
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOrganocatalyst-mediated asymmetric one-pot/two domino/three-component coupling reactions for the synthesis of trans-hydrindanes†
Naoki
Mori
 ,
Toshiki
Tachibana
,
Nariyoshi
Umekubo
and
Yujiro
Hayashi
*
,
Toshiki
Tachibana
,
Nariyoshi
Umekubo
and
Yujiro
Hayashi
*
Department of Chemistry, Graduate School of Science, Tohoku University, Sendai 980-8578, Japan. E-mail: yujiro.hayashi.b7@tohoku.ac.jp
First published on 12th March 2024
Abstract
Highly substituted trans-hydrindanes were synthesized by the three-component coupling reactions of 1,3-diethyl 2-(2-oxopropylidene)propanedioate and two different α,β-unsaturated aldehydes catalyzed by diphenylprolinol silyl ether. The reaction proceeds via two successive independent catalytic domino reactions in a one-pot reaction by a single chiral catalyst. Domino reactions involve Michael/Michael and Michael/aldol reactions to afford trans-hydrindanes with excellent diastereoselectivity and nearly optically pure form.
Introduction
Domino reactions, which can construct several bonds and generate complex molecules in a single reaction vessel, have been shown to be synthetically useful, as all reagents and catalysts can be added at the beginning of the reaction.1 Recently, the field of organocatalyzed-asymmetric domino reactions2 has been developing at a rapid pace.3 In most domino reactions, a single chiral organocatalyst is utilized to generate product with control over several chiral centers. An excellent early and successful example was reported by Enders (Scheme 1, eqn (1)),4 which has been represented schematically in eqn (4) (Scheme 2).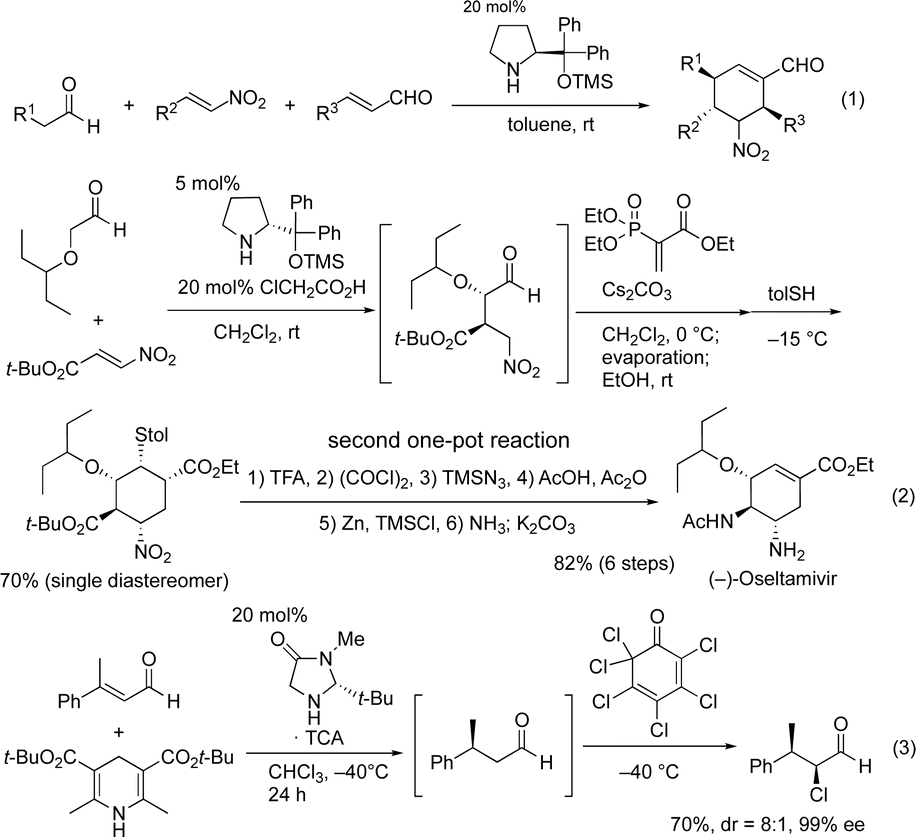 | ||
| Scheme 1 The representative domino and one-pot reactions. | ||
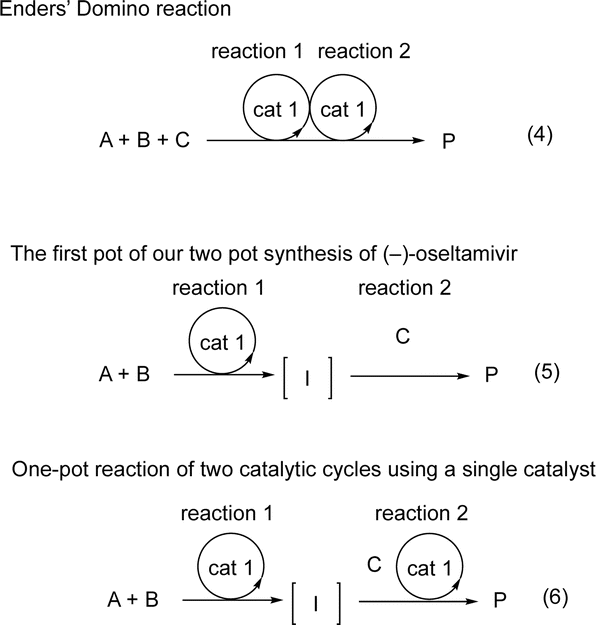 | ||
| Scheme 2 The schematic representation of domino reaction and one-pot reaction. A, B, C = reagents. I = intermediate. P = product. | ||
One-pot reactions have also been shown to be efficient methods for synthesizing complex molecules, in which the construction of multiple bonds in a single reaction vessel is necessary. For a one-pot reaction, consecutive reactions are performed, in which reagents are added sequentially.5 Our group has an interest in organocatalyst mediated one-pot reactions using a single chiral catalyst, and have previously accomplished several total syntheses of biologically active molecules in a pot-economic manner.6 A representative example from our group is the first pot reaction of the two pot synthesis of (−)-oseltamivir (eqn (2)).6b In this synthesis, the first reaction is an organocatalyst mediated asymmetric Michael reaction. The subsequent reactions, however, are non-catalytic, and the observed stereochemistry is achieved via substrate control. This is represented schematically in eqn (5).
