
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRevealing the role of a bridging oxygen in a carbon shell coated Ni interface for enhanced alkaline hydrogen oxidation reaction†
Pengyu
Han‡
,
Xinyi
Yang‡
,
Liqing
Wu‡
,
Hongnan
Jia
and
Wei
Luo
 *
*
College of Chemistry and Molecular Sciences, Wuhan University, Wuhan, Hubei 430072, P. R. China. E-mail: wluo@whu.edu.cn
First published on 6th March 2024
Abstract
Encapsulating metal nanoparticles inside carbon layers is a promising approach to simultaneously improving the activity and stability of electrocatalysts. The role of carbon layer shells, however, is not fully understood. Herein, we report a study of boron doped carbon layers coated on nickel nanoparticles (Ni@BC), which were used as a model catalyst to understand the role of a bridging oxygen in a carbon shell coated Ni interface for the improvement of the hydrogen oxidation reaction (HOR) activity using an alkaline electrolyte. Combining experimental results and density functional theory (DFT) calculations, we find that the electronic structure of Ni can be precisely tailored by Ni–O–C and Ni–O–B coordinated environments, leading to a volcano type correlation between the binding ability of the OH* adsorbate and HOR activity. The obtained Ni@BC with a optimized d-band center displays a remarkable HOR performance with a mass activity of 34.91 mA mgNi−1, as well as superior stability and CO tolerance. The findings reported in this work not only highlight the role of the OH* binding strength in alkaline HOR but also provide guidelines for the rational design of advanced carbon layers used to coat metal electrocatalysts.
Introduction
The anion exchange membrane fuel cell (AEMFC) has been regarded as a promising cost-effective alternative to state-of-the-art proton exchange membrane fuel cells (PEMFCs), because of the development of efficient non-precious metal electrocatalysts for cathodic oxygen reduction reactions (ORR) in alkaline media.1–3 Unfortunately, when the operating environment switched from acidic to alkaline, anodic hydrogen oxidation reaction (HOR) kinetics become very sluggish due to the pH effect.4,5 Even for platinum-group-metals (PGMs), such as Pt and Pd, alkaline HOR activities decrease by about two orders of magnitude when compared to that in acid,6–8 which results in a higher consumption of precious metal to achieve the applicable peak power of AEMFC, thus offsetting advantages obtained by adopting low-cost PGM-free cathodes.9,10 To date, nickel-based catalysts have been regarded as the most promising non-precious metal-based electrocatalysts for alkaline HOR.11,12 However, their activities and stabilities are far lower than those of PGM-based catalysts.13,14 Consequently, several strategies have been endeavouring to improve the HOR performance of Ni, such as alloying,15,16 doping,17,18 constructing heterostructures,19,20 and regulating the metal-support interaction (MSI).21–24 Despite the fact that great efforts have been achieved, it is still a great challenge to precisely design Ni-based electrocatalysts with high activity and stability for alkaline HOR.25,26Recently, encapsulating metal nanoparticles in carbon layers was found to be an effective approach to simultaneously increase the activity and stability of catalysts.27 However, the exact role of coated carbon layers during electrocatalysis still remains debatable.28,29 The conventional view considers that the outer carbon layer of carbon possesses a physical and chemical shielding effect, which protects core active metals and improves the stability.30–32 Moreover, the porous carbon layer with defects and a certain porosity can act as a molecular sieve to prevent the adsorption of poisoning species on the surface of metal active sites, while not interfering with the mass transfer of reactants, thus improving the anti-poisoning ability of the catalyst.28,33,34 Conversely, the other primary view states that the surface on the outer carbon shell should be where real active sites are during electrocatalysis, because it is directly in contact with the electrolyte.35,36 The charge transfer from the metal core to carbon shell will alter the electronic state of the carbon surface, thereby leading to optimal adsorption energy for reaction intermediates and enhanced catalytic activity.37–39 Furthermore, it has been reported that the catalytic performance of carbon coated metal nanoparticle catalysts can be further enhanced by heteroatom doping.40–42 Introducing heteroatoms, such as nitrogen and boron, with great differences of electronegativity into the sp2 lattice of graphitic carbon will alter the local electronic structure of the carbon material and affect the extent of the π electron delocalization, thus breaking the electrical neutrality and creating favorable sites for catalysis.43 However, in most of precursors, metal ions and organic compounds are connected by oxygen atoms, which will result in the inevitable presence of oxygen at the interface between the carbon layer and core metal nanoparticles during pyrolysis.44–46 The oxygen atoms at the interface can act as a bridge to inevitably impact the electronic structure of the carbon layer and metal nanoparticles, but this influence has been investigated rarely.
In this study, carbon coated nickel nanoparticles (Ni@C) were prepared by the pyrolysis of a Ni-BDC metal organic framework (MOF) precursor. X-ray photoelectron spectroscopy (XPS) and X-ray absorption spectroscopy (XAS) results indicate that the carbon layer and Ni nanoparticles are connected by bridging oxygen atoms, thereby leading to the regulated surface electronic state of Ni through Ni–O–C interfaces. Furthermore, boron doped carbon coated Ni nanoparticles (Ni@BC) with an Ni–O–B interface were successfully synthesized by replacing some terephthalic acid ligands with 4-carboxylphenylboronic acid. Experimental results and density functional theory (DFT) calculations demonstrate that the presence of bridging oxygen coordinated environments (Ni–O–C and Ni–O–B) can precisely tailor the electronic structure of Ni, leading to a volcano-type correlation between the binding ability of OH* adsorbate and alkaline HOR activity. The resultant Ni@BC with an optimal d-band center and adsorbed hydroxyl binding energy (OHBE) displays a remarkable HOR performance with an alkaline electrolyte, showing a mass activity of 34.91 mA mgNi−1 at 50 mV and a specific activity of 0.045 mA cmNi−2, outperforming most of the reported Ni-based electrocatalysts.
Results and discussion
Synthesis and characterization of prepared samples
As illustrated in Fig. 1a, carbon coated nickel (Ni@C) and boron doped carbon coated nickel (Ni@BC) with abundant Ni–O–C(B) interfaces were synthesized by a two-step approach. First, the Ni-BDC is assembled from terephthalic acid (H2BDC) and nickel ions (Ni2+) via a solvothermal method, resulting in many Ni–O–C configurations in the structure. After 4-carboxyphenylboronic acid partially replaced the H2BDC to coordinate with Ni2+, the Ni–O–B interface may also be constructed, and the obtained precipitate was denoted as Ni-BDC-BA.47,48 Subsequently, Ni@C and Ni@BC samples were prepared by pyrolyzing Ni-BDC and Ni-BDC-BA precursors at 400 °C in an N2/H2 mixed atmosphere, respectively. During this process, the Ni nanoparticles were gradually confined in the carbon matrix, and Ni–O–C(B) link sites can be transferred to interfacial-O on the surface of nickel.45 Moreover, the number of Ni–O–B motifs can be controlled by varying the feeding molar ratio of terephthalic acid and 4-carboxyphenylboronic acid. Among the samples tested, Ni@BC 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 derived from Ni-BDC-BA 5:5 exhibits the best HOR performance; therefore, it is denoted as Ni@BC when compared with Ni@C and Ni in the text and used for further characterizations. As shown in Fig. S1,† XRD patterns verify the successful synthesis of the Ni-BDC, which corresponded well with simulated patterns. After the ligand was partially replaced, the XRD pattern indicates that the Ni-BDC-BA 5:5 still roughly maintained the structure of the Ni-BDC (Fig. S2†). As shown in Fig. 1b, the XRD patterns of the Ni@C and Ni@BC show the formation of the metallic nickel crystalline phase, and peaks located at 44.5°, 51.8° and 76.4° correspond to the (111), (200), and (220) planes of the face-centered cubic (fcc) Ni (PDF#04-0850). As shown in Fig. S3 and S4,† transmission electron microscopy (TEM) images reveal that the Ni@C and Ni@BC are composed of small-sized nickel nanoparticles distributed on the carbon matrix due to the confined effect of the carbon layers generated in situ.49,50 For the Ni@BC, high-resolution TEM (HRTEM) images shown in Fig. S5† and 1c clearly display the carbon coated nickel structure, and the interface can be apparently observed. The interfacial lattice fringes with interplanar spacings of 0.34 and 0.20 nm can be assigned to the (002) plane of graphite carbon and the (111) plane of fcc Ni (Fig. 1d and e), respectively.51 Furthermore, the high-angle annular dark-field scanning TEM (HAADF-STEM)-energy dispersive X-ray mapping images confirm that O, C and B elements are uniformly distributed around Ni (Fig. 1f), which roughly follows the profile of the Ni@BC structure.
