
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydroalkylation of styrenes enabled by boryl radical mediated halogen atom transfer†
Serena
Pillitteri
 a,
Rajat
Walia
bc,
Erik V.
Van der Eycken
ad and
Upendra K.
Sharma
*a
a,
Rajat
Walia
bc,
Erik V.
Van der Eycken
ad and
Upendra K.
Sharma
*a
aLaboratory for Organic & Microwave-Assisted Chemistry (LOMAC), Department of Chemistry, University of Leuven (KU Leuven), Celestijnenlaan 200F, B-3001 Leuven, Belgium. E-mail: upendrakumar.sharma@kuleuven.be; usharma81@gmail.com
bDepartment of Chemistry, The University of Hong Kong, Pokfulam Road, Hong Kong SAR
cDepartment of Chemistry, City University of Hong Kong, Tat Chee Avenue, Kowloon, Hong Kong SAR
dPeoples' Friendship University of Russia (RUDN University), Miklukho-Maklaya Street 6, 117198 Moscow, Russia
First published on 3rd May 2024
Abstract
In this study, we present an inexpensive, stable, and easily available boryl radical source (BPh4Na) employed in a Halogen Atom Transfer (XAT) methodology. This mild and convenient strategy unlocks the use of not only alkyl iodides as radical precursors but also of the more challenging alkyl and aryl bromides to generate C-centered radicals. The generated radicals were further engaged in the anti-Markovnikov hydroalkylation of electronically diverse styrenes, therefore achieving the formation of C(sp3)–C(sp3) and C(sp3)–C(sp2) bonds. A series of experimental and computational studies revealed the prominent role of BPh4Na in the halogen abstraction step.
The mild and reliable generation of radicals achievable through photoredox catalysis has greatly impacted the way now chemists rethink bond formation and dissociation strategies.1–3 The steady advancement in the field has led to considerable endeavour to find a wide array of alkyl radical precursors, to meet the need for easily synthesizable starting materials and broadly applicable reactivities.4–6 Nevertheless, easily accessed starting materials are often challenging to activate. Radical generation from alkyl and aryl halides in particular, being fundamental building blocks in the organic chemist's toolbox, represents a convenient approach, but the highly negative reduction potential (Ered ≪ −2 V) of these species is a challenge to face in the development of photoredox catalyzed methodologies.7–9 To overcome this inherent limitation, a halogen atom transfer (XAT) step rather than a single electron transfer (SET) step becomes a viable alternative. The feasibility of the XAT process mostly relies on the bond dissociation energy (BDE) difference between the C–X bond and the abstractor-X bond, with the latter generally needing to be stronger than the broken C–X bond (Fig. 1).10 Among the strategies so far developed, the safety and selectivity concerns inherently linked to the first use of tin-containing reagents as halogen abstractors have been overcome by recent elegant photoredox-catalyzed halogen atom transfer approaches, with silicon-based reagents11–15 and α-amino radicals9,16,17 as halogen abstractors. Boron-based radicals, on the other hand, have received far less attention, despite their comparable performance in halogen atom abstraction.18,19 This is mainly due to the intrinsic instability of the transient neutral boryl radicals.20
 | ||
| Fig. 1 Boryl radical mediated halogen atom transfer (XAT) as a viable alternative for the activation of C–X bonds. | ||
To tackle the issue, ligated boryl radical sources were introduced instead. In these species, the boron atom is coordinated to a Lewis base (mostly amines, phosphines and NHC carbenes) and the radical generated after oxidation (and deprotonation) becomes more stable and can be involved in different reaction pathways, such as direct addition to unsaturated bonds, hydrogen atom transfer (HAT), but also XAT.21–24 Despite their appealing reactivity patterns,25,26 scarce reports have appeared reporting C–C bond-forming reactions, where often specific halogen-bearing starting materials were required to achieve halogen abstraction27 or the reactivity was only limited to alkyl iodides,28–30 more reactive in nature.
In these examples, organobromides were found to be unreactive. Given their higher BDE,11 bromine abstraction was found to be kinetically unfavorable.
In light of these synthetic gaps, we wondered if it would be possible to: (a) introduce a bench stable, affordable and easily accessible source of boryl radicals, which would also avoid the need to synthesize the boryl radical source; (b) identify an alternative reaction platform that would effectively expand the generation of C-centred radicals to the more common and abundantly available organobromides; and (c) apply the methodology to the hydroalkylation of styrenes. In this regard, we identified sodium tetraphenylborate (BPh4Na, B1) as an intriguing candidate for the purpose, being also commercially available. Tetraphenylborates have historically been used in the generation of biphenyls through electrochemical,31–34 thermal35 or photoredox methodologies36 and in the formation of aryl radicals as well.37 More recently, Lan, Xia and co-workers employed BPh4Na as a source of diphenylboryl radicals rather than the well-documented biaryl synthesis to accomplish C–O bond homolytic cleavage.38,39 Aware of the central role of the solvent (N,N-dimethylformamide, DMF) in stabilizing the highly reactive diphenyl boryl radical, we wondered if it could be possible to explore the XAT ability of this intermediate.
To assess the feasibility of the devised transformation, we began the optimization studies employing 4-tert-butyl styrene 1 and cyclohexyl bromide 2 as model substrates. Upon initial screening (see ESI† for further details), we found that sodium tetraphenylborate (BPh4Na, B1), in the presence of 4CzIPN (PC1) as the photocatalyst and employing DMF as the solvent, was able to promote the desired hydroalkylation of 1, affording product 3 in 65% yield (Fig. 2, entry 1) after 16 h of irradiation under blue light. Among the photocatalysts tested, iridium based photocatalysts proved to be effective, but provided lower yield (entries 3 and 4). 4CzIPN-tBu (PC2) afforded comparable results to 4CzIPN (entry 5). Nonetheless, 4CzIPN was chosen for further studies due to its easier synthesis and the economical price of starting materials. When analysing the effect of different solvents on the reaction outcome, we noticed the necessity of DMF to stabilize the boryl radical intermediate (entries 6–8).38 Interestingly, a shift in the wavelength (violet light, 390 nm instead of blue light, 456 nm) helped to increase the yield to 83% (entry 9, see also ESI†). Being aware of the challenges associated with the reduction of benzylic radicals,40–42 we hypothesized a final HAT or a proton-coupled electron transfer step and we therefore evaluated the effect of water addition to the reaction mixture. To our delight, adding 5 equiv. of water provided the desired product in high yield (entry 10). A change in the boron source did not lead to any appreciable improvement (entries 11–13). Control experiments (see ESI† for further details) confirmed that light irradiation and photocatalysts are essential for the reaction, which was completely suppressed in their absence. Similarly, in the absence of BPh4Na, product formation was not detected, suggesting the substantial role of the boryl radical source in C-radical generation. In addition, an oxygen-free atmosphere was found to be important for the reaction outcome, since without air exclusion a steep decline in the yield occurred.
 | ||
| Fig. 2 Optimization studies. | ||
Encouraged by the results obtained, we sought to investigate the generality of the method by evaluating a broad variety of substrates. Cyclic alkyl bromides afforded the desired hydroalkylated product in good to excellent yields (3–6, Fig. 3). Similarly, bridged alkyl bromides such as exo-2-bromonorbornane (7) and 1-bromoadamantane (16) were successfully coupled, with the latter substrate demonstrating the feasible involvement of tertiary bromides in the reaction. An acyclic secondary alkyl bromide, 2-bromobutane (8), could also be coupled, despite the lower yield. We then focused on primary alkyl bromides, challenging substrates to engage in the desired reactivity because of their higher reduction potential compared to their secondary and tertiary counterpart (9–15).43 Pleasingly, despite the decreased yield in some cases, a broad variety of substrates were found to be reactive. Non-functionalized alkyl chains, including the short ethyl chain (9), could be successfully incorporated. More importantly, the tolerance for common functionalities was observed, and terminal alkynes (12), esters (13), ethers (15), and silanes (14) could be successfully installed. Free hydroxyl groups could not be employed under our optimized conditions possibly due to their interaction with the boryl radical intermediate.38 Following this hypothesis, the protection of the free hydroxyl group allowed us to obtain the desired product 14. Remarkably, 1-bromo-2-chloroethane was selectively functionalized at the C–Br bond, affording product 11 in 51% yield, providing the pathway for orthogonal functionalization. The protocol was further extended to the installation of O-containing heterocyclic scaffolds (19–21), commonly present in medicinally relevant structures, which were successfully coupled in good to moderate yields, with lower yield observed for the strained oxetane ring (21). The acid-sensitive acetal 22 was also successfully employed, allowing further transformations. The N-containing heterocyclic molecule N-boc piperidine 17 and N-boc pyrrolidine 18 were successfully employed as well. In these cases, the addition of a base was necessary to improve the yield. Pleasingly, acetobromo-α-D-galactose (27) was found to be a competent reaction partner, affording the desired product in 39% yield. The employment of aryl bromides was then also attempted. 2-Bromopyridines (23–25) reacted smoothly and led to the formation of interesting functionalized scaffolds. The selective reactivity of 2-bromo-5-chloropyridine (25) at the C–Br bond in particular forecasts further involvement in more transformations. Aryl bromides such as 1-bromo-4-cyanobenzene underwent the reaction as well, despite the lower yield obtained (26). Csp2–Csp3 bond forging could therefore be unlocked as well. Further studies to broaden the range of aryl halides are currently undergoing in our group. We then sought to study the generality of the vinyl arene counterpart. Differently functionalized styrenes were efficiently alkylated. Despite the lower yield for styrene (28), which afforded the desired product in 37% yield, methyl (29), phenyl (35), acetoxy (38), –F (30–32), cyano (37), methylthio (39) and –BPin (43) substituents were tolerated. 2-Vinylnaphthalene (34) was functionalized as well. Remarkably, the presence of an electron donating group (–OMe, 40–42) on the styrene scaffold was also tolerated. Ortho, meta and para substitutions, both in the case of electron-deficient and in the case of electron-rich substitutions, gave high to moderate yields. Interestingly, 1-phenyl-1,3-butadiene (44) was also alkylated in good yield, providing an alternative to metal-catalyzed functionalization of this scaffold.44 An estrone derivative also successfully engaged in the transformation. Surprisingly, p-Cl and p-CF3 substituted styrenes were not compatible reaction partners, despite their electron-withdrawing nature that should have facilitated the radical attack. In these cases, GC-MS analysis of a reaction mixture that did not contain cyclohexyl bromide revealed that these styrenes underwent decomposition. For such cases, a faster decomposition rate could hinder the desired radical attack. Nevertheless, this catalytic system allows to achieve the alkylation of a broad range of styrenes avoiding the use of Grignard or organometallic reagents, whose employment causes loss in selectivity and functional group tolerance.41
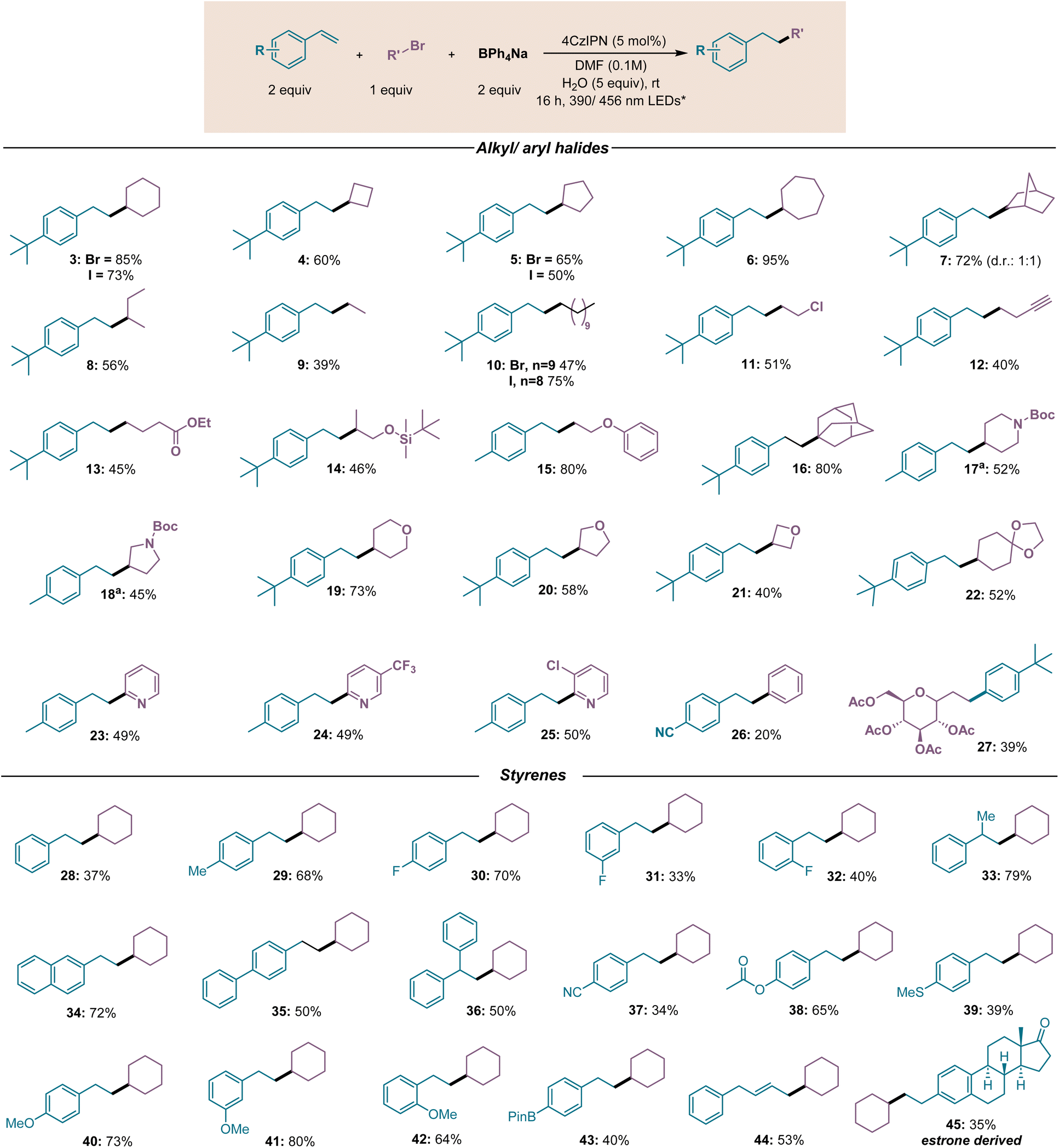 | ||
| Fig. 3 Scope of the developed transformation. Conditions unless otherwise noted: styrene (2 equiv.), alkyl/aryl bromide (1 equiv., 0.3 mmol), BPh4Na (2 equiv.), 4CzIPN (5 mol%), H2O (5 equiv.), DMF (0.1 M). *Irradiation with 390 nm Kessil light (52 W) or with 456 nm Kessil light (50 W), 16 h. a1.5 equiv. of DBU added. | ||
To gain more insights into the underlying mechanism of the reaction, a series of experimental and computational studies were performed. First, the addition of a radical scavenger (2,2,6,6-tetramethyl-1-piperidinyloxy, TEMPO) suppressed the reaction (only traces of the product were detected through GC-MS, see ESI† for more details). In addition, radical clock experiments were performed (Fig. 4C). In the presence of (bromomethyl)cyclopropane (C1), the ring-opened product was formed in 40% yield. Similarly, 6-bromohex-1-ene (C2) led to the formation of a cyclopentyl ring in 39% yield. These experiments therefore confirmed the hypothesis of a radical-based mechanism. We further confirmed by cyclic voltammetry that, among the participants in the described reaction, only BPh4Na is susceptible to oxidation in the redox window of the photocatalyst employed (E1/2ox = + 0.91 V in ACN vs. SCE, E(P*/P˙−) = + 1.35 V and E(P/P˙−) = – 1.21 V vs. SCE in ACN, see ESI† for further details). Fluorescence quenching experiments further showed that the borate initiates the reaction, since it was the only reactant able to quench the excited state of 4CzIPN (Fig. 4B). Despite this evidence, we observed discoloration of the reaction mixture after continuous irradiation, implying photo-bleaching or a structural change of the photocatalyst. We therefore wondered if the need for a 390 nm light could be explained by considering a change in the catalyst structure. As thought, UV-Vis analysis of the reaction mixture showed the decrease of the absorbance at 456 nm, and a blue-shift that would explain the need for a different wavelength to reach the completion of the reaction. Though the catalyst fate was not analysed in more detail, the formation of a new, but still active photocatalytic species cannot be excluded (see ESI† for further details).45
 | ||
| Fig. 4 Mechanistic proposal and investigations. (A) Proposed mechanism. (B) Stern–Volmer analysis. (C) Radical clock experiments. (D) Deuterium labelling experiments. (E) DFT calculations. | ||
After determining the role of BPh4Na in kick starting the reaction, we used computational methods to analyse the halogen atom transfer step. It was found that the interaction between the diphenylboryl radical and cyclohexyl bromide leads to the generation of a cyclohexyl radical through TS1 (ΔGR–Br = +12.5 kcal mol−1, Fig. 4E). Though slightly endergonic, the value is in accordance with the energetic profile calculated for α-amino radicals or NHC-boranes in similar halogen transfer steps.19 Interestingly, Noël et al. explained the lack of reactivity of alkyl bromides with a kinetically disfavoured step compared to alkyl iodides (ΔGR–Br = +17.9 kcal mol−1vs. ΔGR–I = +11.6 kcal mol−1).19 In our case instead, the energy required was found to be lower, thus enabling C–Br homolytic cleavage. Similarly, ΔGR–I was found to be +10.1 kcal mol−1. In light of this result, the use of alkyl iodides was also attempted (Fig. 3, 3, 5, 10). A satisfactory yield was obtained, and the reaction platform proved to be equally effective for alkyl iodides.
Upon radical formation, an exergonic radical attack on 4-tert-butylstyrene then occurs (ΔG = −19.2 kcal mol−1), generating a benzylic radical intermediate (Fig. 4A and E).40,41,46 At this stage, the reactivity observed for electronically diverse styrenes, including non-functionalized or electron-rich styrenes, urged us to investigate the last step of the mechanism. In the case of the latter group of styrenes especially, the reduction potential of the benzylic radical intermediate is high, leading us to hypothesize a hydrogen atom transfer (HAT) step, and not a SET-mediated anion formation, followed by protonation. In line with this hypothesis, we performed control experiments in the presence of DMF-d7 and D2O (Fig. 4D). To our surprise, no appreciable deuteration from the solvent was observed, while the reaction in the presence of D2O afforded the desired product with 80% deuterium enrichment. Considering that a homolytic cleavage of the O–H bond in water is unfavourable (BDE = +118 kcal mol−1),47 we began investigating what in our system could promote a HAT step involving H2O or D2O, with the aid of computational calculations as well. We first performed a control experiment where bromocyclohexane was not added to the reaction mixture. The hydrogenation of 4-tert-butylstyrene was observed. Similarly, if instead of H2O, D2O was added, deuterium incorporation could be detected (Fig. 4D1 and D2). These results suggested that a HAT-mediated radical mechanism could be operating,47 involving BPh4Na (or side products) and H2O. We therefore took into consideration different reaction pathways. It has been demonstrated that a complex between borane and water can act as a HAT mediator, activating water toward a homolytic O–H bond cleavage.48,49 Similarly, water activation toward HAT was presented in a recent study by Studer and co-workers.47 Considering similar mechanistic steps, we have computationally found that, in the presence of a complex between the boryl radical and water, an exergonic process would be operative (ΔG = −65.1 kcal mol−1). Nonetheless, the involvement of borinic acid or HBr (side products of the reaction) as HAT mediators could not be excluded (Fig. 4A). Though not possible to determine the exact nature of the HAT mediator due to the complexity of the reaction mixture, these experiments aided by computational studies suggested that a HAT step is involved, and that the hydrogen comes from water.
An alternative pathway could involve the reduction of the benzyl radical intermediate.50–52 Nevertheless, the anion that would be forming through a radical-polar cross-over could not be trapped with CO2 or other electrophiles (see ESI† for further information).
In conclusion, we have demonstrated the ability of an inexpensive and easily available boryl radical source like sodium tetraphenylborate to enable halogen atom transfer not only from alkyl iodides, but also from alkyl and aryl bromides, commercially available and stable radical precursors. The alkyl/aryl radicals thus generated were further involved in the anti-Markovnikov hydroalkylation of electronically diverse styrenes. A broad scope of substrates, including natural product-derived molecules, was subjected to these mild reaction conditions, achieving the formation of the hydroalkylated products in moderate to good yield.
Overall, the methodology that we present here constitutes a valuable addition to the landscape of halogen atom transfer based methodologies. As a result, we are sure that our findings will serve as the foundation for further studies and development of reactivity patterns for boron centred radical species.
Data availability
All experimental and computational data can be found in the manuscript and in the ESI.† Raw data are available from the authors upon request.Author contributions
The authors confirm contribution to the paper as follows: study conception and design: S. P., U. K. S.; methodology and experiments: S. P.; DFT calculations: R. W.; draft manuscript preparation: S. P., R. W., U. K. S., E. V. d. E.Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. P. (experiments and writing) is thankful to the FWO for obtaining a PhD scholarship (S. P. grant no. 11H0121N). S. P. is grateful to Dr Prabhat Ranjan (AMIBM, Maastricht University) for the valuable scientific discussions. S. P. and U. K. S. (supervision and writing) are thankful to Dr Igor Beckers (KU Leuven) for his valuable suggestions. R. W. (DFT calculations) wishes to thank the Computer Service Center (CSC) at the City University of Hong Kong for its high performance computing (HPC) facilities. The publication has been prepared with the support of the “RUDN University Strategic Academic Leadership Program” (recipient E. V. V. d. E.; writing and supervision).Notes and references
- B. König, Eur. J. Org. Chem., 2017, 2017, 1979–1981 CrossRef.
- J. D. Bell and J. A. Murphy, Chem. Soc. Rev., 2021, 50, 9540–9685 RSC.
- L. Marzo, S. K. Pagire, O. Reiser and B. König, Angew. Chem., Int. Ed., 2018, 57, 10034–10072 CrossRef CAS PubMed.
- S. Crespi and M. Fagnoni, Chem. Rev., 2020, 120, 9790–9833 CrossRef CAS PubMed.
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed.
- D. Ravelli, S. Protti and M. Fagnoni, Chem. Rev., 2016, 116, 9850–9913 CrossRef CAS PubMed.
- D. J. Carlsson and K. U. Ingold, J. Am. Chem. Soc., 1968, 90, 7047–7055 CrossRef CAS.
- I. Ghosh, T. Ghosh, J. I. Bardagi and B. König, Science, 2014, 346, 725–728 CrossRef CAS PubMed.
- T. Constantin, M. Zanini, A. Regni, N. S. Sheikh, F. Juliá and D. Leonori, Science, 2020, 367, 1021–1026 CrossRef CAS PubMed.
- F. Juliá, T. Constantin and D. Leonori, Chem. Rev., 2022, 122, 2292–2352 CrossRef PubMed.
- P. Zhang, C. “Chip” Le and D. W. C. MacMillan, J. Am. Chem. Soc., 2016, 138, 8084–8087 CrossRef CAS PubMed.
- A. ElMarrouni, C. B. Ritts and J. Balsells, Chem. Sci., 2018, 9, 6639–6646 RSC.
- C. Chatgilialoglu, C. Ferreri, Y. Landais and V. I. Timokhin, Chem. Rev., 2018, 118, 6516–6572 CrossRef CAS PubMed.
- H. A. Sakai, W. Liu, C. “Chip” Le and D. W. C. MacMillan, J. Am. Chem. Soc., 2020, 142, 11691–11697 CrossRef CAS PubMed.
- N. Takemura, Y. Sumida and H. Ohmiya, ACS Catal., 2022, 12, 7804–7810 CrossRef CAS.
- Z. Zhang, B. Górski and D. Leonori, J. Am. Chem. Soc., 2022, 144, 1986–1992 CrossRef CAS PubMed.
- H. Zhao, A. J. McMillan, T. Constantin, R. C. Mykura, F. Juliá and D. Leonori, J. Am. Chem. Soc., 2021, 143, 14806–14813 CrossRef CAS PubMed.
- B. Sheeller and K. U. Ingold, J. Chem. Soc., Perkin Trans. 2, 2001, 480–486 RSC.
- L. Capaldo, T. Noël and D. Ravelli, Chem Catal., 2022, 2, 957–966 CrossRef CAS.
- T.-Y. Peng, F.-L. Zhang and Y.-F. Wang, Acc. Chem. Res., 2023, 56, 169–186 CrossRef CAS PubMed.
- B. P. Roberts, Chem. Soc. Rev., 1999, 28, 25–35 RSC.
- C. Wu, X. Hou, Y. Zheng, P. Li and D. Lu, J. Org. Chem., 2017, 82, 2898–2905 CrossRef CAS PubMed.
- G. Duret, R. Quinlan, P. Bisseret and N. Blanchard, Chem. Sci., 2015, 6, 5366–5382 RSC.
- A. Staubitz, A. P. M. Robertson, M. E. Sloan and I. Manners, Chem. Rev., 2010, 110, 4023–4078 CrossRef CAS PubMed.
- S. Kobayashi, T. Kawamoto, S. Uehara, T. Fukuyama and I. Ryu, Org. Lett., 2010, 12, 1548–1551 CrossRef CAS PubMed.
- X. Pan, E. Lacôte, J. Lalevée and D. P. Curran, J. Am. Chem. Soc., 2012, 134, 5669–5674 CrossRef CAS PubMed.
- V. I. Supranovich, V. V. Levin, M. I. Struchkova, A. A. Korlyukov and A. D. Dilman, Org. Lett., 2017, 19, 3215–3218 CrossRef CAS PubMed.
- J. A. Baban and B. P. Roberts, J. Chem. Soc., Perkin Trans. 2, 1988, 1195 RSC.
- T. Kawamoto, S. Uehara, H. Hirao, T. Fukuyama, H. Matsubara and I. Ryu, J. Org. Chem., 2014, 79, 3999–4007 CrossRef CAS PubMed.
- T. Wan, L. Capaldo, D. Ravelli, W. Vitullo, F. J. de Zwart, B. de Bruin and T. Noël, J. Am. Chem. Soc., 2023, 145, 991–999 CrossRef CAS PubMed.
- A. Music, A. N. Baumann, P. Spieß, A. Plantefol, T. C. Jagau and D. Didier, J. Am. Chem. Soc., 2020, 142, 4341–4348 CrossRef CAS PubMed.
- D. H. Geske, J. Phys. Chem., 1959, 63, 1062–1070 CrossRef CAS.
- D. H. Geske, J. Phys. Chem., 1962, 66, 1743–1744 CrossRef CAS.
- S. B. Beil, S. Möhle, P. Enders and S. R. Waldvogel, Chem. Commun., 2018, 54, 6128–6131 RSC.
- C. Gerleve and A. Studer, Angew. Chem., Int. Ed., 2020, 59, 15468–15473 CrossRef CAS PubMed.
- A. Music, A. N. Baumann, F. Boser, N. Müller, F. Matz, T. C. Jagau and D. Didier, Chem.–Eur. J., 2021, 27, 4322–4326 CrossRef CAS PubMed.
- F. Yue, H. Ma, P. Ding, H. Song, Y. Liu and Q. Wang, ACS Cent. Sci., 2023, 9, 2268–2276 CrossRef CAS PubMed.
- W.-D. Li, Y. Wu, S.-J. Li, Y.-Q. Jiang, Y.-L. Li, Y. Lan and J.-B. Xia, J. Am. Chem. Soc., 2022, 144, 8551–8559 CrossRef CAS PubMed.
- D. Didier, Synthesis, 2023, 55, 232–239 CrossRef CAS.
- B. Giese, Angew. Chem., Int. Ed., 1983, 22, 753–764 CrossRef.
- Z. Wu, S. N. Gockel and K. L. Hull, Nat. Commun., 2021, 12, 5956 CrossRef CAS PubMed.
- K. Donabauer and B. König, Acc. Chem. Res., 2021, 54, 242–252 CrossRef CAS PubMed.
- W. Zhang and S. Lin, J. Am. Chem. Soc., 2020, 142, 20661–20670 CrossRef CAS.
- P.-Z. Wang, W.-J. Xiao and J.-R. Chen, Chin. J. Catal., 2022, 43, 548–557 CrossRef CAS.
- S. Grotjahn and B. König, Org. Lett., 2021, 23, 3146–3150 CrossRef CAS PubMed.
- D. D. M. Wayner, D. J. McPhee and D. Griller, J. Am. Chem. Soc., 1988, 110, 132–137 CrossRef CAS.
- J. Zhang, C. Mück-Lichtenfeld and A. Studer, Nature, 2023, 619, 506–513 CrossRef CAS.
- D. A. Spiegel, K. B. Wiberg, L. N. Schacherer, M. R. Medeiros and J. L. Wood, J. Am. Chem. Soc., 2005, 127, 12513–12515 CrossRef CAS PubMed.
- W. Tantawy and H. Zipse, Eur. J. Org. Chem., 2007, 2007, 5817–5820 CrossRef.
- Q.-Y. Meng, T. E. Schirmer, A. L. Berger, K. Donabauer and B. König, J. Am. Chem. Soc., 2019, 141, 11393–11397 CrossRef CAS PubMed.
- K. Donabauer, M. Maity, A. L. Berger, G. S. Huff, S. Crespi and B. König, Chem. Sci., 2019, 10, 5162–5166 RSC.
- S. Grotjahn, C. Graf, J. Zelenka, A. Pattanaik, L. Müller, R. J. Kutta, J. Rehbein, J. Roithová, R. M. Gschwind, P. Nuernberger and B. König, Angew. Chem., Int. Ed., 2024, 63, e202400815 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc01731e |
| This journal is © The Royal Society of Chemistry 2024 |