On the other hand, one-pot reactions, in which a single chiral catalyst can catalyze two independent catalytic cycles by sequential addition of reagents, are also useful reactions to control the absolute configuration of several stereogenic centers using a single chiral catalyst. A successful example of this is MacMillan's one-pot reaction with control over the two stereocenters (eqn (3)),7 its schematic representation is shown in eqn (6). In this reaction, catalyst control must outweigh substrate control in the second reaction. Thus, this is a difficult reaction with few successful examples. Moreover, MacMillan's reaction is a formal addition reaction of HCl. As far as we are aware, there are no examples of this type of reaction (eqn (6)), involving two C–C-bond forming catalytic cycles. In this paper, we will report the first asymmetric one-pot reaction via sequential addition of reagents, which involves two independent C–C– bond forming catalytic cycles, both catalyzed by the same chiral catalyst.
Bicyclo[4.3.0]nonane skeletons are known as hydrindanes,8 and there are several natural products possessing trans-hydrindane skeletons such as isopulo'upone,9 tamariscol,10 and haliclonadiamine11 (Fig. 1). There are a few methods for the synthesis of this skeleton using asymmetric catalytic reactions. An intramolecular Diels–Alder reaction of 2,7,9-decatrienoic acid derivatives or 2,7,9-decatrienals was utilized for the construction of trans-hydrindanes, which was catalyzed by chiral Lewis acids such as titanium, boron and copper with excellent diastereo- and enantio-selectivity (Scheme 3, eqn (7) and (8)).12 Desymmetrization by asymmetric copper-catalyzed intramolecular C–H insertion afforded trans-hydrindanes with moderate selectivity (eqn (9)).13
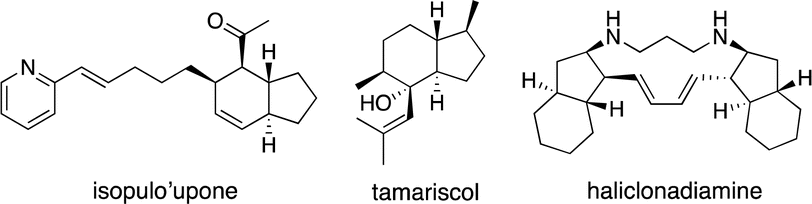 | ||
| Fig. 1 Natural products of trans-hydrindane skeleton. | ||
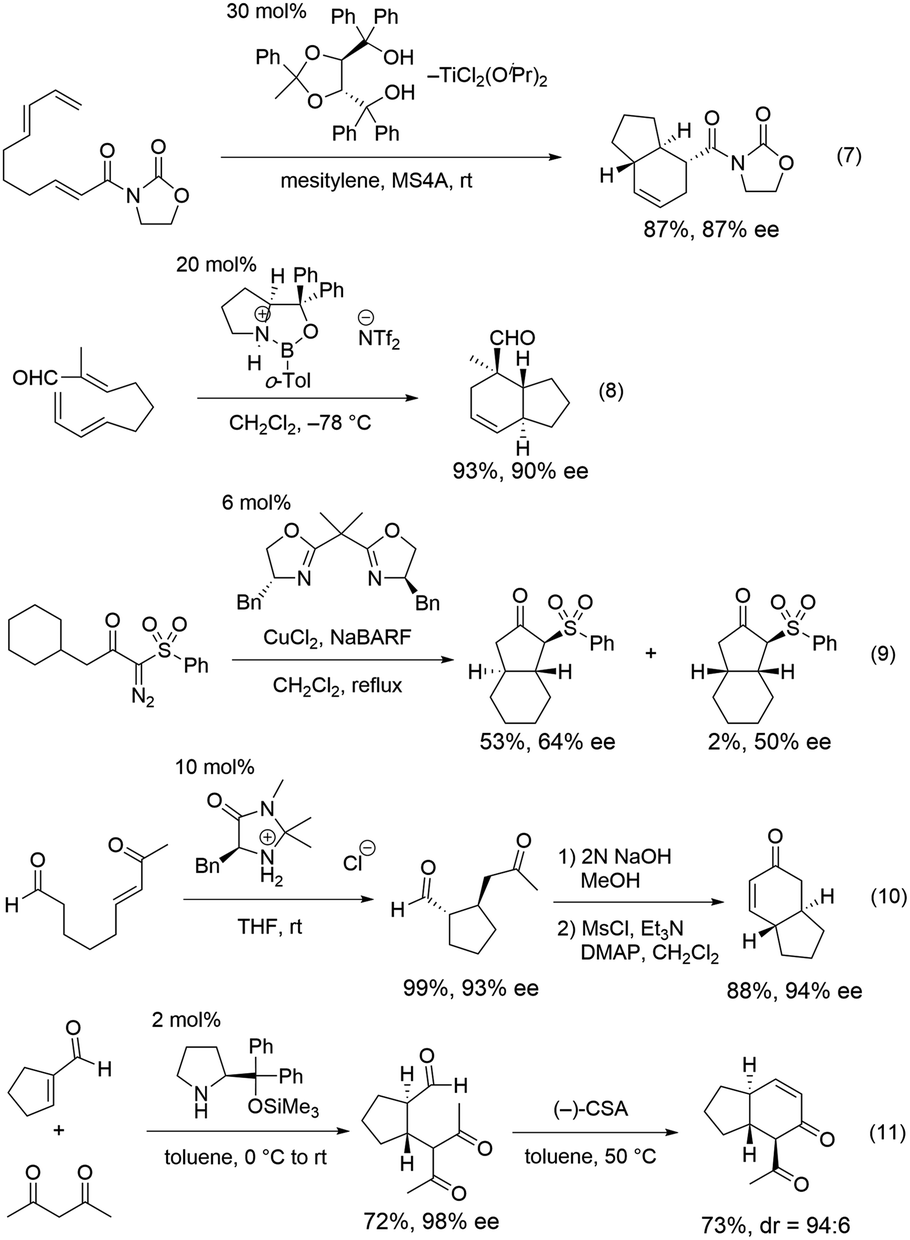 | ||
| Scheme 3 The previous asymmetric catalytic syntheses of trans-hydrindanes. | ||
Additionally, rapid developments in the organocatalytic field have enabled the formation of trans-hydrindanes using organocatalysts. Organocatalyst mediated intramolecular or an intermolecular Michael reaction, followed by an intramolecular aldol reaction afforded the trans-hydrindanes with excellent selectivity (eqn (10) and (11)).14 Although there are excellent catalytic methods for the synthesis of this skeleton, it remains a challenge to efficiently synthesize a scaffold possessing several substituents with excellent optical purity, whilst minimizing the number of pots.
