DOI:
10.1039/D3SC06094B
(Edge Article)
Chem. Sci., 2024,
15, 8545-8556
Supramolecular assembly of amphiphilic platinum(II) Schiff base complexes: diverse spectroscopic changes and nanostructures through rational molecular design and solvent control†
Received
14th November 2023
, Accepted 15th April 2024
First published on 10th May 2024
Abstract
A new class of amphiphilic tetradentate platinum(II) Schiff base complexes has been designed and synthesized. The self-assembly properties by exploiting the potential Pt⋯Pt interactions of amphiphilic platinum(II) Schiff base complexes in the solution state have been systematically investigated. The presence of Pt⋯Pt interactions has further been supported by computational studies and non-covalent interaction (NCI) analysis of the dimer of the complex. The extent of the non-covalent Pt⋯Pt and π–π interactions could be regulated by a variation of the solvent compositions and the hydrophobicity of the complexes, which is accompanied by attractive spectroscopic and luminescence changes and leads to diverse morphological transformations. The present work represents a rare example of demonstration of directed cooperative assembly of amphiphilic platinum(II) Schiff base complexes by intermolecular Pt⋯Pt interactions in solution with an in-depth mechanistic investigation, providing guiding principles for the construction of supramolecular structures with desirable properties using platinum(II) Schiff base building blocks.
Introduction
Supramolecular assembly of square-planar d8 platinum(II) complexes into highly ordered nanostructures has attracted long-standing interest over the past few decades.1–5 Interesting spectroscopic and luminescence properties with drastic color changes and the switching-on of the triplet metal–metal-to-ligand charge transfer (3MMLCT) emission6–8 have been imparted by their propensity to exhibit non-covalent Pt⋯Pt interactions. More importantly, the directional non-covalent Pt⋯Pt interactions can serve as an additional driving force for enhancing the stability of aggregates and in controlling various supramolecular architectures.8 With these intriguing properties of alkynylplatinum(II) polypyridine complexes, this class of complexes has been shown to be used as supramolecular gelators,9–11 in optoelectronic applications,7,12 and as stimuli-responsive materials to solvent,13–15 temperature,4c,13–15 pH,16 and so on through rational molecular design and solvation control. Given the unique advantages of d8 platinum(II) complexes, explorations of new classes of platinum(II) complexes with other readily accessible polydentate π-conjugated ligands with higher rigidity and planarity may provide versatile building motifs for the construction of stimuli-responsive luminescent supramolecular metal–organic materials with good thermal stability.
Salicylaldehyde Schiff bases, which serve as one class of polydentate ligands, are rather simple to synthesize and one of them is able to exhibit reversible photochromic properties.17 Besides, they are found to display excellent ability to coordinate to transition metal centers, such as MnII, PtII, NiII and ZnII, and to show interesting physical and chemical properties for diverse potential applications, such as in catalysis, optoelectronic materials, cell imaging, etc.18–25 However, self-assembly of Schiff base-containing metal complexes is still in its infancy and is mainly focused on zinc(II) Schiff base complexes that made use of Zn⋯O coordination to construct luminescent gels and metal–organic frameworks (MOFs).26,27 There have been examples of solid-state aggregation of platinum(II) Schiff base complexes and their lyotropic liquid crystal characteristics facilitated by π–π, Pt⋯Pt and C–H⋯π interactions according to X-ray crystallography and modeling studies.24,28,29 These results revealed the excellent square-planar geometry of these complexes and a preferred tendency to come close together in the axial direction to form Pt⋯Pt contacts, rendering them potential candidates for supramolecular assembly.24,28,29 Unlike the manipulation of Pt⋯Pt interactions of alkynylplatinum(II) polypyridine and 2,6-bis(benzimidazol-2′-yl)pyridine complexes by multiple external stimuli that has been well-established in the solution state,3a,4,6–8,13,14a,b studies on luminescent Schiff base-containing platinum(II) complexes were mainly confined to research studies on solvatochromic properties and electroluminescent devices.19,21,28b,30 The utilization of their spectroscopic characteristics to assist in an in-depth understanding of self-assembly behaviors in solutions is rather rudimentary and worth further exploring. Besides, it is believed that a thorough mechanistic investigation would play an important role in an understanding of the interplay of the multiple non-covalent interactions of Schiff base-containing platinum(II) complexes. These, together with the lack of reports on investigation of self-assembly architectures in the solution state by Pt⋯Pt interactions, have promoted us to systematically explore new classes of amphiphilic Schiff base-containing platinum(II) complexes and elucidate their mechanism in the assembly process. It would be of great value to provide insights into their self-assembly properties and to reveal guiding principles for the rational molecular design and construction of supramolecular luminescent architectures with desirable properties using platinum(II) Schiff base building blocks for potential applications.
Herein, a new class of amphiphilic tetradentate Schiff base-containing platinum(II) complexes has been designed and synthesized (Scheme 1) with greatly improved solubility for the ease of systematic investigation on the potential exploitation of Pt⋯Pt interactions in the solution state, which have been further supported by computational studies and non-covalent interactions (NCI) analysis. Intriguingly, in addition to a variation of solvent compositions, the extent of these Pt⋯Pt and π–π interactions could be regulated upon changing molecular hydrophobicity, triggering interesting spectroscopic changes and diverse morphological transformations. The close correlation between the length of the alkoxy chains and self-assembled morphologies and the balance between multiple non-covalent interactions has been demonstrated by the realization of the cooperative growth mechanism in the assembly process. The self-assembly behaviors and mechanisms of these platinum(II) Schiff base complexes in water and DMSO–water solutions have been investigated by UV-vis absorption, emission and 1H NMR spectroscopy with their morphologies investigated by transmission electron microscopy (TEM), scanning electron microscopy (SEM) and atomic force microscopy (AFM), and have been shown to involve intermolecular Pt⋯Pt and/or π–π interactions.
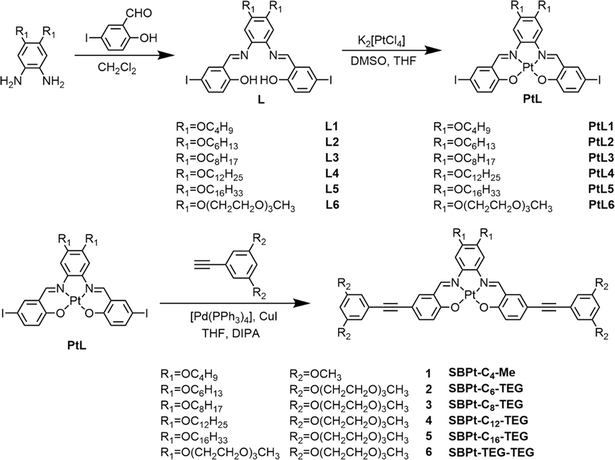 |
| | Scheme 1 Synthetic route for complexes 1–6. | |
Results and discussion
Synthesis and structural characterization
Schiff base-containing platinum(II) complexes 1–6 were prepared in three steps as shown in Scheme 1. The Schiff base ligands, L1–L6, with various lengths of alkoxy chains and triethylene glycol units were synthesized via an aldehyde–amine condensation reaction according to modification of procedures in the literature.19,30 The intermediate platinum(II) complexes, PtL1–PtL6, were obtained by stirring a mixture of K2[PtCl4] in DMSO and L1–L6 with the presence of K2CO3 in distilled THF at 60 °C for 3 days, followed by further purification with recrystallization (detailed in the ESI†). The Sonogashira coupling reactions of PtL1–PtL6 with HC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–Ph–(OCH3)2 or HCC–Ph–(OTEG)2 were carried out with a catalytic amount of copper(I) iodide and Pd(PPh3)4 to afford the target complexes, 1–6, after purification through column chromatography (detailed synthesis in the ESI†). The resulting complexes were characterized using 1H and 13C{1H} NMR spectra, MALDI-TOF mass spectra and elemental analysis (Fig. S1–S12†); the identities of which are established by the full agreement of the data with those expected of the target complexes.
C–Ph–(OCH3)2 or HCC–Ph–(OTEG)2 were carried out with a catalytic amount of copper(I) iodide and Pd(PPh3)4 to afford the target complexes, 1–6, after purification through column chromatography (detailed synthesis in the ESI†). The resulting complexes were characterized using 1H and 13C{1H} NMR spectra, MALDI-TOF mass spectra and elemental analysis (Fig. S1–S12†); the identities of which are established by the full agreement of the data with those expected of the target complexes.
X-ray crystal structure determination
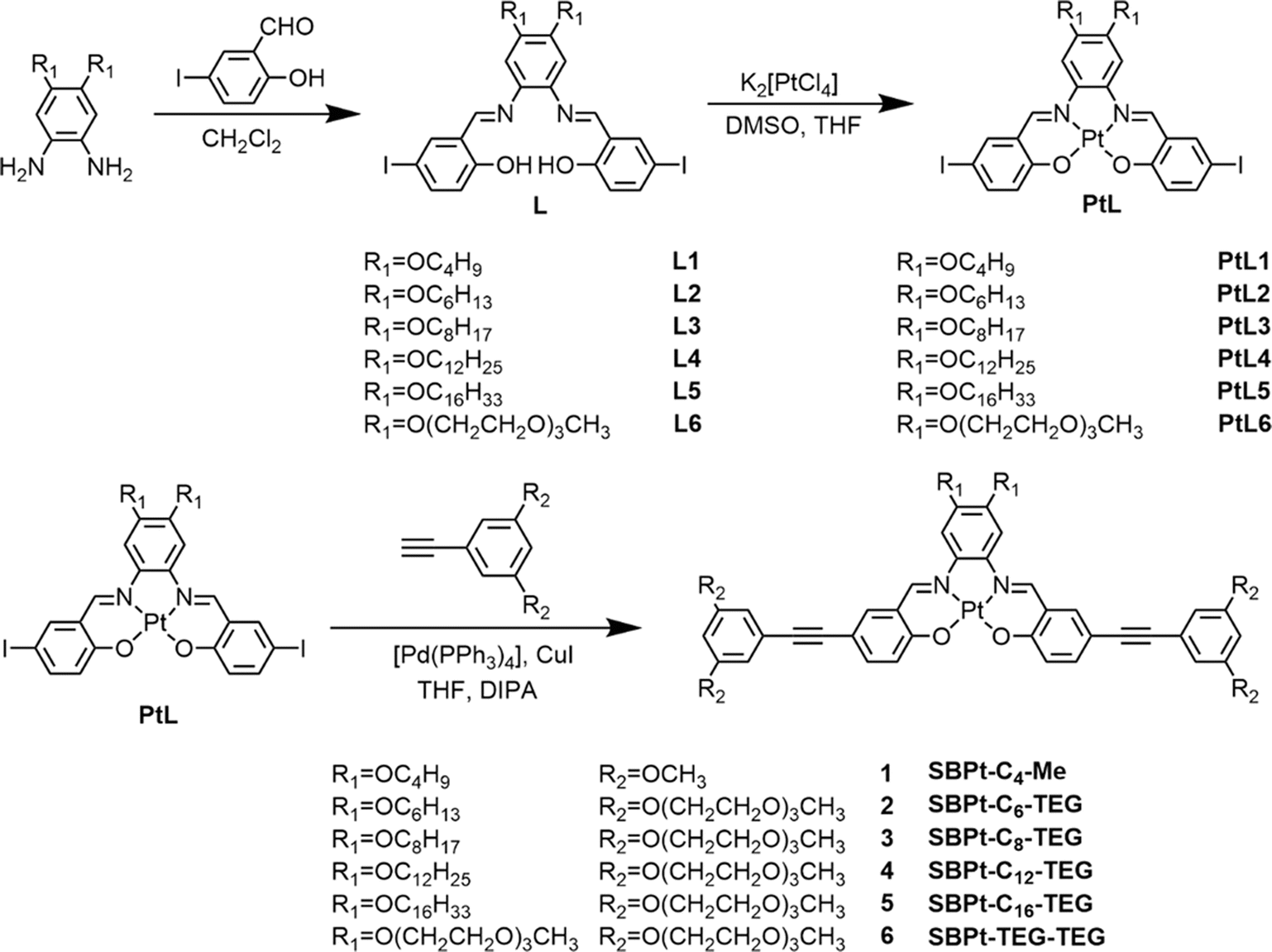
In order to investigate the extent of Pt⋯Pt and π–π stacking interactions and molecular packing model, single crystals of 1 which consists of –OC4H9 chains and methoxy groups have been obtained by slow diffusion of diethyl ether into a concentrated dichloromethane solution of 1.31 A perspective drawing of complex 1 is depicted in Fig. 1a, and the crystal determination data and the selected bond lengths, bond angles and dihedral angles are summarized in Tables S1 and S2 in the ESI,† respectively. It is worth mentioning that the bond lengths of Pt(1)–N(1), Pt(1)–N(2), Pt(1)–O(1) and Pt(1)–O(2) in complex 1 are 1.949(3) Å, 1.963(3) Å, 1.993(2) Å and 1.991(2) Å, respectively and the angles of the N(1)–Pt(1)–O(2) and N(2)–Pt(1)–O(1) are 179.46(10)° and 178.93(11)° respectively, which indicate the square-planar geometry of the individual complex molecule. Interestingly, complex 1 was found to exist in a dimeric form with short Pt⋯Pt contacts (3.354 Å) and π–π stacking distances (3.385 Å) in a head-to-tail anti-parallel arrangement (Fig. 1b and c).29 Meanwhile, compared to the UV-vis spectrum of 1 which exists as non-aggregated species in dichloromethane, the UV-vis absorption spectrum of 1 in the crystalline solid state displays a remarkable and new low-energy absorption tail and a red-shifted emission band, indicating a close relationship between the spectral changes and the Pt⋯Pt and π–π stacking interactions (Fig. 1d and e). Due to the presence of alkynyl groups in the complexes, the ethynylphenyl units of these complexes may be allowed to twist. However, the dihedral angles between plane C1–C6 and plane C1–C15 and between plane C32–C37 and plane C41–C46 are found to be 3.226(153)° and 7.239(164)°, respectively, indicating that the –C6H3(OCH3)3 units in complex 1 are only slightly twisted from the Pt(N2O2) plane and are mainly in a coplanar arrangement with the Pt(N2O2) plane (Table S2†). This is likely to be driven by the gain in stabilization resulting from the enhancement in the extent of Pt⋯Pt and π–π stacking interactions. Besides, the alkoxy chains attached to the phenyl bisimine unit are found to deviate from the Pt(N2O2) plane. It is also a possible factor that the longer alkoxy chains can exert steric influence to increase the interplanar distance in the self-assembled structures. The X-ray crystallography information would lead to a better understanding of the self-assembly of complexes 2–6 in solutions.
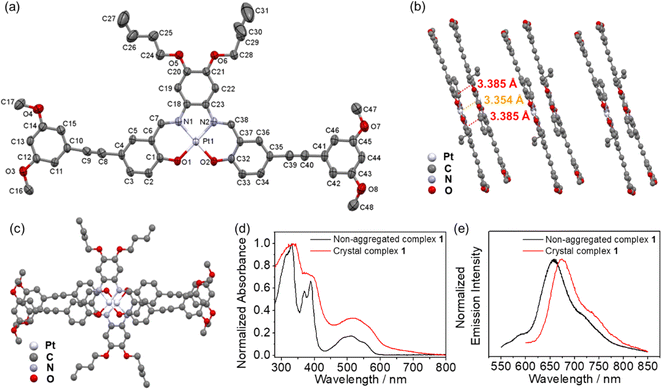 |
| | Fig. 1 (a) Crystal structure of 1 with atomic numbering. Hydrogen atoms have been omitted for clarity. Thermal ellipsoids are shown at the 30% probability level. The crystal packing structure of 1 showing the (b) side view and (c) top view. (d) Normalized UV-vis absorption spectra and (e) normalized emission spectra of 1 in the non-aggregated state in dichloromethane and in the crystal state. | |
UV-vis absorption, emission and solvatochromic properties
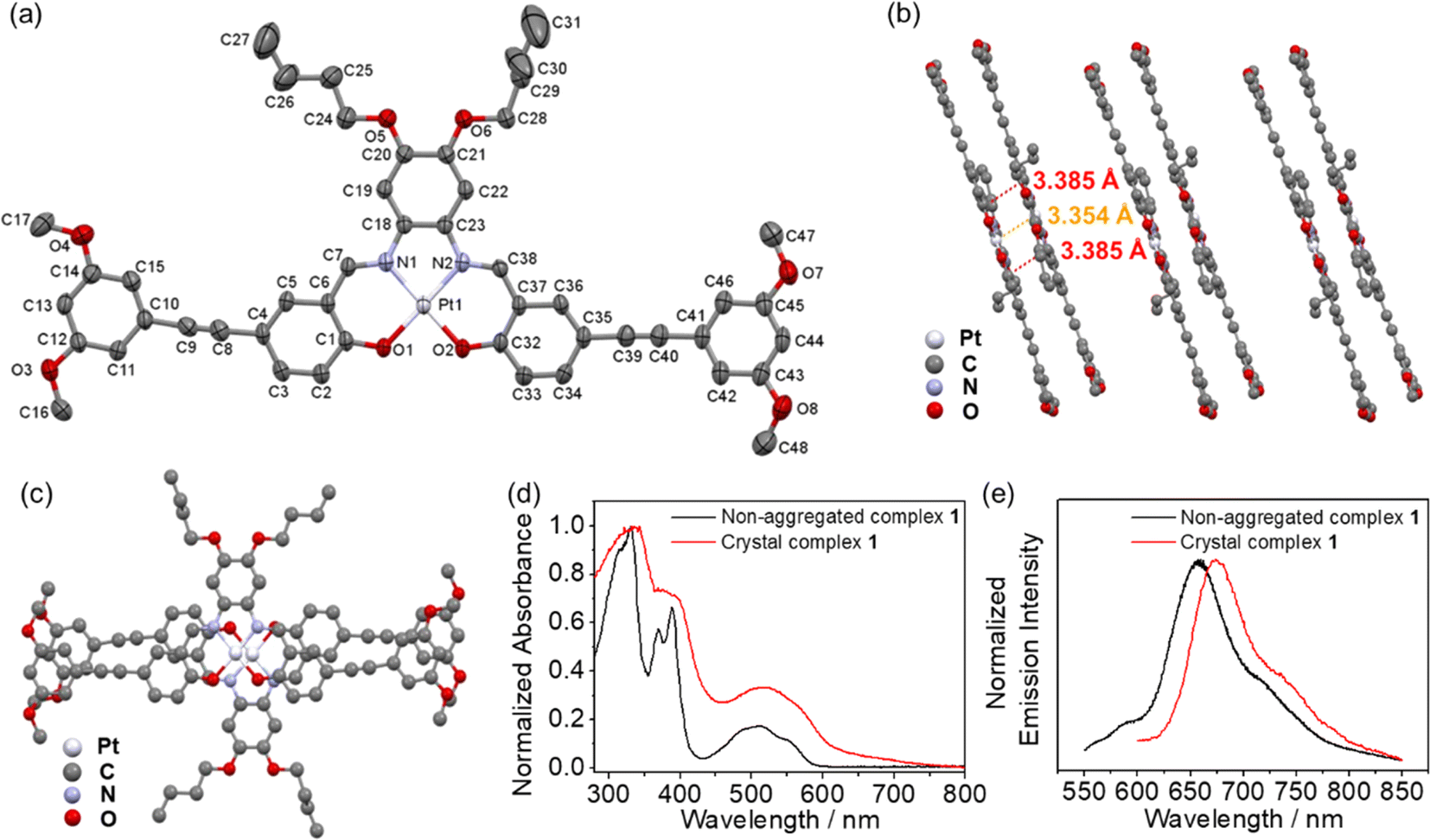
Complexes 1–6 dissolve in DMSO to give orange solutions at 298 K. In degassed DMSO solutions, the UV-vis absorption spectra of 1–6 exhibit intense high-energy absorption bands at 315–395 nm, assignable as intraligand (IL) [π → π*] transitions and low-energy absorption bands at 465–550 nm, attributed to the admixture of metal-to-ligand charge-transfer (MLCT) 1[dπ(Pt) → π*(L)] and ligand-centered (LC) 1[lone pair(phenoxide) → π*(imine)] transitions (Fig. 2a). Similar spectral assignments have also been made.30,32 Upon excitation at λ = 480 nm, complexes 1–6 exhibit phosphorescence at 641 to 654 nm in degassed DMSO solution, which originates from the 3MLCT excited state, mixed with 3LC[lone pair(phenoxide) → π*(imine)] excited state character (Fig. 2b).30,32 It is worth noting that a blue shift in the emission band maximum is only observed for complex 6, which may suggest a different mode of aggregation. This could probably be ascribed to a different stacking mode that is more inclined towards a H-type of aggregation since 6 has two additional hydrophilic TEG chains in place of the hydrophobic alkoxy chains on the central aryl ring of the Schiff base ligand in 2–5. The phosphorescence lifetimes (τ) and quantum yields (Φem) are found to be consistent with the phosphorescence nature, especially the lifetime decay trace displays a clear monoexponential decay in the microsecond regime and the monoexponential decay curve does not change with the excitation wavelength (Fig. 2c and S13†). The corresponding UV-vis absorption and emission data are summarized in Table S3.† Moreover, complexes 2–6 exhibit solvatochromic properties (Fig. S14–S18†), as observed from a bathochromic shift in the emission spectra of 2–6 with increasing solvent polarity under degassed conditions, suggesting the charge transfer character of the excited state.32,33
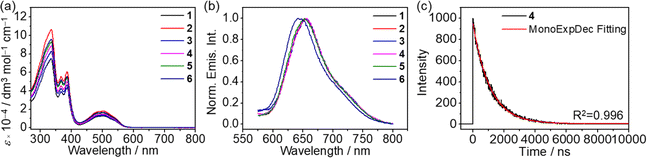 |
| | Fig. 2 (a) Electronic absorption spectra and (b) normalized emission spectra of 1–6 in degassed DMSO solutions (10−5 M) at 298 K. (c) The monoexponential phosphorescence decay trace (detected at the emission band maxima) of 4 in the degassed DMSO solution at 298 K. | |
Computational studies
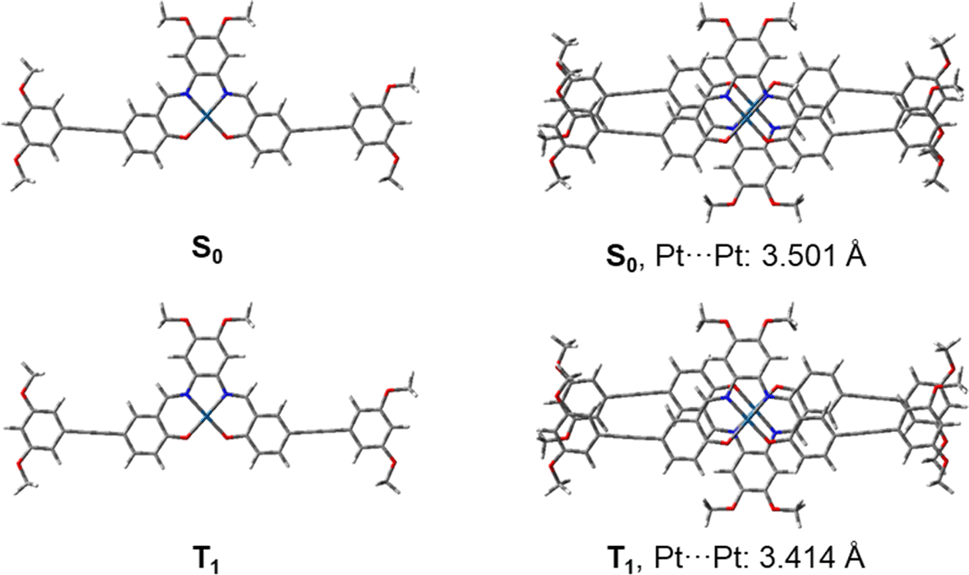
Density functional theory (DFT) optimization and time-dependent density functional theory (TDDFT) calculations were employed to investigate the electronic structure and the nature of the absorption origins of complexes 1–6. By truncating the flexible tails to methoxy groups (R1, R2 = OMe), the model complex 7 was obtained, on which geometry optimization calculations were performed (Fig. S19†). In view of the head-to-tail stacking observed in the X-ray crystal structure, both the monomer and head-to-tail dimer of 7 were analyzed, optimizing the structures shown in Fig. 3. The calculated Pt–N and Pt–O distances, which range from 1.954 to 1.964 Å and 1.976 to 2.001 Å respectively, are in good agreement with the experimental X-ray crystal structure, as are the bond angles around the Pt(II) center. Energies of the frontier molecular orbitals of complex 7 and the dimer 72 are shown in Fig. S20.† Cartesian coordinates can be found in Tables S4–S7.†
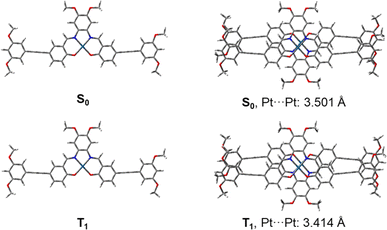 |
| | Fig. 3 Optimized ground state (S0) and lowest triplet excited state (T1) geometries of the monomer and dimer forms of 7. Pt⋯Pt internuclear distances of the dimeric forms are given in ångstroms. | |
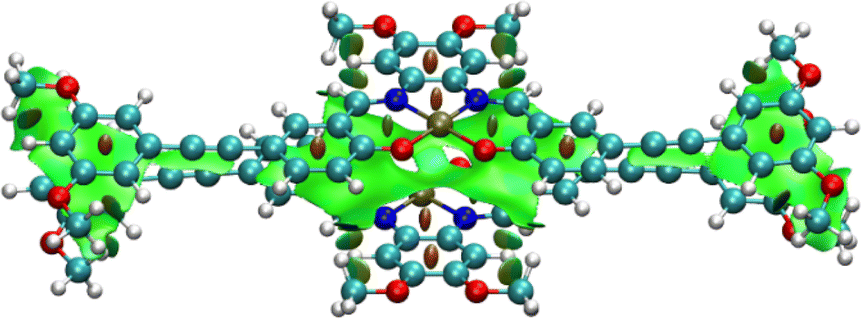
Upon geometry optimization of the ground state dimer of 7, an interplanar distance of less than 3.5 Å is found, implying that π–π interactions exist within the dimer. The optimized structure of the dimer adopts a head-to-tail stacking configuration, with a Pt⋯Pt internuclear distance of 3.501 Å. The interaction between these was further investigated by plotting non-covalent interactions34 (NCI plot) between the two monomers in the dimeric form (Fig. 4). The NCI plot depicts non-covalent interactions as an isosurface plot, using blue for strong attractive interactions, green for weak van der Waals interactions and red for strong repulsive interactions. By symmetry of the head-to-tail dimer, the space between the Pt(II) centers is located in the center of the calculated isosurface. Here, instead of showing one large discontinuity, the isosurface continues in a belt between the Pt(II) centers, dividing the discontinuity into two smaller holes. This demonstrates the presence of Pt⋯Pt interactions in the dimer. Furthermore, this region is colored with slightly more blue than the surrounding green, showing that the Pt⋯Pt interaction is more strongly attractive than the dispersion forces acting on the rest of the complex.
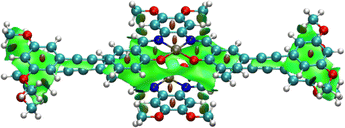 |
| | Fig. 4 Isosurface plot of non-covalent interactions calculated in the dimer of 7, with a Pt⋯Pt internuclear distance of 3.501 Å. | |
The UV-vis absorption bands of 7 have been computed based on the singlet–singlet transitions predicted by TDDFT/SMD(DMSO). These are shown in Fig. S21† and fit well with the experimental spectra (Fig. 2a). The important frontier molecular orbitals involved in transitions are depicted in Fig. S22.† The band near 330 nm (computationally predicted at 334 nm) is attributed to the π → π* intraligand (IL) transition of the Schiff base ligands, while the broader signal at approximately 490 nm (computationally: 479 nm, 503 nm) is assigned to a MLCT transition, namely 1[dπ(Pt) → π*(L)] mixed with IL transitions from the phenoxide lone pair to the π* orbital of the imine. Geometry optimization of the ground state S0 and T1 led to a calculated triplet emission energy with a wavelength of 629 nm,35 which is in good agreement with the experimental emission band maxima of 1–6 at 650 nm.
Geometry optimization calculations have been further performed on the dimeric form of 7 (calculated in H2O solution), followed by TDDFT calculations for the first twenty singlet–singlet transitions. These are summarized in Table S8.† The formation of the dimer introduces an additional absorption band (525 nm) in the longer-wavelength region, which indicates the presence of new electronic transitions in the aggregate form.
Self-assembly of the water-soluble tetradentate platinum(II) Schiff base complexes
Unlike complexes 2–5, only complex 6 is soluble in water to give an orange solution due to the presence of six hydrophilic triethylene glycol chains on the Schiff base ligand. 1H NMR spectroscopic measurement is used to monitor the potential self-assembly behavior of 6, in which the proton NMR signals become broad and upfield-shifted and finally become featureless with increasing D2O content in DMSO-d6 at 298 K, suggesting the formation of intermolecular association via π–π stacking interactions in water (Fig. S23†). Subsequently, the UV-vis absorption of 6 in water was found to not obey Beer's law in the concentration range from 10−6 to 10−3 M (Fig. 5a), with a red-shifted structureless emission band in the emission spectrum of 6 upon increasing the concentration from 1 μM to 100 μM, which is strongly supportive of the formation of Pt⋯Pt and/or π–π stacking interactions (Fig. 5b–d). Furthermore, dynamic light scattering (DLS) studies reveal the formation of aggregates with a hydrodynamic diameter of about 47.6 nm (Table S9†).
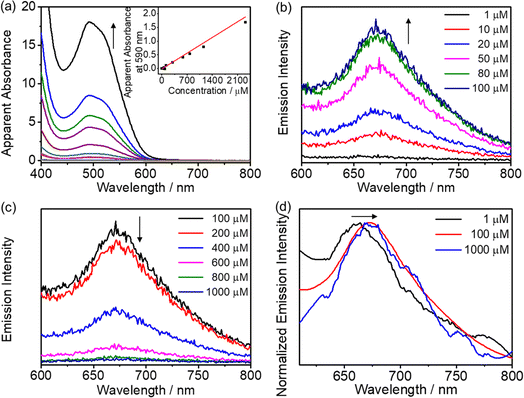 |
| | Fig. 5 (a) Electronic absorption spectra of 6 in aqueous solutions at various concentrations (from 5.73 to 2319 μM). Inset: the plot of absorbance at 590 nm against concentration. Emission spectra of 6 in water at various concentrations from (b) 1 to 100 μM and (c) 100 to 1000 μM. (d) Normalized emission spectra of 6 in water at concentrations of 1 μM, 100 μM and 1000 μM. | |
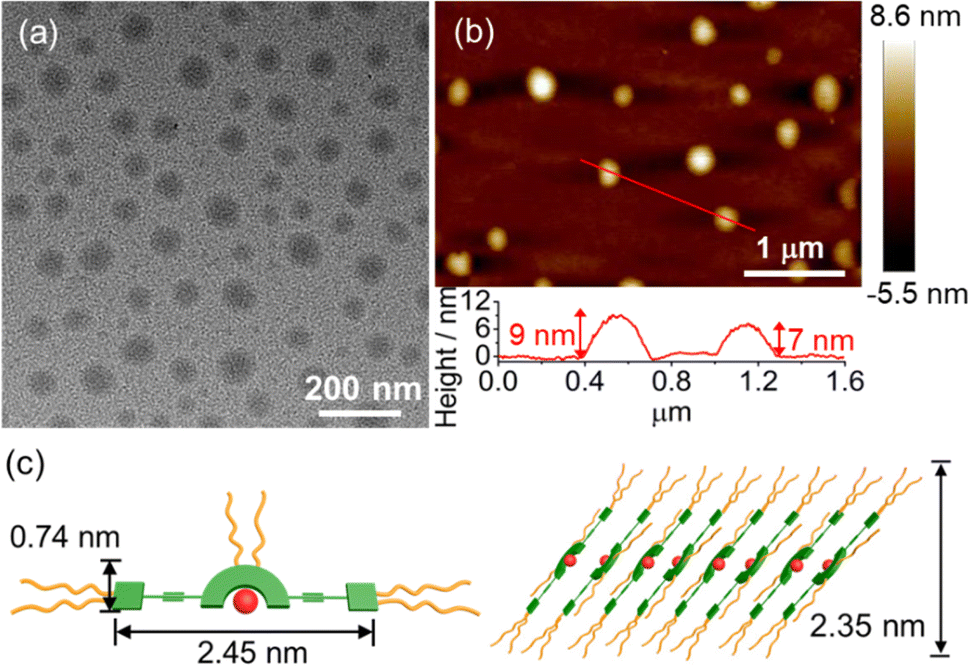
TEM and AFM have been employed to confirm the formation of spherical structures with sizes of about 50 nm and heights of 7–9 nm at a concentration of 2.0 × 10−4 M, which is consistent with the DLS data (Fig. 6). To obtain detailed molecular packing information of the spherical structures, the X-ray diffraction (XRD) pattern of a thin film prepared from 6 in water was obtained and found to show diffraction signals of d-spacing of 0.31 nm, indicating the presence of the Pt⋯Pt and/or π–π stacking interactions between the adjacent molecules in the self-assembly (Fig. S24†). More importantly, d-spacings of 2.35 and 1.17 nm with a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 were found to arise from the first (100) and secondary (200) diffraction planes, respectively, indicating a lamellar packing of the spherical assemblies. According to the crystal structure of 1, the length and width of the rigid π-conjugated part of 6 are estimated to be 2.45 nm and 0.74 nm, respectively. Together with the interdigitation of the flexible TEG chains in water, the layer with a thickness of 2.35 nm is likely to be attributed to the inclined arrangement of the molecules (Fig. 6c). The layers are stacked in a head-to-tail anti-parallel arrangement with the involvement of Pt⋯Pt and/or π–π stacking interactions, which has been further confirmed by 2D NMR studies (vide infra). These results indicate that this class of tetradentate platinum(II) Schiff base complexes obtained through rational introduction of water-solubilizing units might facilitate the formation of highly ordered assembled structures by non-covalent Pt⋯Pt and/or π–π stacking interactions in water.
:2 were found to arise from the first (100) and secondary (200) diffraction planes, respectively, indicating a lamellar packing of the spherical assemblies. According to the crystal structure of 1, the length and width of the rigid π-conjugated part of 6 are estimated to be 2.45 nm and 0.74 nm, respectively. Together with the interdigitation of the flexible TEG chains in water, the layer with a thickness of 2.35 nm is likely to be attributed to the inclined arrangement of the molecules (Fig. 6c). The layers are stacked in a head-to-tail anti-parallel arrangement with the involvement of Pt⋯Pt and/or π–π stacking interactions, which has been further confirmed by 2D NMR studies (vide infra). These results indicate that this class of tetradentate platinum(II) Schiff base complexes obtained through rational introduction of water-solubilizing units might facilitate the formation of highly ordered assembled structures by non-covalent Pt⋯Pt and/or π–π stacking interactions in water.
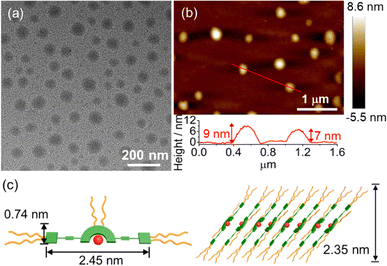 |
| | Fig. 6 (a) TEM and (b) AFM images prepared from a solution of 6 in water ([Pt] = 2.0 × 10−4 M). (c) Schematic diagram of the proposed lamellar structure of 6 in water. | |
Self-assembly of amphiphilic tetradentate platinum(II) Schiff base complexes in mixed solvent compositions
The self-assembly behavior of complexes 2–6 in DMSO–water mixtures has been investigated by 1H NMR spectroscopic studies, suggesting the presence of possible intermolecular association by π–π stacking interactions (Fig. S25–S30†). Further evidence of self-assembly properties comes from the solvent-dependent UV-vis absorption and emission spectroscopy in the concentration regime of 10−5 M. Upon increasing the concentration of 2–6 in DMSO from 10−6 to 10−3 M, the UV-vis absorption spectra are observed to obey Beer's law, which is in line with insignificant scattering signals revealed in DLS studies (Fig. S31–S35 and Table S9†). These observations indicate that 2–6 show no aggregation behaviors and exist in a monodisperse state in the DMSO solutions.
In DMSO–water mixtures, upon increasing the water content to 90% (v/v), the solution remains clear and no precipitation occurs for all the complexes in the same concentration regime of 10−5 M, which prompts subsequent investigation of their solution-state aggregation behaviors. Complexes 2–6 display intriguing and similar UV-vis spectral changes upon addition of water (Fig. 7a, b and S36–S39†). The UV-vis absorption spectra show an increase in the low-energy absorption tail at ∼600 nm upon increasing the water content to a critical point (i.e., 20% water in DMSO, v/v), accompanied by a color change from orange to red (Fig. 7a). These low-energy absorption bands could be assigned to the mixing of ligand-centered 1[lone pair(phenoxide) → π*(imine)] and MMLCT 1[dσ*(Pt) → π*(L)] transitions arising from the formation of the Pt⋯Pt and/or π–π stacking interactions upon reduced solvation. A further increase in the water content from this critical point to 90% (v/v) results in a drop in the low-energy absorption tail, which is not due to the deaggregation of the complexes but a change in the extent of Pt⋯Pt and/or π–π interactions (Fig. 7b). This is supported by the presence of a distinct red-shifted absorption band at ca. 600 nm as observed in the overlaid spectra of 4 in DMSO and 90% water–DMSO (v/v) solutions (Fig. 7c and S40†). Furthermore, the corresponding emission spectra are also observed to show remarkable changes attributed to Pt⋯Pt and/or π–π stacking interactions (Fig. 7d–f and S41–S44†). By taking complex 4 as a representative example, the emission spectra are found to show a red shift from 654 nm to 664 nm with the water content increasing from 0 to 20% (v/v), probably attributed to the increase in the extent of Pt⋯Pt and/or π–π stacking interaction in the solution, while further increasing water content would result in a decrease in emission intensity due to aggregation of these hydrophobic molecules which leads to an increase in the local concentration of the complexes and thus self-quenching, along with quenching due to solvent assisted relaxation. In addition, according to the spectroscopic and computational studies (vide supra), the lowest-energy absorption and emission bands of the complexes are mainly dominated by charge transfer characters. As a result, the red-shift of the low-energy band is unlikely to be a vibronic-structured band, but rather an emergence of a new low-energy structureless band due to the self-assembly of complexes through intermolecular interactions. Further investigations through variable-temperature emission spectral analysis in 20% water–DMSO (v/v) solution at the same concentration were made to verify whether the emission spectral changes are mainly a result of solvatochromism or self-assembly behaviors (Fig. 7g). A reversible blue-shift of the emission bands is found upon increasing temperature with the same solvent composition, which is in line with the spectral changes with increasing water content, indicating that the emission spectral changes are predominantly due to the formation of Pt⋯Pt and/or π–π stacking interactions resulting from supramolecular assembly of the complexes. The monodisperse nature of the complex in 20% water–DMSO (v/v) solution at high temperature is further confirmed by variable-temperature 1H NMR spectroscopy and DLS studies (Fig. S30 and Table S9†). In the meantime, the ground-state supramolecular association of the complex with Pt⋯Pt and/or π–π interactions has been demonstrated by the photoluminescence excitation (PLE) spectra, in which a red-shift and a significant increase in the intensity of the band at 560 nm in 20% water–DMSO (v/v) and 90% water–DMSO (v/v) solvent compositions compared to the DMSO solution are observed (Fig. 7h and S45†). In addition, the lack of bimolecular growth in the time-resolved photoluminescence traces (Fig. 7i) further establishes ground-state aggregate formation rather than excimer formation.36 Notably, the decay traces in the self-assembly studies have been acquired under ambient conditions.
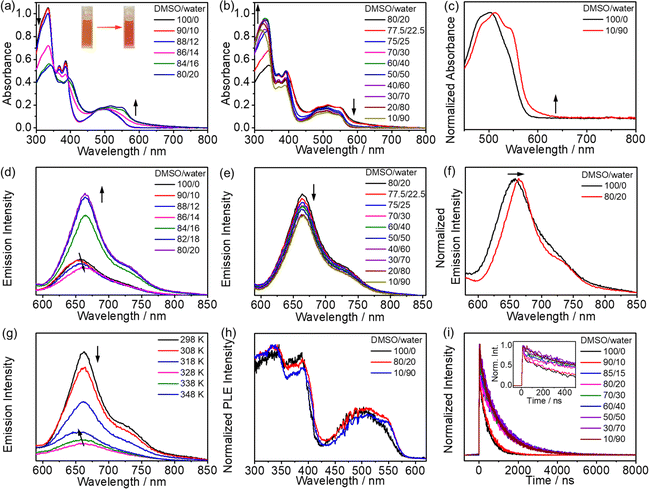 |
| | Fig. 7 (a) UV-vis absorption spectral changes of 4 in DMSO solutions upon increasing water content from 0 to 20% (v/v). (Inset) A photograph showing the solutions of 4 in DMSO solutions (left) and 20% water–DMSO (v/v) solutions (right). (b) UV-vis absorption spectral changes of 4 in DMSO solutions upon increasing water content from 20 to 90% (v/v). (c) Normalized UV-vis absorption spectra of 4 in DMSO solution and 90% water–DMSO (v/v) solution in the concentration regime of 10−5 M. Emission spectral changes of 4 in DMSO solutions upon increasing water content from (d) 0 to 20% and (e) 20 to 90% (v/v) in the concentration regime of 10−5 M. (f) Normalized emission spectra of 4 in DMSO and 20% water–DMSO (v/v) solutions. (g) Variable-temperature emission spectral changes of 4 in 20% water–DMSO (v/v) solutions in the concentration regime of 10−5 M. (h) Normalized photoluminescence excitation (PLE) spectra of 4 in DMSO, 25% water–DMSO and 90% water–DMSO (v/v) solutions. (i) Time-resolved photoluminescence decay traces (detected at the emission band maxima) of 4 in DMSO solutions upon increasing water content from 0 to 90% (v/v). (Inset) The expanded decay traces in the range of 0–500 ns. | |
In aerated solutions, the complex in the lowest-energy excited state is quenched by triplet oxygen. The fast decay that appeared as a spike at early times in aerated pure DMSO and water–DMSO mixtures with low water content is an artefact due to scattered light from the excitation source that became more apparent with the much weaker phosphorescence under aerated conditions. The spike becomes less prominent in high water content, which could be attributed to the aggregation of the complexes by Pt⋯Pt and/or π–π stacking interactions that leads to stronger phosphorescence arising from a larger amount of aggregate formation and rigidification, as well as the lower sensitivity of the aggregates to oxygen quenching. Unlike complexes 2–5, the emission spectra of 6 exhibited a decrease in intensity with increasing water content from 0 to 90% (v/v) in DMSO solution because of a less compact assembly. Therefore, complex 6 tends to adopt a different stacking arrangement in a high polarity solvent environment because of its higher solubility in polar solvents. Notably, by comparing the spectral changes of 2–5, the more hydrophobic the complexes are, the lower the water content is required to reach the maximum growth of the absorption bands and the largest extent of red shift of the emission band, which demonstrates that the smaller overall hydrophobicity of complexes requires a higher water content to induce the formation of Pt⋯Pt and/or π–π stacking interactions.
To address Pt⋯Pt and/or π–π stacking interactions, XRD measurements were performed and the corresponding diffraction signals at a d-spacing of about 3.70 Å were observed, suggesting the existence of weak intermolecular Pt⋯Pt and/or π–π stacking in the assemblies (Fig. S46–S50†). Subsequently, 2D 1H–1H NOESY NMR spectroscopy was performed with the addition of 5% D2O in DMSO-d6 (v/v). Strong NOE signals between Ha and Hb, Ha and Hc, Hc and Hb, and He and Hd are observed, which suggest a close proximity of adjacent molecules through the π–π stacking interactions that bring the protons into vicinity (Fig. 8a and S51–S53†) and also indicated a possible head-to-tail stacking arrangement of adjacent molecules (Fig. 8b and S51–S53†).37 Based on the above results, solvent-induced self-assembly of amphiphilic platinum(II) Schiff base complexes by non-covalent Pt⋯Pt and/or π–π stacking interactions in the solution state is demonstrated and the extent of Pt⋯Pt and/or π–π stacking interactions is found to be induced by the solvent compositions and the molecular hydrophobicity, leading to drastic spectroscopic changes and diverse trends.
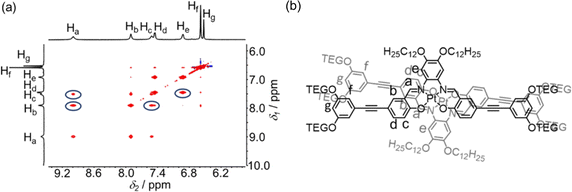 |
| | Fig. 8 (a) Partial 1H–1H NOESY NMR spectrum of complex 4 in DMSO-d6 upon the addition of 5% D2O (v/v) at 298 K. (b) Proposed stacking mode of complex 4 in DMSO upon an increase in H2O content. | |
Studies on the effect of hydrophobicity on self-assembly mechanisms
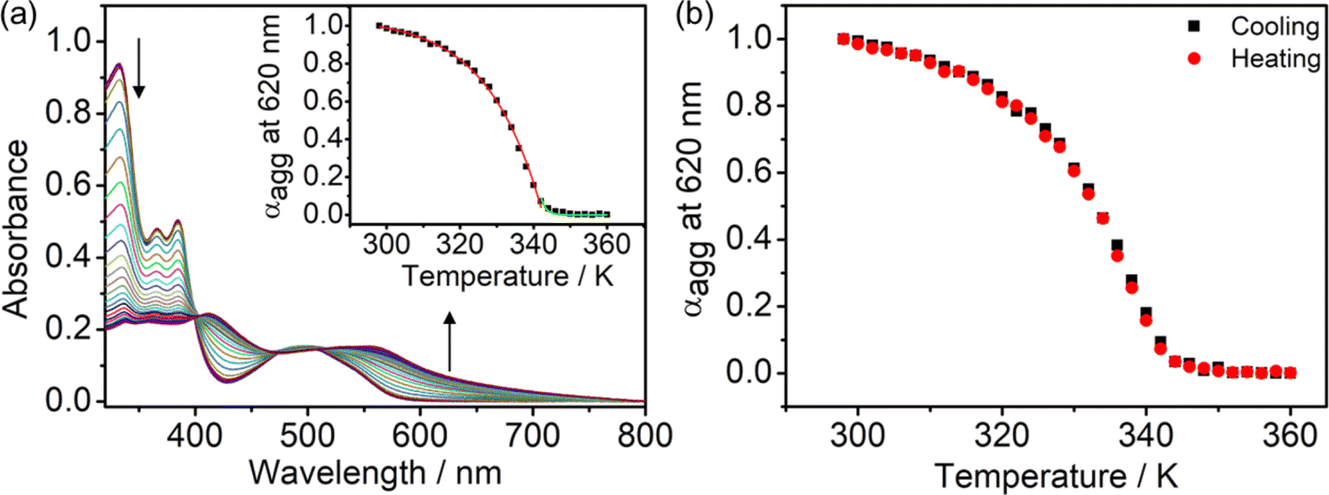
The self-assembly mechanisms of complexes 2–6 have been investigated by variable-temperature UV-vis spectroscopy in DMSO–water mixtures in the same concentration regime of 10−5 M to further study their aggregation processes (Fig. 9a and S54–S57†). By studying the representative complex 4 in a 20% water–DMSO (v/v) mixture with cooling of the solution from 360 to 298 K at a rate of 0.5 K min−1, growth of the absorption band at ∼600 nm is observed, attributed to the formation of Pt⋯Pt and/or π–π stacking interactions. Interestingly, a plot of the degree of aggregation at 620 nm against the temperature of the curve fitting is found to be nonsigmoidal, suggesting the cooperative growth of 4 in the self-assembly process (Fig. 9a, inset). Moreover, no significant hysteresis has been observed in the heating and the cooling processes, suggesting that the formation and the disruption of the intermolecular Pt⋯Pt and/or π–π stacking interactions would occur at the same temperature (Fig. 9b). Complexes 2–6 are all found to undergo a cooperative growth mechanism, and the thermodynamic parameters in the self-assembly process have been obtained according to the temperature-dependent nucleation–elongation model (Fig. S54–S57† and Table 1).37 With 4 as a representative example for illustration purposes, the enthalpy change due to non-covalent interactions during the elongation process (ΔHe) is −66.1 kJ mol−1 and the elongation temperature (Te) is 343 K. The dimensionless equilibrium constant (Ka) of the nucleation process at Te is found to be 1.94 × 10−4, indicating an unfavorable nucleation process with such a relatively small equilibrium constant. Besides, the number-averaged degrees of polymerization at Te and 298 K are found to be 17 and ∼1500, respectively, suggesting that 4 forms a nucleus size of about 17 molecules at the elongation temperature, and elongated to form larger aggregates at a size of around 1500 molecules upon lowering the temperature to 298 K. It is worth noting that the enthalpy change and number-averaged degree of polymerization at room temperature during the aggregation are found to be influenced by the hydrophobicity of the complex, which provided further insights into the self-assembly of this class of complexes. For example, in complexes 2–4, the enthalpy changes of SBPt-C12-TEG (4) (−66.1 kJ mol−1) are found to be more negative than that of SBPt-C6-TEG (2) (−54.6 kJ mol−1) due to a stronger hydrophobic interaction in addition to the Pt⋯Pt and/or π–π stacking interactions upon aggregation of 4, enhancing the stability of the aggregates. The number-averaged degree of polymerization at 298 K of 2–4 (1376 to 1528) was also found to increase with increasing hydrophobicity. While SBPt-C16-TEG (5) possesses the longest alkoxy chain among the complexes, the number-averaged degree of polymerization at room temperature (511) and the enthalpy change (−45.0 kJ mol−1) are not in line with the trend mentioned above. This observation is commonly found in aggregated species with long hydrophobic chains.38 From the previous report,39 the cooperative growth mechanism could be achieved through a subtle balance between multiple non-covalent interactions such as metal–metal, hydrophobic and π–π stacking interactions. Therefore, the long alkoxy chains of 5 might disrupt the delicate balance in the self-assembly, leading to a relatively less favorable aggregation behavior in the DMSO–water mixture due to the predominant contribution from the hydrophobic interactions.39 It is worth noting that complex 5 could form less well-defined fibers compared to the nanospheres formed from other complexes (Fig. 10 and S58–S63†). This might be related to the effect of imbalance of multiple non-covalent interactions and thus leads to discrepancy in the morphologies. With the above-mentioned results on the cooperative mechanisms by temperature-dependent UV-vis spectroscopic analysis, the gradual increase in hydrophobicity would assist in the formation of stable aggregates. However, further enhancement in the hydrophobicity of the complexes may lead to an imbalance of non-covalent interactions.
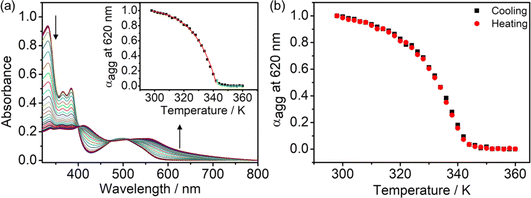 |
| | Fig. 9 (a) UV-vis absorption spectral traces on cooling a solution of 4 in 20% water in DMSO (v/v) at a cooling rate of 0.5 K min−1. (Inset) The degree of aggregation at 620 nm as a function of temperature with the curve fitted in the elongation (red line) and nucleation (green line) regime based on the nucleation–elongation model. (b) Plots of degree of aggregation monitored at 620 nm against temperature for the heating and cooling of 4 in the temperature range from 360 to 298 K. | |
Table 1 Thermodynamic parameters for the self-assembly for 2–6
|
|
ϕ
SAT
|
ΔHe/kJ mol−1 |
T
e/K |
K
a
|
〈Nn(Te)〉 |
〈Nn(298)〉 |
|
2
|
1.180 |
−54.6 |
331 |
1.63 × 10−5 |
39 |
1376 |
|
3
|
1.200 |
−56.9 |
324 |
9.29 × 10−6 |
48 |
1491 |
|
4
|
1.047 |
−66.1 |
343 |
1.94 × 10−4 |
17 |
1528 |
|
5
|
1.153 |
−45.0 |
339 |
1.79 × 10−4 |
18 |
511 |
|
6
|
1.095 |
−61.6 |
337 |
1.06 × 10−4 |
21 |
1022 |
 |
| | Fig. 10 (a) TEM image and (b) AFM image prepared from the solution of SBPt-C12-TEG (4) (2.0 × 10−5 M) in a 20% water–DMSO (v/v) mixture. (c) TEM image and (d) SEM image prepared from the solution of SBPt-C12-TEG (4) (2.0 × 10−5 M) in a 90% water–DMSO (v/v) mixture. (e) TEM image and (f) SEM image prepared from the solution of SBPt-C16-TEG (5) (2.0 × 10−5 M) in a 14% water–DMSO (v/v) mixture. (g) TEM image and (h) SEM image prepared from the solution of SBPt-C16-TEG (5) (2.0 × 10−5 M) in the 90% water–DMSO (v/v) mixture. | |
Molecular hydrophobicity and solvent-induced diverse morphological transformations
With the appealing spectral changes of 2–6, TEM, SEM and AFM were employed to image the morphologies formed under different solvent compositions and the complexes have been found to undergo morphological transformations (Fig. 10 and S58–S63†). For example, complex 4 is found to show a morphological transformation from spherical aggregates with diameters of about 200 nm into nanorods with lengths of about 800 nm with the addition of water from 20 to 90% in DMSO (v/v) (Fig. 10a–d), which is consistent with the DLS results summarized in Table S9.† The aggregation of the spherical structures is ascribed to the formation of Pt⋯Pt and/or π–π stacking interactions in the 20% water–DMSO (v/v) solution, in which maximum growth of the absorption and red shift of the emission band have been reached. Further increasing the water content to 90% (v/v) might have a remarkable effect on its molecular packing under higher polar conditions. It is believed that both hydrophobic and Pt⋯Pt and/or π–π stacking interactions would be enhanced upon increasing water content but eventually the driving force is dominated by the directional Pt⋯Pt interaction, leading to the formation of nanorods. In addition to the solvent compositions, the length of the alkoxy chain also has an effect on the morphologies. Compared to complexes 2–4, the formation of less well-ordered nanofibers of 5 at 14% water–DMSO (v/v) solution is observed (Fig. 10e and f). This distinct morphology might be ascribed to the imbalance of non-covalent interactions that leads to the predominant construction of the less directional hydrophobic interaction for formation of fibrous structures, which is further proved by the least negative enthalpy change of 5. Thus, with the participation of various non-covalent interactions, the change and balance of multiple non-covalent interactions play a pivotal role in self-assembly such that a variety of assembled morphologies can be formed. The nucleation–elongation assembly process of complexes 2–6 as well as corresponding morphological transformations is depicted in Scheme 2 upon addition of water in DMSO solution.
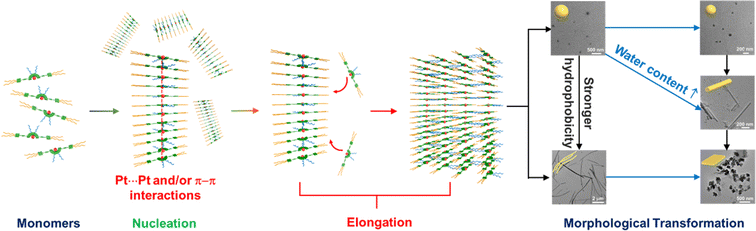 |
| | Scheme 2 Schematic drawing of the proposed cooperative self-assembly process of Schiff base-containing platinum(II) complexes. | |
Conclusion
In conclusion, a new class of amphiphilic tetradentate Schiff base-containing platinum(II) complexes with triethylene glycol units and different lengths of alkoxy chains was designed and synthesized. In water, complex 6 was demonstrated to aggregate and assist the formation of highly ordered spherical structures by non-covalent Pt⋯Pt and/or π–π stacking interactions. In DMSO–water mixtures, the extent of Pt⋯Pt and/or π–π interactions could be regulated by changing the solvent compositions and the hydrophobicity of the complexes, which is accompanied by intriguing spectroscopic changes and morphological transformations in the self-assembly process. In addition, a cooperative growth mechanism, manifested by the balance between various non-covalent interactions, and the correlation between the length of the alkoxy chains, assembly morphologies and self-assembly mechanism were demonstrated. The present work represents a rare example to demonstrate directed cooperative assembly of amphiphilic Schiff base-containing platinum(II) complexes in solution with an in-depth mechanistic investigation, which may provide guiding principles in the rational molecular design and the construction of supramolecular structures with desirable properties using platinum(II) Schiff base building blocks.
Data availability
The datasets supporting this article have been uploaded as part of the ESI.†
Author contributions
V. W.-W. Y. initiated and designed the research; H. Z. conducted the synthesis, characterization, spectroscopic and electron microscopic measurements; J. L., Z. C. and M.-Y. L. performed computational studies; V. W.-W. Y., L. W., H. Z., M. H.-Y. C. and E. K.-H. W. analyzed the data; V. W.-W. Y., L. W., H. Z., M. H.-Y. C., E. K.-H. W., J. L. and Z. C. prepared the manuscript; V. W.-W. Y. and L. W. supervised the work.
Conflicts of interest
The authors declare no competing financial interest.
Acknowledgements
V. W.-W. Y. acknowledges the support from the State Key Laboratory of Supramolecular Structure and Materials of Jilin University and the University of Hong Kong. This work was supported by the Collaborative Research Fund (CRF) (C7075-21G) and the General Research Fund (GRF) (HKU 17303421) of the Research Grants Council (RGC) of the Hong Kong Special Administrative Region, P. R. China, and the CAS-Croucher Funding Scheme for Joint Laboratories on Molecular Functional Materials for Electronics, Switching and Sensing.
References
-
(a) V. M. Miskowski and V. H. Houlding, Inorg. Chem., 1989, 28, 1529 CrossRef CAS;
(b) J. Biedermann, G. Gliemann, U. Klement, K.-J. Range and M. Zabel, Inorg. Chem., 1990, 29, 1884 CrossRef CAS;
(c) W. B. Connick, D. Geiger and R. Eisenberg, Inorg. Chem., 1999, 38, 3264 CrossRef CAS PubMed.
-
(a) V. W.-W. Yam, R. P.-L. Tang, K. M.-C. Wong and K.-K. Cheung, Organometallics, 2001, 20, 4476 CrossRef CAS;
(b) T. J. Wadas, Q.-M. Wang, Y. Kim, C. Flaschenreim, T. N. Blanton and R. Eisenberg, J. Am. Chem. Soc., 2004, 126, 16841 CrossRef CAS PubMed;
(c) I. Eryazici, C. N. Moorefield and G. R. Newkome, Chem. Rev., 2008, 108, 1834 CrossRef CAS PubMed;
(d) K. M.-C. Wong, N. Zhu and V. W.-W. Yam, Chem. Commun., 2006, 3441 RSC.
-
(a) V. W.-W. Yam, K. M.-C. Wong and N. Zhu, J. Am. Chem. Soc., 2002, 124, 6506 CrossRef CAS PubMed;
(b) Y. Chen, K. Li, W. Lu, S. S.-Y. Chui, C.-W. Ma and C.-M. Che, Angew. Chem., Int. Ed., 2009, 48, 9909 CrossRef CAS PubMed;
(c) W. Lu, Y. Chen, V. A. L. Roy, S. S.-Y. Chui and C.-M. Che, Angew. Chem., Int. Ed., 2009, 48, 7621 CrossRef CAS PubMed.
-
(a) V. W.-W. Yam, K. H.-Y. Chan, K. M.-C. Wong and B. W.-K. Chu, Angew. Chem., Int. Ed., 2006, 45, 6169 CrossRef CAS PubMed;
(b) V. W.-W. Yam, Y. Hu, K. H.-Y. Chan and C. Y.-S. Chung, Chem. Commun., 2009, 6216 RSC;
(c) K. H.-Y. Chan, H.-S. Chow, K. M.-C. Wong, M. C.-L. Yeung and V. W.-W. Yam, Chem. Sci., 2010, 1, 477 RSC.
-
(a) C. Yu, K. M.-C. Wong, K. H.-Y. Chan and V. W.-W. Yam, Angew. Chem., Int. Ed., 2005, 44, 791 CrossRef CAS PubMed;
(b) C. Yu, K. H.-Y. Chan, K. M.-C. Wong and V. W.-W. Yam, Proc. Natl. Acad. Sci. U.S.A., 2006, 103, 19652 CrossRef CAS PubMed;
(c) C. Yu, K. H.-Y. Chan, K. M.-C. Wong and V. W.-W. Yam, Chem.–Eur. J., 2008, 14, 4577 CrossRef CAS PubMed;
(d) C. Yu, K. H.-Y. Chan, K. M.-C. Wong and V. W.-W. Yam, Chem. Commun., 2009, 3756 RSC;
(e) M. C.-L. Yeung, K. M.-C. Wong, Y. K. T. Tsang and V. W.-W. Yam, Chem. Commun., 2010, 46, 7709 RSC;
(f) C. Y.-S. Chung, K. H.-Y. Chan and V. W.-W. Yam, Chem. Commun., 2011, 47, 2000 RSC.
-
(a) K. W. Jennette, S. J. Lippard, G. A. Vassiliades and W. R. Bauer, Proc. Natl. Acad. Sci. U. S. A., 1974, 71, 3839 CrossRef CAS PubMed;
(b) K. W. Jennette, J. T. Gil, J. A. Sadownick and S. J. Lippard, J. Am. Chem. Soc., 1976, 98, 6159 CrossRef CAS;
(c) V. M. Miskowski and V. H. Houlding, Inorg. Chem., 1991, 30, 4446 CrossRef CAS;
(d) H.-K. Yip, L.-K. Cheng, K.-K. Cheung and C.-M. Che, J. Chem. Soc., Dalton Trans., 1993, 2933 RSC;
(e) R. H. Herbe, M. Croft, M. J. Coyer, B. Bilash and A. Sahiner, Inorg. Chem., 1994, 33, 2422 CrossRef;
(f) J. A. Bailey, M. G. Hill, R. E. Marsh, V. M. Miskowski, W. P. Schaefer and H. B. Gray, Inorg. Chem., 1995, 34, 4591 CrossRef CAS;
(g) W. B. Connick, R. E. Marsh, W. P. Schaefer and H. B. Gray, Inorg. Chem., 1997, 36, 913 CrossRef CAS;
(h) G. Arena, G. Calogero, S. Campagna, L. M. Scolaro, V. Ricevuto and R. Romeo, Inorg. Chem., 1998, 37, 2763 CrossRef CAS PubMed;
(i) R. Büchner, C. T. Cunningham, J. S. Field, R. J. Haines, D. R. McMillin and G. C. Summerton, J. Chem. Soc., Dalton Trans., 1999, 711 RSC.
-
(a) V. W.-W. Yam, V. K.-M. Au and S. Y.-L. Leung, Chem. Rev., 2015, 115, 7589 CrossRef CAS PubMed;
(b) V. W.-W. Yam, A. K.-W. Chan and E. Y.-H. Hong, Nat. Rev. Chem, 2020, 4, 528 CrossRef CAS.
- S. Y.-L. Leung and V. W.-W. Yam, Chem. Sci., 2013, 4, 4228 RSC.
- J. Zhang and C.-Y. Su, Coord. Chem. Rev., 2013, 257, 1373 CrossRef CAS.
- F. Camerel, R. Ziessel, B. Donnio, C. Bourgogne, D. Guillon, M. Schmutz, C. Iacovita and J.-P. Bucher, Angew. Chem., Int. Ed., 2007, 46, 2659 CrossRef CAS PubMed.
- A. Y.-Y. Tam, K. M.-C. Wong, G. Wang and V. W.-W. Yam, Chem. Commun., 2007, 2028 RSC.
- V. W.-W. Yam, K. H.-Y. Chan, K. M.-C. Wong and N. Zhu, Chem.–Eur. J., 2005, 11, 4535 CrossRef PubMed.
- C. Po, A. Y.-Y. Tam, K. M.-C. Wong and V. W.-W. Yam, J. Am. Chem. Soc., 2011, 133, 12136 CrossRef CAS PubMed.
-
(a) A. Y.-Y. Tam, W. H. Lam, K. M.-C. Wong, N. Zhu and V. W.-W. Yam, Chem.–Eur. J., 2008, 14, 4562 CrossRef CAS PubMed;
(b) A. Y.-Y. Tam, K. M.-C. Wong and V. W.-W. Yam, J. Am. Chem. Soc., 2009, 131, 6253 CrossRef CAS PubMed;
(c) H.-L. Au-Yeung, A. Y.-Y. Tam, S. Y.-L. Leung and V. W.-W. Yam, Chem. Sci., 2017, 8, 2267 RSC;
(d) H. L.-K. Fu, C. Po, S. Y.-L. Leung and V. W.-W. Yam, ACS Appl. Mater. Interfaces, 2017, 9, 2786 CrossRef CAS PubMed;
(e) V. C.-H. Wong, C. Po, S. Y.-L. Leung, A. K.-W. Chan, S. Yang, B. Zhu, X. Cui and V. W.-W. Yam, J. Am. Chem. Soc., 2018, 140, 657 CrossRef CAS PubMed.
-
(a) S. Y.-L. Leung, A. Y.-Y. Tam, C.-H. Tao, H. S. Chow and V. W.-W. Yam, J. Am. Chem. Soc., 2012, 134, 1047 CrossRef CAS PubMed;
(b) S. Y.-L. Leung, W. H. Lam and V. W.-W. Yam, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 7986 CrossRef CAS PubMed;
(c) M. H.-Y. Chan, M. Ng, S. Y.-L. Leung, W. H. Lam and V. W.-W. Yam, J. Am. Chem. Soc., 2017, 139, 8639 CrossRef CAS PubMed.
-
(a) K. M.-C. Wong, W.-S. Tang, X.-X. Lu, N. Zhu and V. W.-W. Yam, Inorg. Chem., 2005, 44, 1492 CrossRef CAS PubMed;
(b) C. Y.-S. Chung and V. W.-W. Yam, Chem.–Eur. J., 2013, 19, 13182 CrossRef CAS PubMed;
(c) M. H.-Y. Chan, S. Y.-L. Leung and V. W.-W. Yam, J. Am. Chem. Soc., 2019, 141, 12312 CrossRef CAS PubMed;
(d) Z. Chen, M. H.-Y. Chan and V. W.-W. Yam, J. Am. Chem. Soc., 2020, 142, 16471 CrossRef CAS PubMed.
- Z. Zeng, J. Wu, Q. Chen, Y. Shi, J. Zheng and C. Xu, Dyes Pigm., 2019, 170, 107649 CrossRef CAS.
- S. Banerjee, M. S. Capper, G. J. Clarkson, H. Huang and P. J. Sadler, Polyhedron, 2019, 172, 157 CrossRef CAS.
- C.-M. Che, S.-C. Chan, H.-F. Xiang, M. C. W. Chan, Y. Liu and Y. Wang, Chem. Commun., 2004, 1484 RSC.
- H.-F. Xiang, S.-C. Chan, K. K.-Y. Wu, C.-M. Che and P. T. Lai, Chem. Commun., 2005, 1408 RSC.
- C.-M. Che, C.-C. Kwok, S.-W. Lai, A. F. Rausch, W. J. Finkenzeller, N. Zhu and H. Yersin, Chem.–Eur. J., 2010, 16, 233 CrossRef CAS PubMed.
- L. Zhou, C.-L. Kwong, C.-C. Kwok, G. Cheng, H. Zhang and C.-M. Che, Chem.–Asian J., 2014, 9, 2984 CrossRef CAS PubMed.
- J. Zhang, L. Wang, A. Zhong, G. Huang, F. Wu, D. Li, M. Teng, J. Wang and D. Han, Dyes Pigm., 2019, 162, 590 CrossRef CAS.
- N. Komiya, T. Muraoka, M. Iida, M. Miyanaga, K. Takahashi and T. Naota, J. Am. Chem. Soc., 2011, 133, 16054 CrossRef CAS PubMed.
- D. Qiao, J.-Y. Wang, L.-Y. Zhang, F.-R. Dai and Z.-N. Chen, Dalton Trans., 2019, 48, 11045 RSC.
- J. K.-H. Hui, Z. Yu and M. J. MacLachlan, Angew. Chem., Int. Ed., 2007, 46, 7980 CrossRef CAS PubMed.
-
(a) I. P. Oliveri, G. Malandrino and S. D. Bella, Dalton Trans., 2014, 43, 10208 RSC;
(b) M. Kuil, I. M. Puijk, A. W. Kleij, D. M. Tooke, A. L. Spek and J. N. H. Reek, Chem.–Asian J., 2009, 4, 50 CrossRef CAS PubMed.
-
(a) P. D. Frischmann, S. Guieu, R. Tabeshi and M. J. MacLachlan, J. Am. Chem. Soc., 2010, 132, 7668 CrossRef CAS PubMed;
(b) K. Chen, M. Hussain, S. S. Razi, Y. Hou, E. A. Yildiz, J. Zhao, H. G. Yaglioglu and M. Di Donato, Inorg. Chem., 2020, 59, 14731 CrossRef CAS PubMed.
- X. Lü, W.-Y. Wong and W.-K. Wong, Eur. J. Inorg. Chem., 2008, 523 CrossRef.
- W.-L. Tong, L.-M. Lai and M. C. W. Chan, Dalton Trans., 2008, 1412 RSC.
- Deposition numbers 2257979 (for 1) contain the ESI† crystallographic data for this paper.
- R. Payal, M. K. Saroj, N. Sharma and R. C. Rastogi, J. Lumin., 2018, 198, 92 CrossRef CAS.
- S. M. Borisov, R. Saf, R. Fischer and I. Klimant, Inorg. Chem., 2013, 52, 1206 CrossRef CAS PubMed.
-
(a) J. Contreras-Garcia, E. R. Johnson, S. Keinan, R. Chaudret, J. P. Piquemal, D. N. Beratan and W. Yang, J. Chem. Theory Comput., 2011, 7, 625 CrossRef CAS PubMed;
(b) E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498 CrossRef CAS PubMed.
- J. M. Younker and K. D. Dobbs, J. Phys. Chem. C, 2013, 117, 25714 CrossRef CAS.
-
(a) B. Stevens and E. Hutton, Nature, 1960, 186, 1045 CrossRef CAS;
(b) J. B. Birks, D. J. Dyson and I. H. Munro, Proc. R. Soc. London, Ser. A, 1963, 275, 575 CrossRef CAS.
-
(a) T. V. Jones, M. M. Slutsky, R. Laos, T. F. A. de Greef and G. N. Tew, J. Am. Chem. Soc., 2005, 127, 17235 CrossRef CAS PubMed;
(b) C. A. Hunter and J. K. M. Sanders, J. Am. Chem. Soc., 1990, 112, 5525 CrossRef CAS.
- P. Jonkheijm, P. van der Schoot, A. P. H. J. Schenning and E. W. Meijer, Science, 2006, 313, 80 CrossRef CAS PubMed.
- M. H.-Y. Chan, S. Y.-L. Leung and V. W.-W. Yam, J. Am. Chem. Soc., 2018, 140, 7637 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b,
Jonathan
Lam
b,
Ziyong
Chen
b,
Jonathan
Lam
b,
Ziyong
Chen