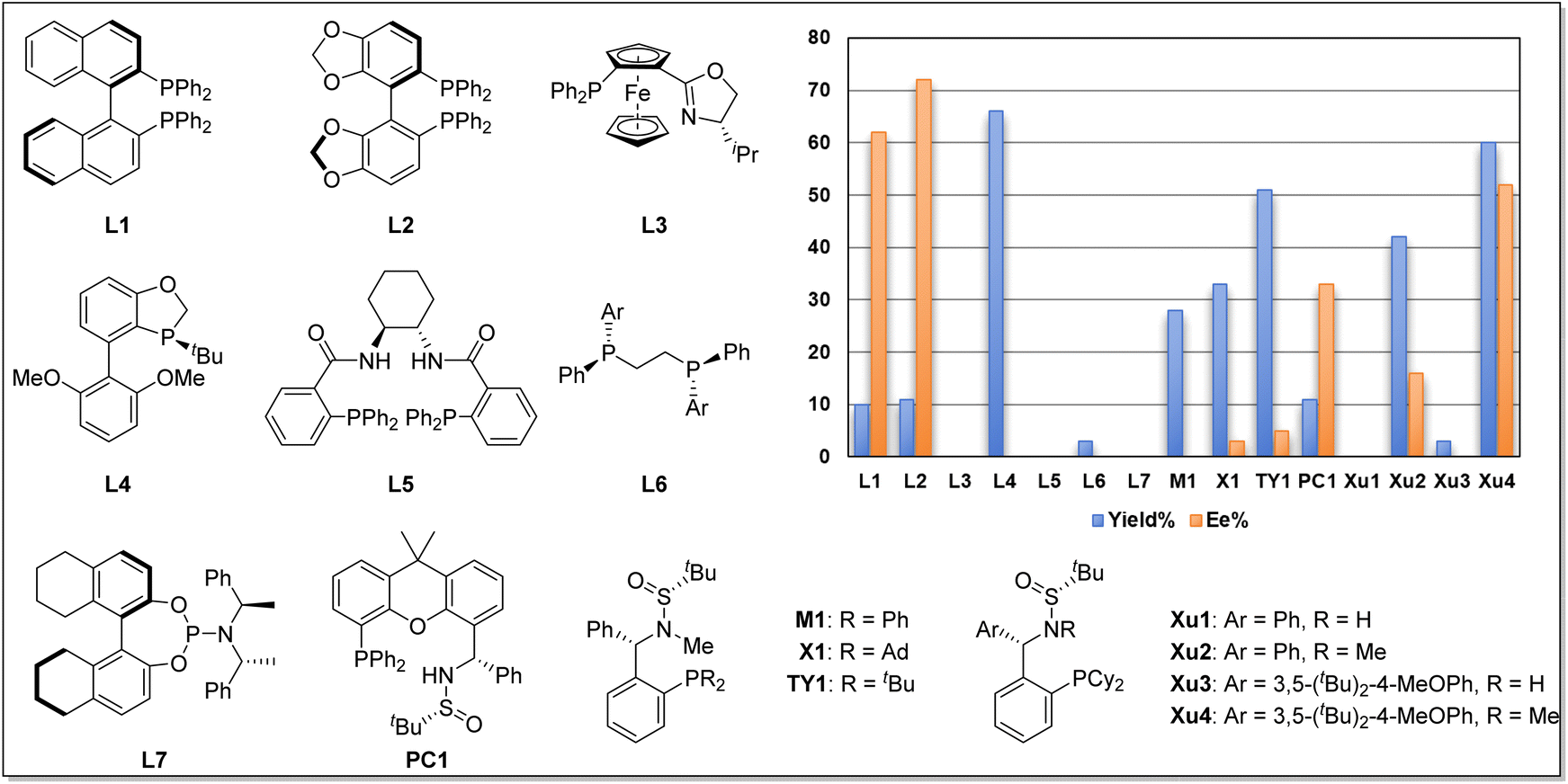 | ||
| Scheme 2 Representative diphosphorus ligands examined in this work. | ||
Having established the optimized conditions, the scope of this reaction was examined by using various o-iodophenol-derived allyl ethers. Different linear and branched alkyl groups on the alkene moiety proceeded smoothly to furnish 3b–3f in good yields with excellent enantioselectivities. Numerous allyl ethers bearing functional groups, such as trimethylsilyl (1g), methoxycarbonyl (1h), chloro (1i and 1j) and fluoro (1k), were compatible with the reaction to form the corresponding products in satisfactory results. To our delight, substrates with various ether, thiol ether and N-heterocycles appended to the alkyl chain were suitable for the reaction to deliver the expected products 3l–3r with 90–93% ee. Particularly noteworthy was the tolerance of the reaction conditions to the more structurally complex contexts. A variety of allyl ethers derived from the core structures of natural products were also suitable substrates, converting to the target products (3s–3u) in excellent yields with outstanding diastereoselectivities.
Subsequently, the effect of substituents on the benzene ring of the o-iodophenol moiety was investigated under the standard reaction conditions (Scheme 3). Substituting the phenyl ring with electron-donating and electron-withdrawing groups at C4 and C5 positions appeared to have limited effects on the results, and 5a–5d were afforded in modest to good yields with excellent ee values. 3,3-Disubstituted indolines and indanes are frequently found in pharmaceuticals, natural alkaloids, and as fascinating building blocks in organic synthesis. Despite progress made in this field, the synthesis of these chiral compounds is still in high demand. Satisfactorily, the present asymmetric C–H functionalization of alkene reaction was also applicable to the substrates employing BocN and C as a tether, delivering the indoline 5e and indanes 5f with good yields and ee values.
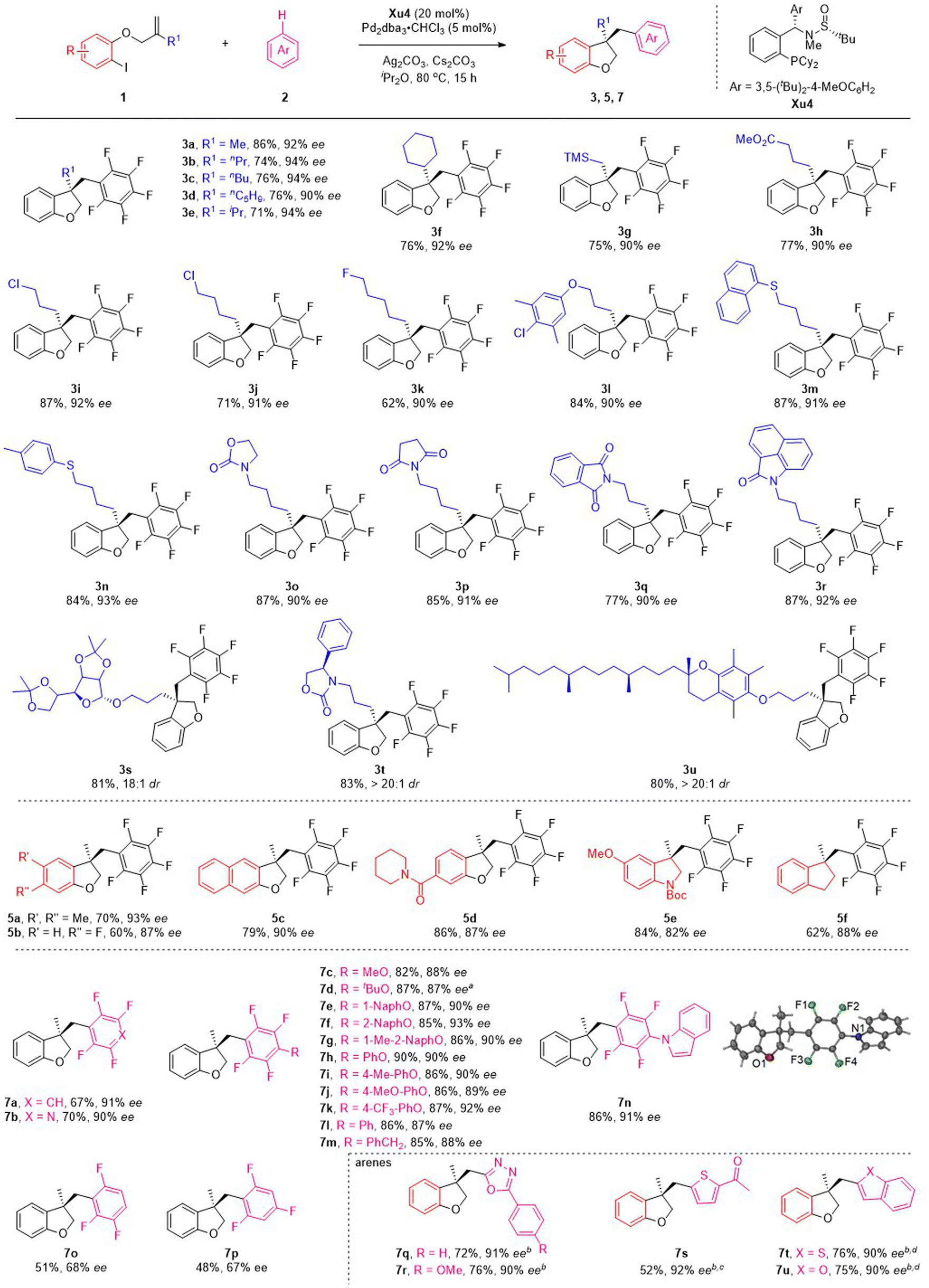 | ||
| Scheme 3 Substrate scope. Conditions: unless otherwise noted, all reactions were performed with 1a (0.3 mmol), 2a (0.9 mmol), Ag2CO3 (0.225 mmol), Cs2CO3 (0.6 mmol), 10 mol% Pd2dba3·CHCl3 and 20 mol% Xu4 in 3.0 mL iPr2O at 80 °C for 15 h. a25 mol% Xu3 was used. bPivOH (0.09 mmol), Pd(η-allyl)Cl2 (10 mol%) and Et2O (3 mL) were used. c100 °C. dtBuONa (3.0 eq.) was used. | ||
To ascertain the scope of this method, a variety of polyfluoroarenes were further investigated (Scheme 3). Both 1,2,4,5-tetrafluorobenzene and 2,3,5,6-tetrafluoropyridine smoothly underwent the C–H functionalization process and transformed to the corresponding products (7a and 7b) in good yields with excellent ee values. For 2,3,5,6-tetrafluoroanisole derivatives, alkyl ethers (such as Me and tBu) and aryl ethers (such as naphthyl and phenyl groups) were also well accommodated under mild conditions, giving the desired products (7c–7h) in 82–90% yields with 87–93% ee. It is noteworthy that electron-donating groups (such as methyl and methoxy groups) and electron-withdrawing groups (such as trifluoromethyl group) on the phenyl ring were all well tolerated, furnishing the desired products (7i–7k) with good to excellent ee. Furthermore, changing the O-substituent to a N- and CH2-substituent on the tetrafluorobenzene ring could also smoothly drive the reaction to form products 7l–7n with satisfactory results. The absolute configuration of the product was confirmed by the X-ray diffraction analysis of 7n. Next, the scope of fluorobenzenes with fewer fluorine atoms was investigated. Unfortunately, decreasing the fluorine atoms could drive the reaction to form products with lower yield and ee (7o–7p), which might be related to the fact that more fluorine atoms can increase the pKa value of substrates. We next investigated several heteroarene substrates. To our delight, oxadiazole 6q and 6r, thiophene 6s, benzothiophene 6t and benzofuran 6u could react smoothly, affording the corresponding products (7q–7u) with high yields (52–76%) and excellent ees (90–92%).
To further demonstrate the reliability of this method, the reaction of 1i and 2a was conducted on a larger scale of 5 mmol, affording the desired products 3i without loss of efficiency and the ee value (Scheme 4). Subsequently, synthetic transformations of 3i were carried out. As shown in Scheme 5, the Cl group could be substituted by different nucleophilic reagents, thus leading to 8 and 9 in 97 and 58% yields, respectively. It's very interesting to find that the substitution of 3i with different equivalents of NaSPh could produce 10 and 11, respectively, in high yields. Notably, 3i have two sites which can conduct nucleophilic substitution reaction. If stronger nucleophilic reagents were used, the direct functionalized of polyfluoroarenes could be selectively achieved to afford 12–14 in good yields. It was found that the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond of 14 was generated through the elimination of the C–Cl bond in the presence of strong base tBuOK.
C bond of 14 was generated through the elimination of the C–Cl bond in the presence of strong base tBuOK.
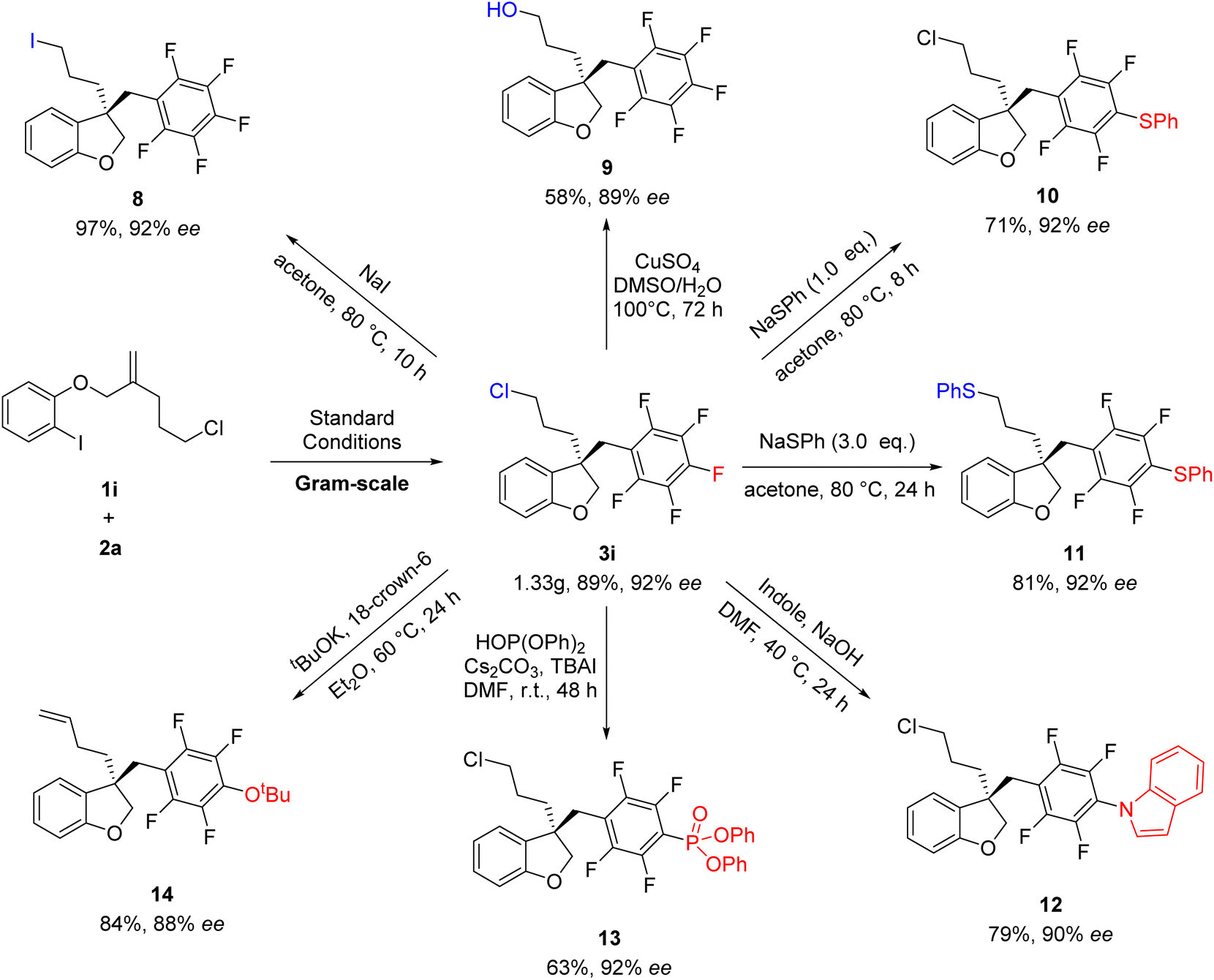 | ||
| Scheme 4 The synthetic transformation of 3i. | ||
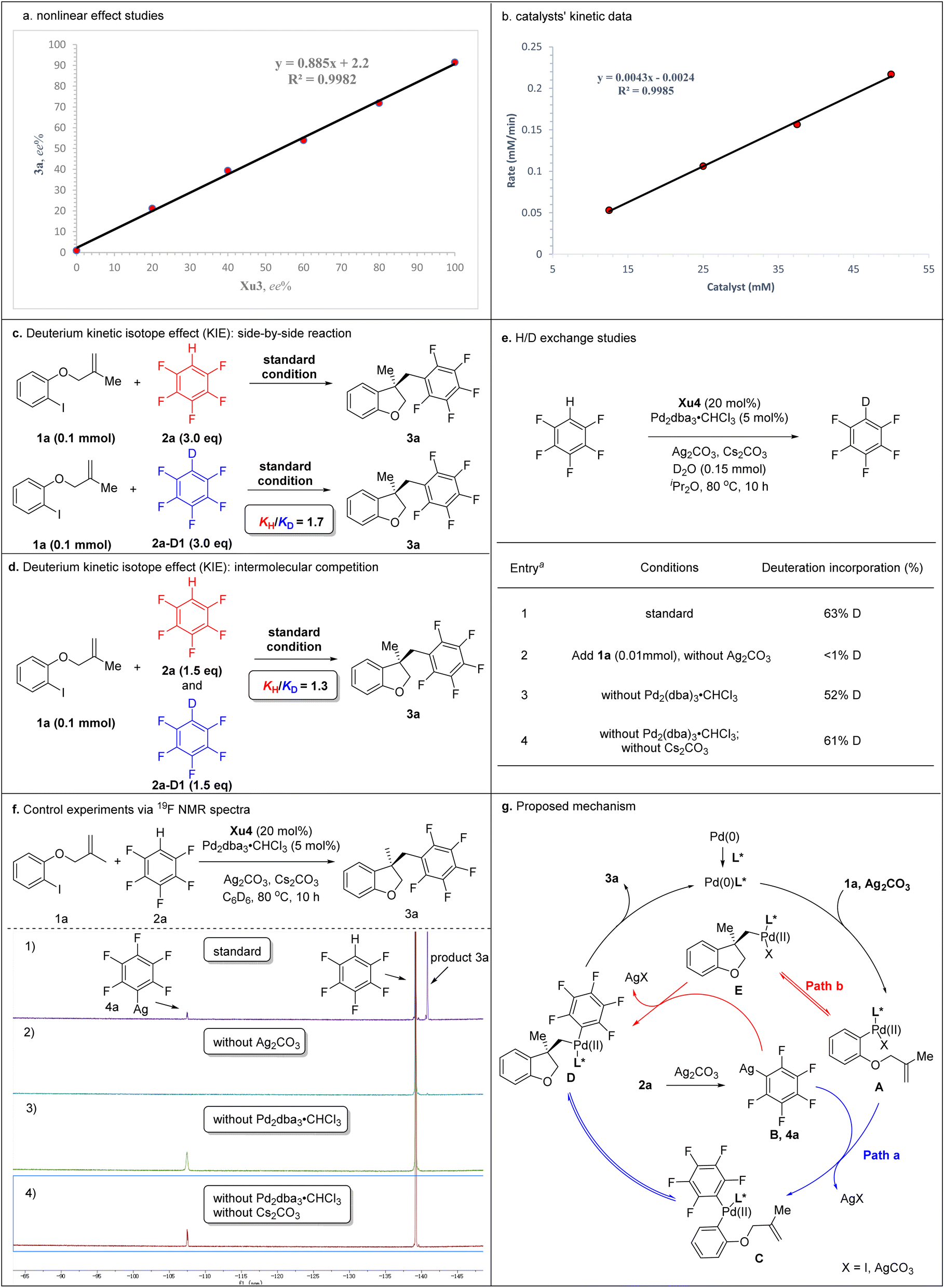 | ||
| Scheme 5 Mechanistic studies and proposed mechanism. | ||
To gain deep insight into the reaction mechanism, several control experiments were carried out (Schemes 5). Nonlinear effect studies on the enantiomeric composition of the chiral ligand Xu3 and product 3a (Scheme 5a) and initial rate experiments (Scheme 5b) indicated that there is a significant first-order dependence on the catalyst. We performed side-by-side experiments with pentafluorobenzene 2a and deuterated pentafluorobenzene 2a-[D1] to measure the initial reaction rate, respectively. The side-by-side experiments provided a KH/KD value of 1.7 (Scheme 5c). The intermolecular competition reaction of 2a and 2a-[D1] in the same pot showed a KH/KD value of 1.3 calculated from the consumption of 2a and 2a-[D1] (Scheme 5d). We also carried out H/D exchange experiments between C6F5H and D2O (5.0 equiv.). Analysis by 2H NMR spectroscopy showed 63% deuterium incorporation under standard conditions (Scheme 5e, entry 1). These results indicated that the C–H activation might not be the rate-determining step in this process. Moreover, adding 1a (0.1 mmol) to the reaction, <1% deuterium incorporation was detected in the absence of Ag2CO3 (Scheme 5e, entry 2). 52% and 61% deuterium incorporation was detected under standard conditions without Pd2dba3·CHCl3 or without Pd2dba3·CHCl3 and Cs2CO3, respectively (Scheme 5e, entries 3 and 4). These results suggested that Ag2CO3 was essential to activate the pentafluorobenzene. We further monitored the reaction via19F NMR spectroscopy. After 10 h, compound 4a was detected based on a diagnostic signal at approximately −107.4 ppm, which matches the C6F5Ag species chemical shift in the literature (Scheme 5f, entry 1).24 In addition, this same intermediate was also formed under standard conditions without Pd2dba3·CHCl3 or without Pd2dba3·CHCl3 and Cs2CO3 (Scheme 5f, entries 3 and 4), which indicated that Ag2CO3 could activate the pentafluorobenzene to afford the C6F5Ag species.
Two possible mechanisms were depicted as shown in Scheme 5g. Oxidative addition of Pd(0) with 1a afforded arylpalladium species A, followed by transmetallization with intermediate B which was generated by the reaction of 2a with Ag2CO3, resulting in the formation of complex C. The subsequent intramolecular Heck-type reaction of intermediate C provided chiral species D, which could undergo reductive elimination to produce product 3a and regenerate the Pd(0) catalyst. Alternatively, the intramolecular Heck-type reaction of intermediate A occurred firstly to generate intermediate E. Then, complex E underwent transmetallization with intermediate B to afford chiral species D. Finally, reductive elimination of D gave 3a and regenerated the Pd(0) catalyst. Notably, the mechanism involving the transformation of ArPdI(II)L* species into positively charged ArPd(II)L* species in the presence of silver salt could not be ruled out.
Conclusions
In summary, with the use of diverse polyfluoro- and heteroarenes as direct arylation coupling partners, Pd/XuPhos complexes are shown to be effective catalysts for asymmetric domino Heck/intermolecular C–H alkylation of unactivated alkenes, in which, a variety of dihydrobenzofuran, indoline and indane compounds are obtained in high performance. Easily accessible substrates, mild conditions, good functional group tolerance and various synthetically transformations of the products make this protocol highly attractive. Additionally, mechanistic studies indicate that C–H activation might not be the rate-determining step in this process. We anticipate that this methodology will inspire the discovery of more novel catalyst systems for handling these valuable and challenging asymmetric transformations.Data availability
All data have been provided in the main text and ESI.†Author contributions
C. F., Q.-P. W. and B. X. carried out the experimental and data-analysis work. Z.-M. Z. and J. Z. designed the reaction, directed the project, and wrote the paper with the assistance of B. X.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was made possible as a result of a generous grant from the National Key R&D Program of China (No. 2021YFF0701600), NSFC (No. 22031004), the Shanghai Municipal Education Commission (No. 20212308), China Postdoctoral Science Foundation (No. 2022M713667) and STCSM (No. 23ZR1445600).Notes and references
- X. Chen, K. M. Engle, D.-H. Wang and J.-Q. Yu, Palladium(II)-catalyzed C–H activation/C–C cross-coupling reactions: versatility and practicality, Angew. Chem., Int. Ed., 2009, 48, 5094 CrossRef CAS PubMed.
- (a) D. A. Colby, R. G. Bergman and J. A. Ellman, Rhodium-catalyzed C–C bond formation via heteroatom-directed C–H bond activation, Chem. Rev., 2010, 110, 624 CrossRef CAS PubMed; (b) K. M. Engle, T.-S. Mei, M. Wasa and J.-Q. Yu, Weak coordination as a powerful means for developing broadly useful C–H functionalization reactions, Acc. Chem. Res., 2012, 45, 788 CrossRef CAS PubMed; (c) Z. Huang, H. N. Lim, F. Mo, M. C. Young and G. Dong, Transition metal-catalyzed ketone-directed or mediated C–H functionalization, Chem. Soc. Rev., 2015, 44, 7764 RSC; (d) T. Gensch, M. N. Hopkinson, F. Glorius and J. Wencel-Delord, Mild metal-catalyzed C–H activation: examples and concepts, Chem. Soc. Rev., 2016, 45, 2900 RSC; (e) J. F. Hartwig, Borylation and silylation of C–H bonds: a platform for diverse C–H bond functionalizations, Acc. Chem. Res., 2012, 45, 864 CrossRef CAS PubMed.
- R. T. Ruck, M. A. Huffman, M. M. Kim, M. Shevlin, W. V. Kandur and I. W. Davies, Palladium-catalyzed tandem Heck reaction/C-H functionalization–preparation of spiroindane-oxindoles, Angew. Chem., Int. Ed., 2008, 47, 4711 CrossRef CAS PubMed.
- T. Piou, L. Neuville and J. Zhu, Activation of a C(sp3)-H bond by a transient salkylpalladium(II) complex: synthesis of spirooxindoles through a palladium-catalyzed domino carbopalladation/C(sp3)-C(sp3) bondforming process, Angew. Chem., Int. Ed., 2012, 51, 11561 CrossRef CAS PubMed.
- J. Ye, Z. Shi, T. Sperger, Y. Yasukawa, C. Kingston, F. Schoenebeck and M. Lautens, Remote C-H alkylation and C-C bond cleavage enabled by an in situ generated palladacycle, Nat. Chem., 2017, 9, 361 CrossRef CAS PubMed.
- Y. Ping, Y. Li, J. Zhu and W. Kong, Construction of quaternary stereocenters by palladium-catalyzed carbopalladationinitiated cascade reactions, Angew. Chem., Int. Ed., 2019, 58, 1562 CrossRef CAS PubMed.
- F. Ye, Y. Ge, A. Spannenberg, H. Neumann and M. Beller, The role of allyl ammonium salts in palladium-catalyzed cascade reactions towards the synthesis of spiro-fused, Nat. Commun., 2020, 11, 5383 CrossRef CAS PubMed.
- B. Xu, D. Ji, L. Wu, L. Zhou, Y. Liu, Z.-M. Zhang and J. Zhang, Palladium/Xu-Phos-catalyzed enantioselective cascade Heck/remote C(sp2) –H alkylation reaction, Chem, 2022, 8, 1 Search PubMed.
- A. D. Marchese, B. Mirabi, C. E. Johnson and M. Lautens, Reversible C–C bond formation using palladium catalysis, Nat. Chem., 2022, 14, 398 CrossRef CAS.
- O. René, D. Lapointe and K. Fagnou, Domino palladium-catalyzed Heck-intermolecular direct arylation reactions, Org. Lett., 2009, 11, 4560 CrossRef PubMed.
- U. K. Sharma, N. Sharma, Y. Kumar, B. K. Singh and E. V. Van der Eycken, Domino Carbopalladation/C–H Functionalization Sequence: An Expedient Synthesis of Bis-Heteroaryls through Transient Alkyl/Vinyl–Palladium Species Capture, Chem.–Eur. J., 2016, 22, 481 CrossRef CAS.
- S. Chen, P. Ranjan, N. Ramkumar, L. V. Meervelt, E. V. Van der Eycken and U. K. Sharma, Ligand-Enabled Palladium-Catalyzed Through-Space C−H Bond Activation via a Carbopalladation/1,4-Pd Migration/C−H Functionalization Sequence, Chem.–Eur. J., 2020, 26, 14075 CrossRef CAS PubMed.
- X.-X. Wu, W.-L. Chen, Y. Shen, S. Chen, P.-F. Xu and Y.-M. Liang, Palladium-catalyzed domino Heck/intermolecular C–H bond functionalization: effcient synthesis of alkylated polyfluoroarene derivatives, Org. Lett., 2016, 18, 1784–1787 CrossRef CAS PubMed.
- K. Ishu, D. Kumar, N. K. Maurya, S. Yadav, D. Chaudharya and M. R. Kuram, Dicarbofunctionalization of unactivated alkenes by palladium-catalyzed domino Heck/intermolecular direct hetero arylation with heteroarenes, Org. Biomol. Chem., 2021, 19, 2243 RSC.
- W. Kong, Q. Wang and J. Zhu, Palladium-catalyzed enantioselective domino heck/intermolecular C–H bond functionalization: development and application to the synthesis of (+)-esermethole, J. Am. Chem. Soc., 2015, 137, 16028 CrossRef CAS.
- Z.-M. Zhang, B. Xu, Y. Qian, L. Wu, Y. Wu, L. Zhou, Y. Liu and J. Zhang, Palladium-catalyzed enantioselective reductive heck reactions: convenient access to 3,3-disubstituted 2,3-dihydrobenzofuran, Angew. Chem., Int. Ed., 2018, 57, 10373 CrossRef CAS.
- Z.-M. Zhang, B. Xu, L. Wu, Y. Wu, Y. Qian, L. Zhou, Y. Liu and J. Zhang, Enantioselective dicarbofunctionalization of unactivated alkenes by palladium-catalyzed tandem heck/Suzuki coupling reaction, Angew. Chem., Int. Ed., 2019, 58, 14653 CrossRef CAS PubMed.
- Z.-M. Zhang, B. Xu, L. Wu, L. Zhou, D. Ji, Y. Liu, Z. Li and J. Zhang, Palladium/XuPhos-catalyzed enantioselective carboiodination of olefin-tethered aryl iodides, J. Am. Chem. Soc., 2019, 141, 8110 CrossRef CAS PubMed.
- L. Zhou, S. Li, B. Xu, D. Ji, L. Wu, Y. Liu, Z.-M. Zhang and J. Zhang, Enantioselective difunctionalization of alkenes by a palladium-catalyzed heck/Sonogashira sequence, Angew. Chem., Int. Ed., 2020, 59, 2769–2775 CrossRef CAS PubMed.
- B. B. Jarvis, S. N. Comezoglu, M. M. Rao, N. B. Pena, F. E. Boettner, G. Forsyth and B. Epling, Isolation of macrocyclic trichothecenes from a large-scale extract of baccharis megapotamica, J. Org. Chem., 1987, 52, 45 CrossRef CAS.
- (a) C. B. Bernard, H. G. Krishnamurty, D. Chauret, T. Durst, B. J. R. Philogene, P. Sanchez-Vindas, C. Hasbun, L. Poveda, L. S. Roman and J. T. Arnason, Insecticidal defenses of piperaceae from the neotropics, J. Chem. Ecol., 1995, 21, 801–814 CrossRef CAS PubMed; (b) D. C. Chauret, C. B. Bernard, J. T. Arnason and T. Durst, Insecticidal neolignans from piper decurrens, J. Nat. Prod., 1996, 59, 152 CrossRef CAS PubMed; (c) K. Ding, Y. Lu, Z. Nikolovska-Coleska, G. Wang, S. Qiu, S. Shangary, W. Gao, D. Qin, J. Stuckey and K. Krajewski, Structure-based design of spiro-oxindoles as potent, specific small-molecule inhibitors of the MDM2-p53 interaction, J. Med. Chem., 2006, 49, 3432 CrossRef CAS PubMed; (d) J. Pang and Z. Xu, Advances in the biological activities and synthesis of 2-arylbenzofurans, Chin. J. Org. Chem., 2005, 25, 25 CAS.
- C. V. Galliford and K. Scheidt, Pyrrolidinyl-spirooxindole natural products as inspirations for the development of potential therapeutic agents, Angew. Chem., Int. Ed., 2007, 46, 8748 CrossRef CAS PubMed.
- (a) S. Liu and J. Zhou, Research progress for biological activity of benzothiazolone compounds, Agrochemicals, 2012, 51, 863 CAS; (b) H. Chen and K. Li, Recent progress on biological activity and synthesis of 2-substituted benzofuran derivatives, J. Pharm. Pract., 2013, 31, 5 CAS; (c) H. Zhang, G. Ouyang, H. Chen, L. Zhang and Y. Wang, Review of oxindole compounds studies, Fine Chem. Intermed., 2016, 46, 9 CAS; (d) A. K. Gupta, M. Bharadwaj, A. Kumar and R. Mehrotra, Spiro-oxindoles as a promising class of small molecule inhibitors of p53-MDM2 interaction useful in targeted cancer therapy, Top. Curr. Chem., 2017, 375, 1 CrossRef CAS PubMed.
- (a) M. D. Lotz, N. M. Camasso, A. J. Canty and M. S. Sanford, Role of Silver Salts in Palladium-Catalyzed Arene and Heteroarene C−H Functionalization Reactions, Organometallics, 2017, 36, 165 CrossRef CAS; (b) W. Li, D. Yuan, G. Wang, Y. Zhao, J. Xie, S. Li and C. Zhu, Cooperative Au/Ag Dual-Catalyzed Cross-Dehydrogenative Biaryl Coupling: Reaction Development and Mechanistic Insight, J. Am. Chem. Soc., 2019, 141, 3187 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2325014. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc00262h |
| This journal is © The Royal Society of Chemistry 2024 |