:5 derived from Ni-BDC-BA 5:5 exhibits the best HOR performance; therefore, it is denoted as Ni@BC when compared with Ni@C and Ni in the text and used for further characterizations. As shown in Fig. S1,† XRD patterns verify the successful synthesis of the Ni-BDC, which corresponded well with simulated patterns. After the ligand was partially replaced, the XRD pattern indicates that the Ni-BDC-BA 5:5 still roughly maintained the structure of the Ni-BDC (Fig. S2†). As shown in Fig. 1b, the XRD patterns of the Ni@C and Ni@BC show the formation of the metallic nickel crystalline phase, and peaks located at 44.5°, 51.8° and 76.4° correspond to the (111), (200), and (220) planes of the face-centered cubic (fcc) Ni (PDF#04-0850). As shown in Fig. S3 and S4,† transmission electron microscopy (TEM) images reveal that the Ni@C and Ni@BC are composed of small-sized nickel nanoparticles distributed on the carbon matrix due to the confined effect of the carbon layers generated in situ.49,50 For the Ni@BC, high-resolution TEM (HRTEM) images shown in Fig. S5† and 1c clearly display the carbon coated nickel structure, and the interface can be apparently observed. The interfacial lattice fringes with interplanar spacings of 0.34 and 0.20 nm can be assigned to the (002) plane of graphite carbon and the (111) plane of fcc Ni (Fig. 1d and e), respectively.51 Furthermore, the high-angle annular dark-field scanning TEM (HAADF-STEM)-energy dispersive X-ray mapping images confirm that O, C and B elements are uniformly distributed around Ni (Fig. 1f), which roughly follows the profile of the Ni@BC structure.
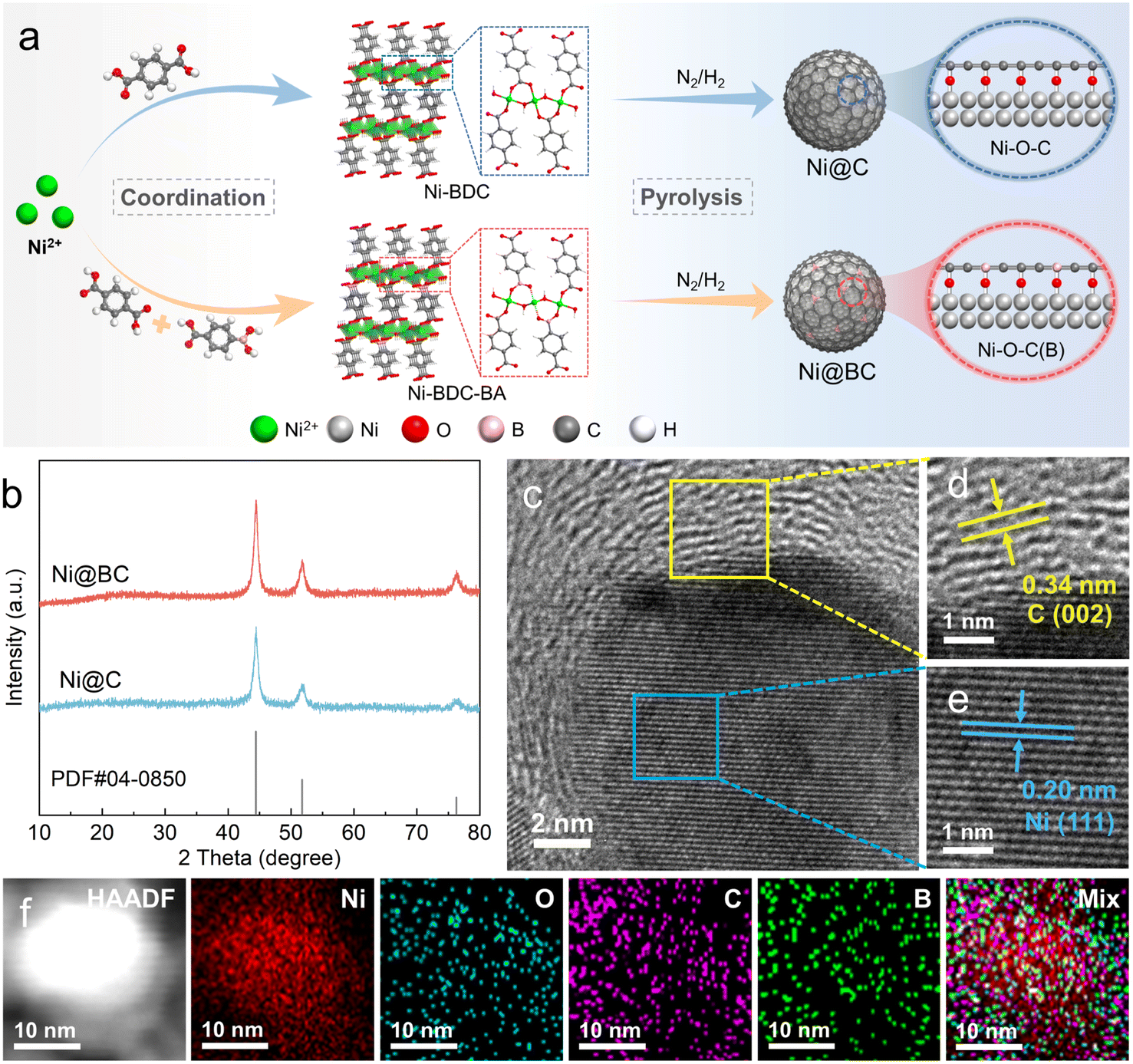 | ||
| Fig. 1 (a) Schematic diagram of the structure and synthesis route of the Ni@C and Ni@BC catalysts. (b) XRD patterns of the Ni@C and Ni@BC. (c)–(e) HRTEM images of the Ni@BC. (f) Energy-dispersive X-ray (EDX) elemental mappings of the Ni@BC. | ||
The XPS was performed to further investigate the valence states and interfacial structures of the Ni@C and Ni@BC. As shown in Fig. 2a, the high-resolution Ni 2p3/2 spectrum of the Ni@C can be deconvoluted to three peaks. The peaks located at 853.1 eV and 855.5 eV can be assigned to metallic Ni (Ni0) and oxidized Ni (Ni2+), respectively.52 Compared with the Ni@C, the Ni0 of the Ni@BC exhibits a negatively shifted binding energy, suggesting the regulation of the electron structure of nickel after the introduction of a boron-containing ligand. The O 1s spectra of the Ni@C and Ni@BC can be shared to three different carbon–oxygen configurations (Fig. 2b). The peaks located at 531.5 and 532.2 eV correspond to C–O–C and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, respectively, and the peak located at 529.8 eV of the Ni@C is attributed to the Ni–O–C bond, suggesting the existence of the Ni–O–C structure at the carbon coated interface.45,53,54 Moreover, it can be found that the Ni–O–C peak of the Ni@BC shifts to a negative binding energy when compared to the Ni@C, which is mainly from the substitution of the partial carbon with boron and formation of the Ni–O–B structure. It is worth noting that the O 1s spectra of the Ni@C and Ni@BC are significantly different to that of NiO. The peaks situated at 529.2, 530.8 and 531.7 eV in the NiO can be assigned to Ni–O, Ni–OH and adsorbed water, respectively, (Fig. S6†).55,56 As shown in Fig. 2c, there are four different carbon bonds (C–C, C–O, CO and O–CO) existing in the Ni@C and Ni@BC samples, whereas the B–C can be observed in the Ni@BC.57–59 For the B 1s spectrum in the Ni@BC, two distinct peaks located at 191.5 and 192.3 eV can be assigned to BC2O and BCO2, respectively (Fig. 2d).22,58
O, respectively, and the peak located at 529.8 eV of the Ni@C is attributed to the Ni–O–C bond, suggesting the existence of the Ni–O–C structure at the carbon coated interface.45,53,54 Moreover, it can be found that the Ni–O–C peak of the Ni@BC shifts to a negative binding energy when compared to the Ni@C, which is mainly from the substitution of the partial carbon with boron and formation of the Ni–O–B structure. It is worth noting that the O 1s spectra of the Ni@C and Ni@BC are significantly different to that of NiO. The peaks situated at 529.2, 530.8 and 531.7 eV in the NiO can be assigned to Ni–O, Ni–OH and adsorbed water, respectively, (Fig. S6†).55,56 As shown in Fig. 2c, there are four different carbon bonds (C–C, C–O, CO and O–CO) existing in the Ni@C and Ni@BC samples, whereas the B–C can be observed in the Ni@BC.57–59 For the B 1s spectrum in the Ni@BC, two distinct peaks located at 191.5 and 192.3 eV can be assigned to BC2O and BCO2, respectively (Fig. 2d).22,58
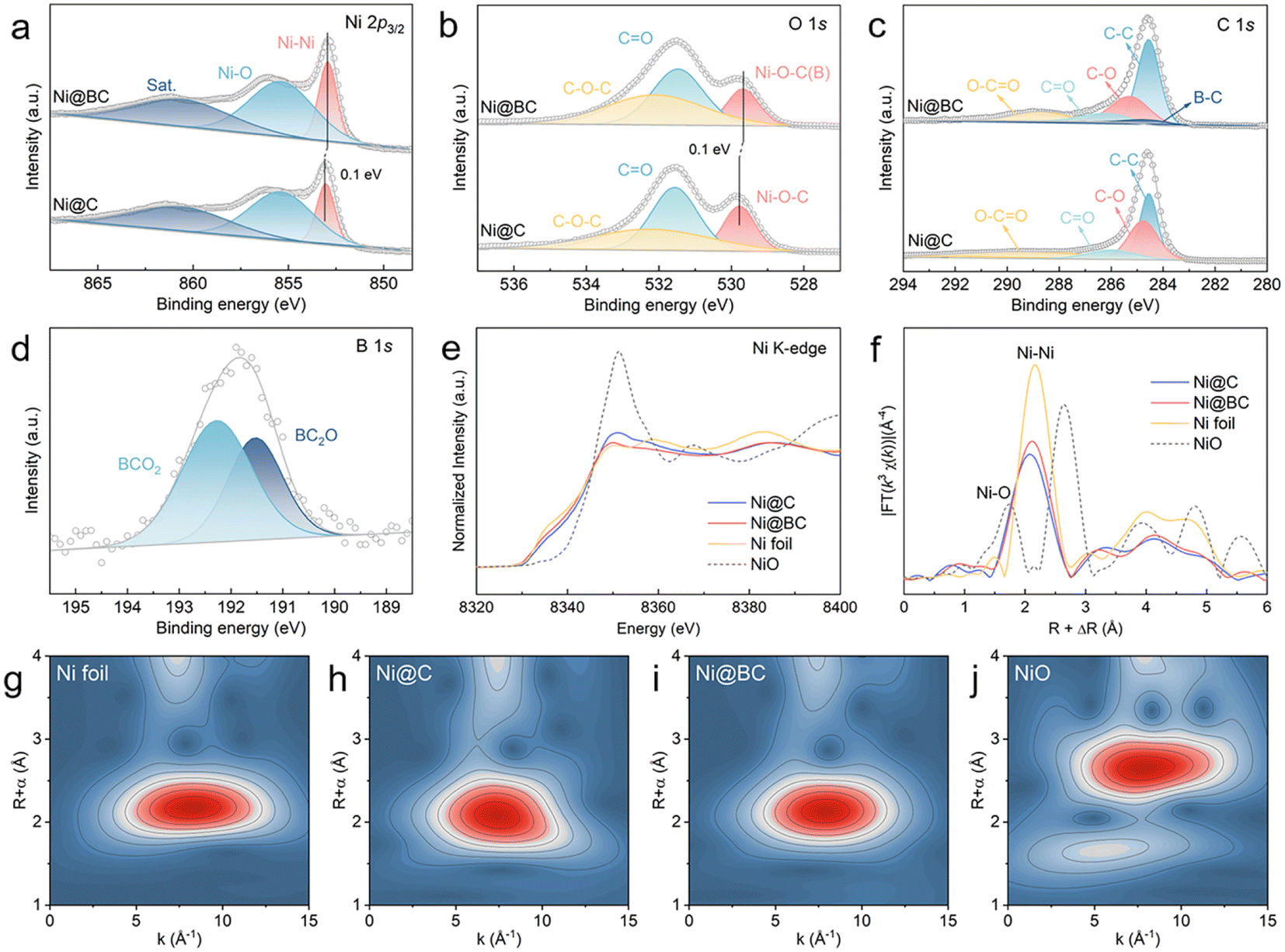 | ||
| Fig. 2 High-resolution XPS spectra of (a) Ni 2p, (b) O 1s and (c) C 1s in the Ni@C and Ni@BC. (d) High-resolution XPS spectra of B 1s in the Ni@BC. (e) Ni K-edge XANES spectra of the Ni@C, Ni@BC, Ni foil and NiO. (f) Fourier-transform EXAFS k3-weighted spectra of the Ni@C, Ni@BC, Ni foil and NiO. (g)–(j) Wavelet transforms of Ni K-edge EXAFS for the Ni foil, Ni@C, Ni@BC and NiO. | ||
The local electronic feature and atomic structure of the samples were further investigated using XAS. As shown in Fig. 2e, the Ni K-edge X-ray absorption near edge structure (XANES) spectra show that the oxidation states of Ni in the Ni@C and Ni@BC are between the Ni foil and NiO, suggesting that Ni species are partial oxidated.23,60 Furthermore, the average oxidation states of all the samples (Fig. S7†) indicate that the valence state of Ni in the Ni@BC is slightly lower than that of the Ni@C sample, which demonstrates that the decreased oxidation state is derived from the construction of the Ni–O–B structure, and this agrees well with XPS results. The extended X-ray absorption fine structure (EXAFS) spectra were further measured to determine the local coordination environment around Ni sites (Fig. 2f). The dominant peaks of the Ni@C and Ni@BC at ∼2.10 Å are ascribed to the metallic Ni–Ni coordination, which shows a slight shift to the left side due to the presence of the Ni–O–C and Ni–O–B structures.61,62 Moreover, weak peaks at about 1.3 Å further indicate that the surface Ni species of the Ni@C and Ni@BC are partially oxidized to produce the Ni–O–C(B) interface. The wavelet transform (WT) analyses were also conducted to further investigate the coordination configuration. As shown in Fig. 2g–j, the maximum intensities of Ni@C and Ni@BC are approximately the same as that of the Ni foil but different to that of the NiO, suggesting that their main components are metallic Ni, and this was consistent with EXAFS results. In addition, the chemical state of carbon was explored by X-ray absorption near edge structure (XANES), as shown in Fig. S8.† Clearly, the C K-edge XANES spectra of Ni@C and Ni@BC show two typical peaks at 285.6 eV (A) and 288.6 eV (B), corresponding to the π* states of the carbon ring (CC) and carbon–oxygen bond (CO), respectively.46,63,64 The occurrence of peak B hints at the presence of the interfacial chemical interaction between Ni and the carbon matrix via the Ni–O–C bond.46 Compared with the Ni@C, the decreased intensity of the A peak and increased intensity of the B peak in the Ni@BC are probably due to the introduction of boron to the graphite carbon structure and enhanced strength of the carbon–oxygen bond. For comparison, the Ni-BDC-BA with different feeding molar ratios were synthesized, and their carbonized products (Ni@BC 8:2 and Ni@BC 2:8) were also characterized as shown in Fig. S9–S16.† The XPS results verify that the element content of boron on the sample surface were constantly grown with the increased 4-carboxyphenylboronic acid feeding (Table S1†). It should be noted that as the added amount of boron increased, the valence state of Ni0 gradually shifts towards a negative binding energy, further demonstrating that the Ni–O–B structure can continuously modulate the surface electron states of Ni (Fig. S17†).
Catalytic performance evaluation
The HOR performances of the obtained samples were evaluated using a rotating disk electrode (RDE) measurement system in a standard three-electrode system with H2-saturated 0.1 M KOH as the electrolyte. For comparison, pure Ni without carbon coated catalysts was also prepared, and its XRD patterns are shown in Fig. S18.† The HOR polarization curves of the Ni, Ni@C and Ni@BC were reordered by linear sweep voltammetry (LSV) with a rotation rate of 2500 revolutions per minute (rpm) at a scan rate of 5 mV s−1. As shown in Fig. 3a, the current density of the Ni@C is significantly enhanced when compared with pure Ni, indicating that the presence of the Ni–O–C interface promotes the HOR activity of Ni. Moreover, the Ni@BC sample exhibits a much superior HOR performance when compared to the Ni@C, which suggests that the Ni–O–B motifs could further optimize the alkaline HOR kinetics of nickel. To avoid the contribution of the non-faradaic current, the steady-state polarization curve of the Ni@BC was also obtained for comparison to determine the hydrogen electrocatalytic response (Fig. S19†). It was observed that these two polarization curves are almost consistent, demonstrating that the anodic current is mainly derived from hydrogen oxidation.52 Typically, HOR kinetic current densities (jk) could be acquired using the Koutecky–Levich equation by eliminating the effect from the mass transfer of hydrogen. As shown in Fig. 3b, the HOR polarization curves of Ni@BC were collected at different rotational speeds from 2500 rpm to 400 rpm. The Koutecky–Levich plot can be constructed at the overpotential of 30 mV, and the calculated slope is calculated to be 13.93 cm2 mA−1 rpm1/2, which is close to the theoretical value of 14.8 cm2 mA−1 rpm1/2 for the two-electron HOR process (Fig. 3b, inset).20 In addition, the Koutecky–Levich plots of Ni@C and Ni are established, and their results are shown in Fig. S20 and S21.† Accordingly, kinetic current densities (jk) of these HOR catalysts can be obtained from the Koutecky–Levich equation and later used for Butler–Volmer fitting (Fig. 3c).6 After normalization by the metal loadings of the catalysts obtained using inductively coupled plasma atomic emission spectroscopy results (Table S2†), the mass activity denoted as jk,m at the overpotential of 50 mV of the Ni@BC is 34.91 mA mgNi−1, which is 2.6-fold and 7.8-fold greater than those of the Ni@C (13.45 mA mgNi−1) and Ni (4.46 mA mgNi−1), respectively, and these surpass most of the previously reported mass activities for the Ni-based HOR catalysts (Fig. 3e and f and Table S3†). The sharply increased HOR activity implies that the construction of the Ni–O–C and Ni–O–B interfaces significantly promotes the alkaline HOR kinetics of nickel. As shown in Fig. S22,† the Tafel slopes of the Ni, Ni@C and Ni@BC were calculated to be 141.0, 122.5 and 79.4 mV dec−1, respectively, suggesting that the HOR process on these catalysts are conducted via a Tafel–Volmer mechanism, with the Volmer step being the rate-determining step.23,50 In addition, the decreased Tafel slope of Ni@BC compared to that of the Ni and Ni@C suggests it has faster HOR kinetics. Conversely, the exchange current density (j0) is an outstanding criterion to estimate the intrinsic activity of electrocatalysts and can be acquired by the nonlinear fitting via the Butler–Volmer equation (Fig. 3c).6 Simultaneously, the value of j0 could also be estimated from the micro-polarization region within a small potential window from −10 to 10 mV according to the simplified Butler–Volmer equation (Fig. 3d),21 and these two results are almost the same. Furthermore, electrochemical active surface area (ECSA) was measured by integrating the charge quantity from the OH desorption from the surface of Ni via cyclic voltammetry (CV) (Fig. S23†).21 After normalizing by ECSA, the exchange current density (j0,s) of the Ni@BC is calculated to be 0.045 mA cmNi−2, which is 1.6 times and 3.5 times higher than that of the Ni@C (0.028 mA cmNi−2) and Ni (0.013 mA cmNi−2), respectively and is also superior to most of the previously recorded Ni-based materials (Fig. 3e–f and Table S3†). This significantly enhanced intrinsic activity demonstrates that the establishment of the Ni–O–C and Ni–O–B motifs are beneficial to optimize the HOR performance of nickel. Meanwhile, the mass activities (j0,m) of these catalysts, namely, the exchange current densities normalized by the corresponding metal mass, are also calculated and shown in Fig. S24† that in fact, the j0,m of the Ni@BC (10.03 mA mgNi−1) is much higher than those of the Ni (2.46 mA mgNi−1) and Ni@C (5.96 mA mgNi−1). Additionally, the samples obtained from other Ni-BDC-BA precursors with different feeding molar ratios were analyzed by electrochemical tests, as shown in Fig. S25–S28.† Among them, the Ni@BC exhibits the best HOR performance with the highest mass activity (jk,m) and the largest specific activity (j0,s) (Fig. S25d†). It is worth noting that a volcanic type relationship can be established when all the prepared samples are compared together (Fig. S29†), where the Ni@BC is located at the peak of the volcano and possesses the best intrinsic activity. This volcanic relationship may imply that the binding energy between the reaction intermediates and surfaces of different catalysts are optimized.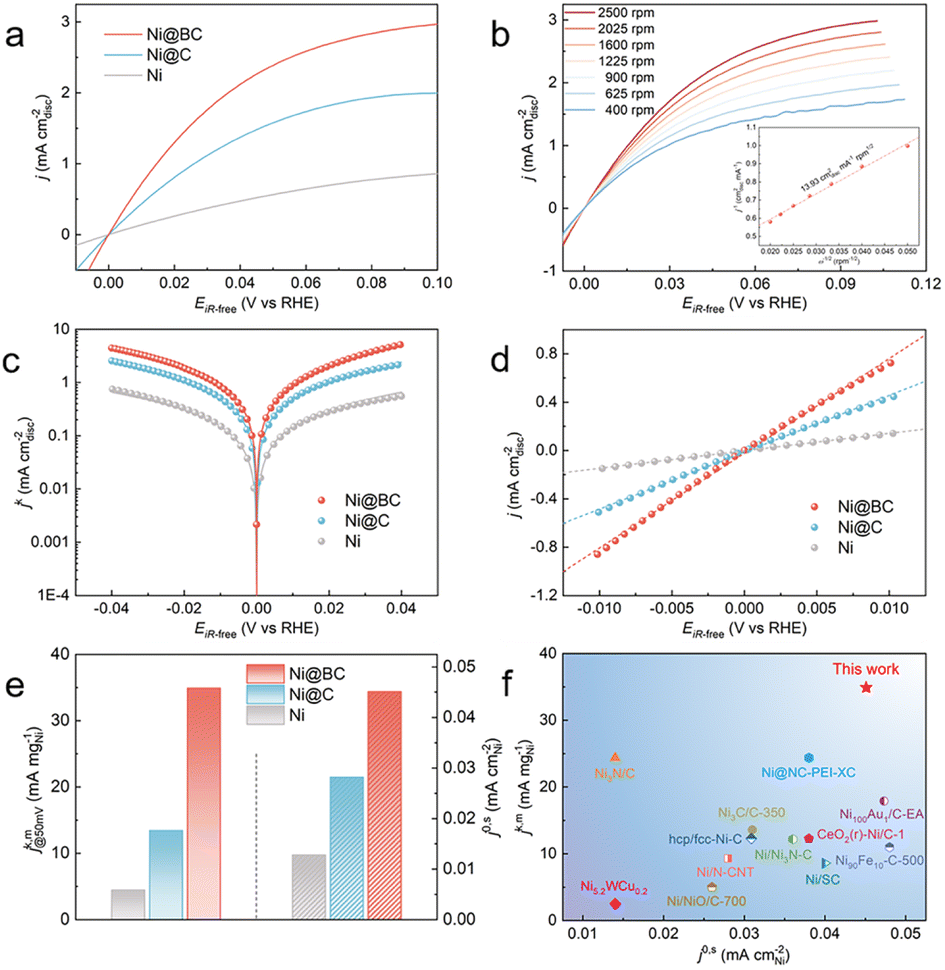 | ||
| Fig. 3 (a) HOR polarization curves of the Ni, Ni@C and Ni@BC in H2-saturated 0.1 M KOH solution measured at the rotational speed of 2500 rpm. (b) Polarization curves of the Ni@BC in H2-saturated 0.1 M KOH solution with a scan rate of 5 mV s−1 at the rotational speeds varying from 2500 to 400 rpm. Inset shows the Koutecky–Levich plot at 30 mV (vs. RHE). (c) Tafel plots derived from the HOR polarization curves of the Ni, Ni@C and Ni@BC normalized kinetic current densities (jk) via Butler–Volmer fittings. (d) Linear fitting curves in the micro-polarization region. (e) Comparison of metal mass-normalized kinetic current densities (jk,m) at an overpotential of 50 mV and ECSA-normalized exchange current densities (j0,s) of the three catalysts. (f) Comparison of mass-normalized jk at 50 mV and ECSA-normalized j0 for Ni@BC with those of the recently reported Ni-based catalysts. | ||
Because the stability is an important parameter to assess the catalysts, the accelerated durability testing (ADT) of the catalysts were carried out by conducting 1000 CV measurements in a H2-saturated KOH electrolyte.65 As shown in Fig. S30a,† LSV curves indicate a significant decrease in the HOR activity for Ni after 1000 CV scans, and the value of j0 is decreased by more than 86%. In contrast, the LSV curves show that the exchange current density of the Ni@BC can remain at about 75%, suggesting that it has good HOR stability (Fig. S30b†). Furthermore, the durability of the samples was evaluated by performing 1000 CV cycles with the potential range between −0.08 V and 0.42 V in Ar-purged 0.1 M KOH solutions, which includes the complete redox process of Ni/Ni2+.20 By comparing the CV and LSV curves before and after the ADT testing (Fig. S32†), it can be observed that the CV and specific activity for the Ni@BC mostly remained stable whereas those for Ni are seriously decreased (Fig. S31†), suggesting the superior stability of the Ni@BC. Moreover, the characterizations conducted after the accelerated durability test demonstrate that the morphology and structure of the Ni@BC are maintained well (Fig. S33†). Presently, carbon monoxide is inevitably contained in most of the commercially available hydrogen; therefore, CO tolerance experiments were conducted.24,66 Fig. S34† shows the polarization curves of the catalysts recorded in H2 (containing 100 ppm CO) saturated 0.1 M KOH electrolyte. Compared with the Ni@BC, the HOR activities of commercial Pt/C and Ni catalysts decreased significantly, indicating the excellent CO tolerance of the Ni@BC. Additionally, the chronoamperometry test was employed to examine the CO tolerance of the catalysts. As shown in Fig. S35,† the Ni@BC shows a much more superior anti-CO poisoning capability than Pt/C. The apparently improved CO tolerance can originate from two possibilities. On the one hand, compared with platinum, the adsorption ability for Ni is stronger,61 which is beneficial to the oxidation of CO in alkaline media.67 Meanwhile, the hydroxyl binding energy of the Ni@BC is further enhanced when compared to that of Ni (vide infra). On the other hand, the carbon shell of the Ni@BC can also prevent the adsorption of CO species on the surface of the inner Ni nanoparticles, protecting them from poisoning.28,68
As the reverse reaction of the HOR, the hydrogen evolution reaction (HER) activity of the Ni@BC was also studied because of its good HOR performance. As shown in Fig. S36a,† the Ni@BC exhibits remarkable HER activity with an overpotential of 65 mV to reach a current density of 10 mA cm−2, which is close to that of Pt/C (43 mV) and superior to that of the Ni@C (91 mV) and Ni (170 mV), indicating its great potential for hydrogen electrocatalysis. Moreover, from their Tafel slopes in Fig. S36b,† the Ni@BC has a smaller slope (87 mV dec−1), than the Ni@C (118 mV dec−1) and Ni (130 mV dec−1), which suggests that the Ni@BC possesses faster HER kinetics. In addition, to identify the real active sites for HOR catalysis, the poisoning test was conducted. As shown in Fig. S37,† the HOR activity of the Ni@BC is decreased severely after introducing KSCN into the KOH electrolyte because the SCN− can poison the surface of the Ni nanoparticles, demonstrating that the active sites for HOR is the Ni core rather than the carbon shell.69
Theoretical calculations and reaction mechanism investigation
To further understand the role of Ni–O–C and Ni–O–B local environment on the superior alkaline HOR activity of the Ni@BC, DFT calculations were conducted (see the ESI† for further details). Herein, the (111) face of fcc Ni was chosen for use in the subsequent theoretical calculations due to the results of former structural characterization and crystal facet stability (Fig. S38†).17 First, the electronic structures of the pristine Ni, Ni@C, and Ni@BC are calculated using Mulliken charges analysis (Fig. 4a). It is clear that the average charge of Ni atoms in the Ni@BC is calculated to be 0.12 eV, higher than that in Ni (0.00 eV), but lower than that in Ni@C (0.16 eV), suggesting that there is moderate electronic interaction, which is consistent with the previous XPS and XANES results.70 In addition, the orbital analysis of the projected density of states (PDOSs) for the pristine Ni, Ni@C, and Ni@BC demonstrates that the d-band center of the Ni atom in the Ni@C is closest to the Fermi level when compared with the pristine Ni, suggesting that there is a strong adsorption for the reaction intermediates.71,72 However, compared to the Ni@C, the d-band center of the Ni atom in the Ni@BC is slightly far away from the Fermi level (Fig. 4b). As shown in Fig. 4c and S39,† it is surprising that the calculated adsorption free energies of the H* (ΔGH*) values of the Ni@C, Ni@BC, and pristine Ni are similar, suggesting that the hydrogen binding energy (HBE) may not play a decisive role in the enhancement of the alkaline HOR performance.73 CO stripping experiments and zeta potential tests are powerful ways to reveal the hydroxyl binding energy (OHBE).61,74 Associated with the CO stripping electrochemical experiments, it can be found that the CO oxidation peak of the Ni@BC is 0.499 V (vs. RHE), which is more negative than that of the Ni catalyst (0.516 V vs. RHE), but more positive than that of Ni@C (0.486 V vs. RHE) (Fig. 4d), which were in good agreement with the theoretical calculation results of the Gibbs free energy of absorbed OH* (ΔGOH*) on the surfaces of the Ni@C, Ni@BC, and pristine Ni (Fig. 4e and S40†). Moreover, as shown in Fig. 4f, the zeta potential of the Ni@BC is −10.9 mV, which is lower than that of the pristine Ni catalyst (−2.26 mV), but higher than that of Ni@C (−20.6 mV). Based on above-mentioned characterizations, it is clear that the OHBE of the Ni@C, Ni@BC, and Ni catalysts is in the order of Ni@C > Ni@BC > Ni, indicating that the moderate OHBE favors the optimal activity.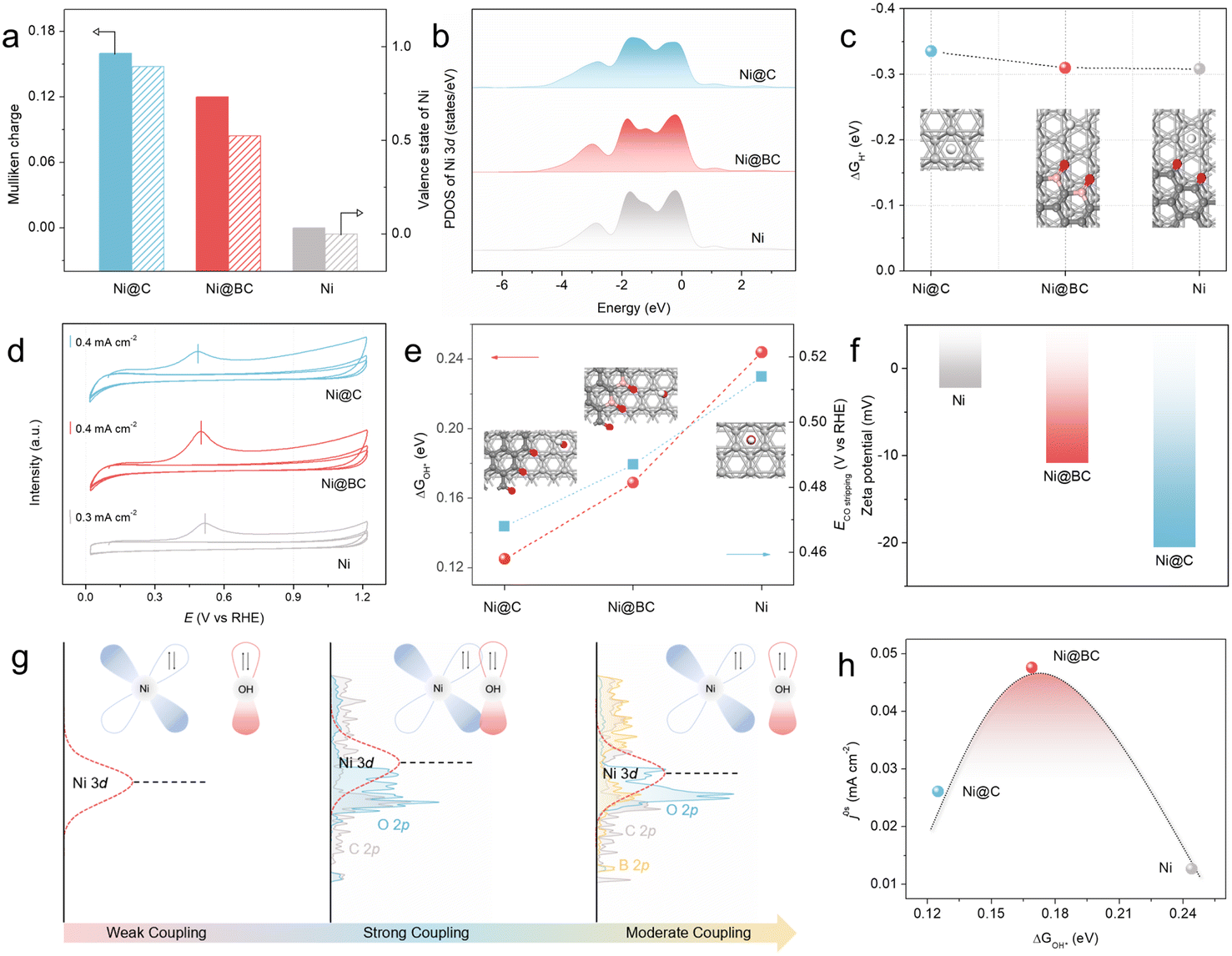 | ||
| Fig. 4 DFT calculations. (a) Mulliken charges based on the DFT calculations and the valence states obtained from XANES results of Ni in the Ni@C, Ni@BC, and pristine Ni. (b) Projected density of states of Ni 3d orbitals in the Ni@C, Ni@BC, and pristine Ni, respectively. (c) Calculated Gibbs free energy of absorbed H* on the surfaces of the Ni@C, Ni@BC, and pristine Ni. Inset: optimized structure models of the adopted adsorption sites of H* on the Ni@C, Ni@BC, and pristine Ni. Ni, C, B, O, and H atoms are denoted by silver, dark gray, pink, red, and white spheres, respectively. (d) CO-stripping measurements on the Ni@C, Ni@BC, and pristine Ni catalysts. (e) Calculated Gibbs free energy of absorbed OH* on the surfaces of the Ni@C, Ni@BC, and pristine Ni and anodic peak positions (Epeak) in the CO-stripping measurements. Inset: optimized structure models of the adopted adsorption sites of OH* on the Ni@C, Ni@BC, and pristine Ni. Ni, C, B, O, and H atoms are denoted by silver, dark gray, pink, red, and white spheres, respectively. (f) Zeta potential of the Ni@C, Ni@BC, and pristine Ni catalysts. (g) Schematic illustration of the adjustment for the adsorption strength of OH* on the surface of the Ni sites in Ni@C, Ni@BC, and pristine Ni. (h) Volcano plot of the HOR intrinsic activity (j0,s) as a function of the ΔGOH* for the Ni@C, Ni@BC, and pristine Ni catalysts. | ||
According to the illustration in Fig. 4g, understanding the role of OHBE and the way of adjustment among the Ni@C, Ni@BC, and Ni can be more intuitive. Compared with the pristine Ni, the introduction of the Ni–O–C local environment to realize the primary regulation on the Ni active sites in the Ni@C can upshift the d-band center of Ni, greatly enhancing the binding energy of OH*, leading to an increased activity. Subsequently, the synergistic effect between the Ni–O–C and Ni–O–B local environments to realize the secondary regulation of the Ni active sites in the Ni@BC can further optimize the d-band center, resulting in a moderate OHBE and much improved activity. More importantly, to summarize the relationship between the electrocatalytic activity and key reaction intermediate, a volcano plot of the HOR intrinsic activity (j0,s) as a function of ΔGOH* can be obtained for the Ni@C, Ni@BC, and pristine Ni catalysts (Fig. 4h). In light of this volcano relationship between j0,s and ΔGOH* for the Ni@C, Ni@BC, and pristine Ni catalysts, it shows that the active catalysts have an optimal binding energy; in other words, a moderate adsorption strength of the intermediates that is neither too strong nor too weak.75
In fact, the adsorption behaviors of H* and OH* on the active sites are competitive, and excessive OHBE will result in high coverage of hydroxyl on the catalyst surface, and this will hinder the adsorption of hydrogen during the alkaline HOR process.76,77 To gain deeper insight into the volcanic relationship of the HOR activity with the introduction of increasing boron (Fig. S29†), calculation models with a slight proportion of the Ni–O–B interface (Ni@BsC) and a large proportion of the Ni–O–B interface (Ni@BrC) were also constructed to investigate the relationship between the alkaline HOR activity and OHBE (Fig. S41–S43†). Based on the DFT calculations, the calculated ΔGH* values of the Ni, Ni@C, Ni@BsC, Ni@BC, and Ni@BrC are a little different from each other, whereas the calculated ΔGOH* values are obviously different (Fig. S44 and S45†). It is essential to correlate the adsorption strength of OH* with the HOR activity.78 As expected, the intrinsic activity (j0,s) shows a volcano plot as a function of the ΔGOH* for Ni@C, Ni@BsC, Ni@BC, Ni@BrC, and pristine Ni catalysts (Fig. S46†). By regulating the proportion of the Ni–O–C and Ni–O–B local environments, there was an optimal OHBE for the active catalyst. Consequently, the regulation of the synergistic effect between the Ni–O–C and Ni–O–B local environments on the Ni active sites in the Ni@BC is realized by optimizing the d-band center and optimizing the binding ability of the OH* adsorbate, which promotes the HOR performance.
Conclusions
In summary, the boron doped carbon layers encapsulating nickel nanoparticles (Ni@BC) with rich bridging oxygen coordinated motifs were successfully synthesized. We found that the coordinated environment of Ni can be precisely regulated by introducing Ni–O–C and Ni–O–B local environments, thereby leading to an optimum binding ability of the OH* adsorbate, which is responsible for the enhanced alkaline HOR performance. The experimental results and theoretical calculations reveal that the presence of the Ni–O–C interface between the carbon layer and Ni nanoparticles can alter the electron density of Ni by the bridging oxygen, thereby shifting up the d-band center of nickel and mainly enhancing its hydroxyl binding energy. Subsequently, the local chemical environment of Ni can be further regulated by the construction of the Ni–O–B coordinated environment, leading to an optimized d-band center and a volcano-type correction between the OHBE and HOR performance, highlighting the key role of the optimal binding ability of the OH* adsorbate in boosting the alkaline HOR kinetics. Specifically, the obtained Ni@BC exhibits a remarkable HOR performance with a mass activity of 34.91 mA mgNi−1 at an overpotential of 50 mV and a specific activity of 0.045 mA cmNi−2 in 0.1 M KOH, as well as superior stability and CO tolerance. This study not only revealed the key role of the bridged oxygen coordinated environment between the metal nanoparticles and coated carbon layer for precise tailoring of the electronic structure of active metal center but also provided a new design principle for rationally regulating the OHBE for use with advanced HOR catalysts.Data availability
All the data supporting this article have been included in the main text and the ESI.†Author contributions
W. L. conceived and supervised the project. X. Y synthesized the catalysts. X. Y. and P. H. performed the catalytic tests and characterization. L. W. performed the density functional theory calculations. H. J. completed the XAFS measurements and analysis. W. L., P. H. and L. W. wrote the manuscript. All the authors discussed the results and assisted during the manuscript preparation. P. H., X. Y. and L. W. contributed equally to this work.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors appreciate the support from the National Key Research and Development Program of China (2021YFB4001200) and the National Natural Science Foundation of China (22272121 and 21972107). The authors thank the Core facility of Wuhan University for ICP-AES and XPS measurements. The authors also thank the Core Research Facilities of College of Chemistry and Molecular Sciences of Wuhan University. The authors thank Beijing Scistar Technology Co. Ltd for XAFS spectroscopy system measurements and analysis. The authors also thank the staff of Beamlines MCD-A and MCD-B (Soochow Beamline for Energy Materials) at NSRL. The numerical calculations have been done on the supercomputing system at the Supercomputing Center of Wuhan University.Notes and references
- Y. Wang, Y. Yang, S. Jia, X. Wang, K. Lyu, Y. Peng, H. Zheng, X. Wei, H. Ren, L. Xiao, J. Wang, D. A. Muller, H. D. Abruña, B. J. Hwang, J. Lu and L. Zhuang, Nat. Commun., 2019, 10, 1506 CrossRef PubMed.
- Y. Yuan, J. Wang, S. Adimi, H. Shen, T. Thomas, R. Ma, J. P. Attfield and M. Yang, Nat. Mater., 2020, 19, 282–286 CrossRef CAS PubMed.
- G. Chen, H. Zhong and X. Feng, Chem. Sci., 2021, 12, 15802–15820 RSC.
- Y. Cong, B. Yi and Y. Song, Nano Energy, 2018, 44, 288–303 CrossRef CAS.
- Z. Yao, T. Tang, Z. Jiang, L. Wang, J. Hu and L. Wan, ACS Nano, 2022, 16, 5153–5183 CrossRef CAS PubMed.
- W. Sheng, H. A. Gasteiger and Y. Shao-Horn, J. Electrochem. Soc., 2010, 157, B1529–B1536 CrossRef CAS.
- J. Zheng, W. Sheng, Z. Zhuang, B. Xu and Y. Yan, Sci. Adv., 2016, 2, e1501602 CrossRef PubMed.
- C. Yang, Y. Li, J. Yue, H. Cong and W. Luo, Chem. Sci., 2023, 14, 6289–6294 RSC.
- H. A. Firouzjaie and W. E. Mustain, ACS Catal., 2020, 10, 225–234 CrossRef CAS.
- X. Wang, X. Li, D. Kong, L. Zhao, Y. Cui, Y. Wang, T. Cai, Q. Xue, Z. Yan and W. Xing, Nano Energy, 2022, 104, 107877 CrossRef CAS.
- L. Su, Y. Jin, X. Fan, Z. Liu and W. Luo, Sci. China: Chem., 2023, 66, 3262–3268 CrossRef CAS.
- L. Gao, Y. Wang, H. Li, Q. Li, N. Ta, L. Zhuang, Q. Fu and X. Bao, Chem. Sci., 2017, 8, 5728–5734 RSC.
- C. Huang, M. Feng, Y. Peng, B. Zhang, J. Huang, X. Yue and S. Huang, Adv. Funct. Mater., 2023, 33, 2300593 CrossRef CAS.
- Y. Men, D. Wu, Y. Hu, L. Li, P. Li, S. Jia, J. Wang, G. Cheng, S. Chen and W. Luo, Angew. Chem., Int. Ed., 2023, 62, e202217976 CrossRef CAS PubMed.
- M. Wang, H. Yang, J. Shi, Y. Chen, Y. Zhou, L. Wang, S. Di, X. Zhao, J. Zhong, T. Cheng, W. Zhou and Y. Li, Angew. Chem., Int. Ed., 2021, 60, 5771–5777 CrossRef CAS PubMed.
- S. Qin, Y. Duan, X. Zhang, L. Zheng, F. Gao, P. Yang, Z. Niu, R. Liu, Y. Yang, X. Zheng, J. Zhu and M. Gao, Nat. Commun., 2021, 12, 2686 CrossRef CAS PubMed.
- X. Zhao, X. Li, L. An, K. Iputera, J. Zhu, P. Gao, R. Liu, Z. Peng, J. Yang and D. Wang, Energy Environ. Sci., 2022, 15, 1234–1242 RSC.
- Y. Men, X. Su, P. Li, Y. Tan, C. Ge, S. Jia, L. Li, J. Wang, G. Cheng, L. Zhuang, S. Chen and W. Luo, J. Am. Chem. Soc., 2022, 144, 12661–12672 CrossRef CAS PubMed.
- Y. Duan, X. Zhang, F. Gao, Y. Kong, Y. Duan, X. Yang, X. Yu, Y. Wang, S. Qin, Z. Chen, R. Wu, P. Yang, X. Zheng, J. Zhu, M. Gao, T. Lu, Z. Yu and S. Yu, Angew. Chem., Int. Ed., 2023, 62, e202217275 CrossRef CAS.
- P. Han, X. Yang, L. Wu, H. Jia, J. Chen, W. Shi, G. Cheng and W. Luo, Adv. Mater., 2024, 36, 2304496 CrossRef CAS PubMed.
- Z. Zhuang, S. A. Giles, J. Zheng, G. R. Jenness, S. Caratzoulas, D. G. Vlachos and Y. Yan, Nat. Commun., 2016, 7, 10141 CrossRef CAS PubMed.
- F. Yang, X. Bao, Y. Zhao, X. Wang, G. Cheng and W. Luo, J. Mater. Chem. A, 2019, 7, 10936–10941 RSC.
- X. Zhao, X. Li, L. An, L. Zheng, J. Yang and D. Wang, Angew. Chem., Int. Ed., 2022, 61, e202206588 CrossRef CAS PubMed.
- T. Wang, M. Wang, H. Yang, M. Xu, C. Zuo, K. Feng, M. Xie, J. Deng, J. Zhong, W. Zhou, T. Cheng and Y. Li, Energy Environ. Sci., 2019, 12, 3522–3529 RSC.
- J. Liu, B. Zhang, Y. Fo, W. Yu, J. Gao, X. Cui, X. Zhou and L. Jiang, Chem. Eng. J., 2023, 464, 142692 CrossRef CAS.
- E. S. Davydova, F. D. Speck, M. T. Y. Paul, D. R. Dekel and S. Cherevko, ACS Catal., 2019, 9, 6837–6845 CrossRef CAS.
- L. Shen, J. Ying, K. I. Ozoemena, C. Janiak and X. Yang, Adv. Funct. Mater., 2022, 32, 2110851 CrossRef CAS.
- J. M. Yoo, H. Shin, D. Y. Chung and Y. Sung, Acc. Chem. Res., 2022, 55, 1278–1289 CrossRef CAS PubMed.
- J. A. Varnell, E. C. M. Tse, C. E. Schulz, T. T. Fister, R. T. Haasch, J. Timoshenko, A. I. Frenkel and A. A. Gewirth, Nat. Commun., 2016, 7, 12582 CrossRef CAS PubMed.
- M. Karuppannan, Y. Kim, S. Gok, E. Lee, J. Y. Hwang, J. Jang, Y. Cho, T. Lim, Y. Sung and O. J. Kwon, Energy Environ. Sci., 2019, 12, 2820–2829 RSC.
- Y. Hu, X. Guo, T. Shen, Y. Zhu and D. Wang, ACS Catal., 2022, 12, 5380–5387 CrossRef CAS.
- H. Xu, J. Wan, H. Zhang, L. Fang, L. Liu, Z. Huang, J. Li, X. Gu and Y. Wang, Adv. Energy Mater., 2018, 8, 1800575 CrossRef.
- Y. Hu, T. Shena, X. Zhao, J. Zhang, Y. Lu, J. Shen, S. Lu, Z. Tu, H. L. Xin and D. Wang, Appl. Catal., B, 2020, 279, 119370 CrossRef CAS.
- X. Wang, J. Fang, X. Liu, D. Wei, Y. Yin, H. Wei, J. Zhang, Y. Zhang, X. Zhang, W. Zhu and Z. Zhuang, Appl. Catal., B, 2023, 327, 122442 CrossRef CAS.
- Y. Hu, J. O. Jensen, W. Zhang, L. N. Cleemann, W. Xing, N. J. Bjerrum and Q. Li, Angew. Chem., Int. Ed., 2014, 53, 3675–3679 CrossRef CAS PubMed.
- X. Yan, C. Dong, Y. Huang, Y. Jia, L. Zhang, S. Shen, J. Chen and X. Yao, Small Methods, 2019, 3, 1800439 CrossRef.
- D. Deng, L. Yu, X. Chen, G. Wang, L. Jin, X. Pan, J. Deng, G. Sun and X. Bao, Angew. Chem., Int. Ed., 2013, 52, 371–375 CrossRef CAS.
- Y. Shen, Y. Zhou, D. Wang, X. Wu, J. Li and J. Xi, Adv. Energy Mater., 2018, 8, 1701759 CrossRef.
- Q. Yang, P. Jin, B. Liu, L. Zhao, J. Cai, Z. Wei, S. Zuo, J. Zhang and L. Feng, J. Mater. Chem. A, 2020, 8, 9049–9057 RSC.
- Z. Chen, R. Wu, Y. Liu, Y. Ha, Y. Guo, D. Sun, M. Liu and F. Fang, Adv. Mater., 2018, 30, 1802011 CrossRef PubMed.
- J. Deng, P. Ren, D. Deng and X. Bao, Angew. Chem., Int. Ed., 2015, 54, 2100–2104 CrossRef CAS PubMed.
- M. Jia, C. Choi, T. Wu, C. Ma, P. Kang, H. Tao, Q. Fan, S. Hong, S. Liu, Y. Soo, Y. Jung, J. Qiu and Z. Sun, Chem. Sci., 2018, 9, 8775–8780 RSC.
- W. Xue, Q. Zhou, X. Cui, S. Jia, J. Zhang and Z. Lin, Nano Energy, 2021, 86, 106073 CrossRef CAS.
- S. Liu, Y. Fan, Y. Wang, S. Jin, M. Hou, W. Zeng, K. Li, T. Jiang, L. Qin, Z. Yan, Z. Tao, X. Zheng, C. Shen, Z. Liu, T. Ahmad, K. Zhang and W. Chen, Nano Lett., 2022, 22, 9107–9114 CrossRef CAS.
- X. Zhang, Z. Li, X. Sun, L. Wei, H. Niu, S. Chen, Q. Chen, C. Wang and F. Zheng, ACS Mater. Lett., 2022, 4, 2097–2105 CrossRef CAS.
- Z. Yang, W. Lai, B. He, J. Wang, F. Yu, Q. Liu, M. Liu, S. Zhang, W. Ding, Z. Lin and H. Huang, Adv. Energy Mater., 2023, 13, 2300881 CrossRef CAS.
- Y. Li, F. Ma, L. Zheng, Y. Liu, Z. Wang, P. Wang, Z. Zheng, H. Cheng, Y. Dai and B. Huang, Mater. Horiz., 2021, 8, 2842–2850 RSC.
- Y. Li, G. Zhai, Y. Liu, Z. Wang, P. Wang, Z. Zheng, H. Cheng, Y. Dai and B. Huang, Chem. Eng. J., 2022, 437, 135363 CrossRef CAS.
- Y. Zhu, J. Zhang, Q. Qian, Y. Li, Z. Li, Y. Liu, C. Xiao, G. Zhang and Y. Xie, Angew. Chem., Int. Ed., 2022, 61, e202113082 CrossRef CAS PubMed.
- X. Wang, Y. Tong, X. Li, L. Zhao, Y. Cui, Y. Wang, H. Hu, T. Cai, M. Wu, H. Hu, Q. Xue, Z. Yan and W. Xing, Chem. Eng. J., 2022, 445, 136700 CrossRef CAS.
- Y. Gao, H. Peng, Y. Wang, G. Wang, L. Xiao, J. Lu and L. Zhuang, ACS Appl. Mater. Interfaces, 2020, 12, 31575–31581 CrossRef CAS.
- L. Su, D. Gong, N. Yao, Y. Li, Z. Li and W. Luo, Adv. Funct. Mater., 2021, 31, 2106156 CrossRef CAS.
- P. Ge, S. Li, L. Xu, K. Zou, X. Gao, X. Cao, G. Zou, H. Hou and X. Ji, Adv. Energy Mater., 2019, 9, 1803035 CrossRef.
- Y. Yang, J. Liu, S. Guo, Y. Liu and Z. Kang, J. Mater. Chem. A, 2015, 3, 18598–18604 RSC.
- Z. Liu, C. Zhang, H. Liu and L. Feng, Appl. Catal., B, 2020, 276, 119165 CrossRef CAS.
- Y. Yan, J. Lin, T. Xu, B. Liu, K. Huang, L. Qiao, S. Liu, J. Cao, S. C. Jun, Y. Yamauchi and J. Qi, Adv. Energy Mater., 2022, 12, 2200434 CrossRef CAS.
- Z. Fan, W. Kai, J. Yan, T. Wei, L. Zhi, J. Feng, Y. Ren, L. Song and F. Wei, ACS Nano, 2011, 5, 191–198 CrossRef CAS PubMed.
- X. Yu, P. Han, Z. Wei, L. Huang, Z. Gu, S. Peng, J. Ma and G. Zheng, Joule, 2018, 2, 1610–1622 CrossRef CAS.
- C. Wang, Z. Guo, W. Shen, Q. Xu, H. Liu and Y. Wang, Adv. Funct. Mater., 2014, 24, 5511–5521 CrossRef CAS.
- W. Ji, C. Zhan, D. Li, Y. Xu, Y. Zhang, L. Wang, L. Liu, Y. Wang, W. Chen, H. Geng and X. Huang, J. Mater. Chem. A, 2021, 9, 26323–26329 RSC.
- Y. Duan, Z. Yu, L. Yang, L. Zheng, C. Zhang, X. Yang, F. Gao, X. Zhang, X. Yu, R. Liu, H. Ding, C. Gu, X. Zheng, L. Shi, J. Jiang, J. Zhu, M. Gao and S. Yu, Nat. Commun., 2020, 11, 4789 CrossRef CAS.
- P. Shen, P. Cao, Y. Yu, L. Wang, L. Zhao, L. Yang, Y. Lin and K. Xu, Small, 2023, 19, 2303142 CrossRef CAS.
- K. Feng, J. Zhong, B. Zhao, H. Zhang, L. Xu, X. Sun and S. Lee, Angew. Chem., Int. Ed., 2016, 55, 11950–11954 CrossRef CAS PubMed.
- T. Kou, M. Chen, F. Wu, T. J. Smart, S. Wang, Y. Wu, Y. Zhang, S. Li, S. Lall, Z. Zhang, Y. Liu, J. Guo, G. Wang, Y. Ping and Y. Li, Nat. Commun., 2020, 11, 590 CrossRef CAS PubMed.
- W. Ni, A. Krammer, C. Hsu, H. M. Chen, A. Schüler and X. Hu, Angew. Chem., Int. Ed., 2019, 58, 7445–7449 CrossRef CAS PubMed.
- L. Han, P. Ou, W. Liu, X. Wang, H. Wang, R. Zhang, C. Pao, X. Liu, W. Pong, J. Song, Z. Zhuang, M. V. Mirkin, J. Luo and H. L. Xin, Sci. Adv., 2022, 8, eabm3779 CrossRef CAS.
- Y. Dong, Z. Zhang, W. Yan, X. Hu, C. Zhan, Y. Xu and X. Huang, Angew. Chem., Int. Ed., 2023, 62, e2023117 Search PubMed.
- Y. Hu, J. Zhang, T. Shen, Y. Lu, K. Chen, Z. Tu, S. Lu and D. Wang, Small Methods, 2021, 5, 2100937 CrossRef CAS.
- H. Zhang, Z. Ma, J. Duan, H. Liu, G. Liu, T. Wang, K. Chang, M. Li, L. Shi, X. Meng, K. Wu and J. Ye, ACS Nano, 2016, 10, 684–694 CrossRef CAS PubMed.
- F. Yang, P. Han, N. Yao, G. Cheng, S. Chen and W. Luo, Chem. Sci., 2020, 11, 12118–12123 RSC.
- X. Wang, Y. Tong, W. Feng, P. Liu, X. Li, Y. Cui, T. Cai, L. Zhao, Q. Xue, Z. Yan, X. Yuan and W. Xing, Nat. Commun., 2023, 14, 3767 CrossRef PubMed.
- L. Wang, Z. Xu, C. H. Kuo, J. Peng, F. Hu, L. Li, H. Y. Chen, J. Wang and S. Peng, Angew. Chem., Int. Ed., 2023, 62, e202311937 CrossRef CAS PubMed.
- J. Kim, H. Jung, S. M. Jung, J. Hwang, D. Y. Kim, N. Lee, K. S. Kim, H. Kwon, Y. T. Kim, J. W. Han and J. K. Kim, J. Am. Chem. Soc., 2021, 143, 1399–1408 CrossRef CAS PubMed.
- Y. Yang, F. Y. Gao, X. L. Zhang, S. Qin, L. R. Zheng, Y. H. Wang, J. Liao, Q. Yang and M. Gao, Angew. Chem., Int. Ed., 2022, 61, e202208040 CrossRef CAS PubMed.
- I. T. McCrum and M. T. M. Koper, Nat. Energy, 2020, 5, 891–899 CrossRef CAS.
- H. Huang, K. Liu, F. Yang, J. Cai, S. Wang, W. Chen, Q. Wang, L. Fu, Z. Xie and S. Xie, Angew. Chem., Int. Ed., 2023, 62, e2023157 Search PubMed.
- Y. Feng, S. Lu, L. Fu, F. Yang and L. Feng, Chem. Sci., 2024, 15, 2123–2132 RSC.
- R. Subbaraman, D. Tripkovic, K. C. Chang, D. Strmcnik, A. P. Paulikas, P. Hirunsit, M. Chan, J. Greeley, V. Stamenkovic and N. M. Markovic, Nat. Mater., 2012, 11, 550–557 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc00043a |
| ‡ These authors have contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |