
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn silico screening of LRRK2 WDR domain inhibitors using deep docking and free energy simulations†
Evgeny
Gutkin‡
 a,
Filipp
Gusev‡
ab,
Francesco
Gentile‡
de,
Fuqiang
Ban
c,
S. Benjamin
Koby
a,
Chamali
Narangoda
a,
Olexandr
Isayev
*ab,
Artem
Cherkasov
*c and
Maria G.
Kurnikova
*a
a,
Filipp
Gusev‡
ab,
Francesco
Gentile‡
de,
Fuqiang
Ban
c,
S. Benjamin
Koby
a,
Chamali
Narangoda
a,
Olexandr
Isayev
*ab,
Artem
Cherkasov
*c and
Maria G.
Kurnikova
*a
aDepartment of Chemistry, Mellon College of Science, Carnegie Mellon University, Pittsburgh, PA 15213, USA. E-mail: olexandr@olexandrisayev.com; kurnikova@cmu.edu
bComputational Biology Department, School of Computer Science, Carnegie Mellon University, Pittsburgh, PA 15213, USA
cVancouver Prostate Centre, The University of British Columbia, Vancouver, BC, Canada. E-mail: acherkasov@prostatecentre.com
dDepartment of Chemistry and Biomolecular Sciences, University of Ottawa, Ottawa, ON, Canada
eOttawa Institute of Systems Biology, Ottawa, ON, Canada
First published on 11th April 2024
Abstract
The Critical Assessment of Computational Hit-Finding Experiments (CACHE) Challenge series is focused on identifying small molecule inhibitors of protein targets using computational methods. Each challenge contains two phases, hit-finding and follow-up optimization, each of which is followed by experimental validation of the computational predictions. For the CACHE Challenge #1, the Leucine-Rich Repeat Kinase 2 (LRRK2) WD40 Repeat (WDR) domain was selected as the target for in silico hit-finding and optimization. Mutations in LRRK2 are the most common genetic cause of the familial form of Parkinson's disease. The LRRK2 WDR domain is an understudied drug target with no known molecular inhibitors. Herein we detail the first phase of our winning submission to the CACHE Challenge #1. We developed a framework for the high-throughput structure-based virtual screening of a chemically diverse small molecule space. Hit identification was performed using the large-scale Deep Docking (DD) protocol followed by absolute binding free energy (ABFE) simulations. ABFEs were computed using an automated molecular dynamics (MD)-based thermodynamic integration (TI) approach. 4.1 billion ligands from Enamine REAL were screened with DD followed by ABFEs computed by MD TI for 793 ligands. 76 ligands were prioritized for experimental validation, with 59 compounds successfully synthesized and 5 compounds identified as hits, yielding a 8.5% hit rate. Our results demonstrate the efficacy of the combined DD and ABFE approaches for hit identification for a target with no previously known hits. This approach is widely applicable for the efficient screening of ultra-large chemical libraries as well as rigorous protein–ligand binding affinity estimation leveraging modern computational resources.
Introduction
The Critical Assessment of Computational Hit-Finding Experiments (CACHE) Challenge1 was organized to identify state-of-the-art computer-aided drug design methodologies for discovering active compounds to various types of protein targets, following in the tradition of other community efforts.2,3 This work describes the methodology and results of the first phase of our 1st place submission for the CACHE Challenge #1. The identification of small molecule compounds with significant binding affinity for a specific molecular target is a primary objective during the early stages of drug discovery.4,5 Computational approaches that search large databases of chemical compounds for potential hits, known as virtual screening, have emerged as efficient tools for accelerating hit identification.6–12 In structure-based virtual screening (SBVS), hits are identified based on the interactions of ligands with a target of interest. This is typically achieved using molecular docking programs that place ligands into a binding site of the target protein and then rank them by scoring functions that predict protein–ligand binding free energy.13The absence of experimentally validated ligands presents a major difficulty for drug discovery. Since docking typically requires only the structure of the protein binding pocket, SBVS is routinely used in cases where no binders are known for a given target protein. However, the recent exponential expansion of the sizes of the available chemical libraries poses a significant challenge for SBVS. Advancements in automated chemical synthesis and the surge of available chemicals have led to the emergence of ultra-large virtual databases that include billions of compounds. For instance, Enamine REAdily accessibLe (REAL) database14 currently contains over 38 billion compounds, with 5.5 billion drug-like compounds that can be synthesized at high success rate (>80%). It is noteworthy that the “make-on-demand” libraries are expanding without compromising chemical diversity, with novel scaffolds being consistently incorporated.11 At the same time, the current capabilities of docking software and the amount of available computational resources rarely allow for SBVS campaigns to surpass hundreds of millions of molecules threshold,15 with several exceptions.10,16,17 Therefore, much faster screening protocols are required to enable an efficient screening of currently available chemical space.
Recently, Gentile et al.18 developed the Deep Docking (DD) method to screen billions of molecules using a deep learning machine learning (ML) model capable of predicting docking scores. DD utilizes a multilayer perceptron (MLP) model which is trained using docking scores and circular chemical fingerprints. MLP model training uses active learning (AL). In this iterative approach, an ML model is first trained on the results of structural docking of a small subset of molecules and then used to predict docking scores for the rest of the chemical library. Next, a sample of ML predicted virtual hits is structurally docked and used for training set augmentation and ML model retraining. The latter step is repeated iteratively until the model converges or a pre-defined number of iterations is reached. AL iteratively improves the ML model performance and ability to identify top-scoring molecules. Screening an ultra-large database using DD was demonstrated to provide enrichment factors of up to 6000 for top-ranked hits with an approximate 50× speed acceleration compared to brute force docking. For example, the application of DD for the SBVS of ZINC15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 19 database against the SARS-CoV-2 main protease yielded several novel experimentally validated inhibitors.18
19 database against the SARS-CoV-2 main protease yielded several novel experimentally validated inhibitors.18
The second major challenge for SBVS stems from the limited accuracy of modern scoring functions, resulting in a large share of false positives among top-scoring ligands and thus the limited success rate of virtual screening campaigns.13 While the assessment of docking results by expert medicinal chemists can reduce20 this deficiency, this approach is not feasible for large libraries; thus, automated ways of discarding false positives from docking hits are needed. One common approach to reduce the number of artifacts is to dock each molecule using multiple programs, then re-ranking the molecules based on a consensus docking score.21,22
The predictive power of SBVS can be further improved by rescoring docking hits using absolute binding free energy (ABFE) simulations based on classical molecular dynamics (MD) simulations.23–25 This method utilizes a thermodynamic pathway between the two end-point states: the protein–ligand complex and the free protein and ligand in solvent. To achieve the most accurate results, all-atom simulations in explicit solvent are required.24–26 For example, benchmarking of ABFE simulations on congeneric series of ligands binding to eight protein targets demonstrated an agreement between calculations and experiment with a root mean squared error of 1.1 kcal mol−1 and a weighted average R2 of 0.55.24
In this work, we combined DD and ABFE simulations for the in silico screening of Enamine REAL (4.1B compounds) for potential inhibitors of leucine rich repeat kinase 2 (LRRK2) targeting the WD40 repeat (WDR) domain. Mutations in LRRK2 are known as the most common genetic cause of the familial form of Parkinson's disease (PD).27 Therefore, inhibitors of LRKK2 are considered promising therapeutic agents for the treatment of PD. LRRK2 is a large (286 kDa), multi-functional protein which contains seven unique domains: three N-terminal repeat domains, the GTPase domain, the dimerization domain, the kinase domain, and the C-terminal WDR domain.28 Pathogenic mutations of LRRK2 were suggested to induce the kinase domain to adopt a closed active conformation which promotes the binding of LRRK2 to microtubules. In vitro motility assays demonstrated that LRKK2 mutants inhibit kinesin and dynein movement along microtubules, suggesting that these mutants act as roadblocks for the motors.28 Kinase inhibitors that stabilize the open conformation of LRRK2 were shown to reduce the formation of LRRK2 filaments and recover the motor motility. However, most kinase inhibitors stabilize the closed conformation of the kinase domain and do not inhibit the oligomerization of LRKK2 on microtubules.28
The WDR domain is in close proximity to the kinase domain and can play a role in modulating LRRK2 kinase activity. The WDR domain is a closed protein solenoid domain consisting of repeating units forming a circularized beta-propeller structure.29,30 The structural model of the microtubule bound LRRK2 filaments suggested that the WDR domain mediates protein dimerization and formation of the LRRK2 filaments.28 Therefore, targeting the WDR domain is a promising yet underutilized strategy to antagonize the pathogenic LRRK2 filament formation. Identifying small molecule compounds binding to the WDR domain of LRRK2 can thus open the pathway towards the design of efficient therapeutics for the treatment of PD associated with LRKK2 malfunction.
Our workflow for in silico hit identification included the following steps: first, DD was performed for the Enamine REAL database to the LRRK2 WDR domain. Two sets of ligands from top-scoring docked molecules for reranking with ABFE simulations were selected: (1) molecules with the best consensus docking score, and (2) molecules chosen based on visual inspection of docked poses. For both sets, 7 ns MD simulations were performed to obtain equilibrated ligand poses in the protein binding pocket. ABFE for these poses were then computed using an automated MD TI protocol developed in our previous work.9 The molecules were then re-ranked by computed ABFE and top-scoring molecules were submitted for experimental testing.
