
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFrom propenolysis to enyne metathesis: tools for expedited assembly of 4a,8a-azaboranaphthalene and extended polycycles with embedded BN†
Federica
Rulli‡
 a,
Guillem
Sanz-Liarte‡
b,
Pol
Roca
b,
Nina
Martínez
a,
Víctor
Medina
b,
Raimon
Puig de la Bellacasa
a,
Alexandr
Shafir
*bc and
Ana B.
Cuenca
*ac
a,
Guillem
Sanz-Liarte‡
b,
Pol
Roca
b,
Nina
Martínez
a,
Víctor
Medina
b,
Raimon
Puig de la Bellacasa
a,
Alexandr
Shafir
*bc and
Ana B.
Cuenca
*ac
aBISi-Bonds/CRISOL Group, Dept. of Organic and Pharmaceutical Chemistry. Institut Químic de Sarrià, Universitat Ramon Llull, Vía Augusta 390, 08017 Barcelona, Spain. E-mail: anabelen.cuenca@iqs.url.edu
bBISi-Bonds Group, Dept. Química Biológica. Institut de Química Avançada de Catalunya, IQAC-CSIC, C/Jordi Girona 20, 08034 Barcelona, Spain. E-mail: alexandr.shafir@iqac.csic.es
cCentro de Innovación en Química Avanzada (ORFEO-CINQA), Barcelona, Spain
First published on 7th March 2024
Abstract
The synthesis of BN-containing molecules, which have an interesting isosteric relationship to their parent all-C cores, has drawn a great deal of attention as an avenue to alter and tune molecular function. Nevertheless, many cores with embedded BN are still hard to synthesize, and thus, further effort is required in this direction. Herein, we present an integrated approach to BN-containing polycycles rooted in an exceptionally clean B–N condensation of amines with a tri-allylborane. Having released propene as the only byproduct, the resulting BN precursors are seamlessly telescoped into BN-containing polycyclic cores via a set of additional methodologies, either developed here ad-hoc or applied for the first time for the synthesis of BN-cycles. As the “sharpening stone” of the process, BN-embedded naphthalene, which has previously only been obtained in low yield, can now be synthesized efficiently through propenolysis, ring-closing metathesis and a new high-yielding aromatization. As a more advanced application, an analogously obtained BN-containing bis-enyne is readily converted to BN-containing non-aromatic tetra-, penta- and hexacyclic structures via ring-closing enyne metathesis, followed by the Diels–Alder cycloaddition. The resulting air-sensitive structures are easily handled by preventive hydration (quaternization) of their B–N bridge; reverting this hydration restores the original Bsp2–Nsp2 structure. In the future, these structures may pave the way to BN-anthracenes and other π-extended BN-arenes.
Introduction
The notion of BN-isosterism refers to the kinship existing between an organic molecule and a derivative containing a boron-nitrogen (BN) pair in place of a carbon–carbon (CC) unit. Such BN derivative tends to be isostructural and isoelectronic with its all-carbon parent as a consequence of an electron counting formalism, with combinations B–N (3 + 5) and C–C (4 + 4) contributing a sum total of 8 valence electrons.1 Although B/N replacement at mutually remote C-sites may produce, among others, interesting 1,3 or 1,4-BN-isosteres, the lion's share of research in this field is focused on 1,2-isosterism, which maps carbon–carbon single, double or even triple bonds onto their equivalent boron–nitrogen pairs. Hence, the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond in ethylene maps onto an isosteric H2N–BH2 aminoborane (A, Fig. 1A), while the aromatic benzene ring is recast as the unsaturated 6-membered 1,2-azaborinine cycle (B, Fig. 1B), as demonstrated in the pioneering study launched by the Dewar laboratory in the late 1950's.2
C bond in ethylene maps onto an isosteric H2N–BH2 aminoborane (A, Fig. 1A), while the aromatic benzene ring is recast as the unsaturated 6-membered 1,2-azaborinine cycle (B, Fig. 1B), as demonstrated in the pioneering study launched by the Dewar laboratory in the late 1950's.2
 | ||
| Fig. 1 (A) BN-isosterism illustrated through BN-mapping of ethylene, and (B–D) some common unsaturated ring structures. | ||
