
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNear-infrared visualisation of single microparticle electrochemistry for batteries†
Xinyue
Wang
,
Si-Cong
Wang
,
Junjie
Ma
,
Ruo-Chen
Xie
* and
Wei
Wang
 *
*
State Key Laboratory of Analytical Chemistry for Life Science, School of Chemistry and Chemical Engineering, ChemBIC (Chemistry and Biomedicine Innovation Center), Nanjing University, Nanjing, 210023, China. E-mail: crxie@nju.edu.cn; wei.wang@nju.edu.cn
First published on 10th May 2024
Abstract
While optical microscopy of single particle electrochemistry has proven insightful for future nanoparticle-based batteries, little is explored for micron-sized particles of more practical interest. This is largely hindered by the currently limited methodology. Accordingly, we report transmission optical microscopy using near-infrared light for accessing intra-particle electrochemistry in virtue of strong light penetration as compared to visible light. Using near-infrared (λ > 730 nm) bright-field microscopy, the redox electrochemistry of single LiCoO2 microparticles can be readily measured based on the measurements of optical contrast changes during electrochemical cycling. Further using the established methodology, we discover that the solid-state diffusion inside most single microparticles is distinctly directional, instead of in an isotropic manner from outer to inner as observed for the other particles. This phenomenon is also observed using dark field scattering microscopy with near-infrared light, suggesting non-uniform crystal inner structures responsible for the geometrically asymmetric heterogeneity of charge transfer kinetics within each single particle. These results indicate potential opportunities offered by the near-infrared optical methodology for operando studying practical battery materials.
Introduction
Apart from electron microscopy and electrochemical microscopy,1 advanced optical microscopic techniques have been largely facilitating in-depth understanding of single particle electrochemistry for the fundamentals of future nanoparticle-based batteries.2–8 The microscopic method features an operando nature, high throughput (hundreds to thousands at a time) and desired spatiotemporal resolutions (i.e. frequently nanometers and milliseconds).3 Consequently, based on material changes in solely optical contrast and/or luminescence, the methodology allows physical insights into the chemical composition and transformation of battery electrode materials at the single nanoparticle level. For instance, surface plasmon resonance microscopy allows the study of phase transition and lithium ion diffusion within single LiCoO2 nanoparticles.9 Dark-field microscopy allows electrochemically active or inactive zones to be revealed within single Prussian blue nanocubes,10 as well as determining the precise depth of their surface charging layers.11 When tracing the product, H2 bubbles, of water splitting for fuel cells, both fluorescence microscopy12 and interference reflection microscopy13 have been shown to comprehensively unravel the detailed mechanisms and reaction kinetics of the battery reactions. Alternatively, reaction kinetics can be optically assessed based on locally induced metal hydroxide precipitation.14 In addition, when combined with well-designed nanoparticle arrays, these optical microscopy methods can be readily used to efficiently screen nanocatalysts for optimal battery electrodes under relevant reaction conditions.15Although intensive studies have focused on nano-sized electroactive materials, of wide practical interest are the commercial battery-containing particles which are predominantly micron-sized. Visualising the electrochemistry of these particles can directly contribute to the understanding and development of material design of commercial electrodes.16 The particle dimensions are much larger than the diffraction limit of light and thus optical characterisation of one such single particle should not be technically challenging. However, studies have shown that the revealed ion dynamics is limited to hundreds of nanometers deep into the microparticle surface when using visible light.17 This technical limitation could greatly hinder the measurements of intra-particle chemistry, which is key to understanding overall electrode processes.
To overcome the limitation, we originally show in this work that by measuring transmission (and scattering) light with wavelengths in both visible and near-infrared ranges, an appropriate wavelength of incident light is crucial for accessing single microparticle electrochemistry. This is not quite the concept of spectroscopic characterisation18 but more related to the penetration depth of light that has been well adopted in the fields of bio-imaging. Second, we subsequently report how near-infrared (far-field and wide-field) microscopy with different imaging modes allows interesting exploration of single microparticle electrochemistry that is potentially useful for battery fields.
Results and discussion
Fig. 1a shows the schematic set-up of applying bright-field microscopy to study the redox electrochemistry of single lithium cobalt oxide microparticles. Experimentally, the particles are spin-coated and immobilised on an ITO transparent electrode with a low surface coverage of ca. 1 particle per 100 μm2, which minimises possible multiple scattering from adjacent particles. These supported particles are immersed in a 1 M LiNO3 solution with a pH of 10. The pH is selected due to the experimentally measured excellent electrochemical cyclability which is not seen with a lower-pH (i.e. less than 9) electrolyte due to stability-degrading H+ intercalation19 (see Fig. S1†). A monochromatic collimated light beam is emitted and passes through the immersed particles. The transmitted light is collected by a CCD camera. AFM imaging shows that the LiCoO2 particles have a quasi-oblate and quite irregular shape with a projection-area-equivalent diameter of around 2.7 ± 1.5 μm and a height of about 600 ± 450 nm (Fig. 1b). These lead to an average volume-equivalent diameter of 1.8 ± 0.8 μm for the single microparticles.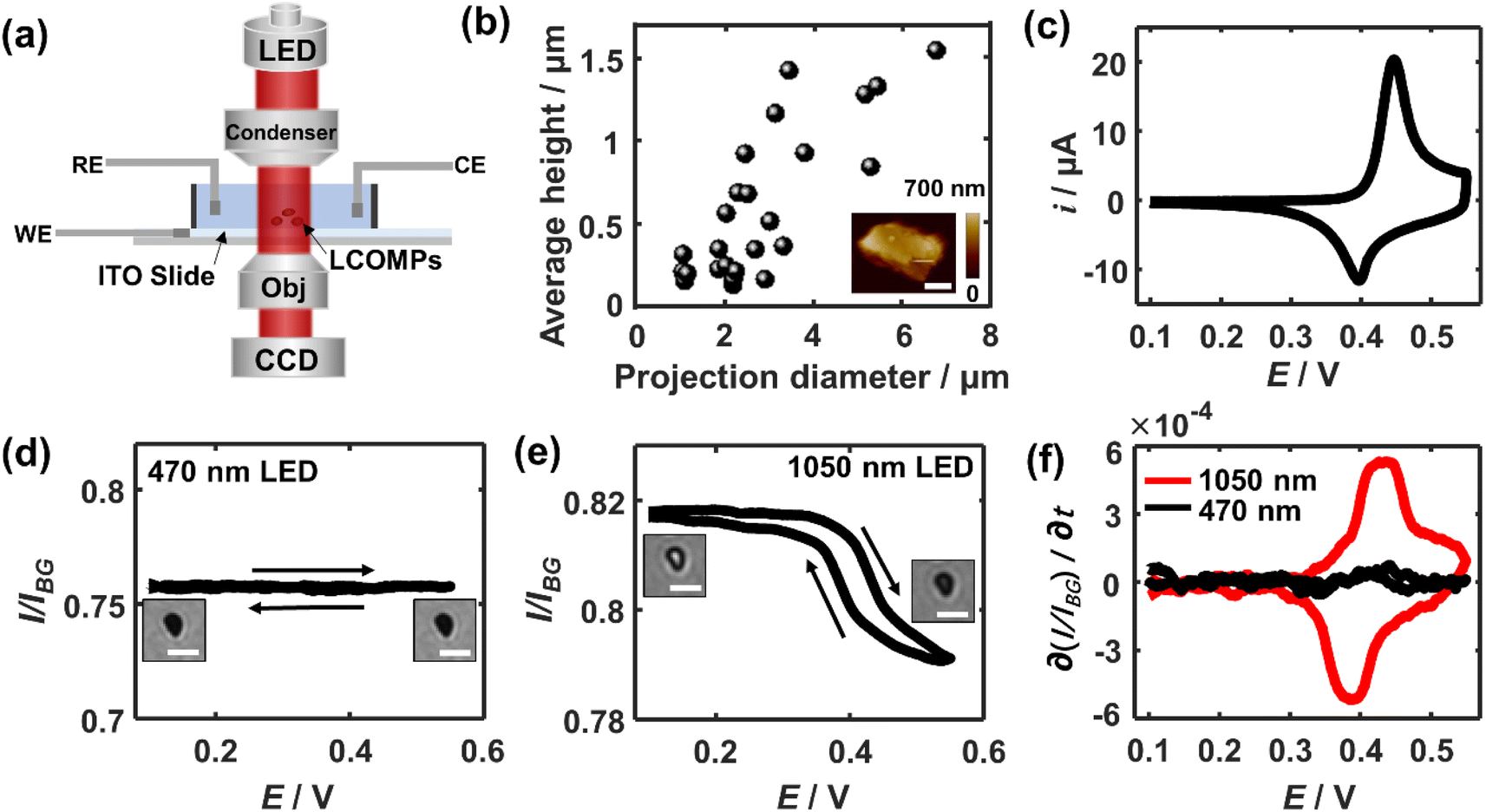 | ||
| Fig. 1 (a) Schematics of transmission optical microscopy for single microparticle electrochemistry with a three-electrode system. The reference electrode is a porous Ag/AgCl wire throughout the work. (b) Geometry distribution of the studied microparticles (N = 26). Inset shows a representative particle imaged using AFM (scale bar 1 μm). (c) Cyclic voltammetry of LiCoO2 microparticle ensembles immobilised on an ITO electrode. Scan rate: 5 mV s−1. Optical transmittance measured as a function of applied potentials measured using (d) 470 nm light and (e) 1050 nm light (scale bar 5 μm). (f) Overlaid optical voltammograms using the light of the two wavelengths. | ||
Prior to the study of the optical responses of the microparticles, the electrochemical behaviour of the particle ensembles is first examined voltammetrically. As the potential is scanned positively (vs. Ag/AgCl) from OCP, a current rise is seen from a potential of +0.4 V and peaked at +0.45 V (Fig. 1c), whereas a reductive current peak at +0.40 V occurs during the backward scan. Both the potentials and the shapes of the pair of the observed voltammetric peaks agree with reported redox electrochemistry between LiCoO2 (reduced state) and the deinserted cobalt oxide Li1−xCoO2 (oxidised state)9 despite the redox features at further positive potentials that are not involved in this work due to the accompanied solvent breakdown that would acidify the local solution. During this measured process, the x is reported to change with a magnitude of 0.25, corresponding to a single insulator-metal phase transition of the material, while it maintains the hexagonal CoO6 structure.20–22
| LiCoO2 ⇌ Li0.75CoO2 + 0.25Li+ + 0.25e− |
To exemplify the near-infrared opto-electrochemistry of the battery material, we focus on the electrochemistry within this potential window. Accompanied by this reaction, the conductivity variation of the solid structure already implies potentially significant changes in chromatic characteristics.
We thus next optically investigate their electrochemical behaviour at the single particle level. First, a common visible wavelength of 470 nm is used for the illumination light. Fig. 1d shows that the particles appear completely dark as compared to the background ITO. As the electrode is polarised in the same potential window as above, constantly little change in the optical contrast of the microparticles is seen. However, when the light wavelength is switched to 1050 nm, while the periphery region of the microparticle stays dark due to the scattering of the edge leading to significant intensity loss, its central region becomes immediately bright as seen in Fig. 1e. The depths of field of optical microscopy are estimated to be 1.2 μm and 2.6 μm for the used 470 nm and 1050 nm light, respectively (see ESI section 2†). Both depths are larger than the thickness of most microparticles (<1.2 μm). Therefore, the observations suggest that it is the varying penetration of the incident light that causes the differences of light transmittance through the particle.
As the longer-wavelength light accesses the intraparticle structure, the light transmittance through the single microparticle when subjected to cyclic voltammetry is examined. Specifically, when the potential is swept to more positive than ca. 0.35 V, the optical contrast of the particle decreases in an increasingly drastic way until the potential reaches around +0.43 V. Conversely, the particle becomes gradually bright again and the intensity variation is the steepest at ∼0.39 V during the backward scan. Quantitatively, the average transmission light intensity over the particle region is measured and subtracted from that from the background ITO. Since optical intensity is directly proportional to the charges stored in the particle,9,11 differentiating the intensity curve with respect to time gives an equivalent electrochemical current in the optical term. Thus, further by converting the timescale to the potential scale, the optical CV for the single LiCoO2 particle is obtained. The depicted oxidation peak at ca. 0.43 V and the reduction peak at 0.39 V from the optical CV are both broadly consistent with that electrochemically measured for the particle ensembles (Fig. 1f). The minor discrepancy in the peak potentials and the shape of the voltammetric wave from the electrical CV may reflect the differences in the behaviour between single particles and the particle ensembles. The oxidation state dependent LiCoO2 absorption has been found to arise from the d–d transition of cobalt 3d orbitals, which is only allowed for the oxidised state but not for the reduced state.23 The spectroscopic difference results from the change of the electronic structure upon lithium insertion or deinsertion. As such, these results evidence the quantitative optical measurements that are enabled by using longer-wavelength light in the near-infrared region.
It follows that the optical principle of the near-infrared visualisation of the LiCoO2 microparticles resides in understanding the wavelength dependence of the particle transmittance. Therefore, a closer look at the detailed wavelength dependence is conducted. Experimentally, as the potential cycle proceeds continuously, six different light wavelengths ranging from 470 nm all the way up to 1050 nm are sequentially used, each for 3 cycles, to image the very same single microparticles. The measured light intensity of the particle regions is divided by the background intensity, resulting in I/IBG (or called transmittance) to quantify the optical changes. Consequently, the temporal variations of transmittance through a representative microparticle are plotted as a function of light wavelength (Fig. 2a). As a result, it is clear that the magnitude of the optical intensity change between the maxima and the minima during the potential cycling of the single particle significantly and constantly decreases when using a shorter light wavelength (Fig. 2b). By contrast, the optical changes almost completely vanish in the cases of 530 nm and 470 nm. This observed variation applies to all the particles in the optical field of view as presented in ESI Fig. S2†, with SEM co-localisation that confirms each as a single particle. By contrast, Fig. 2c shows that both the oxidative (Q+) and reductive charges (Q−) transferred between the electrode and the particle ensembles measured from the electrical CV show negligible decline (less than 5% from the experiments using 470 nm to 1050 nm) during the electrochemical cycling and excellent cyclability (Q+/Q− ∼0.91 after the 18 consecutive cycles). It is also carefully checked by reversing the sequential use of the different lights that consequently, the monotonic optical intensity variation as a function of the light wavelength is found to be not affected by the frequent exposure (see ESI Fig. S3†). As such, it is confirmed that a longer wavelength of light is favoured preferred for accessing particle redox electrochemistry.
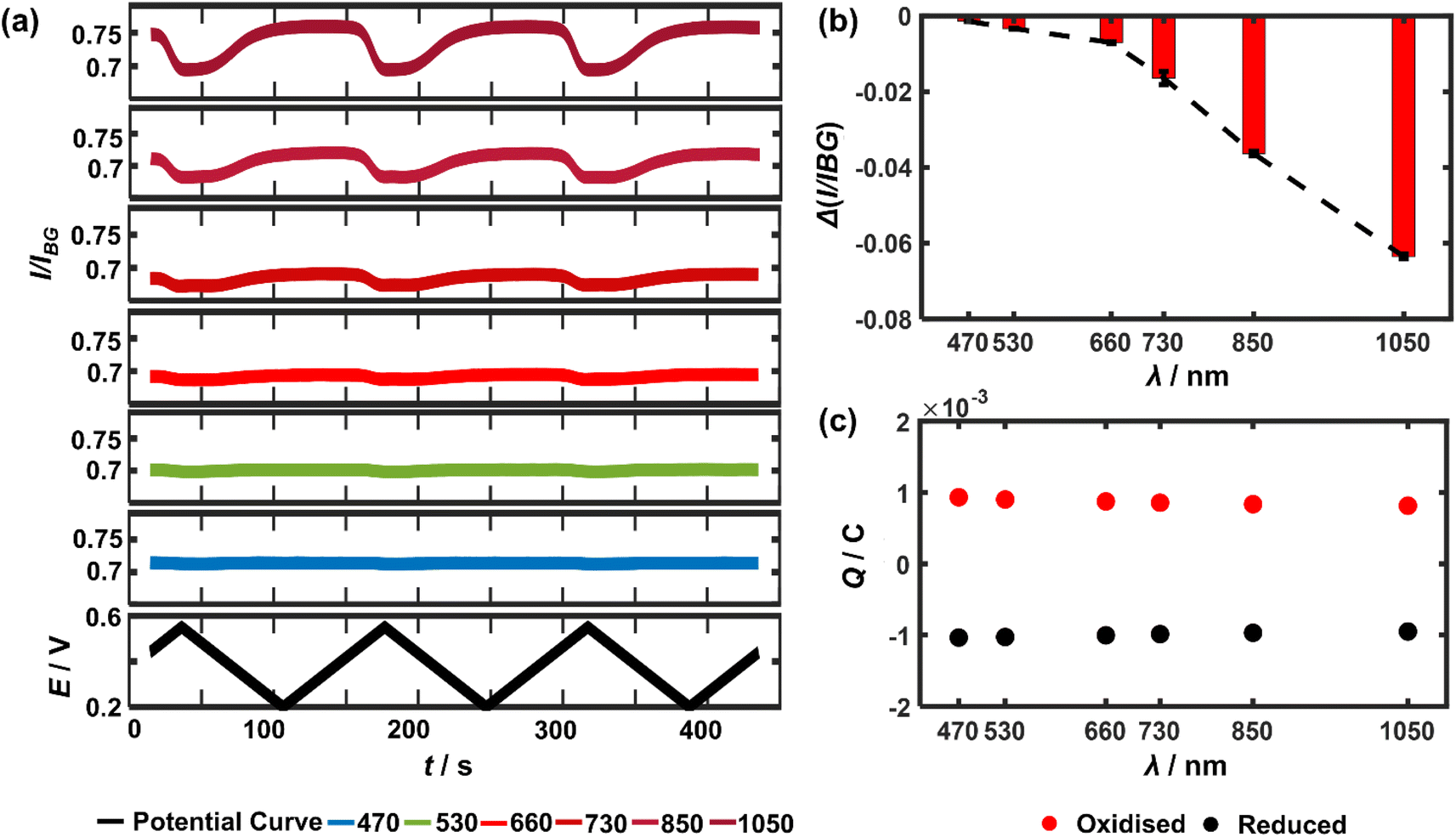 | ||
| Fig. 2 (a) Optical transmittance measured as a function of polarisation time using light with a range of wavelengths between 470 nm and 1050 nm (continuously switched from one wavelength to another). Black curve presents the applied potential ramps. (b) Maximum optical changes upon delithiation of the particle as a function of the applied light wavelength. (c) Electrical charges transferred during (red dots) delithiation and (black dots) lithiation processes respectively, from the ensembles of the immobilised particles as a function of different wavelengths showing good reproducibility. | ||
To further understand the visualisation, the optical patterns of a single LiCoO2 microparticle when exposed to different light wavelengths are examined by comparing the images of the reduced state and oxidised state, respectively. Fig. 3a shows markedly that as the reduced single LiCoO2 particle exhibits increasingly higher intensity especially with a longer light wavelength than 660 nm, there is only a very slight transmittance increase for the oxidised state, which stays quite evenly dark until the use of 1050 nm. This means that the deintercalated lithium ions from the cobalt dioxide structure significantly increase the material opacity. This is consistent with its insulator-metal phase transition upon particle oxidation, where the Li+ ion deintercalation increases the electrical conductivity of the structure.9,23,24 On the other hand, as the particle dimensions decrease to a projection-area-equivalent diameter of ∼400 nm and a thickness of less than 150 nm (see Fig. S4†), Fig. S5† shows that whilst the use of 1050 nm light expectedly reveals the differences in the optical contrasts between the reduced state and oxidised state, the measurements using visible light also show significant differences between the two oxidation states, albeit to a lesser extent. This in turn reflects the role of the penetration depth of light as a relative property to particle thickness for visualising the opaque material.
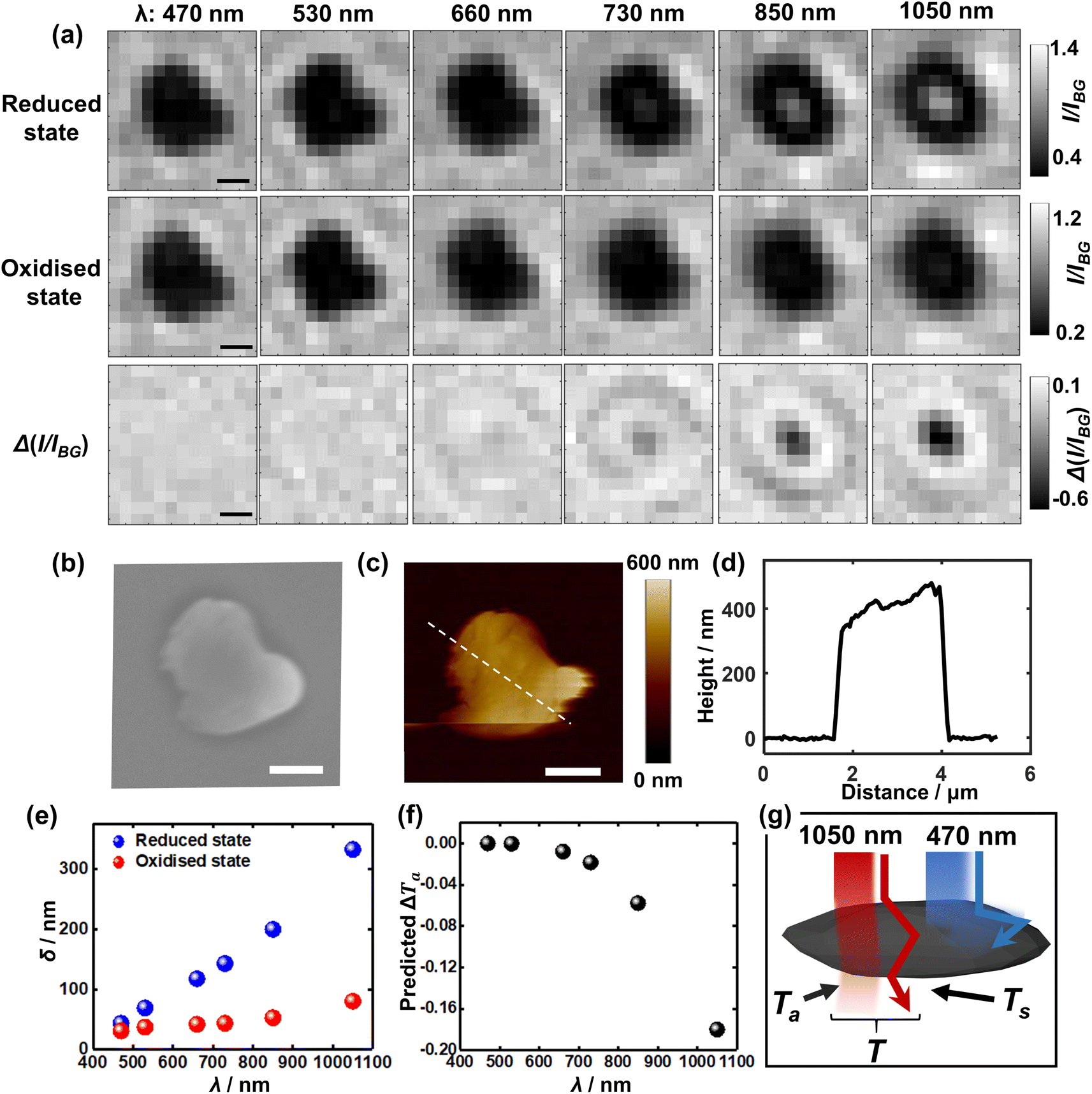 | ||
| Fig. 3 (a) Bright field images of both (upper row) the reduced and (middle row) oxidised states as a function of incident wavelength. The lower row presents the differential images upon delithiation (the middle row subtracts from the upper row) for each wavelength. (b) SEM and (c) AFM of the very same microparticle as above. All scale bars: 1 μm. (d) Height profile along the drawn line in (c). (e) Penetration depth (δ) of the reduced and oxidised states respectively23 and (f) the calculated contribution of decreased transmittance upon delithiation solely due to chromatic absorption (ΔTa) as a function of incident wavelength. (g) Proposed optical principle of the superior near-infrared light accessibility as compared to visible light via two contributions of the remaining light upon (chromatic) absorption (Ta) and scattering (Ts) by the absorbing large particle. | ||
On the basis of the optical observations above at the two different oxidation states of the single Li1−xCoO2 microparticles, the measured wavelength dependence of light transmittance (and its change upon particle oxidation) is quantitatively justified. The optical transmittance is considered to result from light scattering and chromatic absorption of the micron-sized solids. For clarity, in the following we only focus on the discussion of the transmittance through the central part of the particle without considering edge effects.
The total transmittance can be described by
| T = Ta + Ts | (1) |
As described by the Beer–Lambert law, the transmittance (Ta) depends upon solely chromatic absorption of a uniform material medium27 (here not involving scattering).
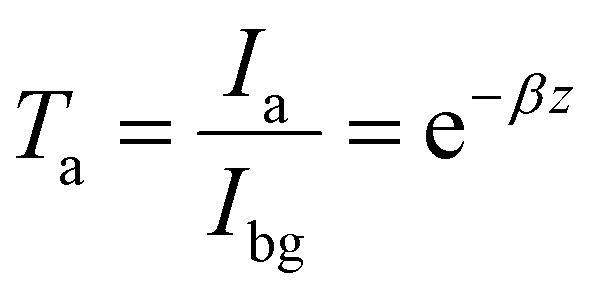 | (2) |
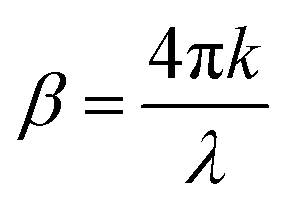 .27 The power absorption coefficient is also the reciprocal of the penetration depth (δ, in μm):
.27 The power absorption coefficient is also the reciprocal of the penetration depth (δ, in μm):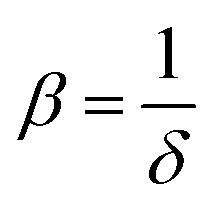 | (3) |
Referring to the datasets provided in the literature,23 the power absorption coefficients at different light wavelengths for both reduced and oxidised states of LiCoO2 are given in Table S1.† Note that these data are measured for local areas of single macrocrystals, thus involving no scattering and being applicable here to account for solely Ta. The coefficients are shown to be all at the order of 1–10 μm−1, indicating the highly absorbing properties of the particles in our work, consistent with their black appearance.
Consequently, Fig. 3e shows the penetration depth as a function of the light wavelength for the solid. It is seen that for the reduced state of LiCoO2, the penetration depth witnesses a steep rise and reaches over 140 nm for a 730 nm light, whereas the penetration stays below 100 nm into the oxidised (Li0.75CoO2) material surface even when using 1050 nm light. This latter is, as mentioned before, due to a relatively strong absorption by the oxidised state of light excited by the d–d transition of cobalt 3d orbitals, which peaks at 780 nm, whereas this d–d electronic transition is lacking in the reduced LiCoO2 state.23
As seen from the SEM (Fig. 3b) and AFM (Fig. 3c and d) results, the average thickness of the LiCoO2 microparticle with a projection-area-equivalent diameter of ca. 1.2 μm is measured to be around 400 nm. Although this thickness is three times as large as the penetration depth (140 nm) of 730 nm light, the penetration depth is defined as the depth at which the incident light declines to 1/e of its original intensity. This means that a thicker material is still likely to be accessed, especially further considering the additional scattering effects, which augment light transmittance (which will be discussed next). As such, the measured thickness of the microparticles is consistent with the fact that 730 nm (approximately the lower limit of near-infrared wavelengths) is just adequate to measure the redox electrochemistry of the microparticle. Quantitatively, the differential transmittance related to material absorption ΔTa is predicted for different wavelengths using the equation
| ΔTa = TO − TR = e−βOz − e−βRz | (4) |
The contribution from the microparticle scattering is then discussed. It is recognised that the scattering light transmittance is proportional to the scattering cross section of a particle:
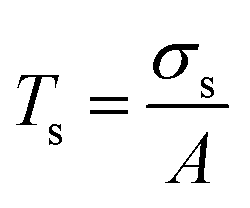 | (5) |
Thus far, both contributions to the transmittance variation from particle scattering and the transmittance left from particle absorption have been inferred to exhibit monotonically positive dependence on the light wavelength. Mathematically, the sum of two monotonic functions of the same sign does not alter its monotonicity. Consequently, the final trend of light transmittance decline is expected to also monotonically ascend with a longer wavelength. This is consistent with that observed in Fig. 2 and 3.
These findings indicate that the successful optical measurements result from the much stronger penetration effects of near-infrared light as compared to visible light. We also show that this near-infrared imaging is applicable to other electrochromic battery materials such as LiFePO4 microparticles,33 indicating the universality of the methodology (see Fig. S6†). The allowed access by near-infrared light is the key prerequisite for optically differentiating the intraparticle redox electrochemistry of single absorbing microparticles especially with a thicker dimension. It is thus demonstrated that the widely accepted concept in the bioimaging field – using strong penetrating near-infrared light to study thicker biological (living) tissues – has been successfully extended to the battery field for the first time via this work.
Building on the strong penetration capacity of near-infrared light and the spatial resolution offered by optical microscopy, we then look closer into the microparticle during the electrochemical regulation and seek to measure in more detail how the structure evolves spatially using 1050 nm light. Fig. 4 shows the mapping of the optical intensity as a function of applied potentials for the entire projection area of representative particles. Evidently, a minor fraction of the population (15 out of 40 particles, refer also to Fig. S7†) shows symmetric optical changes in the whole central bright regions of the particle (Fig. 4a) for both the forward positive polarisation and the backward negative polarisation. Given the optical intensity variation arising from delithiation-dependent light transmittance, this may be explained that the solid state diffusion should proceed at a similar rate inside the particle from all directions if there are no significant differences between the crystal structures (e.g. defects and electrochemically inactive zones) along each direction and at the micrometer scale the particle is evenly (or at least symmetrically) thick. Indeed, only a fraction (30%) of individual microparticles are seen to have a fairly even thickness (i.e. STD divided by mean ∼10%), whereas the others are irregular (STD/mean > 30%) or directionally uneven (see Fig. S8†).
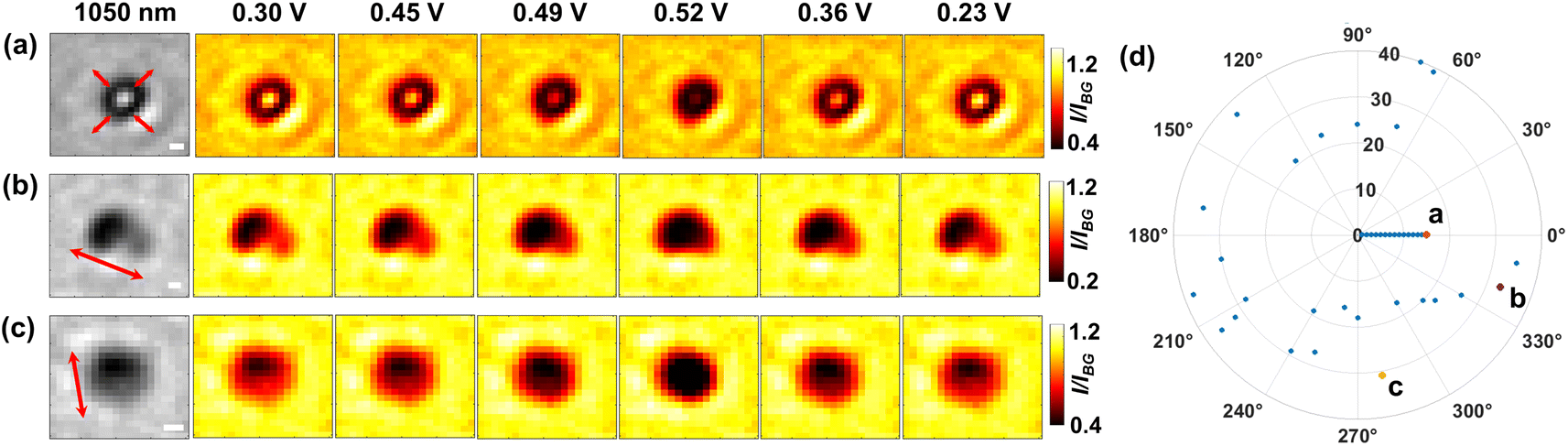 | ||
| Fig. 4 Initial bright-field images at 1050 nm of three representative microparticles (leftmost images of a, b and c) at the open circuit potential. Pseudo-coloured images show the potential-dependent transmission light through the single microparticles of representative (a) isotropic and (b and c) anisotropic (clearly directional) optical changes as labelled by the red arrows. Scale bar: 1 μm. (d) Conversion anisotropicity (characterised by the preferential angle along which the directional optical intensity decline follows during the delithiation process) distribution of the microparticles (Ntot = 40, corresponding to the value of the outmost radius of the plot). | ||
Interestingly, by contrast, most (i.e. 24 in total) of the particles exhibit clear anisotropic optical changes along particular directions as labelled by the arrows in Fig. 4b and c. Specifically, the transmitted light initially (E = 0.30 V) concentrates predominantly on the right part of the particle (Fig. 4b), which suggests that this particular particle may be thinner for this half part. As the potential is scanned more positively to 0.52 V, during which the particle is subject to oxidation, the right half of the particle becomes increasingly less transmissive sooner from the upper-left sub-zone, and later for the lower-right corner (see Video S1†). Since the transmittance is positively related to the lithium content, the observed contrast change indicates that the lithium-poor phase (Li0.75CoO2) is gradually formed by directional lithium diffusion propagating towards the lower-right corner. Similar changes are also evidenced for the reverse potential sweep, corresponding to the gradual reduction to the lithium-rich phase (LiCoO2) along the opposite direction. These observations are very interesting as they indicate that the ‘unit cells’ constituting a single microparticle are likely to be not uniform (i.e. different lattice parameters, crystal orientations and defects) across the structure, which causes the heterogeneity of charge transfer kinetics within the chemical structure. Accordingly, structurally asymmetric differences in these unit cell properties would lead to anisotropic formation dynamics of phase domain boundaries (between the lithium-rich and lithium poor phases), which directs the intraparticle diffusion.17
We also find a single microparticle with characteristic optical changes consistent with the previous work from Merryweather et al.,17 although there is only one such particle in our case. The particle shows mixed behaviour (see Fig. S9†) that the oxidative delithiation follows a symmetric evolution of optical contrast, but the reductive lithiation witnesses a directional phase front propagation. The observations are attributed to differing kinetic mechanisms (i.e. diffusion limited or charge transfer limited processes) of the two redox processes.17 Nevertheless, our statistics show that apart from this special case, all 24 microparticles show preferential and opposite directions of the lithium diffusion for lithiation and delithiation processes. To quantify the directions of the reversible optical changes, the directional angles along which the optical intensity decline follows during the delithiation process are measured and plotted as a function of the sequential number of individual microparticles (randomly numbered, N = 40). Consequently, the directions are found, as shown in Fig. 4d, to vary completely randomly from particle to particle. This implies possible electrochemical heterogeneity of the crystal structures across different microparticles. In comparison with preceding work,17 although the same phase transitions are studied, these dissimilar observations could arise from the polycrystallinity of the particles and, besides the structural factors discussed above, also likely from the absence of conductive agents that could lead to poor particle-substrate electrical contacts.34
Last, to examine the further applicability of near-infrared optical microscopy and to also complement the understanding of the optical responses of single microparticles, another optical means of dark field microscopy based on particulate scattering is briefly investigated. Experimentally, the single LiCoO2 particles are obliquely illuminated and imaged during cyclic voltammetry using light wavelengths from 470 nm to 850 nm. Unfortunately, the dark field condenser is not compatible with the 1050 nm light but the following results show that the 850 nm light is sufficient for penetrating whole microparticles. The initial scattering light intensity of the same single particle is recorded as a function of the light wavelength for both reduced and oxidised states. Fig. 5a shows that when exposed to 470 nm light the LiCoO2 particle (reduced state) is only bright at the surface and not much inwards. However, as the incident light wavelength increases, the particle becomes gradually brighter and then the scattered light reaches the maximum and meanwhile appears much more uniform over the particle region for the 850 nm light. Similar plots of quantitative optical changes to Fig. 2 of single microparticles obtained using dark field microscopy and an optical field of view containing a number of such particles (N = 29) are provided collectively in Fig. S10.† The observed trend strongly suggests that the light interaction with the particle occurs deeply inside the solid structure, which is thus consistent with the penetration effects of the opaque material (in the visible region) inferred from the results using bright-field microscopy. Again by contrast, the scattering only penetrates slightly more into the oxidised particle using the 850 nm light than using the 470 nm light. This is consistent with the almost unchanged optical contrast observed in the BFM results, confirming the vital use of near-infrared light for visualising the intraparticle electrochemistry of microparticles.
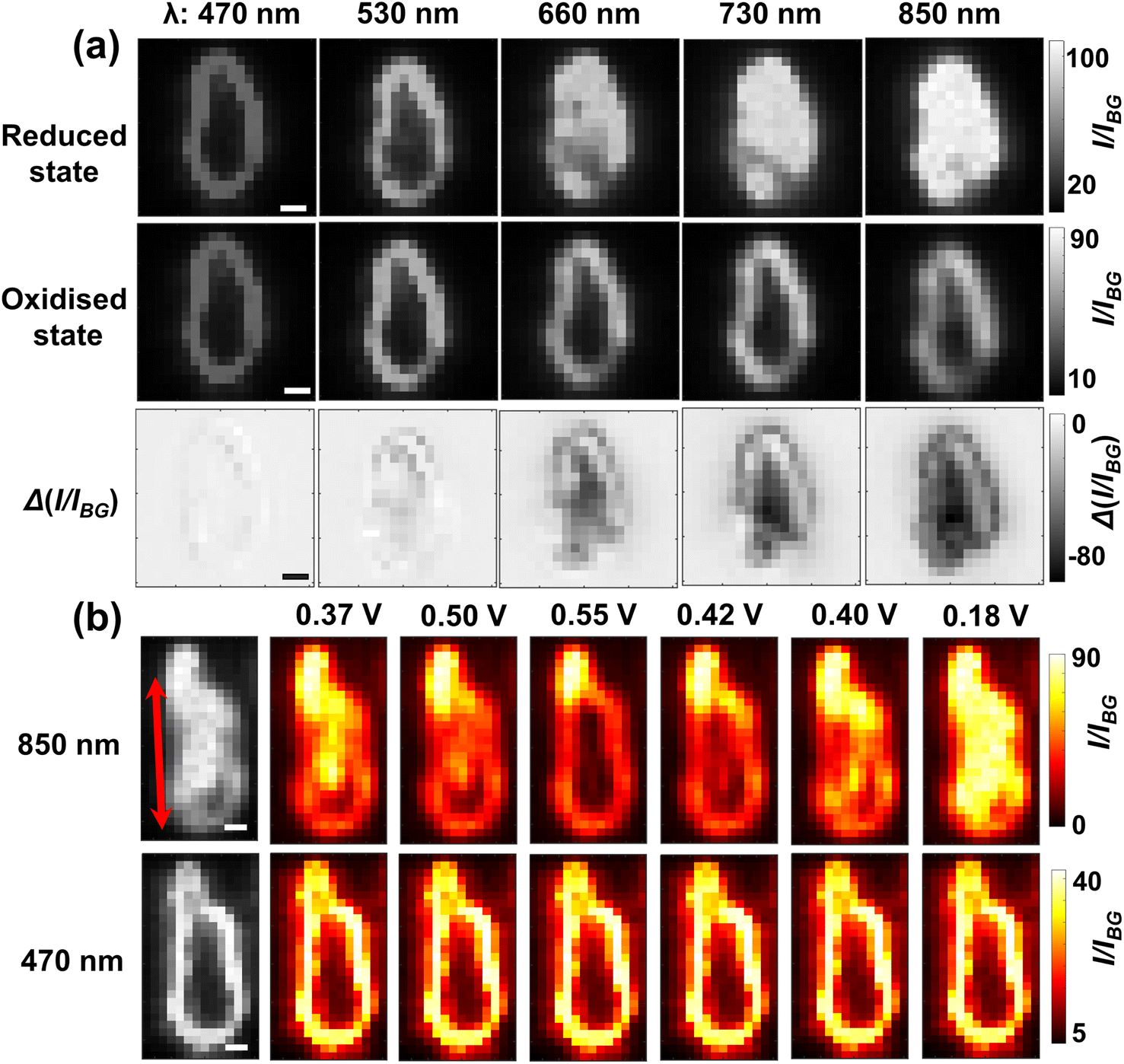 | ||
| Fig. 5 (a) Dark-field particle images of both (upper row) reduced and (middle row) oxidised states as a function of the wavelength of the light. The lower row presents the scattering intensity decrease upon delithiation for each wavelength. (b) Initial bright-field images of a representative microparticle (leftmost images) at the open circuit potential. Pseudo-coloured images show the potential-dependent transmission light over the single microparticle using 850 nm and 470 nm light, respectively. Red arrows represent the reversible directions of the solid state diffusion (scale bar 1 μm). | ||
The imaging capacity of dark field scattering microscopy with 850 nm near-infrared light is also further checked for solid-state lithium ion diffusion. Fig. 5b shows, consistent with the results from the BFM experiments, that for most particles (ca. 60%) the potential-determining scattering intensity variation appears to be clearly directional with large heterogeneity (see Fig. S11†). Although the spatial evolution seems complicated, it is evident that the upper part of this particle shows significantly more drastic changes than the lower part of the particle, Video S2.† Notably, there are also subareas (the lower right corner) of many microparticles showing significantly smaller scattering changes than other regions despite longer times and larger overpotentials (Fig. S12†), indicating the presence of thermodynamically less active zones. This thus supports a non-uniform crystal structure as the origin of directional Li+ diffusion. Clear directional changes of more microparticles are provided in Fig. S13.† This is in contrast to the imaging using 470 nm, where no obvious changes are observed throughout the potential window. The latter fact also suggests that the brightness of the particle outline comes from the scattering mainly from the particle edges and thus carries limited information regarding the intercalation states of the overall structure. However, it is critically noted that when accessing deeper zones of the material, the spatial resolution would be diminished according to the Abbe diffraction limit (resolution ∝ 1/λ); a doubling in the penetration depth of monochromatic light would lead to a half good spatial resolution. Despite this, the simple use of a near-infrared light source permits access to the structural variation of micron-sized particles. As such, the imaged lithium ion dynamics using both DFM and BFM has well demonstrated the universal capacity of near-infrared light in imaging microparticle redox electrochemistry.
Conclusions
This work proposes the use of near-infrared light sources for visualising redox electrochemistry of single micron-sized lithium cobalt oxide particles. The study shows that both the electrochemical behaviour of single LiCoO2 microparticles and intraparticle lithium ion diffusion can be readily measured, despite the imaging modes of bright-field optical contrast or dark field scattering. This is attributed to the strong penetration through the large absorbing particles of near-infrared light. In particular, the visualised solid-state diffusion has been discovered to have preferential directions for the majority of the particles, possibly due to structurally asymmetric crystal structures that cause directional, rather than homogeneous, formation dynamics of phase domain boundaries between lithium-rich and lithium-poor phases. This part of lithium ion battery science features complex multidimensional data, which means opportunities of being efficiently elucidated with the aid of artificial intelligence.35 As such, this near-infrared optical methodology is demonstrated to offer potential opportunities for in situ accessing the redox electrochemistry of practical battery particles as well as other intraparticle kinetic analyses36,37 at the single particle level.Data availability
The authors declare that all the data are available within the article and its ESI† or from the corresponding author upon reasonable request.Author contributions
Xinyue Wang performed the experiments. Xinyue Wang and Ruo-chen Xie analyzed the data and wrote the manuscript. Si-cong Wang and Junjie Ma helped with experiments and data analysis. Wei Wang conceptualised and supervised the project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is financially supported by the Natural Science Foundation of Jiangsu Province (BK20220768) and the National Natural Science Foundation of China (21925403).Notes and references
- C. Luan, J. Angona, A. Bala Krishnan, M. Corva, P. Hosseini, M. Heidelmann, U. Hagemann, E. Batsa Tetteh, W. Schuhmann and K. Tschulik, Angew. Chem., Int. Ed., 2023, 62, e202305982 CrossRef CAS PubMed.
- S. E. Kleijn, S. C. Lai, M. T. Koper and P. R. Unwin, Angew. Chem., Int. Ed., 2014, 53, 3558–3586 CrossRef CAS PubMed.
- J.-F. Lemineur, H. Wang, W. Wang and F. Kanoufi, Annu. Rev. Anal. Chem., 2022, 15, 57–82 CrossRef PubMed.
- M. Saqib, Y. Fan, R. Hao and B. Zhang, Nano Energy, 2021, 90, 106539 CrossRef CAS.
- C. Zeng, J. Liang, C. Cui, T. Zhai and H. Li, Adv. Mater., 2022, 34, 2200777 CrossRef CAS PubMed.
- M. A. Sani and K. Tschulik, Curr. Opin. Electrochem., 2023, 37, 101195 CrossRef.
- Z. Nilsson, M. Van Erdewyk, L. Wang and J. B. Sambur, ACS Energy Lett., 2020, 5, 1474–1486 CrossRef CAS.
- R. C. Evans, A. Ellingworth, C. J. Cashen, C. R. Weinberger and J. B. Sambur, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 12666–12671 CrossRef CAS PubMed.
- D. Jiang, Y. Jiang, Z. Li, T. Liu, X. Wo, Y. Fang, N. Tao, W. Wang and H.-Y. Chen, J. Am. Chem. Soc., 2017, 139, 186–192 CrossRef CAS PubMed.
- W. Jiang, W. Wei, T. Yuan, S. Liu, B. Niu, H. Wang and W. Wang, Chem. Sci., 2021, 12, 8556–8562 RSC.
- B. Niu, W. Jiang, B. Jiang, M. Lv, S. Wang and W. Wang, Nat. Commun., 2022, 13, 2316 CrossRef CAS PubMed.
- R. Hao, Y. Fan, M. D. Howard, J. C. Vaughan and B. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 5878–5883 CrossRef CAS PubMed.
- J.-F. Lemineur, P. Ciocci, J.-M. Noël, H. Ge, C. Combellas and F. Kanoufi, ACS Nano, 2021, 15, 2643–2653 CrossRef CAS PubMed.
- L. Godeffroy, P. Ciocci, N. Ortiz Peña, D. Alloyeau, J. M. Noël, J. F. Lemineur and F. Kanoufi, Angew. Chem., Int. Ed., 2023, 135, e202304950 CrossRef.
- X. Shan, I. Díez-Pérez, L. Wang, P. Wiktor, Y. Gu, L. Zhang, W. Wang, J. Lu, S. Wang and Q. Gong, Nat. Nanotechnol., 2012, 7, 668–672 CrossRef CAS PubMed.
- G. Feng, J. Guo, H. Tian, Z. Li, Y. Shi, X. Li, X. Yang, D. Mayerich, Y. Yang and X. Shan, Adv. Energy Mater., 2022, 12, 2103484 CrossRef CAS.
- A. J. Merryweather, C. Schnedermann, Q. Jacquet, C. P. Grey and A. Rao, Nature, 2021, 594, 522–528 CrossRef CAS PubMed.
- I. T. Lucas, A. McLeod, J. Syzdek, D. Middlemiss, C. Grey, D. Basov and R. Kostecki, Nano Lett., 2015, 15, 1–7 CrossRef CAS PubMed.
- X. Gu, J.-l. Liu, J.-h. Yang, H.-j. Xiang, X.-g. Gong and Y.-y. Xia, J. Phys. Chem. C, 2011, 115, 12672–12676 CrossRef CAS.
- G. Wang, Q. Qu, B. Wang, Y. Shi, S. Tian, Y. Wu and R. Holze, Electrochim. Acta, 2009, 54, 1199–1203 CrossRef CAS.
- C. A. Marianetti, G. Kotliar and G. Ceder, Nat. Mater., 2004, 3, 627–631 CrossRef CAS PubMed.
- H. Xia, L. Lu, Y. S. Meng and G. Ceder, J. Electrochem. Soc., 2007, 154, A337 CrossRef CAS.
- H. L. Liu, T. Ou-Yang, H. Tsai, P. Lin, H. Jeng, G. Shu and F. Chou, New J. Phys., 2015, 17, 103004 CrossRef.
- M. Ménétrier, I. Saadoune, S. Levasseur and C. Delmas, J. Mater. Chem., 1999, 9, 1135–1140 RSC.
- M. Jonasz, Principles, Methods and Application of Particle Size Analysis, ed. J. P. M. Syvitski, Cambridge University Press, Cambridge, 1991, pp. 143–162 Search PubMed.
- J. Dave, IBM J. Res. Dev., 1969, 13, 302–313 Search PubMed.
- F. Kanoufi, Encyclopedia of Electrochemistry, pp. 1–80, DOI:10.1002/9783527610426.bard030108.
- A. Cox, A. J. DeWeerd and J. Linden, Am. J. Phys., 2002, 70, 620–625 CrossRef.
- I. B. Slima, K. Karoui and A. B. Rhaiem, Ionics, 2023, 29, 1731–1739 CrossRef CAS.
- H. C. van de Hulst and V. Twersky, Phys. Today, 1957, 10, 28–30 CrossRef.
- G. W. Kattawar and G. N. Plass, Appl. Opt., 1967, 6, 1377–1382 CrossRef CAS PubMed.
- F. Seyedheydari, Electromagnetic Response of Metal-Semiconductor-Oxide Particles in the Near-Infrared Regime, 2023.
- S. Furutsuki, S.-C. Chung, S.-i. Nishimura, Y. Kudo, K. Yamashita and A. Yamada, J. Phys. Chem. C, 2012, 116, 15259–15264 CrossRef CAS.
- W. Wei, T. Yuan, W. Jiang, J. Gao, H.-Y. Chen and W. Wang, J. Am. Chem. Soc., 2020, 142, 14307–14313 CrossRef CAS PubMed.
- Z.-W. Zhu, J.-Y. Qiu, L. Wang, G.-P. Cao, X.-M. He, J. Wang and H. Zhang, J. Electrochem., 2022, 28, 2219003 Search PubMed.
- Y. Lei, G. Zhang, Q. Zhang, L. Yu, H. Li, H. Yu and Y. He, Nat. Commun., 2021, 12, 4483 CrossRef CAS PubMed.
- Q. Wang, H. Yu, X. Chen and Y. He, Adv. Funct. Mater., 2023, 2300487 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedure, Sections 1–14, including Fig. S1–S12, Table S1 and discussion on the depth of field; Videos S1 and S2. See DOI: https://doi.org/10.1039/d4sc00072b |
| This journal is © The Royal Society of Chemistry 2024 |