Methods
Ligand–protein docking
132679525 molecules, in SMILES format.14 Structures of small molecules were prepared for docking to the protein by processing SMILES with OpenEye tautomers tool31 to calculate one dominant ionization state per compound followed by translating them into 3D low-energy structures with OpenEye omega tool.32 For docking with AutoDock-GPU,33 input files were generated with OpenBabel.34
39 within a 40 × 40 × 40 points grid with 0.375 Å spacing. Parameters for AutoDock-GPU were set as 5 million energy evaluations per Lamarckian genetic algorithm run for 10 independent runs using Solis-Wets local search, results were clustered based on a root-mean-square deviation (RMSD) threshold of 2 Å, and the best solution was chosen as the best scoring pose from the most populated cluster.40 ICM maps were generated with ICM's GUI. For ICM docking the thoroughness value was 3.
000 molecules. The same process was then repeated using ICM with sample sizes of 120000 molecules.
Molecular dynamics simulations
The simulations were performed using the GPU-accelerated pmemd.cuda module of AMBER20.45–48 A simulation time step of 2 fs was used with hydrogen atoms constrained via SHAKE. A Langevin thermostat and Berendsen barostat were used to control temperature and pressure, respectively. Long-range electrostatics were calculated using the Particle Mesh Ewald method with a cutoff distance of 10 Å.
Only the WDR domain monomer chain A (residues A2141 to A2496) and crystal water molecules were included in the simulation. The missing loop regions of the protein (residues 2159–2163, 2254–2258, 2306–2313, 2396–2407, 2463–2467, and 2478–2487) were modeled using the ModLoop web server.49,50 The remaining missing atoms and hydrogens were added using the tleap program of AMBERTools18. The protein was solvated with water extending 10 Å in all directions and Cl− ions were added to neutralize the system.
The system energy was minimized for 3000 steps and heated at constant volume from 0.1 K to 300 K in 250 ps MD. The protein Cα atoms were restrained using a harmonic potential with a force constant (k) of 20 kcal mol−1 Å−2 during the above stages. The system was then equilibrated for 40 ns in the NPT ensemble. The restraints on the protein were gradually released during equilibration and only the Cα atoms of the end residues (residues 2141–2142 and 2495–2496) were restrained with k = 1 kcal mol−1 Å−2 for the final 25 ns. A production run of 100 ns was then carried out in NVT ensemble with the Cα atoms of the end residues restrained.
52 and ligand atomic charges were derived using the RESP51 method. GPU-accelerated MD simulations were performed using the pmemd.cuda module of AMBER20. The simulation protocol included the following steps: (1) 2000 steps of minimization with gradient descent method; (2) 100 ps of heating at constant volume from 1 K to 298 K (3) 300 ps of density equilibration in NPT ensemble; (4) 7 ns of production simulation in NVT. Harmonic RMSD restraints were imposed on heavy atoms of the protein and the ligand during minimization and heating and were gradually removed during density equilibration; no restraints were used during production simulations.
To identify unstable ligands, the average RMSD of ligand heavy atoms with respect to the average ligand structure (RMSDavg) was calculated for each protein–ligand complex using the cpptraj package.53 The first 2 ns of the MD production simulation was discarded and the average structure was obtained from the last 5 ns of the simulation. The RMSDavg was computed for each trajectory frame and its arithmetic mean was obtained. The MD trajectories of a subset of ligands with different average RMSDavg values were visually inspected. Based on this visual inspection, ligands with the average RMSDavg over 3 Å were discarded due to the instability of binding mode. For each ligand with average RMSDavg in the equilibrium MD simulation not exceeding 3 Å, a trajectory frame with the minimum RMSDavg was selected as a representative structure and used in further analysis and TI simulations.
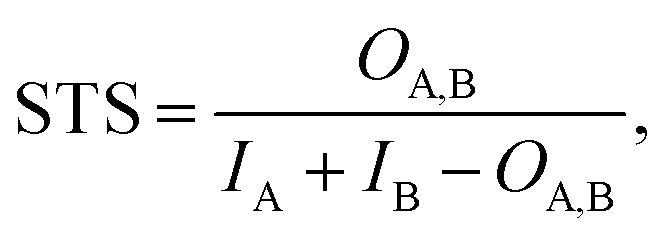 | (1) |
| STD = 1 − STS | (2) |
The matrix of STDs was used to perform clustering with the DBSCAN algorithm as implemented in the scikit-learn library.55 Ligands assigned to the same cluster were visually validated.
During TI simulations of the protein–ligand complex, the position and orientation of the ligand with respect to the protein was restrained using the virtual bond approach.56 The carbonyl carbons of Cys2302 and Leu2303, and the backbone oxygen of Leu2303 were used for the virtual bond restraints as the protein atoms P1, P2 and P3, respectively (Fig. S1†). The majority of ligands provided by DD and expert selection contained an amide group. The requisite ligand atoms for the virtual bond restraints were selected by the following algorithm:
1. The amide group of the ligand with the oxygen closest to the carbonyl carbon of residue Cys2302 (P1) was selected.
2. The following two angles were compared: P1–O–C versus P1–O–N, where O, C and N are oxygen, carbon and nitrogen atoms of the ligand amide group, respectively. The atom combination with an angle less than 150°, greater than 30° and closest to 90° were selected as L1 and L2.
3. If the L1 and L2 were O and C, then N was selected as L3. If L1 and L2 were O and N, then the alpha carbon of the ligand amide group (Cα) was selected as L3.
4. If both atom combinations had angles that were greater than 150° or less than 30°, then N, Cα, and O were selected as L1, L2 and L3.
For the majority of ligands, this algorithm was successful for the selection of atoms for the virtual bond; however, some ligands had no amide groups, had amide groups with unsuitable angles, or amide groups too far on the periphery of the ligand. In these cases, the representative structure was visually inspected and restraint atoms were selected such that they:
1. Included rigid bonds, ideally sp2 hybridized carbons or otherwise constrained species.
2. The angles P2–P1–L1, P1–L1–L2 and L1–L2–L3 were close to 90°.
3. Were located close to the center of mass of the ligand.
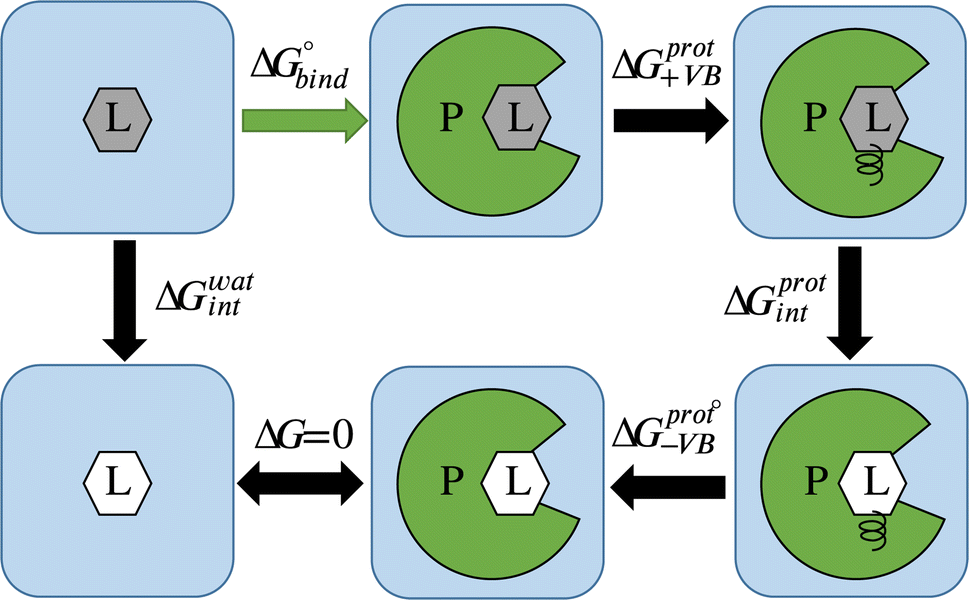 | ||
| Fig. 1 Thermodynamic cycle for alchemical ABFE simulations. The ligand (L) in its interacting and non-interacting form is shown by a gray and white hexagon correspondingly, and the protein (P) is shown by green. ΔG0bind corresponds to the ABFE. ΔGwatint and ΔGprotint correspond to the free energy of turning off interactions of the ligand with the environment in solvent and in complex with the protein, respectively. ΔGprot+VB and ΔGprot−VB correspond to the addition and removal of the virtual bond restraints. | ||
Average gradients were calculated from the last 4.5 ns of the production simulations using the alchemlib python library.57 Errors were estimated using the bootstrap method. The free energies for both the free ligand, protein, and restraints were obtained by the trapezoid rule. The free energy of adding virtual bond restraints for the non-interacting ligand was calculated using the Boresch formula.56 The final values of ABFE were obtained according to the following equation (see Fig. 1 for the representation of free energy terms):
| ΔG = ΔG0bind = ΔGwatint − ΔGprotint − ΔGprot+VB − ΔGprot−VB. | (3) |
Selecting final list of ligands for submission to CACHE
For molecules with computed ABFE, the solubility property (logS) was predicted using an XGBoost58 model trained on alvaDesc molecular descriptors.59 Reference data was extracted from the literature,60 and the dataset was curated according to a well-known protocol.61,62 All models were trained within two nested five-fold cross validation loops and all splits were random. Final scoring was done by a simple averaging of five predictions from the external CV loop. The internal CV loop was used to perform hyperparameter search and variable selection for the corresponding fold using the protocol described in our previous work.62
Molecules with a predicted logS lower than −5.5 were discarded from further selection. The remaining molecules were ranked based on ABFE, and the 150 molecules with the lowest ABFE were submitted to Enamine for price estimation and availability. Synthetic accessibility was verified for 127 molecules by the vendor. Considering the updated price estimate and the funding available from the CACHE Challenge, the final set of 76 molecules with the lowest ABFE was ordered for experimental validation.
Results
Overview
This paper reports a multistep workflow for identifying inhibitors to the LRRK2 WDR domain (see Fig. 2A). Using neural network-accelerated docking (Deep Docking), we were able to screen the Enamine REAL library of ∼4.1B compounds. We selected ∼18 M compounds, which were then re-docked with two docking packages to generate a consensus score. Compounds having high RMSD between the two packages were filtered out, resulting in the Consensus2 set (Fig. 2B and Methods section). The compounds were then docked with the third package to generate the Consensus3 set using an analogous procedure (Fig. 2B and Methods section). Drug-likeness filtering, best-first clustering, and selection of the top 100 compounds from each set resulted in the 199 compound Consensus set (see Methods section for details). In parallel, visual inspection of compounds was applied to the results of all three docking packages, resulting in the selection of 605 unique compounds, denoted as the Expert set.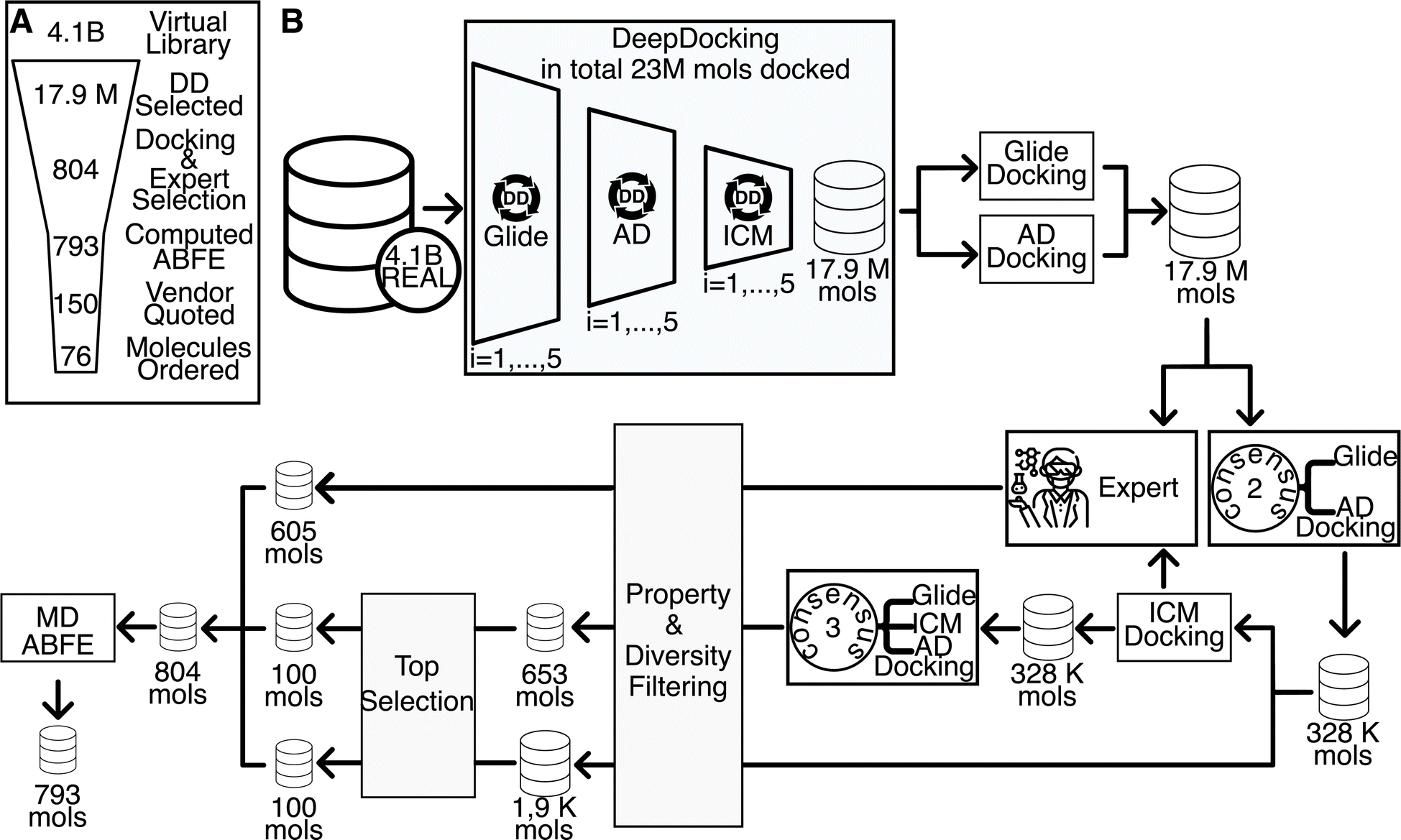 | ||
| Fig. 2 Overview of hit identification pipeline. (A) Broad overview of the computational pipeline. Each number within the funnel relates to the number of molecules after the corresponding step has been taken. (B) In-depth breakdown of the computational pipeline: (1) Deep Docking virtual screening of the initial 4.1B library leading to the focused library for conventional docking, (2) conventional docking via multiple docking programs, (3) filtering by consensus of 2 and 3 docking programs, (4) in-parallel to step 3—Expert selection of prioritized molecules, (5) poverty & diversity filtering and docking-score based prioritization, (6) assessment of absolute binding free energy via molecular dynamics simulations. See Methods section for detailed descriptions of each step. | ||
The docked structure of the 804 compounds selected as described above were equilibrated with molecular dynamics simulations in aqueous solution and standard thermodynamic conditions. All unstable protein–ligand complex structures were discarded as described below. Representative structures for each protein–ligand complex were extracted from the final 5 ns of the equilibrium trajectory and used for ABFE simulations. In total, 793 ABFE simulations were completed (see Fig. 2A). 77 molecules had a ΔG of less than −6.5 kcal mol−1. The top 76 compounds based on computed ΔG were purchased for experimental characterization, of which 59 compounds were successfully synthesized. The compounds not synthesized were weighted towards our top compounds, with 4 of our top 10 not synthesized while only 2 of our bottom 20 compounds failed synthesis. The mean ABFE of synthesized compounds was −7.70 kcal mol−1 while the mean ABFE of compounds that were not synthesized was −8.28 kcal mol−1.
Seven compounds were identified as hit molecules by the CACHE Challenge organizers based on their binding affinities measured via surface plasmon resonance (SPR); however, two of these compounds failed subsequent orthogonal validation via19F nuclear magnetic resonance (NMR) or isothermal titration calorimetry (ITC), resulting in a final list of five hit compounds. Thus, we achieved an overall hit rate of 8.5%. Four hit compounds were obtained from the Expert set while one came from Consensus set, which approximately matches their relative frequencies within the synthesized set of compounds (Expert set: 50 compounds; Consensus set: 9 compounds). Experimental validation performed by the CACHE Challenge organizers will be detailed in a follow up publication. Overall results of the CACHE Challenge #1 can be found here: https://cache-challenge.org/results-cache-challenge-1.
Binding mode and protein–ligand interactions of hit compounds
Five compounds were experimentally identified as active compounds by the CACHE Challenge organizers via SPR, 19F NMR and ITC. Structures of these ligands, interactions of their MD representative poses with the WDR domain binding pocket, and their computed and experimental ABFEs are presented in Fig. 3. Hits 1–3 and 5 have similar binding modes exhibiting an L-shaped pose (see “Binding pose analysis” section). These ligands contain an indole ring connected to an amide group that occupies approximately the same region of the binding pocket and form specific interactions with the protein residues. The nitrogen of the indole ring and the oxygen of the amide group form hydrogen bonds with the backbone oxygen of Lys2415 and the hydroxyl group oxygen of Thr2416. The indole ring forms cation–π interactions with the guanidino group of Arg2456 and hydrophobic interactions with Ile2355 and Met2301. Hits 1, 2, and 5 also contain an oxamide group which forms hydrogen bonds with the backbone of Met2155 and Leu2202. Hit 3 does not contain an oxamide group but has a hydroxyl group that forms a hydrogen bond with Val2357. In contrast to the other hit compounds, Hit 3 is significantly shifted with respect to its initial pose during equilibrium MD simulations with an RMSD of the representative pose with respect to the initial pose (ligand RMSD) of 3.4 Å (see “Equilibration of solvated protein–ligand complexes via MD simulations” section).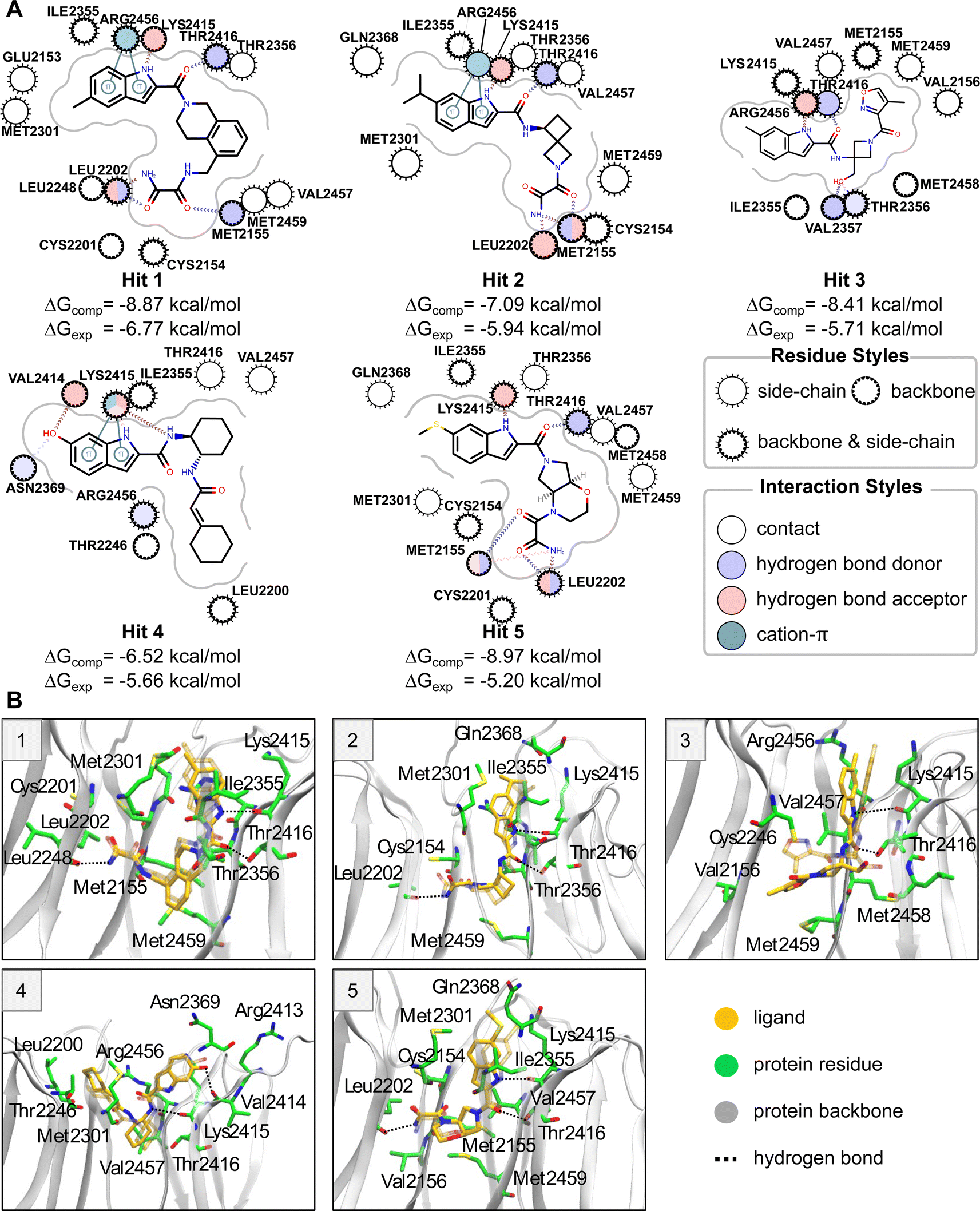 | ||
| Fig. 3 (A) Protein–ligand interaction maps for hit compounds. Protein residues are shown as circles. Protein–ligand interactions are shown as lines. The residues and interactions are colored depending on the interaction type according to the legend. (B) Simulated representative structure (ligand shown in opaque yellow) of five hit compounds superimposed on the initial docked structure using protein Cα atoms. The initial position of the protein is not shown. The initial ligand position is shown in transparent yellow. The simulated protein is shown by a transparent cartoon representation. The protein residues that interact with the ligand in the simulated structure are shown in green stick representation. Protein–ligand hydrogen bonds are shown as dotted lines. | ||
Hit 4 deviates in its binding pose when compared to the rest of the hits and has the highest computed ABFE among all hit compounds. This ligand adopts a U-shaped conformation; however, it is not within the U-shaped cluster (see “Binding pose analysis” section), as its binding pose is located closer to the periphery of the binding site. The conformation and orientation of Hit 4 are relatively stable in equilibrium MD simulations. Both the nitrogens of indole ring and amide group can form hydrogen bonds with the backbone of Lys2415. The indole ring also forms cation–π interactions with the amino group of the Lys2415 sidechain. The hydroxyl group connected to the indole ring forms a hydrogen bond with the backbone oxygen of Val2414, and the cyclohexane moiety forms hydrophobic interactions with Leu2200.
Docking
No known inhibitor has been reported for the human WDR domain of LRRK2, whose apo structure has been resolved. A large solvent-exposed doughnut-like binding site (see Fig. 4A) was predicted by Molecular Operating Environment (MOE) 2020 Site Finder, where many residues are possibly involved in substrate binding of protein–protein interactions in nature. However, Ile2355, Met2301, Val2457 and Met2459 are considered as possible important hydrophobic residues for small molecule binding (Fig. 4B).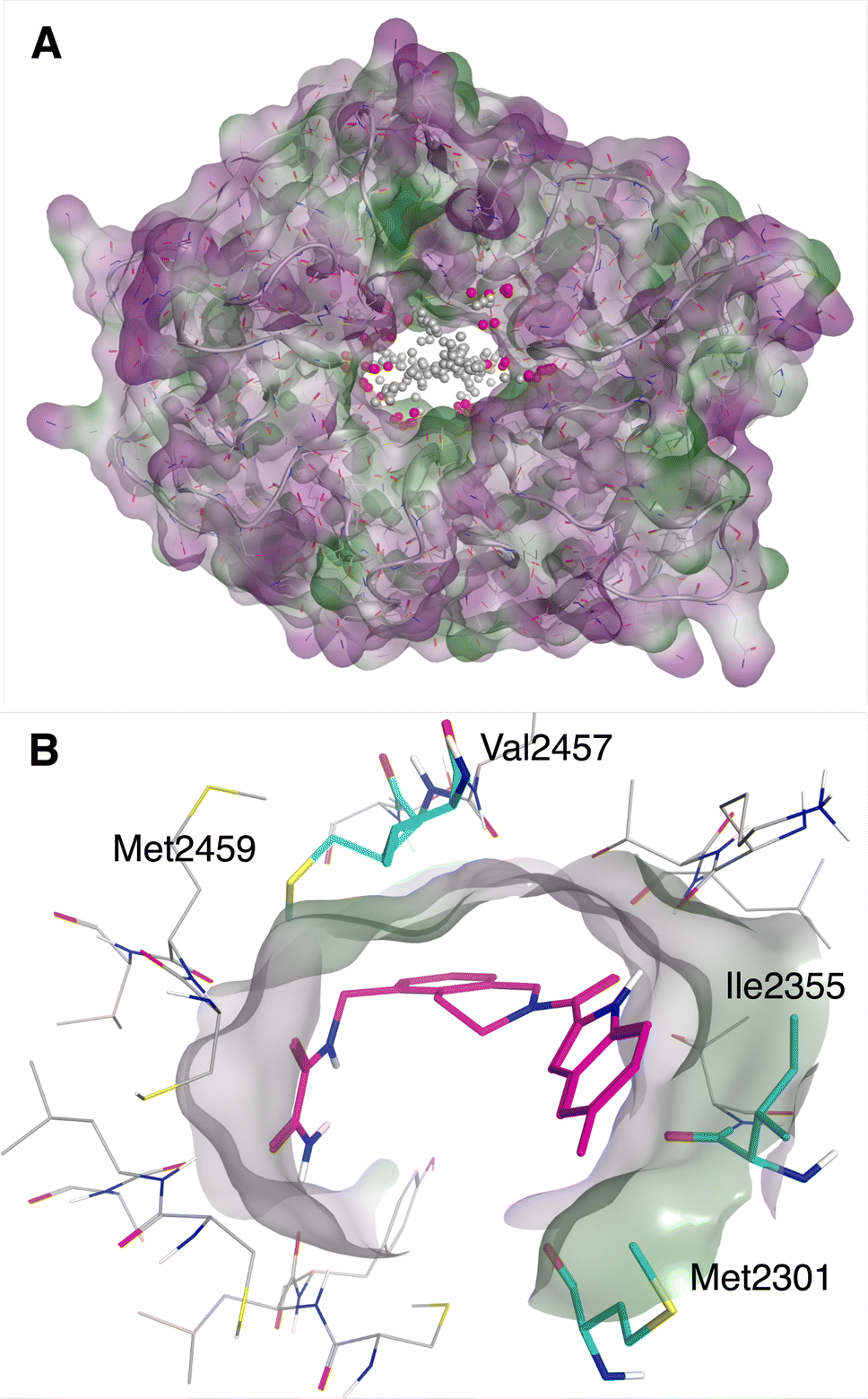 | ||
| Fig. 4 (A) The binding site of WDR domain of LRRK2 predicted by MOE Site finder. (B) Putatively important hydrophobic residues for ligand binding, in complex with one of the virtual hits. | ||
Docking grids centered on residues Ile2355 and Met2301 of the WDR domain were generated (Fig. 4B). The sequential Deep Docking process (Fig. 2B) reduced the size of Enamine REAL library from 4132679525 to 17885310 molecules (231-fold reduction). In total, 23088153 molecules were docked to the LRRK2 target as part of the training (0.56% of the library). Consequently, the ∼17.9 million molecules predicted as hits by Deep Docking were docked to LRRK2 with Glide SP and AutoDock-GPU, and filtered based on the RMSD value between the poses generated by the programs. In total, 59187165 molecules were structure docked to LRRK2 as both DD training and consensus docking procedures (1.4% of the Enamine REAL library).
Notably, applying consensus filtering drastically reduced the number of potential candidate molecules from Enamine REAL, 328392 molecules passed the Consensus2 filter. Out of these, 100000 top scoring molecules were selected and the drug-likeness filter was applied followed by the best-first clustering of the top 5000 molecules. This reduced compound set contained 1891 molecules (Fig. 2B). The same protocol was applied to the Consensus3 molecular set resulting in 653 molecules. The top 100 molecules from each reduced compound set were selected and merged, resulting in 199 unique molecules considered for further evaluation (these molecules will be referred to as the Consensus set). In parallel, 605 unique molecules from the results of the three individual docking programs were selected via expert inspection of their predicted binding poses; compounds selected must have good hydrophobic interactions (see Fig. 4B) with Ile2355 and Met2301, and possibly good hydrophobic interactions with Val2457 and Met2459 (these molecules will be referred to as the Expert set). Thus, 804 molecules in total were sent for further evaluation by MD and ABFE simulations. Overall, the whole process took approximately 14 days in total, using 100 Tesla V100 GPUs for Autodock-GPU docking and machine learning and 1000 Intel® Xeon® Silver 4116 CPU@2.10 GHz cores for Glide and ICM docking and conformer generation. Brute-force screening of the entire library using the fastest screening tool (Autodock-GPU) and the same computational resources would have taken approximately 950 days just for the docking simulations.
Equilibration of solvated protein–ligand complexes via MD simulations
We equilibrated each protein–ligand complex obtained from docking with 7 ns of MD in explicit solvent in order to obtain more physically realistic starting poses for MD TI simulations and to eliminate ligands with unstable binding poses from further consideration (see Methods for details). To identify ligands with significant instabilities observed during equilibrium MD simulations, we computed the mean RMSD of a ligand with respect to the average structure of the ligand extracted from the final 5 ns portion of production MD simulations, RMSDavg. We then excluded all ligand with the mean RMSDavg > 3 Å from further evaluation (11 molecules in total); thus, 793 molecules advanced to ABFE simulations by MD TI.To further characterize protein and ligand stability during equilibrium MD simulations, we computed the RMSD of: (1) Cα atoms of protein residues that form the inner surface of the central cavity of WDR domain with respect to the minimized structure used for docking (protein RMSD), and (2) heavy atoms of the ligand from MD representative structure with respect to its docked structure (ligand RMSD). For all ligands selected for MD simulations, the average protein RMSD was below 1 Å, implying the structure of the protein binding site was stable during equilibrium simulations. In contrast, the stability of the ligand docked pose varied significantly for different ligands. The distribution of the ligand RMSD is depicted in Fig. 5A. Approximately 35% of ligands remained relatively stable with respect to the docked pose during MD (ligand RMSD ≤ 2 Å), while 45% of ligands had ligand RMSD from 2 Å to 4 Å, and 20% of ligands had ligand RMSD above 4 Å. On average, ligands from the Consensus set demonstrated smaller deviations from the docked pose compared to ligands from the Expert set. The ratio of ligands that deviated significantly from their docked pose was considerably larger for the Expert set than the Consensus set (Fig. 5A). For the Expert set, 47% and 23% of ligands had ligand RMSD from 2 Å to 4 Å and above 4 Å, respectively. For the Consensus set, these proportions were 31% and 10%, respectively. To estimate the conformational stability of ligands, we computed the best-fit RMSD of the MD representative pose of a ligand to its docked pose (conformational RMSD). For most ligands, conformations of docked poses remained relatively stable (see Fig. S2†). Only 6% ligands from the Expert set and 4% ligands from the Consensus set had conformational RMSD greater than 2 Å. Therefore, deviations from docked poses are mainly due to rigid-body translations and rotations of the ligand. Examples of molecules with ligand RMSD of less than 2 Å, ranging from 2 Å to 4 Å, and greater than 4 Å are shown in Fig. 5B–D.
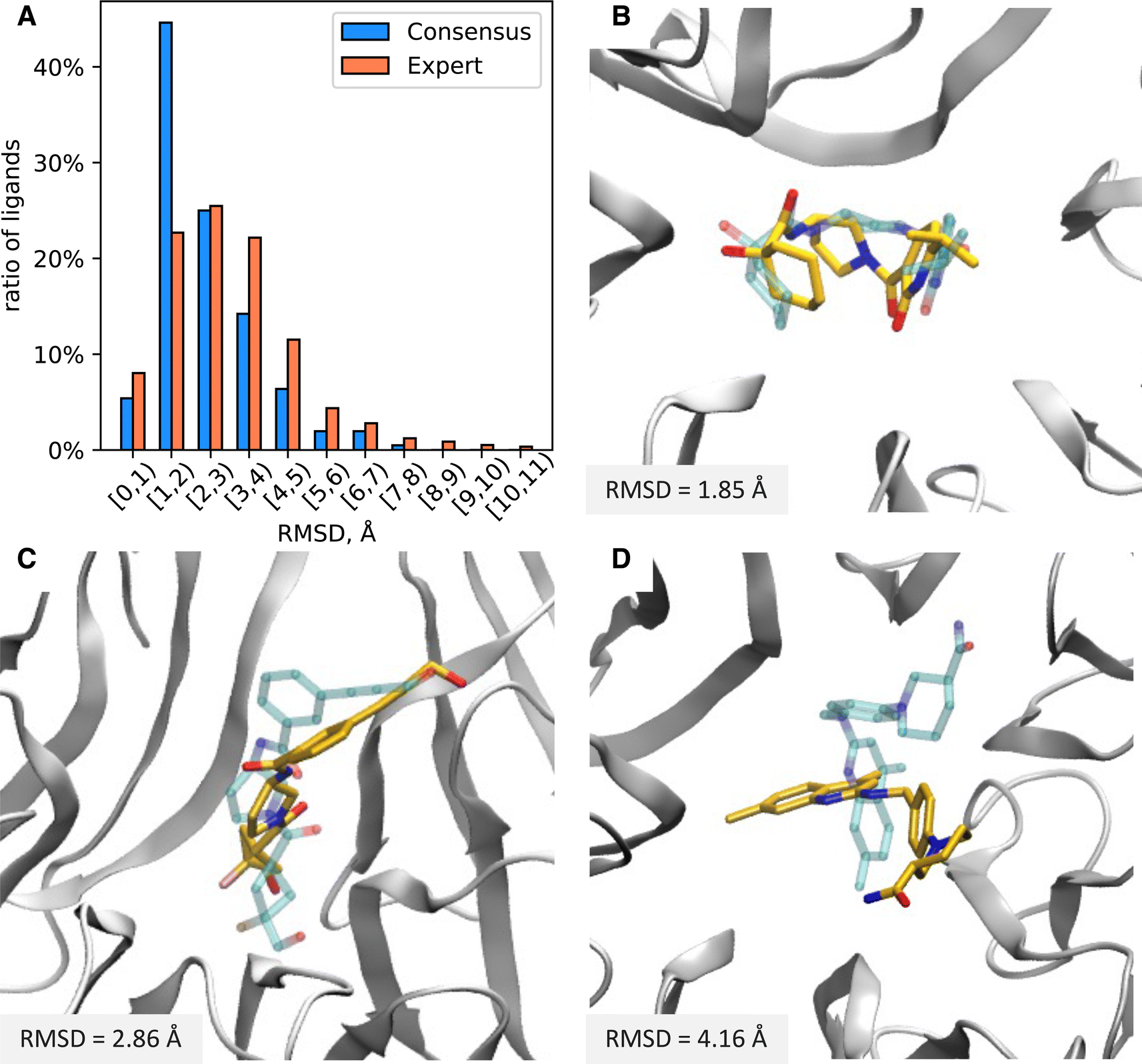 | ||
| Fig. 5 (A) Distribution of ligand RMSD of the MD representative poses with respect to the docked pose. (B–D) Structure of ligands with various ligand RMSDs in the binding pocket of the WDR domain. The docked pose of the ligand is shown by transparent blue. The MD representative pose of the ligand is shown by yellow. The protein is presented by the gray cartoon. The ligand RMSDs are displayed in the lower left corner of each panel. | ||
Computing absolute binding free energies using MD TI simulations
MD TI simulations were performed on 793 molecules from the Enamine REAL dataset, with a mean ΔG of −2.11 kcal mol−1 (Fig. 6A). 77 molecules had a ΔG of less than −6.5 kcal mol−1, and 5 molecules had a ΔG of less than −10 kcal mol−1. Molecules from the Expert set performed similarly to those from the Consensus set, with the former having a slightly wider distribution and a mean of −2.30 kcal mol−1vs. −2.00 kcal mol−1. Of the 76 molecules selected for experimental validation, 61 were selected from the Expert set and 16 were selected from the Consensus set. Computed ABFEs showed no correlation with consensus docking scores (see Fig. 6B). The significant ratio of molecules with positive ABFE and the absence of correlation between ABFEs and consensus docking scores suggests that docking provided a considerable number of false positive hits. This justifies the use of ABFE simulations to prioritize molecules for experimental testing.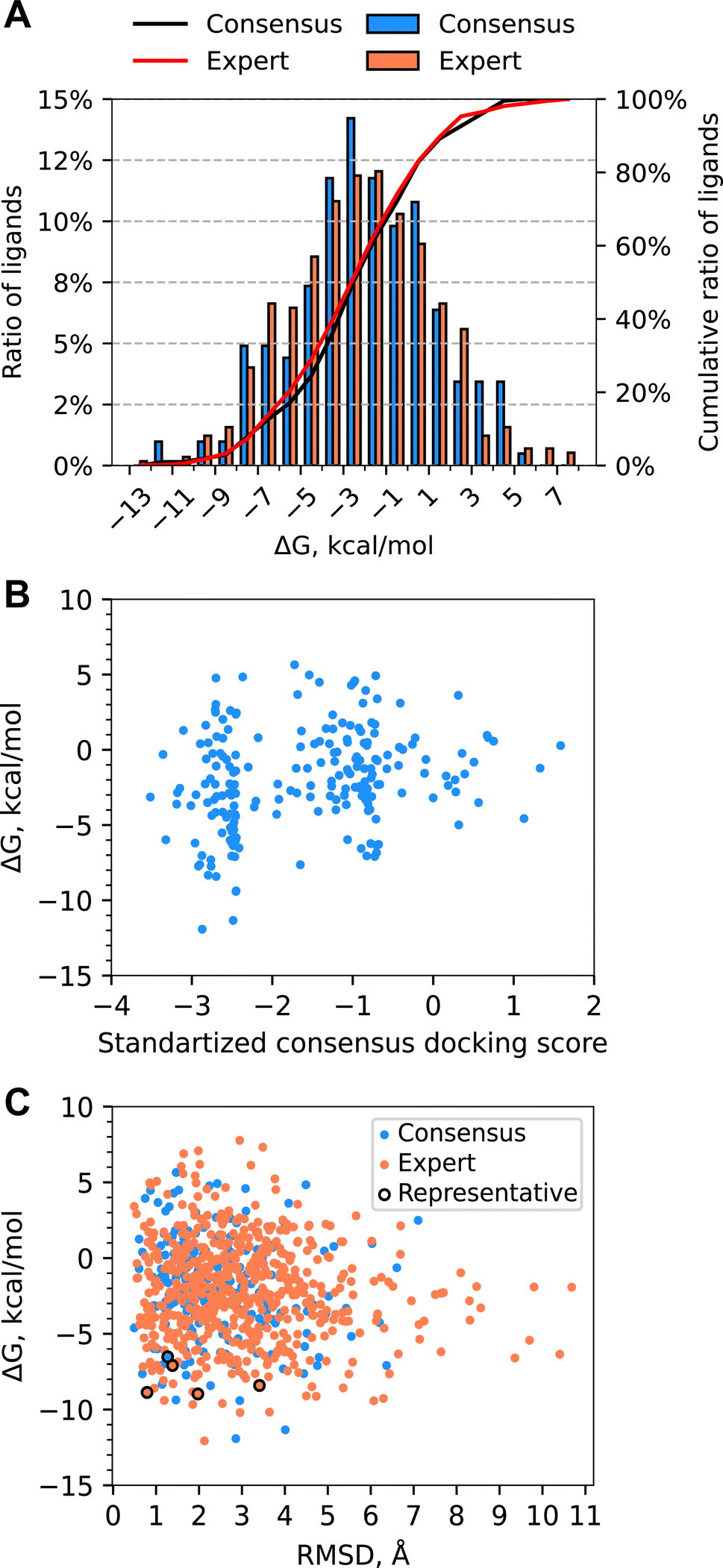 | ||
| Fig. 6 (A) Distribution of ABFE calculated with MD TI for all molecules for which free energy simulations were performed. The Expert set was selected by visual inspection of docked ligands. The Consensus set was selected by consensus docking score. There were no overlaps between the Expert and Consensus sets. (B) Scatter plot of computed ABFE and consensus docking scores for molecules from the Consensus set. (C) Scatter plot of computed ABFE and ligand RMSD of the MD representative poses with respect to the docked pose. | ||
While the ligand RMSD had no correlation with computed ABFE (Fig. 6C), the ligands with the lowest ABFE (ΔG of less than −10 kcal mol−1) had ligand RMSD from 2 to 4 Å for both sets. Interestingly, for the Expert set, the ligand RMSDs of ligands with the highest ABFE were also within this range. The spread of the ABFE distribution for ligands with ligand RMSD above 4 Å was smaller than for the rest of the ligands; however, the median and average ΔGs were quite similar.
Overall, system preparation, equilibrium MD simulations and MD TI simulations took approximately 40 days in total, using Intel Xeon E5-2620 CPUs for QM calculations for ligand charges and 100 Tesla V100 GPUs for MD simulations. The total computational cost of the QM calculations for all ligands was around 8400 CPU-hours. The total simulation time for equilibrium MD and MD TI simulations for all ligands was around 100 μs and the corresponding computational cost was around 13000 GPU-hours.
Binding pose analysis
Upon visual analysis of the equilibrium MD trajectories and representative poses of ligands with the most negative computed ABFE, we identified several ligands with a similar binding mode. To further investigate ligand binding mode similarities, we performed clustering of MD representative poses for all ligands with computed ABFEs (see Methods section for details).The clustering resulted in ∼18% of ligands distributed between the four most populated clusters and ∼5% of ligands distributed between 17 clusters each of which included no more than five ligands. The rest of the molecules (∼77%) were identified as singletons. The clustering results are therefore indicative of significant diversity within the MD representative poses. Descriptive statistics of ligand ABFEs for the four most populated clusters is provided in Table 1. The name of each cluster corresponds to the MD representative pose ligand shape in the binding pocket. Remarkably, the ABFE distributions for L- and C1-shaped ligands are significantly shifted towards more negative ABFE values compared to the ABFE distribution for all ligands.
| Cluster | N | m(ΔG), kcal mol−1 | μ(ΔG) ± σ, kcal mol−1 |
|---|---|---|---|
| L-shaped | 20 | −5.53 | −5.88 ± 3.02 |
| C1-shaped | 14 | −2.94 | −3.70 ± 2.07 |
| C2-shaped | 48 | −1.48 | −1.71 ± 3.35 |
| U-shaped | 53 | −0.72 | −1.17 ± 3.51 |
Molecules contained in the L-shaped cluster are composed of two sections. The first is long and rod-like and runs parallel to the interior of the protein. This section typically exhibits hydrogen bonds between a carbonyl oxygen and Thr2416 hydroxyl group, and an indole nitrogen and Lys2415 backbone oxygen. The second section runs perpendicular to the first section, forming the bend of the “L”. This section typically exhibits an oxamide group with hydrogen bonding between amide hydrogens and the backbone of both Met2155 and Leu2202. Using these hydrogen bonds as a reference, we identified an additional 16 ligands that exhibited hydrogen bonding to at least three of these residues in at least 40% of the frames of the equilibrium MD trajectory. These 16 ligands had an average ΔG of −2.84 ± 0.54 kcal mol−1. These results suggest that, while ligands exhibiting the L-shaped pose have significant similarity when considering hydrogen bonding interactions, these interactions by themselves are not enough to predict binding affinity. 8 out of 76 ligands submitted exhibit an L-shaped binding pose.
Conclusions
Computer-aided drug design aims to predict how to target and modulate any protein with small-molecule binders. With the bold launch of the CACHE Challenge, this process can be rigorously benchmarked through a series of competitions. In this paper, we report our computational results of stage one of the CACHE Challenge #1. Using the challenging LRRK2 WDR domain with no known molecular inhibitors as a target, we rapidly identified a series of chemical starting points binding to the WDR domain through a combination of ML and physics-based computational methods. Our approach directly aims to improve prediction methods and discover new small-molecule binders for an arbitrary target. The outcomes of the CACHE Challenge will be a major technological step towards achieving the goal of Target 2035, a global initiative to identify pharmacological probes for all human proteins.63We virtually screened 4.1B compounds from the Enamine REAL library to identify binders of the central cavity of the WDR domain of LRRK2. Our computational workflow combined Deep Docking for the initial scoring of protein–ligand binding and ABFE simulations for the rescoring of docked poses. Deep Docking afforded the screening of this multi-billion compound chemical library within a reasonable timeframe, requiring only around 59 M compounds (1.4% of the library) to be docked with three programs (AutoDock-GPU, Glide SP, and ICM) while the rest of the compounds were deprioritized based on the ML-predicted docking score. Consensus filtering based on the deviation between docking poses obtained with different docking programs further reduced the number of compounds from 59 M to 238 K. The application of a drug-likeness filter followed by best-first clustering and the selection of the top compounds based on their docking scores resulted in the Consensus set of 199 compounds. In parallel, the Expert set of 605 molecules was selected based on the visual inspection of the docked poses.
Short equilibrium MD simulations were performed on each selected compound in complex with the protein to eliminate potential inaccuracies caused by docking. We found that a significant number of ligands changed their poses in the binding pocket as a result of MD simulations. On average, 45% of ligands had a ligand RMSD of 2–4 Å, and around 20% had more than 4 Å.
ABFE simulations utilizing MD TI were performed on the stable compounds with RMSDavg < 3 Å. ABFEs were computed for 793 molecules and then used to re-rank the molecules for the selection of the 76 compounds for the experimental testing. The distribution of Consensus and Expert set ABFEs were similar, with values ranging from approximately −13 to 8 kcal mol−1 and average ABFE of approximately −2 kcal mol−1. For both sets, a considerable ratio of ligands with favorable docking scores had weak predicted binding affinity. The ABFEs showed no correlation with either docking scores or the ligand RMSD. The poses of ligands with the lowest ABFEs for both sets significantly shifted during MD simulations (ligand RMSD > 2 Å).
Overall 76 compounds were submitted for experimental validation, with 59 compounds tested successfully and 5 compounds identified as hits, for an overall hit rate of 8.5%. Of the 23 participants in the CACHE Challenge #1, our submission was awarded 1st place. To the best of our knowledge, our method was the only one that utilized the combination of Deep Docking and ABFE simulations. Cumulatively these results suggested the importance of combining ultrahigh-throughput computational screening tools such as Deep Docking with advanced methods such as MD TI in structure-based virtual screening campaigns, especially for targets with no previously known ligands.
Data availability
Challenge results and raw data is publicly available at https://cache-challenge.org/results-cache-challenge-1.Author contributions
Evgeny Gutkin: free energy methodology and software, visualization, investigation, formal analysis, writing – original draft ; Filipp Gusev: methodology, software, visualization, investigation, formal analysis, writing – original draft; Francesco Gentile methodology, software, investigation, formal analysis, writing – original draft; Fuqiang Ban: investigation, formal analysis, writing – review & editing; S. Benjamin Koby: visualization, investigation, formal analysis, writing – original draft; Chamali Narangoda: investigation, writing – review & editing; Olexandr Isayev: conceptualization, formal analysis, writing – review & editing, supervision, funding acquisition; Artem Cherkasov: conceptualization, writing – review & editing, supervision, funding acquisition; Maria Kurnikova: conceptualization, formal analysis, writing – review & editing, supervision, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
O. I. acknowledges support by the NSF grant CHE-2154447. M. K. is supported by grants NSF DMS-1563291, MCB-1818213, NIH R01NS083660. Part of the computation for this work was performed at the San Diego Supercomputer Center (SDSC) and the Pittsburgh Supercomputing Center (PSC) through allocation CHE200122 from the Advanced Cyberinfrastructure Coordination Ecosystem: Services & Support (ACCESS) program, which is supported by National Science Foundation grants #2138259, #2138286, #2138307, #2137603, and #2138296 and Frontera computing project at the Texas Advanced Computing Center (NSF OAC-1818253) award.References
- S. Ackloo, R. Al-Awar, R. E. Amaro, C. H. Arrowsmith, H. Azevedo and R. A. Batey,
et al., CACHE (Critical Assessment of Computational Hit-finding Experiments): a public-private partnership benchmarking initiative to enable the development of computational methods for hit-finding, Nat. Rev. Chem, 2022, 6(4), 287–295 CrossRef PubMed
.
- J. Schimunek, P. Seidl, K. Elez, T. Hempel, T. Le and F. Noe,
et al., A community effort in SARS-CoV-2 drug discovery, Mol. Inf., 2023, e202300262 Search PubMed
- M. L. Boby, D. Fearon, M. Ferla, M. Filep, L. Koekemoer and M. C. Robinson,
et al., Open science discovery of potent noncovalent SARS-CoV-2 main protease inhibitors, Science, 2023, 382(6671), eabo7201 CrossRef CAS PubMed
- M. D. Parenti and G. Rastelli, Advances and applications of binding affinity prediction methods in drug discovery, Biotechnol. Adv., 2012, 30(1), 244–250 CrossRef CAS PubMed
- A. V. Sadybekov and V. Katritch, Computational approaches streamlining drug discovery, Nature, 2023, 616(7958), 673–685 CrossRef CAS PubMed
- T. S. Chisholm, M. Mackey and C. A. Hunter, Discovery of High-Affinity Amyloid Ligands Using a Ligand-Based Virtual Screening Pipeline, J. Am. Chem. Soc., 2023, 145(29), 15936–15950 CrossRef CAS PubMed
- J. Kuan, M. Radaeva, A. Avenido, A. Cherkasov and F. Gentile, Keeping pace with the explosive growth of chemical libraries with structure-based virtual screening, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2023, 13(6), e1678 CAS
- F. Gentile, J. C. Yaacoub, J. Gleave, M. Fernandez, A. T. Ton and F. Ban,
et al., Artificial intelligence-enabled virtual screening of ultra-large chemical libraries with deep docking, Nat. Protoc., 2022, 17(3), 672–697 CrossRef CAS PubMed
- F. Gusev, E. Gutkin, M. G. Kurnikova and O. Isayev, Active Learning Guided Drug Design Lead Optimization Based on Relative Binding Free Energy Modeling, J. Chem. Inf. Model., 2023, 63(2), 583–594 CrossRef CAS PubMed
- C. Gorgulla, A. Boeszoermenyi, Z. F. Wang, P. D. Fischer, P. W. Coote and K. M. Padmanabha Das,
et al., An open-source drug discovery platform enables ultra-large virtual screens, Nature, 2020, 580(7805), 663–668 CrossRef CAS PubMed
- J. Lyu, S. Wang, T. E. Balius, I. Singh, A. Levit and Y. S. Moroz,
et al., Ultra-large library docking for discovering new chemotypes, Nature, 2019, 566(7743), 224–229 CrossRef CAS PubMed
- A. A. Sadybekov, A. V. Sadybekov, Y. Liu, C. Iliopoulos-Tsoutsouvas, X. P. Huang and J. Pickett,
et al., Synthon-based ligand discovery in virtual libraries of over 11 billion compounds, Nature, 2022, 601(7893), 452–459 CrossRef CAS PubMed
- E. H. B. Maia, L. C. Assis, T. A. de Oliveira, A. M. da Silva and A. G. Taranto, Structure-Based Virtual Screening: From Classical to Artificial Intelligence, Front. Chem., 2020, 8, 343 CrossRef CAS PubMed
- Enamine, REAL Database 2021, available from: https://enamine.net/compound-collections/real-compounds/real-database Search PubMed.
- B. J. Bender, S. Gahbauer, A. Luttens, J. Lyu, C. M. Webb and R. M. Stein,
et al., A practical guide to large-scale docking, Nat. Protoc., 2021, 16(10), 4799–4832 CrossRef CAS PubMed
- A. Acharya, R. Agarwal, M. B. Baker, J. Baudry, D. Bhowmik and S. Boehm,
et al., Supercomputer-Based Ensemble Docking Drug Discovery Pipeline with Application to Covid-19, J. Chem. Inf. Model., 2020, 60(12), 5832–5852 CrossRef CAS PubMed
- D. M. Rogers, R. Agarwal, J. V. Vermaas, M. D. Smith, R. T. Rajeshwar and C. Cooper,
et al., SARS-CoV2 billion-compound docking, Sci. Data, 2023, 10(1), 173 CrossRef CAS PubMed
- A. T. Ton, F. Gentile, M. Hsing, F. Ban and A. Cherkasov, Rapid Identification of Potential Inhibitors of SARS-CoV-2 Main Protease by Deep Docking of 1.3 Billion Compounds, Mol. Inf., 2020, 39(8), e2000028 CrossRef PubMed
- T. Sterling and J. J. Irwin, ZINC 15 – Ligand Discovery for Everyone, J. Chem. Inf. Model., 2015, 55(11), 2324–2337 CrossRef CAS PubMed
- A. Fischer, M. Smiesko, M. Sellner and M. A. Lill, Decision Making in Structure-Based Drug Discovery: Visual Inspection of Docking Results, J. Med. Chem., 2021, 64(5), 2489–2500 CrossRef CAS PubMed
- D. R. Houston and M. D. Walkinshaw, Consensus docking: improving the reliability of docking in a virtual screening context, J. Chem. Inf. Model., 2013, 53(2), 384–390 CrossRef CAS PubMed
- F. Ban, K. Dalal, H. Li, E. LeBlanc, P. S. Rennie and A. Cherkasov, Best Practices of Computer-Aided Drug Discovery: Lessons Learned from the Development of a Preclinical Candidate for Prostate Cancer with a New Mechanism of Action, J. Chem. Inf. Model., 2017, 57(5), 1018–1028 CrossRef CAS PubMed
- T. S. Lee, B. K. Allen, T. J. Giese, Z. Guo, P. Li and C. Lin,
et al., Alchemical Binding Free Energy Calculations in AMBER20: Advances and Best Practices for Drug Discovery, J. Chem. Inf. Model., 2020, 60(11), 5595–5623 CrossRef CAS PubMed
- W. Chen, D. Cui, S. V. Jerome, M. Michino, E. B. Lenselink and D. J. Huggins,
et al., Enhancing Hit Discovery in Virtual Screening through Absolute Protein-Ligand Binding Free-Energy Calculations, J. Chem. Inf. Model., 2023, 63(10), 3171–3185 CrossRef CAS PubMed
- M. Feng, G. Heinzelmann and M. K. Gilson, Absolute binding free energy calculations improve enrichment of actives in virtual compound screening, Sci. Rep., 2022, 12(1), 13640 CrossRef CAS PubMed
- H. Zhang, C. Yin, H. Yan and D. van der Spoel, Evaluation of Generalized Born Models for Large Scale Affinity Prediction of Cyclodextrin Host–Guest Complexes, J. Chem. Inf. Model., 2016, 56(10), 2080–2092 CrossRef CAS PubMed
- D. N. Wojewska and A. Kortholt, LRRK2 Targeting Strategies as Potential Treatment of Parkinson’s Disease, Biomolecules, 2021, 11(8), 1101 CrossRef CAS PubMed
- C. K. Deniston, J. Salogiannis, S. Mathea, D. M. Snead, I. Lahiri and M. Matyszewski,
et al., Structure of LRRK2 in Parkinson's disease and model for microtubule interaction, Nature, 2020, 588(7837), 344–349 CrossRef CAS PubMed
- C. Xu and J. Min, Structure and function of WD40 domain proteins, Protein Cell, 2011, 2(3), 202–214 CrossRef CAS PubMed
- T. F. Smith, C. Gaitatzes, K. Saxena and E. J. Neer, The WD repeat: a common architecture for diverse functions, Trends Biochem. Sci., 1999, 24(5), 181–185 CrossRef CAS PubMed
- QUACPAC 2.2.2.1, OpenEye, Cadence Molecular Sciences, Santa Fe, NM, http://www.eyesopen.com Search PubMed.
- P. C. Hawkins, A. G. Skillman, G. L. Warren, B. A. Ellingson and M. T. Stahl, Conformer generation with OMEGA: algorithm and validation using high quality structures from the Protein Databank and Cambridge Structural Database, J. Chem. Inf. Model., 2010, 50(4), 572–584 CrossRef CAS PubMed
- D. Santos-Martins, L. Solis-Vasquez, A. F. Tillack, M. F. Sanner, A. Koch and S. Forli, Accelerating AutoDock4 with GPUs and Gradient-Based Local Search, J. Chem. Theory Comput., 2021, 17(2), 1060–1073 CrossRef CAS PubMed
- N. M. O'Boyle, M. Banck, C. A. James, C. Morley, T. Vandermeersch and G. R. Hutchison, Open Babel: An open chemical toolbox, J. Cheminf., 2011, 3(1), 33 Search PubMed
- P. Zhang, Y. Fan, H. Ru, L. Wang, V. G. Magupalli and S. S. Taylor,
et al., Crystal structure of the WD40 domain dimer of LRRK2, Proc. Natl. Acad. Sci. U. S. A., 2019, 116(5), 1579–1584 CrossRef CAS PubMed
- Maestro, Schrödinger, LLC, New York, NY, 2023.
- R. A. Friesner, J. L. Banks, R. B. Murphy, T. A. Halgren, J. J. Klicic and D. T. Mainz,
et al., Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy, J. Med. Chem., 2004, 47(7), 1739–1749 CrossRef CAS PubMed
- R. Abagyan, M. Totrov and D. Kuznetsov, Icm – a New Method for Protein Modeling and Design - Applications to Docking and Structure Prediction from the Distorted Native Conformation, J. Comput. Chem., 1994, 15(5), 488–506 CrossRef CAS
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew and D. S. Goodsell,
et al., AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility, J. Comput. Chem., 2009, 30(16), 2785–2791 CrossRef CAS PubMed
- S. Forli, R. Huey, M. E. Pique, M. F. Sanner, D. S. Goodsell and A. J. Olson, Computational protein-ligand docking and virtual drug screening with the AutoDock suite, Nat. Protoc., 2016, 11(5), 905–919 CrossRef CAS PubMed
- F. Gentile, M. Fernandez, F. Ban, A. T. Ton, H. Mslati and C. F. Perez,
et al., Automated discovery of noncovalent inhibitors of SARS-CoV-2 main protease by consensus Deep Docking of 40 billion small molecules, Chem. Sci., 2021, 12(48), 15960–15974 RSC
- D. A. Case, T. E. Cheatham 3rd, T. Darden, H. Gohlke, R. Luo and K. M. Merz Jr,
et al., The Amber biomolecular simulation programs, J. Comput. Chem., 2005, 26(16), 1668–1688 CrossRef CAS PubMed
- J. A. Maier, C. Martinez, K. Kasavajhala, L. Wickstrom, K. E. Hauser and C. Simmerling, ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB, J. Chem. Theory Comput., 2015, 11(8), 3696–3713 CrossRef CAS PubMed
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, Comparison of Simple Potential Functions for Simulating Liquid Water, J. Chem. Phys., 1983, 79(2), 926–935 CrossRef CAS
- D. A. Pearlman, D. A. Case, J. W. Caldwell, W. S. Ross, T. E. Cheatham and S. Debolt,
et al., Amber, a Package of Computer-Programs for Applying Molecular Mechanics, Normal-Mode Analysis, Molecular-Dynamics and Free-Energy Calculations to Simulate the Structural and Energetic Properties of Molecules, Comput. Phys. Commun., 1995, 91(1–3), 1–41 CrossRef CAS
- S. Le Grand, A. W. Götz and R. C. Walker, SPFP: Speed without compromise—A mixed precision model for GPU accelerated molecular dynamics simulations, Comput. Phys. Commun., 2013, 184(2), 374–380 CrossRef CAS
- A. W. Gotz, M. J. Williamson, D. Xu, D. Poole, S. Le Grand and R. C. Walker, Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 1. Generalized Born, J. Chem. Theory Comput., 2012, 8(5), 1542–1555 CrossRef CAS PubMed
- R. Salomon-Ferrer, A. W. Gotz, D. Poole, S. Le Grand and R. C. Walker, Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 2. Explicit Solvent Particle Mesh Ewald, J. Chem. Theory Comput., 2013, 9(9), 3878–3888 CrossRef CAS PubMed
- A. Fiser, R. K. Do and A. Sali, Modeling of loops in protein structures, Protein Sci., 2000, 9(9), 1753–1773 CrossRef CAS PubMed
- A. Fiser and A. Sali, ModLoop: automated modeling of loops in protein structures, Bioinformatics, 2003, 19(18), 2500–2501 CrossRef CAS PubMed
-
Baily C. J., Cieplak P., Cornell W. D. and Kollman P. A., A well-behaved electrostatic potential-based method using charge restraints for deriving atomic char, 1993 Search PubMed
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman and et al.Gaussian 16, Revision C.01, Wallingford, CT, Gaussian, Inc, 2019 Search PubMed
- D. R. Roe and T. E. Cheatham 3rd, PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data, J. Chem. Theory Comput., 2013, 9(7), 3084–3095 CrossRef CAS PubMed
- EON 2.4.0., OpenEye, Cadence Molecular Sciences, Inc., Santa Fe, NM, http://www.eyesopen.com Search PubMed.
- F. Pedregosa, G. Varoquaux, A. Gramfort, V. Michel, B. Thirion and O. Grisel,
et al., Scikit-learn: Machine Learning in Python, J. Mach. Learn. Res., 2011, 12, 2825–2830 Search PubMed
- S. Boresch, F. Tettinger, M. Leitgeb and M. Karplus, Absolute binding free energies: a quantitative approach for their calculation, J. Phys. Chem. B, 2003, 107(35), 9535–9551 CrossRef CAS
- O. Beckstein, D. Dotson, Z. Wu, D. Wille, D. Marson, I. Kenney and et al.alchemistry/alchemlyb: 2.0.0 (2.0.0), 2022.
-
T. Chen and C. Guestrin, XGBoost: A Scalable Tree Boosting System, Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, San Francisco, California, USA, Association for Computing Machinery, 2016, p. 785–794 Search PubMed
-
A. Mauri, alvaDesc: A Tool to Calculate and Analyze Molecular Descriptors and Fingerprints, Ecotoxicological QSARs, ed. Roy K., Springer US, New York, NY, 2020. p. 801–820 Search PubMed
- M. C. Sorkun, A. Khetan and S. Er, AqSolDB, a curated reference set of aqueous solubility and 2D descriptors for a diverse set of compounds, Sci. Data, 2019, 6(1), 143 CrossRef PubMed
- D. Fourches, E. Muratov and A. Tropsha, Trust, But Verify: On the Importance of Chemical Structure Curation in Cheminformatics and QSAR Modeling Research, J. Chem. Inf. Model., 2010, 50(7), 1189–1204 CrossRef CAS PubMed
- A. Cichońska, B. Ravikumar, R. J. Allaway, F. Wan, S. Park and O. Isayev,
et al., Crowdsourced mapping of unexplored target space of kinase inhibitors, Nat. Commun., 2021, 12(1), 3307 CrossRef PubMed
- S. Müller, S. Ackloo, A. Al Chawaf, B. Al-Lazikani, A. Antolin and J. B. Baell,
et al., Target 2035 – update on the quest for a probe for every protein, RSC Med. Chem., 2022, 13(1), 13–21 RSC
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc06880c |
| ‡ These authors contributed equally and share co-first authorship. |
| This journal is © The Royal Society of Chemistry 2024 |