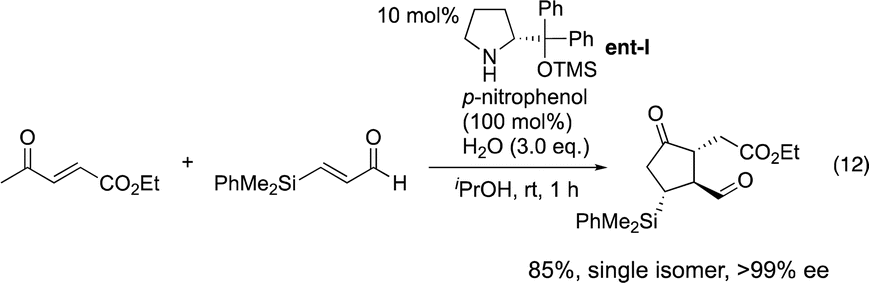
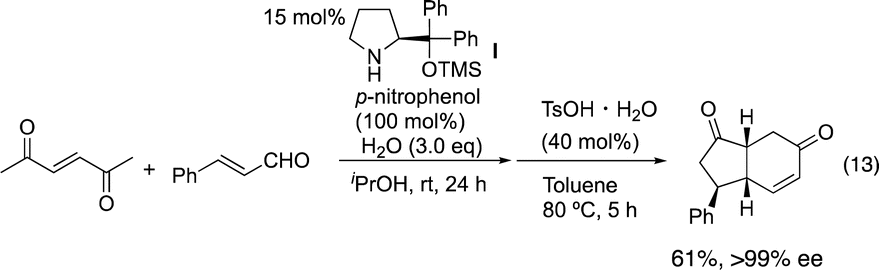
Our group15 and Jørgensen's group16 developed diphenylprolinol silyl ether I (Fig. 2),17 independently in 2005. We reported that this amine catalyzes the Michael reaction of ketones and α,β-unsaturated aldehydes,18 which was then used in the asymmetric synthesis of cyclopentanone derivatives (eqn (12))6g and one-pot synthesis of cis-hydrindanes via Michael/Michael/aldol condensation reaction (eqn (13)).19
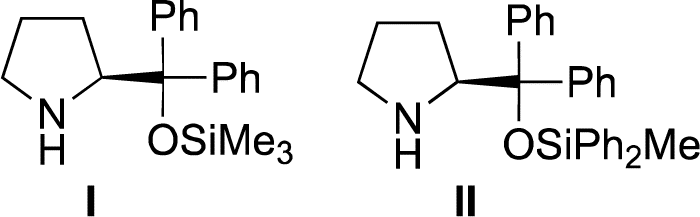 | ||
| Fig. 2 The catalysts examined in this study. | ||
Our continuous interest in the synthesis of hydrindanes lead us to develop a one-pot synthesis of highly substituted trans-hydrindanes with high diastereo- and enantioselectivities via two independent catalytic cycles catalyzed by the same chiral organocatalyst.
Results and discussion
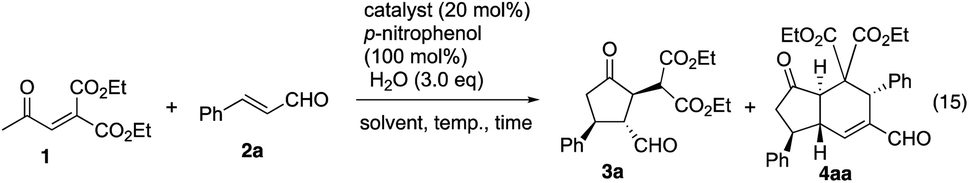
In the synthesis of cyclopentanone derivatives via an asymmetric domino Michael/Michael reaction, we employed ethyl 4-oxo-2-pentenoate (eqn (12)). Expecting the same domino Michael/Michael reaction, we used 1,3-diethyl 2-(2-oxopropylidene)propanedioate (1)20 instead of ethyl 4-oxo-2-pentenoate (eqn (14)).21 The reaction of 1 and cinnamaldehyde (2a) proceeded as expected. However, the desired product 3a was obtained in 32% yield along with the side product 4aa in 18% yield. 4aa possesses a highly substituted trans-hydrindane structure, which is generated by further reaction of 3a with cinnamaldehyde (2a) (vide infra). This is a three-component coupling reaction of 1 with two molecules of α,β-unsaturated aldehyde 2a. If two different α,β-unsaturated aldehydes can be employed, it would generate a variety of trans-hydrindanes. As 4aa was generated by the two successive reactions such as the reaction (1 + 2a → 3a) and the reaction (3a + 2a → 4aa), we first investigated each reaction separately, and then combined the two reactions to realize the one-pot reaction.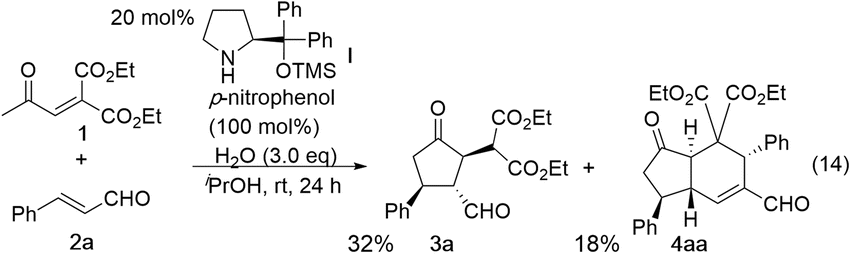
We investigated the first reaction of 1 and 2a for the synthesis of 3a using an equal amount of each reagent alongside catalyst I (Table 1). For future application to a one-pot operation, it is not possible to use an excess of reagent 1 to help prevent the three-component self-coupling reaction that forms 4aa. Although i-PrOH was the best solvent in the previous reaction of ethyl 4-oxo-2-pentenoate (eqn (12)), the present reaction afforded the product 3a in a low yield (entry 1). As 1 has a highly activated double bond with three electron withdrawing groups, it is a reactive Michael acceptor and trimethylsilyl ether catalyst I might react with 1 as a Michael donor. In order to prevent this possible side reaction, we used a more sterically hindered diphenylmethylsilyl ether II as a catalyst which resulted in a higher yield (entry 2). Next, different solvents were investigated such as i-PrOH, CH3CN, CH2Cl2, THF and toluene (entries 2–6). Toluene was found to be a suitable solvent, affording 3a in 65% yield and the over-reaction product 4aa in 9% yield. The generation of 4aa was completely suppressed when the reaction was conducted at lower temperature (0 °C), providing the desired product 3a in good yield (75%, entry 7). It was also found that 3a was obtained in a nearly optical pure form. The yield of 3a slightly decreased in the absence of water (entry 8).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Solvent | Time [h] | Yield of 3a [%] | Yield of 4aa [%] |
| a Unless otherwise shown, the reaction was performed by employing 1 (0.5 mmol), 2a (0.5 mmol), organocatalyst (0.1 mmol), p-nitrophenol (0.5 mmol), water (1.5 mmol), in solvent (1.0 mL) at room temperature. b 1 (2.3 mmol), 2a (2.3 mmol), organocatalyst (0.47 mmol), p-nitrophenol (2.3 mmol), water (7.0 mmol), and solvent (4.7 mL) were employed. c Reaction was carried out at 0 °C. d Enantiomeric excess (ee) was determined to be >99% by HPLC analysis on a chiral column material. e The reaction was carried out in the absence of water. | |||||
| 1 | I | iPrOH | 6 | 32 | 18 |
| 2 | II | iPrOH | 6 | 53 | 22 |
| 3 | II | CH3CN | 14 | 42 | 16 |
| 4 | II | CH2Cl2 | 20 | 49 | 20 |
| 5 | II | THF | 20 | 46 | 19 |
| 6 | II | Toluene | 6 | 65 | 9 |
| 7b | II | Toluenec | 24 | 75d | Trace |
| 8 | II | Toluenec,e | 24 | 70 | Trace |
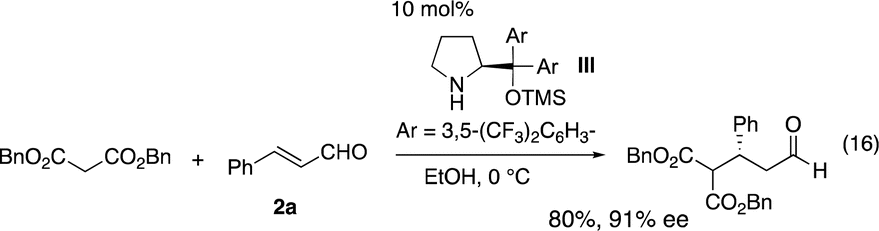
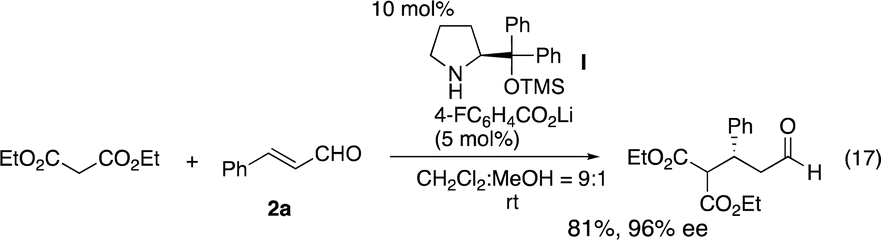
Once a good result was obtained in the first reaction, the second reaction was investigated using the isolated 3a and 3-(p-methoxyphenyl)prop-2-enal (2b). Malonate is known to be a good Michael donor in the reaction of α,β-unsaturated aldehyde catalyzed by diarylprolinol silyl ether, but the best reaction conditions vary depending on the paper. While Jørgensen reported the Michael reaction of malonates and α,β-unsaturated aldehyde catalyzed by diarylprolinol silyl ether with trifluoromethyl substituents (eqn (16)),22 Ye and coworkers reported that the same reaction was catalyzed by a combination of diphenylprolinol silyl ether with lithium 4-fluorobenzoate (eqn (17)).23
For a one-pot reaction, it is desirable if both the first and second reaction can be catalyzed by the same catalyst. Thus, the reaction was examined with catalyst II. After the optimization of the reaction conditions, (see Table S1†) p-nitrophenol and t-BuOH24 were found to be the best additive and the solvent, respectively (eqn (18)). It should be noted that the reaction conditions are rather different from those previously reported (eqn (16) and (17)).
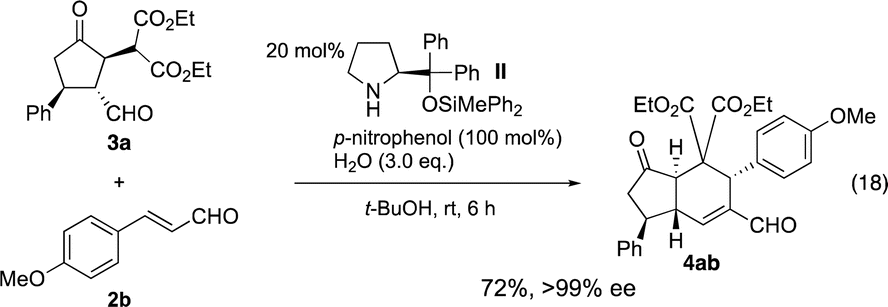
Both the first reaction and the second reaction were optimized. In the first reaction, we used 1 and 2 as a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio, and we can combine these two reactions in one-pot to afford the trans-hydrindane 4ab with excellent enantioselectivity (eqn (19)).
:1 ratio, and we can combine these two reactions in one-pot to afford the trans-hydrindane 4ab with excellent enantioselectivity (eqn (19)).
After this one-pot synthesis of 4ab was established, the generality of the reaction was investigated (Table 2). First, we examined the effect of β-aryl group (R1) of the α,β-unsaturated aldehyde 2 in the first reaction, in which cinnamaldehyde (2a, R2 = Ph) was employed as a second α,β-unsaturated aldehyde 2′. Not only electron rich p-methoxyphenyl but also electron deficient p-chloro-, p-bromo, and p-trifluoromethyl phenyl substituents are suitable to afford the trans-hydrindanes 4 in a nearly optically pure form. The (E)-4-methoxystyryl substituent also gave product 4 even though the yield was low. (E)-5-Phenylpent-2-enal, (E)-5-(trimethylsilyl)pent-2-en-4-ynal and 2-propenal were found to not be suitable for this reaction (see the ESI†).
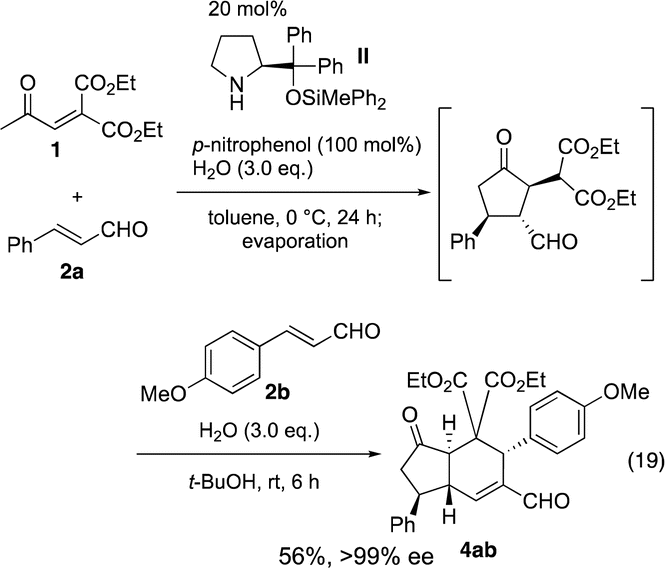
|
a Unless otherwise shown, the first reaction was performed by employing 1 (0.5 mmol), 2 (0.5 mmol), organocatalyst (0.1 mmol), p-nitrophenol (0.5 mmol), water (1.5 mmol), in toluene (1.0 mL), and the second reaction was performed by employing 2′ (0.5 mmol), water (1.5 mmol), in t-BuOH (1.0 mL).
b Products 4 were obtained as a single diastereomer.
c Enantiomeric excess (ee) was determined by HPLC analysis on a chiral column material.
d It took 5 d for the first reaction.
e
t-BuOH/toluene (1:1) was used in the second reaction.
|
|---|
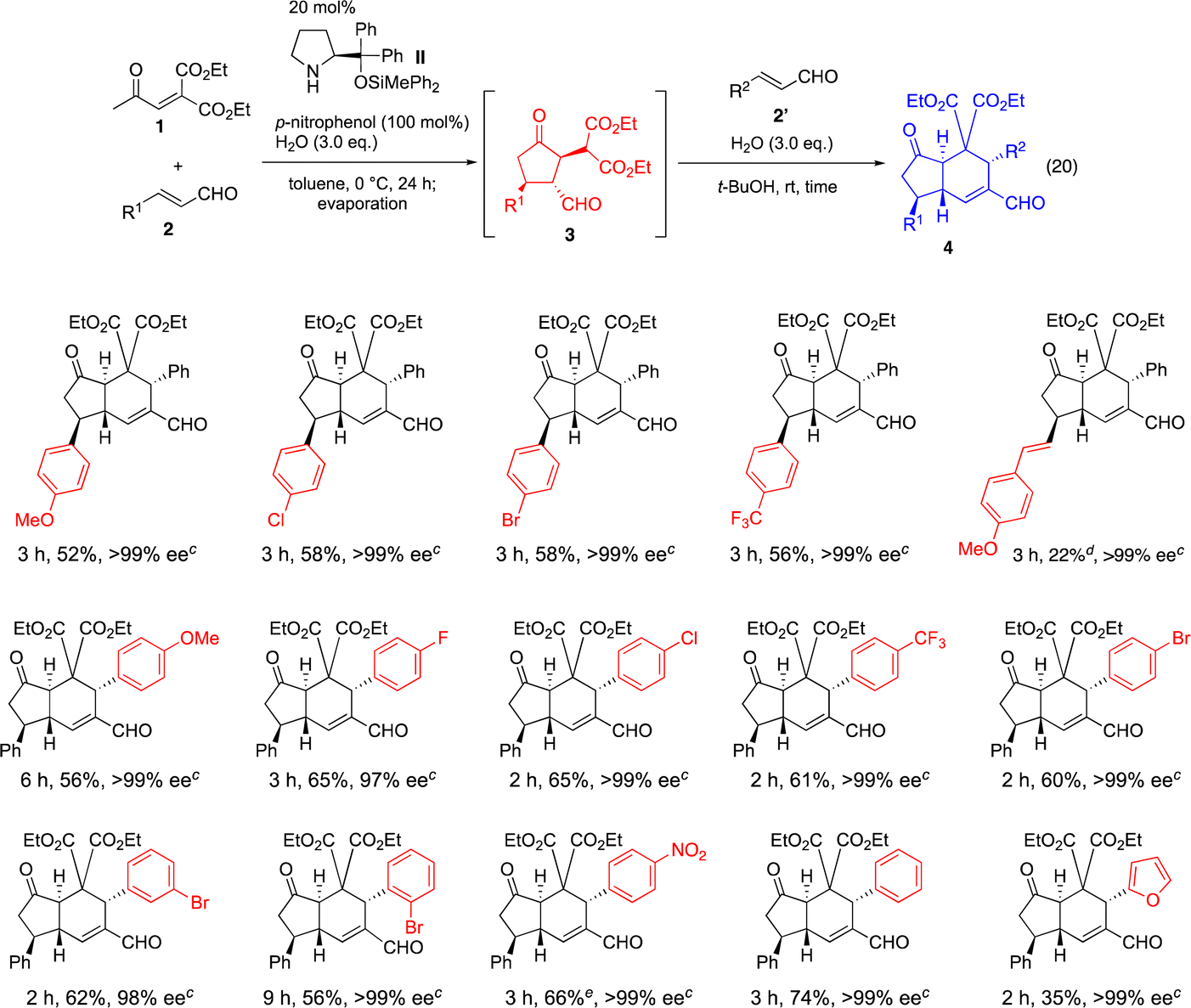
|
Next, we investigated the β-substituent (R2) of the α,β-unsaturated aldehyde 2′ in the second reaction, in which cinnamaldehyde (2a, R1 = Ph) was employed as a first α,β-unsaturated aldehyde 2. For the β-aryl substituent of the α,β-unsaturated aldehyde, not only electron rich p-methoxyphenyl group but also electron withdrawing p-fluorophenyl, p-chlorophenyl, p-trifluoromethylphenyl, p-bromophenyl, m-bromophenyl, o-bromophenyl and p-nitrophenyl are suitable substituents to afford the trans-hydrindanes with excellent enantioselectivity. Neutral aryl groups such as a phenyl group and heteroaryl groups such as a 2-furyl group can also be employed successfully. On the other hand, (E)-5-phenylpent-2-enal, (2E,4E)-5-phenylpenta-2,4-dienal, (E)-5-(trimethylsilyl)pent-2-en-4-ynal and 2-propenal resulted in no reaction or the formation of a complex mixture (see the ESI†).
It should be noted that the reaction is highly diastereoselective and enantioselective. In all cases examined, the product 425 was obtained with excellent diastereoselectivity and nearly optically pure form.
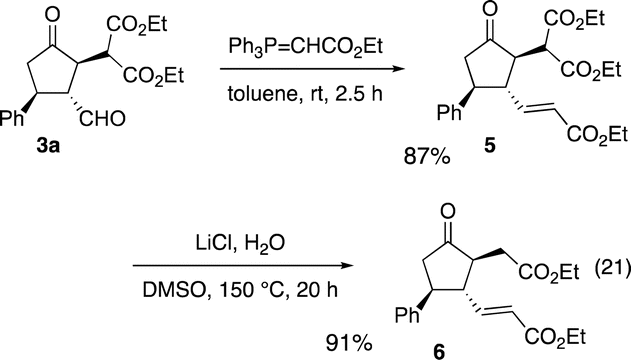
The absolute configuration of the first domino reaction product 3a was determined by comparison of the optical rotation with known compound 6,6g which was synthesized as shown in eqn (21): 3a was treated with Ph3P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCO2Et to afford 5 in good yield, and the reaction of 5 with LiCl afforded diester 6.
CHCO2Et to afford 5 in good yield, and the reaction of 5 with LiCl afforded diester 6.
The reaction proceeds as follows (Scheme 4): in the first domino reaction of 1 and α,β-unsaturated aldehyde, catalyst II reacts with α,β-unsaturated aldehyde to generate an iminium ion, which reacts with 1 to generate an enamine and intramolecular Michael reaction proceeds to afford 3. In the second domino reaction, catalyst II reacts with another α,β-unsaturated aldehyde to generate an iminium ion, which reacts with 3via Michael reaction to afford an enamine. Intramolecular aldol reaction successively proceeds to afford the trans-hydrindane.
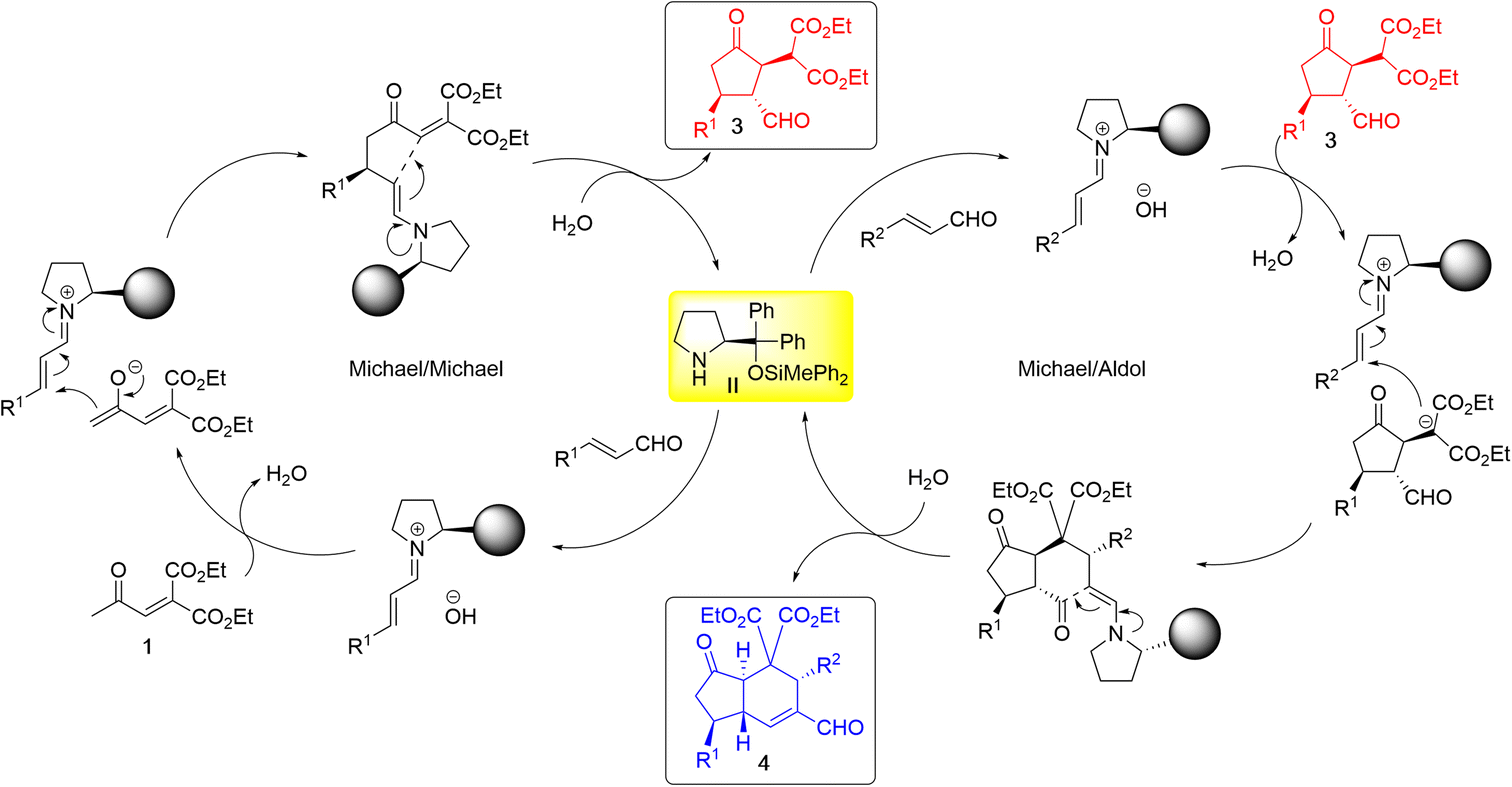 | ||
| Scheme 4 The reaction mechanism. | ||
Conclusions
In summary, we established a one-pot synthesis of highly substituted trans-hydrindanes with excellent diastereoselectivity and a nearly optical pure form. There are several noteworthy features in this reaction. There are four chiral centers, and one single diastereomer was obtained among the possible 16 isomers. This is a rare one-pot reaction composed of two independent domino reactions catalyzed by the same single chiral catalyst resulting in the formation of C–C bonds, in which the reaction proceeds via sequential addition of the reagents. The first domino reaction involves an asymmetric Michael/Michael reaction of iminium ion and enamine, respectively, catalyzed by diphenylprolinol silyl ether. The second domino reaction involves a diastereoselective Michael/aldol reaction of iminium ion and enamine, respectively, and is also catalyzed by diphenylprolinol silyl ether. Diphenylprolinol silyl ether catalyst II performs four roles. The first domino reaction involves, [1] an iminium ion generation, as well as, [2] generation of an enamine. While in the second cycle, [3] an iminium ion is generated, followed by, [4] generation of an enamine. This is a three-component coupling reaction of 1,3-diethyl 2-(2-oxopropylidene)propanedioate (1) with two different α,β-unsaturated aldehydes. The reaction proceeded using a synthetically efficient 1:1:1 ratio of these three components, enabling a library of trans-hydrindanes to be easily prepared by changing the two α,β-unsaturated aldehydes.
Data availability
General information, detailed experimental procedures, characterization data for compounds, and NMR, HPLC, IR spectra are available in the ESI.†Author contributions
N. M., T. T., and N. U. performed the experiments. Y. H. conceived the concept and prepared the manuscript with feedback from N. M., T. T., and N. U.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JSPS KAKENHI Grant Number JP19H05630.Notes and references
- For reviews, see: (a) L. F. Tietze, Chem. Rev., 1996, 96, 115 CrossRef CAS PubMed; (b) Domino Reactions, ed. L. F. Tietze, Wiley-VCH, Weinheim, 2014 Search PubMed, for recent examples, see: (c) Y. Chen, X. Liu, W. Luo, L. Lin and X. Feng, Synlett, 2017, 28, 966 CrossRef CAS; (d) F. E. Held, A. A. Guryev, T. Fröhlich, F. Hampel, A. Kahnt, C. Hutterer, M. Steingruber, H. Bahsi, C. von Bojničić-Kninski, D. S. Mattes, T. C. Foertsch, A. Nesterov-Mueller, M. Marschall and S. B. Tsogoeva, Nat. Commun., 2017, 8, 15071 CrossRef PubMed; (e) B. W. Grau, S. Bönisch, A. Neuhauser, F. Hampel, A. Görling and S. B. Tsogoeva, ChemCatChem, 2019, 11, 3982 CrossRef CAS.
- Selected reviews on organocatalysis: (a) Asymmetric Organocatalysis 1, ed. B. List, Thieme, Stuttgart, Germany, 2012 Search PubMed; (b) H. Jiang, Ł. Albrecht, G. Dickmeiss, K. L. Jensen and K. A. Jørgensen, Comprehensive Enantioselective Organocatalysis: Catalysts, Reactions, and Applications, ed. P. I. Dalko, Wiley-VCH, Weinheim, Germany, 2013, p. 33 Search PubMed.
- For review, see: (a) C. Grondal, M. Jeanty and D. Enders, Nat. Chem., 2010, 2, 167 CrossRef CAS PubMed; (b) Ł. Albrecht, H. Jiang and K. A. Jørgensen, Angew. Chem., Int. Ed., 2011, 50, 8492 CrossRef; (c) H. Pellissier, Adv. Synth. Catal., 2012, 354, 237 CrossRef CAS; (d) C. M. R. Volla, I. Atodiresei and M. Rueping, Chem. Rev., 2014, 114, 2390 CrossRef CAS PubMed.
- D. Enders, M. R. M. Huttl, C. Grondal and G. Raabe, Nature, 2006, 441, 861 CrossRef CAS PubMed.
- (a) Y. Hayashi, Chem. Sci., 2016, 7, 866 RSC; (b) Y. Hayashi, J. Org. Chem., 2021, 86, 1 CrossRef CAS PubMed; (c) Y. Hayashi, Acc. Chem. Res., 2021, 54, 1385 CrossRef CAS PubMed.
- (a) H. Ishikawa, T. Suzuki and Y. Hayashi, Angew. Chem., Int. Ed., 2009, 48, 1304 CrossRef CAS PubMed; (b) H. Ishikawa, T. Suzuki, T. Uchimaru and Y. Hayashi, Chem.–Eur. J., 2010, 16, 12616 CrossRef CAS PubMed; (c) Y. Hayashi and S. Umemiya, Angew. Chem., Int. Ed., 2013, 52, 3450 CrossRef CAS; (d) Y. Hayashi, D. Sakamoto and D. Okamura, Org. Lett., 2016, 18, 4 CrossRef CAS; (e) Y. Hayashi and S. Ogasawara, Org. Lett., 2016, 18, 3426 CrossRef CAS PubMed; (f) Y. Hayashi, S. Koshino, K. Ojima and E. Kwon, Angew. Chem., Int. Ed., 2017, 56, 11812 CrossRef CAS PubMed; (g) N. Umekubo, Y. Suga and Y. Hayashi, Chem. Sci., 2020, 11, 1205 RSC; (h) T. Terumuma and Y. Hayashi, Nat. Commun., 2022, 13, 7503 CrossRef PubMed; (i) G. Kawauchi, Y. Suga, S. Toda and Y. Hayashi, Chem. Sci., 2023, 14, 10081 RSC.
- Y. Huang, A. M. Walji, C. H. Larsen and D. W. C. MacMillan, J. Am. Chem. Soc., 2005, 127, 15051 CrossRef CAS.
- (a) P. Jankowski, S. Marczak and J. Wicha, Tetrahedron, 1998, 54, 12071 CrossRef CAS; (b) N. A. Eddy and P. Ichalkaranje, Molecules, 2016, 21, 1358 CrossRef.
- A. Spinella, L. A. Alvarez and G. Cimino, Tetrahedron, 1993, 49, 3203 CrossRef CAS.
- J. D. Connolly, L. J. Harrison and D. S. Rycroft, Tetrahedron Lett., 1984, 25, 1401 CrossRef CAS.
- E. Fahy, T. F. Molinski, M. K. Harper, B. W. Sullivan and D. J. Faulkner, Tetrahedron Lett., 1988, 29, 3427 CrossRef.
- (a) N. Iwasawa, J. Sugimori, Y. Kawase and K. Narasaka, Chem. Lett., 1989, 18, 1947 CrossRef; (b) G. Zhou, Q.-Y. Hu and E. J. Corey, Org. Lett., 2003, 5, 3979 CrossRef CAS PubMed.
- T. A. Brouder, C. N. Slattery, A. Ford, U. B. R. Khandavilli, E. Skořepová, K. S. Eccles, M. Lusi, S. E. Lawrence and A. R. Maguire, J. Org. Chem., 2019, 84, 7543 CrossRef CAS PubMed.
- (a) M. T. H. Fonseca and B. List, Angew. Chem., Int. Ed., 2004, 43, 3958 CrossRef; (b) Y. Stöckl, W. Frey, J. Lang, B. Claasen, A. Baro and S. Laschat, Synthesis, 2019, 51, 1123 CrossRef.
- Y. Hayashi, H. Gotoh, T. Hayashi and M. Shoji, Angew. Chem., Int. Ed., 2005, 44, 4212 CrossRef CAS PubMed.
- M. Marigo, T. C. Wabnitz, D. Fielenbach and K. A. Jørgensen, Angew. Chem., Int. Ed., 2005, 44, 794 CrossRef CAS PubMed.
- For reviews, see: (a) C. Palomo and A. Mielgo, Angew. Chem., Int. Ed., 2006, 45, 7876 CrossRef CAS; (b) A. Mielgo and C. Palomo, Chem.–Asian J., 2008, 3, 922 CrossRef CAS PubMed; (c) L.-W. Xu, L. Li and Z.-H. Shi, Adv. Synth. Catal., 2010, 352, 243 CrossRef CAS; (d) K. L. Jensen, G. Dickmeiss, H. Jiang, L. Albrecht and K. A. Jørgensen, Acc. Chem. Res., 2012, 45, 248 CrossRef CAS PubMed; (e) H. Gotoh and Y. Hayashi, Diarylprolinol Silyl Ethers: Development and Application as Organocatalysts, in Sustainable Catalysis, ed. P. J. Dunn, K. K. Hii, M. J. Krische and M. T. Williams, John Wiley & Son, Hoboken, NJ, 2013, ch. 13, p. 287 Search PubMed; (f) B. S. Donslund, T. K. Johansen, P. H. Poulsen, K. S. Halskov and K. A. Jørgensen, Angew. Chem., Int. Ed., 2015, 54, 13860 CrossRef CAS PubMed; (g) G. J. Reyes-Rodriguez, N. M. Rezayee, A. Vidal-Albalat and K. A. Jørgensen, Chem. Rev., 2019, 119, 4221 CrossRef CAS PubMed.
- N. Umekubo and Y. Hayashi, Angew. Chem., Int. Ed., 2018, 57, 1958 CrossRef PubMed.
- (a) N. Umekubo, R. Iwata and Y. Hayashi, Chem. Lett., 2020, 49, 867 CrossRef CAS; (b) D. Kalaitzakis, M. Sofiadis, V. Tsopanakis, T. Montagnon and G. Vassilikogiannakis, Org. Biomol. Chem., 2020, 18, 2817 RSC.
- Preparation of 2-(2-oxopropylidene)malononitrile and 4,4-bis(phenylsulfonyl)but-3-en-2-one, which possess methyl ketone and electron withdrawing groups, was unsuccessful, see the ESI†.
- D. Ma employed (Z)-triethyl 3-hydroxybuta-1,3-diene-1,1,4-tricarboxylate as a similar Michael acceptor, see: A. Ma and D. Ma, Org. Lett., 2010, 12, 3634 CrossRef CAS PubMed.
- S. Brandau, A. Landa, J. Franzén, M. Marigo and K. A. Jørgensen, Angew. Chem., Int. Ed., 2006, 45, 4305 CrossRef CAS PubMed.
- Y. Wang, P. Li, X. Liang and J. Ye, Adv. Synth. Catal., 2008, 350, 1383 CrossRef CAS.
- The beneficial effect of t-BuOH as a solvent is unclear.
- Decarboxylation of 4ab and its derivatives was tried but unsuccessful, see the ESI†.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc00193a |
| This journal is © The Royal Society of Chemistry 2024 |