BN isosteric replacement thus allows for even a small molecular core to be mapped onto a range of BN isosteres to potentially give diverse physical and chemical properties. This has sparked a great deal of interest from groups working in areas ranging from medicinal chemistry3 to new organic electronic materials, as the mapping of BN onto polycyclic aromatic (or heteroaromatic) ring structures (PAH) can lead to cores with improved optoelectronic properties.4 Taking naphthalene as the smallest PAH archetype, the energy levels of π-type molecular orbitals have been altered by mapping the all-carbon core onto several possible BN-isosteres,5 including the historically important bicyclic azaborine 1 (Fig. 1C).6 Potentially very interesting but much less explored are the BN isosteres of fully or partially saturated polycyclic molecules, such as the decaline-type derivative 1′ (Fig. 1D).7
The need for new synthetic tools to unlock the full potential of BN-isosterism is reflected in game-changing advances from several laboratories for the production of BN-containing mono- and polycyclic aromatic cores,1,8 including impactful N-directed electrophilic borylation protocols,9 ring-closing metathesis (RCM) of diolefinic aminoboranes,10 or late-stage diversification of BN heterocycles.8b Nevertheless, efforts are ongoing to find more efficient ways to produce certain classes of polycyclic BN-containing cores, with challenges including the poor yield (see below) of the synthesis of BN-containing naphthalene 1 (shown in Fig. 1C), difficulties in accessing BN-containing acene-type cores, or the synthesis – and handling – of non-aromatic BN-containing cycles.
We now show how such aromatic and non-aromatic polycycles can be prepared and handled through a combination of virtually quantitative proto-deallylation of the B-allyl moiety, norbornene-aided aromatization, core extension via ring-closing enyne metathesis/Diels–Alder manifold, and product handling via interim B–N hydration.
Results and discussion
Leveraging propenolysis with B(allyl)3 for clean formation of aminoboranes
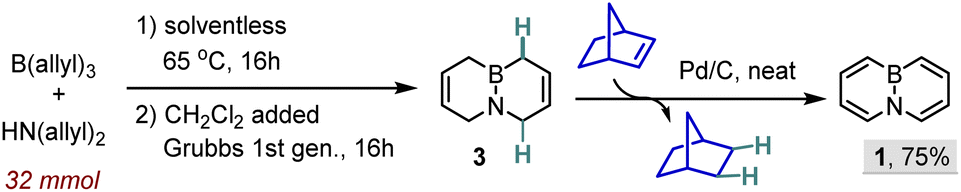
Following the group's recent foray into the development of BN-inspired methods (a collaborative effort with the Liu laboratory),11 we became interested in exploiting the proto-deallylation reactivity of tri-allylboranes, which was originally documented by Mikhailov and Tutorskaya in the 1950s,12 as an entry point into 6-membered BN-heterocycles. BN-naphthalene 1 was chosen as a test case due to the shortcomings of current routes to access this compound.13 While the reported synthesis of 1 relies on a robust ring-closing metathesis (RCM) of tetra-allyl aminoborane 2 to bicycle 3 (see Scheme 1), the process suffers from the rather inefficient aromatization of 3 to 1. Specifically, while the Pd-catalyzed dehydrogenation of 3 in cyclohexene leads to a hard-to-separate mixture of 1 and BN-tetralin 4,13b the alternative entails an even lower-yielding, albeit cleaner, week-long oxidation of 3 with dichloro-dicyano-1,4-benzoquinone (DDQ, Scheme 1).13c In our hands, the latter route meant that the considerable effort (and cost) to produce a 4.8 g batch of 3 ultimately resulted in a meager ∼600 mg of 1. Importantly, even the synthesis of the initial precursor 2 leaves a wide margin for improvement, particularly considering the cost and waste removal issues stemming from the use of the allyl-tin reagent13b or labor-intensive (filtration, distillations) sequence based on the allyl Grignard reagent.13c | ||
| Scheme 1 Current approach to prepare BN-naphthalene 1. | ||
With 1 serving as a building block for a wide range of BN targets, from simple BN-mapped naphthalene derivatives to extended BN-perylenes14 to emissive core candidates (Samsung Display Ltd),15 we envisage that improving its synthesis might have a positive impact in a number of areas in the field.
Hence, as an alternative path to 2, we proceeded to expose B(allyl)3 to a solution of N,N-diallylamine in CH2Cl2 at 0 °C, resulting in a species tentatively identified (via1H NMR, see ESI†) as the acid–base adduct 5 (Scheme 2A). When heated to 45 °C, this adduct gradually evolved to tetra-allyl aminoborane 2, reaching complete conversion after ∼48 h. Seeking to shorten the reaction time, we opted for a solvent-free approach in hopes of enabling a higher-temperature process and doing away with the subsequent solvent switch in the RCM step. Gratifyingly, dropwise addition of N,N-diallylamine to neat triallylborane at 0 °C, followed by heating the resulting adduct to 65 °C, led to visible bubbling, and after ∼6 h, the formation of a clear liquid, shown by 1H NMR to be the remarkably clean neat tetra-allyl species 2 (Scheme 2A).
 | ||
| Scheme 2 (A) One-pot two-stage sequence to convert B(allyl)3 to bicycle 3. (B) Comparison of DFT barriers for 4- and 6-membered transition states for the proto-deallylation step. | ||
This efficient reaction of an N–H substrate with organoborane appears to be unique for B(allyl)3 and does not extend to non-allylic trialkylboron derivatives.12 In the original 1961 report, this reactivity difference was rationalized through the low B–Callyl bond energy.12b It is important to note, however, that while any NH amine-organoborane adduct could in principle undergo proto-dealkylation via a 4-membered transition state (i.e. NH proton transferred to B–C carbon), allylic boranes are uniquely capable of propene elimination via a 6-membered transition state. In line with this reasoning, DFT modelling revealed a barrier of ∼40 kcal mol−1 for converting 5 to 2via a 4-membered transition state, which would be common to allyl- and alkyl boranes. However, this activation energy was nearly halved (ΔG‡ = 21 kcal mol−1) for the propene elimination via a 6-membered TS, which we believe to be the real reason for the enhanced reactivity of B(allyl)3 adducts in this process (see Scheme 2B).
As propylene gas was the only by-product, the freshly obtained tetra-allyl precursor 2 was employed directly in the subsequent metathesis step. In a 16 mmol run, newly formed 2 was simply dissolved in CH2Cl2 and treated with the Grubbs first-generation catalyst for 16 h, leading to bicycle 3 in a 72% overall yield (1.53 g) upon distillation.
Having established one-pot access to 3, we briefly turned our attention to the final aromatization step, in which losses of up to 80–90% (!) in yield are generally incurred. The large quantities of undesired BN-tetralin 4 formed when using the Pd/C-cyclohexene system13b (see Scheme 1) not only divert a large share of the precursor away from the target but also hamper the product purification. Since 4 presumably arises from the shuttling of an H2 molecule between two unsaturated rings in substrate 3 rather than H2 being passed off to the sacrificial acceptor, we wondered whether this side process might be suppressed by uncovering a more efficient H2 acceptor. To put this into practice, the catalytic aromatization of 3 with Pd/C was tested in the presence of a range of olefins (6.5 equiv.) at 70 °C for 24 h (Scheme 3). While simple cyclic and linear olefins still led to significant amounts of undesired 4, the use of norbornene led to BN-naphthalene being formed with an unprecedented 92![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :8 selectivity (see ESI Fig. S1†). We note that the chemoselectivity of reaction tracks with the enthalpy of hydrogenation (ΔH°) of the acceptor in the order cyclohexene (−28 kcal mol−1) < 1-hexene ≈ t-Bu-ethene (−30 kcal mol−1) < norbornene (−33 kcal mol−1).16
:8 selectivity (see ESI Fig. S1†). We note that the chemoselectivity of reaction tracks with the enthalpy of hydrogenation (ΔH°) of the acceptor in the order cyclohexene (−28 kcal mol−1) < 1-hexene ≈ t-Bu-ethene (−30 kcal mol−1) < norbornene (−33 kcal mol−1).16
 | ||
| Scheme 3 Evaluation of hydrogen acceptors in the Pd-catalyzed aromatization of 3 to 1. a Conducted in neat olefin acceptor. b Using cyclohexane as solvent. | ||
Following additional optimization, the aromatization of 3 was conducted at 110 °C in the presence of catalytic Pd/C and 3.5 equiv. of norbornene (neat), affording 1 in an 76% yield, a major improvement over the prior art. The streamlined process was then carried out on a multi-gram (32 mmol) scale (Scheme 4). A neat 1:1 mixture of B(allyl)3 and diallylamine was heated to 65 °C for 16 h; the as-obtained 2 was then diluted with CH2Cl2 and treated with the Grubbs first-generation catalyst to afford 3.1 g of 3 (72% over 2 steps) upon distillation. Finally, the aromatization over Pd/C in norbornene (3.5 equiv.) produced 2.33 g of target BN-naphthalene 1 upon column chromatography (75%), representing an overall yield of 54% based on starting B(allyl)3.
 | ||
| Scheme 4 Streamlined multi-gram synthesis of BN-naphthalene 1. | ||
Integrated approach towards polycyclic BN-containing cores
Seeing how the proto-deallylation reaction enabled the facile and clean assembly of aminoborane 2, this process was exploited to accelerate the discovery of new BN-containing polycyclic cores. B(allyl)3 was allowed to react with neat N,N-dipropargyl amine, leading after ∼6 h to clean formation of neat aminoborane 6 containing two 1,7-enyne groups (Scheme 5A). This product offers an interesting opportunity to access polycyclic BN products via the ring-closing enyne metathesis reaction (RCEYM).17 Indeed, exposing 6 to the first-generation Grubbs catalyst (sealed tube, 3 days) afforded the expected bicyclic di-vinyl aminoborane 7, which was isolated in 40% overall yield via low-pressure bulb-to-bulb distillation as a colorless moisture-sensitive low-melting crystalline solid. The structure of the compound was confirmed, in part, by 1H NMR resonances for the newly formed vinyl group at 6.35, 4.98 and 4.92 ppm. | ||
| Scheme 5 (A) Access to bis-enyne aminoborane 6 and enyne metathesis ring closure towards 1,3-diene core 7. (B) Access to internal bis-alkyne 9 and its RCEYM to form bicycle 11 (GH2 = Grubbs–Hoveyda second-generation catalyst). | ||
Encouraged by this result, we also sought to extend this approach to substituted bicyclic BN-cores by applying the RCM reaction to more challenging internal alkynes. To this end, doubly arylated precursor 8 was prepared via Sonogashira coupling and subsequently deprotected to dipropargylamine 9 (Scheme 5B). This compound reacted smoothly with B(allyl)3 to afford new aminoborane 10 in good purity. As anticipated, enyne metathesis in this case proved to be more challenging, with initial attempts using the Grubbs first-generation catalyst leading to little-to-no conversion. Nevertheless, we were pleased to find that switching to the more active Grubbs–Hoveyda second-generation catalyst and raising the temperature to 45 °C led to the selective formation of the target bicyclic structure 11, as confirmed by GC-MS and 1H NMR analyses (Scheme 5B).
With the new bis-diene aminoborane 7 in hand, we proceeded to explore double Diels–Alder reaction as a way to access new BN-doped polycyclic cores.18 Our initial test involved heating 7 with 2.4 equiv. of acetylene dicarboxylate (12) as dienophile in toluene at 80 °C (Scheme 6A). After 12 h, the GC-MS analysis of the mixture showed nearly complete consumption of 7 and formation of a new major species with m/z = 355, which would be consistent with the mono Diels–Alder adduct. A small peak with m/z = 525 was also observed, hinting at the possible formation of the target double-Diels–Alder product 13. Fortunately, increasing the amount of dienophile to 4.0 equiv. and temperature to 110 °C led, after 15 h, to the formation of the target tetracyclic aminoborane 13 as the major product (Scheme 6A). We note that given the hydroscopic nature of the product, unequivocal determination of the stereochemistry of the compound (i.e., cis- vs. trans-adduct) via NMR was hampered by the difficult purification of the crude mixture. After a number of unsuccessful attempts, we wondered whether product manipulation might be facilitated by temporary quaternization of the boron atom. The addition of H2O to a solution of crude 13 in Et2O led to the precipitation of hydrate 14 with a B–N bridge present in HN⋯B(OH) form.19 Compound 14 proved to be air-stable and could now be readily purified using reversed-phase (C18) column chromatography or simply by repeated washing.
 | ||
| Scheme 6 Synthesis of non-aromatic BN-containing tetra-, penta- and hexacycles via double Diels–Alder reaction. (A) Synthesis of BN-tetracycle 13 and its interim hydration via B-quaternized derivative 14. (B) Selected region of the 1H NMR spectrum of 14 showing the olefinic resonance and diastereotopic B–CHH′ group. (C) ORTEP diagram of the X-ray structure of 14 at 50% thermal ellipsoids; most H atoms have been omitted for clarity. (D) TGA analysis trace for 14 showing the possible water loss at ∼162 °C. (E) Synthesis of BN-containing non-aromatic pentacycle 16via hexacycle 15. (F) X-ray structure of 16; all H atoms have been omitted for clarity. (G) Diagram showing the trans-endo, endo approach of maleic anhydride during the Diels–Alder step. | ||
The clean 1H NMR spectrum of 14 now clearly revealed the presence of a single diastereomer with a symmetry element at the B–N bridge, as evidenced by a single characteristic set of diastereotopic B–CHH signals (0.78 and 0.11 ppm, Jgem = 13.7 Hz), along with a sole olefinic resonance at 5.61 ppm (2H) (see Scheme 6B). Given that the hydrated (H)N–B(OH) bridge precludes the possibility of a C2 symmetry axis, the two halves of bicycle 14 must be related by a mirror symmetry (Cs), which is only possible if 14—and, by extension, 13—is a Diels–Alder cis-adduct. This conclusion was confirmed by single crystal X-ray diffraction analysis of 14, which not only showed the cis-adduct configuration but also revealed the trans disposition of H and OH groups on the hydrated B–N bridge (Scheme 6C).20
At this point, we wondered whether the BN-hydration could be reversed to recover pure adduct 13. Initial attempts to chemically remove the water molecule, including with Burgess reagents, met with little success. However, thermogravimetric analysis (TGA) of 14 revealed a well-defined endothermic weight loss of ∼3% at 162 °C, in line with the release of a water molecule (Scheme 6D). Translating this knowledge to the preparatory scale, a sample of 14 was heated under vacuum to ∼160 °C, which indeed resulted in the formation of BN-tetracycle 13 in >90% purity. This transformation was accompanied a change in the 11B NMR chemical shift from ∼0.0 ppm for 14 (quaternized borane) to 40 ppm for 13. The latter value is typical for related Bsp2 aminoboranes, including 3 (41.2 ppm) and 7 (40.6 ppm). We believe that this approach, which we dubbed “interim hydration,” may prove useful for the purification of other moisture-sensitive non-aromatic cyclic aminoboranes through temporary protection of the BN unit via the reversible addition of water or other protic agents.
Continuing with our exploration, the double Diels–Alder addition between 7 and maleic anhydride led to the formation of hexa-cyclic species 15, as evidenced by a GC-MS peak with m/z = 381 (Scheme 6E). Although the compound was not isolated, subsequent reaction with H2O led not only to the hydration of the BN bond but also the hydrolytic opening of two anhydride rings. The 1H NMR spectrum of resulting species was complicated by the apparent lack of any symmetry and included two olefinic CH resonances at 5.60 and 5.42 ppm, along with three discernible COOH resonances in the 11–13 ppm region. Fortunately, X-ray-quality crystals of the product could be grown via slow diffusion of Et2O into a MeOH solution.20 The solid-state structure revealed that this lack of symmetry was due to one of the carboxylic acid moieties undergoing condensation with the B–OH group to form a 6-membered mixed B–O–C(O) anhydride ring, resulting in an overall pentacyclic core (16, Scheme 6F). The 3D structure also revealed a trans-endo-endo configuration, indicating that the initial Diels–Alder cycloaddition took place with two dienophile molecules approaching opposing sides of bicyclic precursor 7 in an endo configuration.21
Having successfully applied the newly developed sequence to polycyclic targets 13/14 and 15/16, a number of future applications can now be envisioned. For example, this opens the door to BN isosteres of certain natural polycycles and of course, to the preparation of BN-containing polycyclic aromatic compounds, including access to new BN-tetracenes via 4-fold aromatization. En route to the latter goal, preliminary experiments show that heating 13 with Pd/C in decane (180 °C, 48 h)22 produces new species 17 with aromatized external rings (Scheme 7), as observed from two characteristic 1H NMR doublets at 7.81 and 7.20 ppm. The product was found to be hydroscopic, with 1H NMR spectra typically presenting varying amounts of BN-hydrate, observed as a second set of doublets at 7.66 and 7.16 ppm. Similarly to the isolation of a BN-hydrate shown earlier, purification via reversed-phase column chromatography (C18) produced a mixture of 17 and 17·H2O, which could be fully converted to the rather clean hydrate form (see ESI†) through the addition of water. In line with our interim hydration strategy, the dehydration of 17·H2O at 160 °C under vacuum led to clean 17, which was characterized by a peak with m/z = 522 (M + H, ESI+).23 The 1H NMR spectrum of the product showed, in addition to aforementioned aromatic doublets (2H each), two broad singlets arising from N–CH2 (4.38 ppm, 4H) and B–CH2 (2.28 ppm, 4H) groups of central rings.
 | ||
| Scheme 7 Semi-aromatization of tetracycle 13 (un-optimized). | ||
Conclusions
In conclusion, this study contributes to the area of BN-isosterism through an integrated synthetic approach to a series of BN-containing cyclic cores. Owing to the highly efficient assembly of BN-containing precursors using the proto-deallylation of B(allyl)3, which produces propylene as the only by-product, the B–N formation step is seamlessly integrated with the subsequent olefin or enyne metathesis stages. When combined with a newly developed high-performing aromatization, the approach gives rise to the first efficient synthesis of BN-naphthalene 1. Furthermore, the manifold in which the proto-deallylation leads into the enyne metathesis/Diels–Alder ring core extension is shown to be a potentially versatile entry point into interesting new non-aromatic B–N embedded systems containing 4, 5 or 6 six-membered rings. It is also shown that the addition of water converts air-sensitive non-aromatic B–N cores to BN-hydrates suitable for handling and purification, following which the water molecule can be removed to recover original polycycles, thus converting this interim hydration strategy into a potentially interesting synthetic tool. Although beyond the scope of this work, based on our preliminary results with partial aromatization (product 17), one can easily imagine further applications of strategies presented here, including the potential conversion of tetracyclic cores such as 13 into new π-extended cores, such as hitherto unreported BN-containing tetracene.24Data availability
The datasets supporting this article have been uploaded as part of the ESI.†Author contributions
The manuscript was written through contributions of all authors. F. R. and G. S. were primarily responsible for designing and conducting most of the experimental work, hypothesis validation and data analysis. P. R. contributed to initial experiments on proto-deallylation and enyne metathesis. V. M. was responsible for the preparation and metathesis of the enyne 10, while N. M. carried out a part of method development for the synthesis of 1. R. P. B. helped design, validate and supervise synthetic efforts towards BN-naphthalene. A. S. and A. C. were responsible for project conceptualization and overall coordination, supervised the experimental work and wrote the first draft of the manuscript. A. S. was also responsible for DFT calculations.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported through MICINN grants (PID2020-113661GB-I00 and PDC2023-145801-I00) and AGAUR (2021 SGR 00520). We thank CSIC for the JAE-Intro scholarship to V. M. and the Generalitat de Cataluña (AGAUR) for the Investigo contract to G. S.-L. (2023 INV-2 00014G1). We thank Sonia Pérez from the «Josep Carilla» Thermal Analysis and Calorimetry service at IQAC for conducting thermogravimetric analyses used in this work.Notes and references
- The earlier efforts and concepts have been reviewed here:
(a) M. J. D. Bosdet and W. E. Piers, Can. J. Chem., 2009, 87, 8–29 CrossRef
; (b) P. G. Campbell, A. J. V. Marwitz and S. Y. Liu, Angew. Chem., Int. Ed., 2012, 51, 6074–6092 CrossRef CAS PubMed
- For some of the earliest synthesis of cyclic 1,2-BN derivatives, see:
(a) M. J. S. Dewar, V. P. Kubba and R. Pettit, J. Chem. Soc., 1958, 3073–3076 RSC
- For selected examples see:
(a) D. H. Knack, J. L. Marshall, G. P. Harlow, A. Dudzik, M. Szaleniec, S. Y. Liu and J. Heider, Angew. Chem., Int. Ed., 2013, 52, 2599–2601 CrossRef CAS PubMed
-
(a) X. Y. Wang, J. Y. Wang and J. Pei, Chem.–Eur. J., 2015, 21, 3528–3539 CrossRef CAS PubMed
- For an enlightening discussion of this phenomenon, see for example: Z. Liu, J. S. A. Ishibashi, C. Darrigan, A. Dargelos, A. Chrostowska, B. Li, M. Vasiliu, D. A. Dixon and S. Y. Liu, J. Am. Chem. Soc., 2017, 139, 6082–6085 CrossRef CAS PubMed
-
(a) M. J. S. Dewar, G. J. Gleicher and B. P. Robinson, J. Am. Chem. Soc., 1964, 86, 5698–5699 CrossRef CAS
- For an interesting recent application of the partially saturated BN cycle 1′ in group transfer, see: J. B. Geri, E. Y. Aguilera and N. K. Szymczak, Chem. Commun., 2019, 55, 5119–5122 RSC
- The state of affairs in preparation of BN-cyclic cores is the subject of authoritative recent reviews, including:
(a) Z. X. Giustra and S. Y. Liu, J. Am. Chem. Soc., 2018, 140, 1184–1194 CrossRef CAS PubMed
- For selected examples, see:
(a) T. Hatakeyama, S. Hashimoto, S. Seki and M. Nakamura, J. Am. Chem. Soc., 2011, 133, 18614–18617 CrossRef CAS PubMed
- For some key references on the use of RCM reaction for the synthesis of BN-cyclic structures, see:
(a) A. J. Ashe and X. Fang, Org. Lett., 2000, 2, 2089–2091 CrossRef CAS PubMed
- Y. Liu, R. Puig De La Bellacasa, B. Li, A. B. Cuenca and S. Y. Liu, J. Am. Chem. Soc., 2021, 143, 14059–14064 CrossRef CAS PubMed
-
(a) B. M. Mikhailov and F. B. Tutorskaya, Dokl. Akad. Nauk SSSR, 1958, 123, 479–482 CAS
- For an earlier synthesis combining RCM and Matteson homologation, see:
(a) X. Fang, H. Yang, J. W. Kampf, M. M. B. Holl and A. J. Ashe, Organometallics, 2006, 25, 513–518 CrossRef CAS
-
(a) A. Abengózar, M. A. Fernández-González, D. Sucunza, L. M. Frutos, A. Salgado, P. García-García and J. J. Vaquero, Org. Lett., 2018, 20, 4902–4906 CrossRef PubMed
-
S. Ko, S. Shin, E. Ahn, J. Lyu, J. Lee and J. Han, EP, 3940805A1, Samsung Display Co., Ltd, 2022 Search PubMed
- Experimental enthalpy of hydrogenation data as taken from the National Institute of Standards (NIST) Chemistry WebBook, https://webbook.nist.gov/chemistry/.
-
(a) H. Villar, M. Frings and C. Bolm, Chem. Soc. Rev., 2007, 36, 55–66 RSC
- For an elegant complementary approach in which the BN core acts as the dienophile in a Diels–Alder reaction, rather than diene, see: P. F. Zhang, F. D. Zhuang, Z. H. Sun, Y. Lu, J. Y. Wang and J. Pei, J. Org. Chem., 2020, 85, 241–247 CrossRef CAS PubMed
- An analogous hydrate form of the aminoborane 3 was previously reported by Fang et al. in ref. 13c.
- Deposition numbers CCDC 2297111 (for 16) and CCDC 2297112 (for 14) contain the supplementary crystallographic data for this paper. This data is provided free of charge by the joint Cambridge Structural Database (CSD).
- Unlike the cis addition observed in the case of 13, the addition of the two molecules of maleic anhydride to the same side of the diene 7 (prod. 15) is disfavored by steric repulsion between the two inward-disposed (endo) dienophiles.
- For prior examples of similar decane-based acceptorless dehydrogenation in the BN context, see:
(a) E. R. Abbey, L. N. Zakjarov and S. Y. Liu, J. Am. Chem. Soc., 2010, 132, 16340–16342 CrossRef CAS PubMed
- We note that hydration during the HPLC-MS(ESI+) analysis of the sample cannot be ruled out, since both 17 and 17·H2O ionize to the same cation (m/z = 522), either via protonation in the case (17+H), or via loss of OH- from the hydrated form (17·H2O-OH).
- For a discussion and examples of known BN-tetracenes, see: J. S. A. Ishibashi, A. Dargelos, C. Darrigan, A. Chrostowska and S. Y. Liu, Organometallics, 2017, 36, 2494–2497 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2297111 and 2297112. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc06676b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |