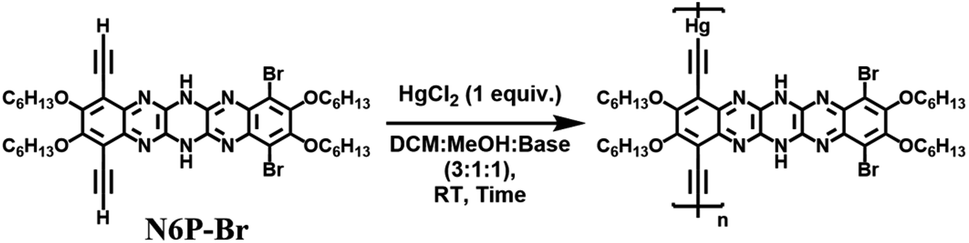
DOI:
10.1039/D3SC06552A
(Edge Article)
Chem. Sci., 2024,
15, 4054-4067
Pyrazinacene conjugated polymers: a breakthrough in synthesis and unraveling the conjugation continuum†
Received
6th December 2023
, Accepted 29th January 2024
First published on 2nd February 2024
Abstract
Pyrazinacenes are next generation N-heteroacenes and represent a novel class of stable n-type materials capable of accepting more than one electron and displaying intriguing features, including prototropism, halochromism, and redox chromism. Astonishingly, despite a century since their discovery, there have been no reports on the conjugated polymers of pyrazinacenes due to unknown substrate scope and lack of pyrazinacene monomers that are conducive to condensation polymerization. Breaking through these challenges, in this work, we report the synthesis of previously undiscovered and highly coveted conjugated polymers of pyrazinacenes. In order to understand the intricacies of conjugation extension within the acene and along the polymer backbone, a series of electronically diverse four pyrazinacene conjugated polymers were synthesized. Polymers synthesis required optimizing a few synthetic steps along the 12-step synthetic pathway. The generated pyrazinacene monomers are not amenable to the popular condensation polymerizations involving Pd or Cu catalysts. Gratifyingly, Pd and Cu free dehydrohalogenation polymerization of the monomer with HgCl2 resulted in high molecular weight organometallic conjugated pyrazinacene polymers within a few minutes at room temperature. The dual role played by the Hg(II) during the polymerization, combined with the self-coupling of the RHgCl (intermediate), is at the core of successful polymerization. Notably, the self-coupling of intermediates challenges the strict stoichiometric balance typically required for step-growth polymerization and offers a novel synthetic strategy to generate high molecular weight conjugated polymers even with imbalanced monomer stoichiometries. A combination of electrochemical studies and DFT-B3LYP simulations indicated that the presence of the reduced pyrazine ring promotes interacene π-conjugation through the metal center, in contrast to completely oxidized tetrazaazaanthracene. The extension of conjugation results in ca. 2 eV lower reduction potential for polymers compared to the monomer, placing the LUMO energy levels of these polymers on par with some of the best-known n-type polymers. Also, the presence of NH protons in the pyrazinacene polymers show ionochromism and red-shift UV-vis absorption maximum by ca. 100 nm. This work not only shows a way to realize highly desirable and elusive pyrazinacene conjugated polymers but also paves the way for a library of n-type conjugated polymers that can undergo multi-electron reduction.
1. Introduction
The delocalization of π-electrons of the repeat unit along the conjugated polymer backbone lowers the band gap and leads to a red-shift in the absorption spectra of the polymer and enables intrachain charge transport relative to individual monomeric units. Furthermore, the polymer's chain length and architecture offer several indispensable properties, including processability, mechanical flexibility, self-healing, and stretchability. These properties not only simplify device fabrication but also enhance device performance and lifetime.1–13 Thus, polymerization typically transforms an ineffectual monomeric unit into a solution-processable organic semiconductor material with tunable optoelectronic and mechanical properties. Polymerization has been shown to have a positive impact not only on small aromatic molecules but also on fused acenes. The development of polymeric acenes has resulted in significant enhancements in properties for electronic and optoelectronic applications when compared to the properties of individual acene units.14,15 Polymeric acenes also overcome challenges associated with pure acenes, such as solubility and stability. For example, Bao et al. showed conjugated polymers of pentacene are air-stable and solution-processable, unlike pentacene small molecules.16,17 This breakthrough has inspired the emergence of various functionalized acene oligomers and polymers that are both soluble and stable.5,8,18–23 In terms of electronic structure, recent studies have demonstrated that acene-based polymers exhibit non-linear band gap alterations as the size of the carbocyclic acene unit changes, and a zero-band gap conjugated polymer of pentacene was synthesized.24 Thus, the polymerization of acenes generates functional polymers with several indispensable features that are not accessible in small molecules.
While carbocyclic acenes primarily demonstrate p-type characteristics, significant efforts have been channeled into the creation of n-type acenes by replacing C–H groups with electronegative nitrogen atoms. Nitrogen atoms are intricately integrated into the aromatic framework of N-heteroacenes. The strategic placement of nitrogen atoms within the acenes serves to stabilize the frontier molecular orbitals (FMO), increase the electron affinity, and invert the electronic behavior of acenes, i.e., instead of hole transport, azaacenes show electron transport behavior. Pyrazinacenes represent the next generation of N-heteroacenes having high nitrogen content and are composed of linearly fused pyrazine units, including dihydropyrazine groups. The linearly fused pyrazine rings enable pyrazinacenes to undergo multi-electron reduction and contribute to the stabilization of the reduced states. Additionally, the presence of multiple nitrogen atoms in the acene leads to the tautomerization of NH protons in these compounds. The NH protons on the dihydropyrazines can be deprotonated in the presence of anions, resulting in changes in the band gap, energy levels, and optoelectronic properties. Electron-deficient pyrazinacenes interact with anions through hydrogen bonding and anion–π interactions, which are fundamental in many biological processes and hence can be used as markers, sensors, and dyes in medicinal applications.25–35 The presence of electron-deficient pyrazine rings also makes pyrazinacenes less susceptible to degradation through oxidation or dimerization, making them more stable than their carbocyclic acene counterparts. Consequently, pyrazinacenes represent a novel class of stable n-type materials capable of accepting more than one electron and displaying intriguing features, including prototropism, halochromism, and redox chromism.36–43 Considering the array of applications in various fields, the conjugated polymers of pyrazinacenes will be of immense interest and useful in multiple areas including organic electronics, energy conversion and storage, electrochemical transistors, catalysis, and sensors. Surprisingly, to date, there have been no reports on the conjugated polymers of pyrazinacenes.
The key reasons for no reports on the synthesis of conjugated polymers of pyrazinacenes, even after a century since their first discovery, are twofold. First, the lack of pyrazinacene monomers that are conducive to condensation polymerization, resulting in soluble conjugated polymers, has been a significant obstacle. Second, the intricacies of pyrazinacene substrates' scope and the stability of linearly fused pyrazine rings when subjected to condensation polymerizations remain unknown. Breaking through these challenges, in this work, we report the synthesis of previously undiscovered and highly coveted conjugated polymers of pyrazinacenes (Fig. 1). As mentioned earlier, the polymerization of pyrazinacene chromophores is expected to enhance the performance of the materials towards various applications due to the extension of π-conjugation along the chain and molecular wire effect. As the pyrazinacene monomer is polymerized through the terminal fused ring of the acene, the intricacies of conjugation extension within the acene and along the polymer backbone will determine the polymer properties. Therefore, in this work, we focused on (i) establishing the synthetic pathway for generating soluble and solution-processable conjugated polymers of pyrazinacenes via condensation polymerization; (ii) varying the number of fused rings in pyrazinacene as well as substituents on the rings to study their impact on photophysical, and electrochemical properties, as a means to uncover the intricacies of extension of conjugation within the acene and along the chain.
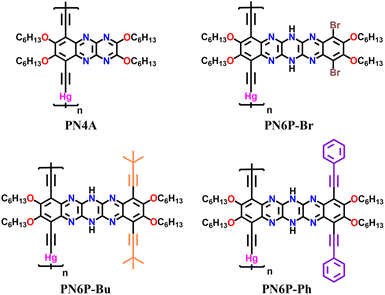 |
| | Fig. 1 Pyrazinacene polymers synthesized and studies in this work. | |
2. Results and discussion
2.1 Synthesis of monomers
The nucleophilic aromatic substitution (SNAr) reaction between dichlorodicyanopyrazine and aryl diamines is one of the efficient and quicker ways to generate pyrazinacenes containing multiple linearly fused pyrazine rings. A few hexaazapentacene-based pyrazinacenes have been synthesized through this approach by Miao and Hill groups.39,44 However, none of these derivatives have solubilizing chains, so the self-condensation polymerization of these pyrazinacenes would result in insoluble oligomers beyond two repeat units. To impart solubility to the polymers, hexaazapentacene monomers containing four hexyloxy chains were synthesized as shown in Scheme 1. Installing hexyloxy chains onto the hexazaacene monomers has increased the required number of synthetic steps, but this is imperative to generate soluble polymers. Synthesis of the desired pyrazinacene monomers with pendant solubilizing chains is synthetically challenging because chemical transformations of pyrazinacenes containing multiple linearly fused-pyrazine are highly structure specific; thus, synthesizing the desired monomers required screening of reaction conditions at a couple of synthetic steps (vide infra). Compounds 2–5 were synthesized following typical synthetic conditions reported for analogous compounds in the literature.45–47 The Sonogashira coupling of dibromo dihexyloxy benzothiadiazole (5) with TIPS acetylene, followed by the reduction of thiadiazole resulted in the phenylene diamine compound 7. Nucleophilic aromatic substitution reaction (SNAr) by 7 onto 2,3-dichloro-5,6-dicyanopyrazine in 1,4-dioxane gave dihydrotetraazacenedicyano compound 8. Oxidized 8 was subjected to another nucleophilic aromatic substitution reaction by dibromo dihexyloxy-phenylene diamine (10) to obtain the desired dihydrohexaazaacene compound 11 in 39% yield. The position of the hydrogens along the azaacene backbone can vary, giving rise to tautomers having different optical and electrochemical properties. Single crystal X-ray structure of 11 confirms that the hydrogens were on the middle pyrazine ring (Fig. S5†). Compound 11 was air-stable and did not oxidize to all pyrazine rings in the ambient atmosphere even though it contains an antiaromatic dihydropyrazine ring. This exceptional stability can be attributed to several factors, including the extended conjugation by the nitrogen lone pairs of the dihydropyrazine via enamine formation, an increased number of Clar rings, and delocalization of antiaromaticity. Desilylation of 11 gave the dialkynyl azaacene monomer N6P-Br in 68% yield. Attempts to oxidize the dihydropyrazine to pyrazine using PbO2 was unsuccessful due to the electronic stabilization by delocalization and the presence of different substituents around N6P-Br. The bromo substituents on 11 were useful to functionalize the azaacenes further and generate electronically diverse monomers. With this in mind, 11 was subjected to various Sonogashira cross-coupling reaction conditions with trimethylsilylacetylene, which were unsuccessful (Table S1†). The Bunz48,49 group have previously used Stille cross-coupling reactions to derivatize azaacenes. Taking cues from their work, 11 was subjected to Stille cross-coupling reaction with two alkynyl stannyl derivatives (R: t-Bu; Ph) in the presence of bis(benzonitrile)palladium(II) dichloride and P(t-Bu)3 (Scheme 1). We were gratified to see that both derivatization reactions successfully resulted in azaacenes containing t-butyl acetylene (compound 13) and phenyl acetylene (compound 14). The observation that azaacenes were susceptible to Stille rather than Sonogashira cross-coupling indicated that the presence of amine base and/or Cu(I) salt in the Sonogashira coupling might have interfered with the hydrogens of the dihydropyrazine leading to no or undesired reaction. Desilylation of 13 and 14 gave two electronically diverse azaacene monomers, N6P-Bu and N6P-Ph (Scheme 2). Alkyne substituents in both these monomers are expected to extended π-conjugation of the acene.
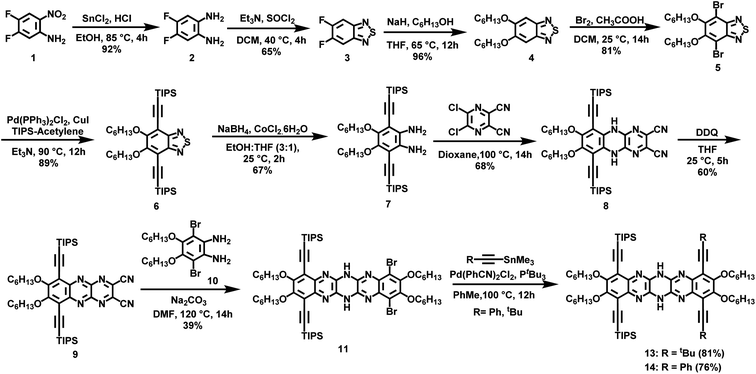 |
| | Scheme 1 Synthesis of TIPS protected pyrazinacene monomer precursors. | |
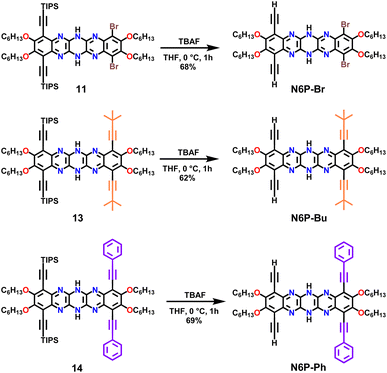 |
| | Scheme 2 Synthesis of pyrazinacene (hexaazapentacene) monomers. | |
To understand the effect of the number of fused rings in the azaacene repeat unit as well as the presence of the reduced pyrazine (dihydropyrazine) ring on the polymer's optical and electrochemical properties, compound 9 was used as comparison. 9 was subjected to a desilylation reaction using TBAF, but the starting material decomposed due to the strong nature of the fluoride source. Next, KF in the presence of AgNO3 in MeOH/THF was used as a mild fluoride source for desilylation (Scheme 3). Surprisingly, this resulted in expected desilylation as well as unexpected nucleophilic aromatic substitution by methoxy groups at the carbons containing cyano groups (17). This unexpected outcome was leveraged to install two additional solubilizing pendant chains onto 9, as shown in Scheme 3. For this, 9 was subjected to nucleophilic aromatic substitution reaction by hexyloxy groups to generate 18, which was desilylated using KF/AgNO3 to generate tetrazaanthracene dialkynyl monomer (N4A).
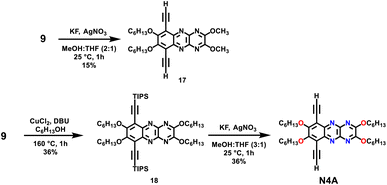 |
| | Scheme 3 Synthesis of tetrazaanthracene (N4A) monomer. | |
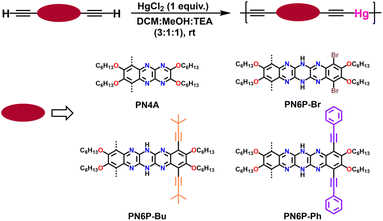
2.2 Synthesis of polymers
N6P-Br was used to optimize the polymerization conditions since its acene core is similar to the other two monomers (N6P-Bu and N6P-Ph) and requires two less synthetic steps. N6P-Br was subjected to Glaser–Hay polymerization because of its mild reaction conditions, but unfortunately, this resulted in only oligomers (Table 1). Synthesized azaacenes are air-stable; nonetheless, to see if the presence of oxygen in the reaction might be the reason for the lower degree of polymerization, the monomer was subjected to Eglinton50 polymerization under nitrogen, but the reaction was unsuccessful. Continuing our efforts to replace oxygen, a modified Glaser–Hay polymerization conditions reported by the Swager51 group that involved Pd(II) and Cu(I) catalysts and hydroquinone as an internal oxidant was tested (Table S2†). Unfortunately, the modified Glaser Hay polymerization conditions with the internal oxidant was also unsuccessful. Sonogashira polymerization of N6P-Br with diiodobenzene also did not yield any results. The limited success of the attempted polymerizations can be attributed to the presence of N–H protons on the dihydropyrazine ring of the pyrazinacene, which can interact with amine base and copper catalyst and interfere with the polymerization, similar to what we had observed for the Sonogashira cross-coupling of compound 11 with alkynes. To avoid polymerization reactions that involved amine base and copper, compound 11 was subjected to Stille condensation polymerization with bis(tributylstannyl) butadiyne52 (Table S3†). To our surprise, even this reaction also resulted in only oligomers.
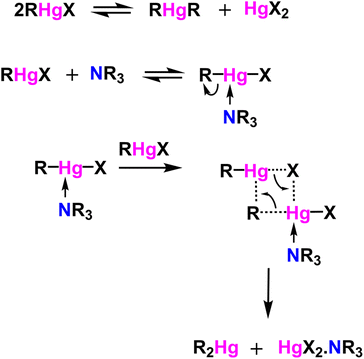
Table 1 Attempted alkyne–alkyne coupling polymerization conditionsb
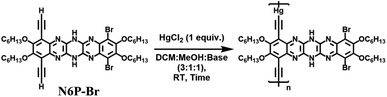
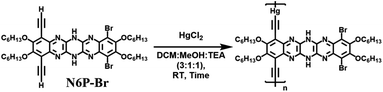
We scouted the literature for mild polymerization conditions for alkynes that do not involve amine base and Cu catalyst and result in conjugated polymers. Dehydrohalogenation–polymerization of the terminal diacetylenes with HgCl2 in the presence of triethylamine to generate conjugated Hg(II) containing rigid rod polymetallynes caught our attention.53,54 Mercury forms di-coordinated Hg(II) complexes (d10 configuration) with a linear geometry suitable for generating linear rigid rod conjugated polymers. A typical polymerization mechanism is shown in Scheme 4. The advantage of this reaction is that Hg(II) plays two roles: (i) it helps with the deprotonation of terminal acetylene similar to that of copper through eta complex, and (ii) it forms the σ-acetylide co-ordination compound with the terminal alkynes thus generating Hg(II) containing polymetallynes.55–59 Several Hg(II) alkynyl small molecule complexes and a few polymers have been synthesized through a dehydrohalogenation approach by reacting Hg(II) halide precursors and active terminal alkynes in the presence of a base.53,54,58,60 Both experimental and theoretical studies have shown that the mercury metal center is involved in extending the π-delocalization along the polymer backbone. A handful of Hg(II) containing conjugated polymetallynes have been synthesized to take advantage of the dπ–pπ conjugation and the resultant spin–orbit coupling between the mercury and organic spacer to generate highly phosphorescent polymers.58 Since this polymerization does not require a copper catalyst and generates Hg(II) containing small molecule complexes and polymers in good yield at room temperature, these polymerization conditions were tested on pyrazinacenes. It is gratifying to report that the polymerization of N6P-Br in the presence of HgCl2, triethylamine, in a mixture of DCM and MeOH gave organometallic conjugated pyrazinacene polymer (PN6P-Br) with a decent number average molecular weight (14 kDa).
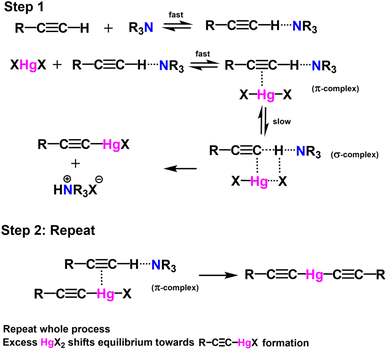 |
| | Scheme 4 Typical dehydrohalogenation reaction mechanism of terminal alkynes with HgCl2. | |
The effect of the amine base on polymerization was screened by changing the amine base and keeping the other reaction conditions the same (Table 2). All the studied amines except pyridine gave similar molecular weight polymers (Mn: ca. 11 kDa), whereas pyridine gave a lower molecular weight polymer (Mn: 9 kDa). Next, triethylamine was kept as the amine, and the equivalence of HgCl2 was changed from 0.5 to 1.5 equivalents with respect to the dialkyne monomer (Table 3). The change in the degree of polymerization as the reactant's stoichiometric ratio is varied does not follow the step-growth polymerization model. The seminal Carothers equation shows that a strict stoichiometry of monomers (A2 and B2) is required for a step-growth polymerization to realize high molecular weight polymers. A slight imbalance in the stoichiometry leads to a drastic reduction in the polymer molecular weight. Excess monomer present in the polymerization mixture ends up as a terminal group, thus hindering further condensation of growing chains resulting in low molecular weight polymers/oligomers. Contrary to that, polymer molecular weight increased with the increase in HgCl2 equivalence until 1.25 and reached a plateau. The key reason for the difference between conventional step-growth polymerization and HgCl2 containing organometallic polymerization is that the RHgCl self-condenses to generate R2Hg and HgCl2.56,57 The concentration of RHgCl increases in the reaction mixture with the increase in HgCl2 equivalence; RHgCl can react with terminal alkyne as shown in Scheme 4 to generate dialkynylmercury, which will elongate the polymer chain. Alternatively, two RHgCl molecules can self-react to generate dialkynylmercury (RHgR) and HgCl2 as shown in Scheme 5, which will also elongate the polymer chain and regenerate the (HgCl2) monomer respectively.
Table 2 Amine base optimization for dehydrohalogenation polymerizationa
|
|
| Entry |
Base |
Time (h) |
M
n/Mw (kDa) |
X
n/Xw |
Đ
|
|
Where Mn is the number average molecular weight, Mw is the weight average molecular weight, Xn is number average degree of polymerization, Xw is weight average degree of polymerization and Đ is the polydispersity index. All reactions were performed with 10 mg of N6P-Br (1 eq.) at RT. All GPCs were reported after washing with MeOH/diethyl ether. Polymer molecular weights were determined using gel permeation chromatography (tetrahydrofuran as the eluent) against polystyrene standards.
|
| 1 |
Diisopropylamine |
1 |
12/18 |
11/16 |
1.5 |
| 2 |
Tetramethylethylenediamine |
1 |
11/14 |
10/13 |
1.2 |
| 3 |
Diisopropylethylamine |
1 |
12/18 |
11/16 |
1.5 |
| 4 |
Pyridine |
1 |
9/11 |
8/10 |
1.2 |
| 5 |
Piperidine |
1 |
11/16 |
10/15 |
1.4 |
| 6 |
Triethylamine |
1 |
14/21 |
13/19 |
1.5 |
Table 3 HgCl2 equivalents optimization for dehydrohalogenation polymerizationa
|
|
| Entry |
HgCl2 (equiv.) |
Time (h) |
M
n/Mw (kDa) |
X
n/Xw |
Đ
|
|
Where Mn is the number average molecular weight, Mw is the weight average molecular weight, Xn, is number average degree of polymerization, Xw, is weight average degree of polymerization and Đ is the polydispersity index. All reactions were performed with 10 mg of N6P-Br (1 eq.) at RT. All GPCs were reported after washing with MeOH/diethyl ether. Polymer molecular weights were determined using gel permeation chromatography (tetrahydrofuran as the eluent) against polystyrene standards.
|
| 1 |
0.50 |
1 |
8/13 |
7/12 |
1.2 |
| 2 |
0.75 |
1 |
14/25 |
13/23 |
1.8 |
| 3 |
1.0 |
1 |
14/21 |
13/19 |
1.5 |
| 4 |
1.25 |
1 |
20/45 |
18/41 |
2.3 |
| 5 |
1.50 |
1 |
19/31 |
17/28 |
1.6 |
 |
| | Scheme 5 Typical self-condensation reaction of organomercury intermediate (RHgCl) into R2Hg and HgCl2. | |
In order to have better control over resultant polymer molecular weight and soluble polymer yield, optimized polymerization conditions (triethylamine, methanol, and DCM as solvents in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:3 ratio and 1 equivalent of HgCl2) were used for all the four monomers (Scheme 6). The polymerization mixture was concentrated, precipitated in methanol, and filtered. The crude polymer was subjected to soxhlation with methanol for all polymers. Diethyl ether was used as the second purification solvent for PN6P-Br and PN4A, whereas acetonitrile was used as the second purification solvent for PN6P-Bu and PN6P-Ph. The precipitate left in the thimble was dried and used for characterization. The synthesized polymers were soluble in typical organic solvents used for conjugated polymers, such as tetrahydrofuran, chloroform, and dichlorobenzene. The polymer molecular weights were determined using gel permeation chromatography (tetrahydrofuran as the eluent) against polystyrene standards. The number average molecular weights of the copolymers are in the range of 15 to 22 kDa.
:1:3 ratio and 1 equivalent of HgCl2) were used for all the four monomers (Scheme 6). The polymerization mixture was concentrated, precipitated in methanol, and filtered. The crude polymer was subjected to soxhlation with methanol for all polymers. Diethyl ether was used as the second purification solvent for PN6P-Br and PN4A, whereas acetonitrile was used as the second purification solvent for PN6P-Bu and PN6P-Ph. The precipitate left in the thimble was dried and used for characterization. The synthesized polymers were soluble in typical organic solvents used for conjugated polymers, such as tetrahydrofuran, chloroform, and dichlorobenzene. The polymer molecular weights were determined using gel permeation chromatography (tetrahydrofuran as the eluent) against polystyrene standards. The number average molecular weights of the copolymers are in the range of 15 to 22 kDa.
 |
| | Scheme 6 Synthesis of pyrazinacene polymers using dehydrohalogenation polymerization with HgCl2. | |
2.3 UV-vis absorption and emission properties
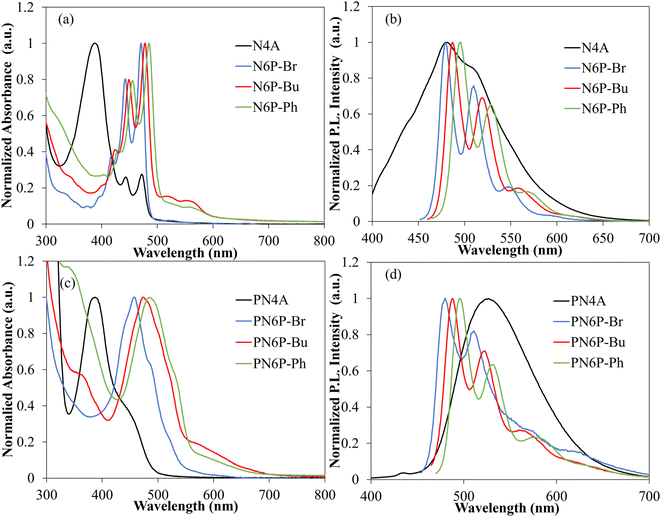
An increase in the number of fused rings from three (N4A) to five (N6P-series) has increased the absorption maximum of monomers by ca.100 nm (Table 4). This trend is also observed in the polymers as well, the absorption maximum of hexaazapentacene polymers (PN6Ps: PN6P-Br, PN6P-Bu, PN6P-Ph) is ca.100 nm red-shifted compared to tetraazaanthracene polymer (PN4A). This suggests that even though the middle dihydropyrazine ring is formally antiaromatic (4nπ electrons), it does not isolate hexazapentacene into two electronically independent halves, each containing quinoxalines. Moreover, both the absorption and emission spectra of hexaazapentacene monomers show a fine structure typically observed in acenes,42 which also supports the extended conjugation in the acene through the dihydropyrazine (Fig. 2). Bunz group and others have suggested that the azaacenes containing dihydropyrazines exhibit enamine conjugations involving the nitrogen lone pairs of dihydropyrazine. Also, using nucleus-independent chemical shift calculations it has been shown that the antiaromaticity in these azaacenes is reduced due to delocalization.43 Thus, the dihydropyrazine in the hexaazapentances, even though it is in the reduced form, extends the conjugation across the hexaazaapentance resulting in higher UV-vis absorption maximum. Within the hexaazapentacene monomers, installing t-butylacetylene and phenylacetylene in the place of bromide has slightly red-shifted the absorption maximum due to extension of π-conjugation. The absence of significant changes to the absorption peak pattern indicates that the hexaazaacene core is still the effective chromophore in these substrates and the alkynyl substituents have minimal effect on the major electronic transitions.
Table 4 Comprehensive optical properties of polymers and monomersa
| Polymer/monomer |
M
n/Mw (kDa) |
X
n/Xw |
Yieldb (%) |
λ
absmax THF (nm) |
λ
ex
(nm) |
λ
emmax THF (nm) |
Stokes shift (nm) |
|
Where Mn is the number average molecular weight, Mw is weight average molecular weight, Xn is number average degree of polymerization and Xw is weight average degree of polymerization. Polymer molecular weights were determined using gel permeation chromatography (tetrahydrofuran as the eluent) against polystyrene standards.
After Soxhlet extraction.
Excitation wavelength for reported emission spectra.
|
| PN4A |
22/35 |
26/38 |
94 |
386 |
386 |
527 |
141 |
| N4A |
|
|
|
388 |
390 |
481 |
93 |
| PN6P-Br |
15/37 |
14/34 |
89 |
458 |
437 |
480 |
22 |
| N6P-Br |
|
|
|
470 |
442 |
479 |
9 |
| PN6P-Bu |
21/36 |
19/33 |
93 |
474 |
455 |
488 |
14 |
| N6P-Bu |
|
|
|
477 |
450 |
487 |
10 |
| PN6P-Ph |
20/36 |
18/32 |
90 |
485 |
460 |
496 |
11 |
| N6P-Ph |
|
|
|
485 |
455 |
495 |
10 |
 |
| | Fig. 2 Normalized UV-Vis absorption and emission spectra of monomers (a and b) and polymers (c and d) in tetrahydrofuran. Excitation wavelengths are reported in Table 2. | |
All the hexaazapentacene monomers showed clear vibronic transitions but upon polymerization the absorption spectra became broader with less defined vibronic transitions. Even though the absorption maximum of PN6Ps is near the corresponding monomers, the polymers have an ill-defined vibronic transition in the red-region of the spectra (ca. 530 and 580 nm) (Fig. 2). This indicates that the π-electrons of azaacene repeat unit can delocalize beyond a single repeat unit and confirms the extension of π-conjugation along the polymer backbone through the metal center. Thus, Hg(II), albeit weaker than the conventional fully organic backbone, extends the π-conjugation along the polymer backbone. Emission spectra of hexaazapentacenes is approximately a mirror image of the UV-vis absorption spectra with very small Stokes shift (ca. 10 nm) typically observed in rigid molecular frameworks having acene-type structures. Both the alkyne and hexyloxy substituents on N4A have a significant effect on the N4A absorption and emission spectra. Unlike hexaazapentacene monomers, N4A has broad peaks in both absorption and emission spectra and displayed ca. 140 nm Stokes shift. The deviation from typical acene-like optical transitions in N4A indicates that the effective chromophore responsible for optical transitions in N4A has extended beyond the acene core. The emission spectra of PN4A and PN6Ps is similar to that of corresponding monomers.
A significant advantage of the dihydrohexaazaapentacenes is that the acidic protons on the reduced pyrazine ring bestows anion-dependent UV-vis absorption and emission properties compared to the oxidized tetrazaanthracene. The impact of F− and OH− anions on the UV-vis absorption and emission properties of monomers and polymers are studied. In the presence of both these anions, all the dihydrohexaazaapentancene monomers and polymers showed significant changes to the UV-vis absorption (Fig. 3). Whereas, fully oxidized tetrazaanthracene monomer and polymer showed no significant changes to the UV-vis absorption properties in the presence of studied anions confirming that: (i) the NH protons are responsible for observed spectral shifts; and (ii) interaction of anions with the Hg(II), if it exists, is not the reason for the observed spectral changes. In general, the introduction of F−, and OH− anions into the PN6Ps solution suppressed the intensity of the existing peaks (parent peaks) and three new major vibronic transitions (range: 500–600 nm) appeared in the red-region of the spectra. Polymer solutions change color from yellow to pink or purple depending on the polymer and ions added. The spectral changes i.e., the amount of suppression of parent peaks and the intensity of the new transitions are higher for OH− compared to F−. The spectral shifts are generally associated with the deprotonation of dihydropyrazine NH protons in the presence of F− or OH−. Similar shifts and color changes have been reported for the monoanion formation in pyrazines.36 The deprotonated PN6Ps are stable against oxidation by ambient oxygen (purging with air) and persist in solution. The formation of a nonsymmetric diamagnetic Meisenheimer complex/C–F bond via a nucleophilic attack61 was ruled out as the shift in UV-Vis spectra could be reversed back to the original state after work-up with water. Emission spectra of dihydrohexaazapentacenes monomers and polymers in the presence of F− and OH− shifted from the parent emission peak by 80 to 100 nm similar to that of UV-Vis absorption peaks (Fig. S9 and S11†). PN4A showed no new emission peaks in the presence of studied anions instead the intensity of the emission peak is suppressed.
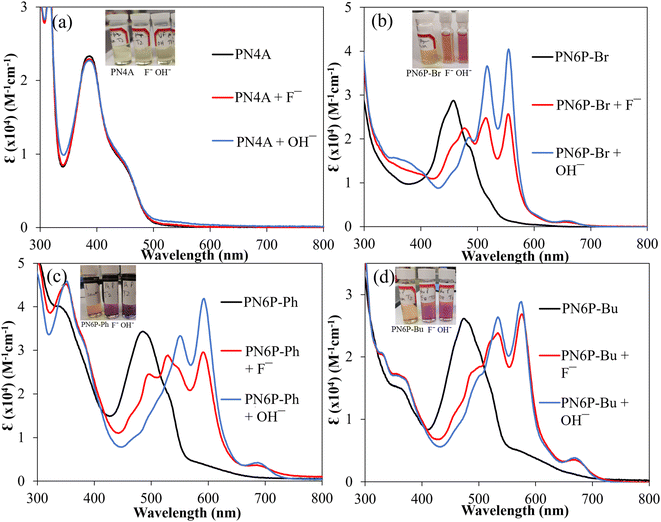 |
| | Fig. 3 UV-vis absorption spectra of polymers before and after the addition of 10 equivalents of fluoride and hydroxide anions in tetrahydrofuran recorded after 30 min of anion addition (inset shows color change of solutions after addition of anions). | |
2.4 Cyclovoltammetry and DFT-B3LYP simulations
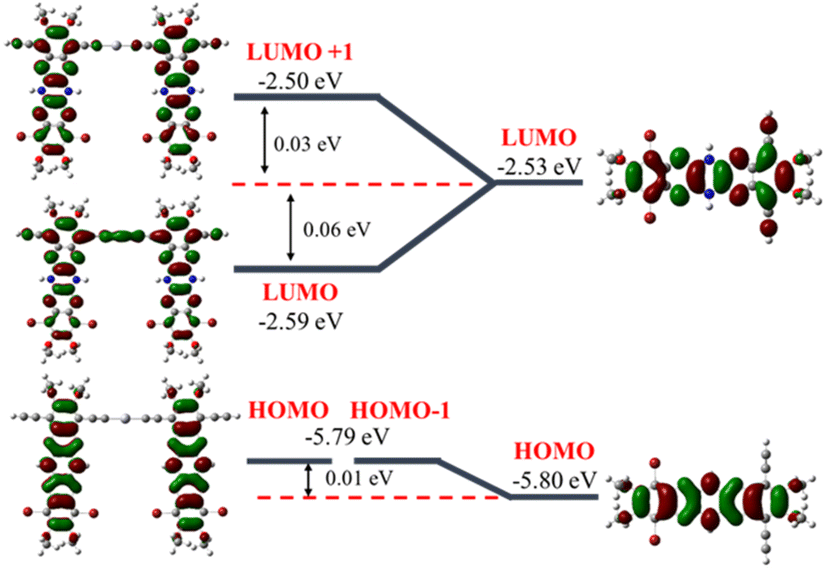
Cyclic voltammetry of polymers and monomer-precursors is performed (Fig. 4) to understand the role of Hg(II) on polymers electrochemistry and obtain frontier orbital energy levels. Alkyne-protected monomers i.e., monomer-precursors (11, 13, 14, and 18) are used instead of monomers for electrochemical studies to avoid undesired electrochemical reactions. Electrochemical properties are affected by the number of pyrazine units and the substituents. All the small molecules and polymers did not show any oxidation peaks highlighting the electron deficient nature of the azaacenes studied here. Unlike the UV-vis absorption data the N4A-precursor (18), which contains lower number of fused rings than the hexaazaacene-precursors (11, 13 and 14), reduces at smaller negative potentials highlighting the importance of presence of fully oxidized pyrazines over hexaazapentacenes with a dihydropyrazine for accepting electrons. The N4A-precursor, without the dihydropyrazine ring, show two well-defined quasi reversible reduction peaks, while the hexaazacene-precursors containing a dihydropyrazine ring undergo reduction at larger negative potentials. The hexaazapentacene-precursors showed a clear substitution-dependent electrochemical behavior. N6P-Bu-precursor (13) and N6P-Ph-precursor (14) showed two reduction peaks similar to that of N4A-precursor (18) but at slightly larger negative potentials. On the other hand, the N6P-Br-precursor (11) showed only one reduction peak at a potential similar to that of N6P-Ph-precursor. The presence of two (oxidized) pyrazine rings in the N4A precursor make it more electron deficient, help stabilize the FMOs and favor redox process at lower negative potential. Whereas the lone pair of electrons on the nitrogens of the reduced dihydropyrazine make hexaazapentacenes electron rich via enamine conjugation, thus destabilizing the FMOs and pushing the reduction potentials to larger negative potentials. Simulations of molecular orbitals provide relative energy levels and help in understanding the electronic structure of the repeat units. More importantly, DFT simulations provide a detailed account of the distribution of the electron density of the orbitals across the repeat units including the metal center (Fig. 5). In order to rationalize the electrochemical reduction behavior, frontier molecular orbitals of monomers and dimers (containing one Hg(II) at the center) are simulated (Fig. 6, S13 and S14†). The molecular geometries were optimized using density functional theory (DFT at genecp level, Stuttgart RSC 1997 ECP was used for Hg, and B3LYP/6-311g(d,p) was used for all other atoms). Indeed, the simulated FMOs of N4A are more stabilized than the hexaazaacenes. The LUMO of N4A has the electron density distributed across all the pyrazine nitrogens, whereas the nitrogens in the middle pyrazine ring of hexaazapentacene monomers have no electron density and act as nodal points. The difference in the distribution of electron density along the azaacenes stem from the oxidation state of pyrazine rings and could be the reason for lower reduction potentials of N4A. Within the hexaazapentacene monomers, installing t-butylacetylene and phenylacetylene in the place of bromide has further extended the electron delocalization of acene core in the LUMO of N6P-Bu and N6P-Ph, possibly facilitating two redox processes in the alkyne substituents containing substrates. It is worth noting that the LUMOs of the hexaazapentacene precursors are higher than that of similar azaacene compounds reported in the literature, showing that the addition of different substituents, including alkoxy substituents, significantly affects the electron affinity.44,62,63
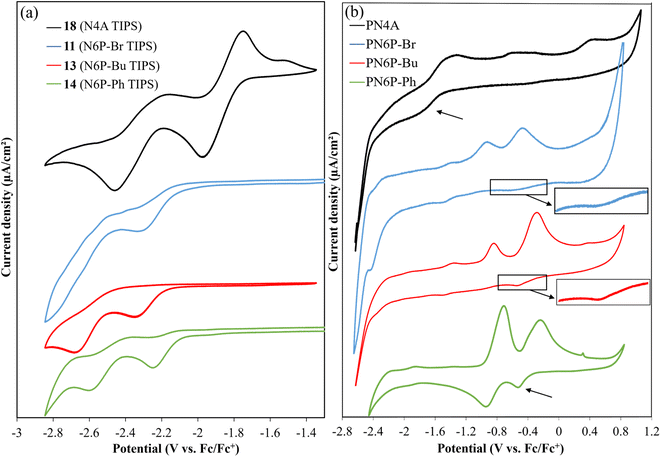 |
| | Fig. 4 Cyclic voltammograms of monomer precursors (left) and polymers (right). Glassy carbon, platinum wire, and Ag/Ag+ wire (0.01 M AgNO3 in 0.1 M TBAPF6 solution of MeCN) were used as working, counter, and reference electrodes respectively. Monomer precursors were dissolved in 0.1 M TBAPF6 solution of THF and polymer films were drop coated onto glassy carbon electrode and immersed in 0.1 M TBAPF6 solution of MeCN. | |
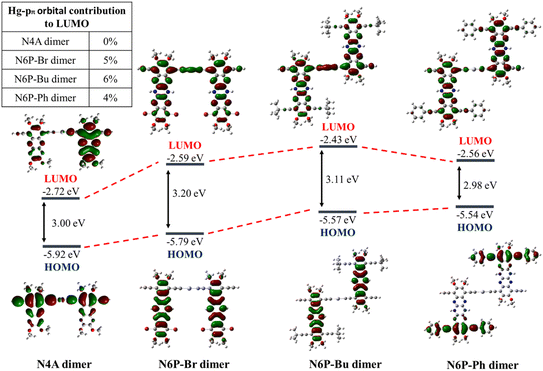 |
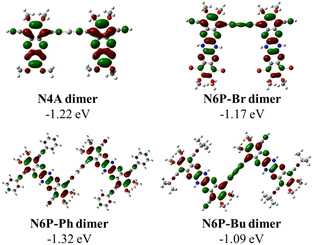
| | Fig. 5 DFT-B3LYP simulated HOMO and LUMO orbitals of dimers and the inset shows the Hg(II) pπ orbital contribution to the LUMO of the dimers. | |
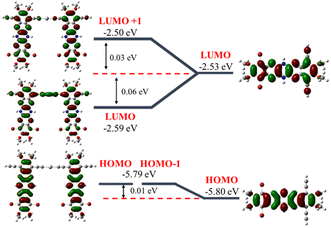 |
| | Fig. 6 DFT-B3LYP simulations of frontier molecular orbitals of N6P-Br monomer (right) and dimer (left) to compare the change in HOMO and LUMO energy levels upon bridging the monomers through Hg(II). | |
As a pleasant surprise, contrary to what has been observed in monomers, the hexaazaacene polymers (PN6Ps) showed reduction waves at smaller negative potentials than the PN4A without any reduced pyrazine rings (Table 5). PN6Ps retained the electrochemical behavior observed in monomer-precursors. All the PN6Ps, including the PN6P-Br, showed two reduction peaks. More importantly, a remarkable drop in the reduction potentials by ca. 2 eV for hexaazapentacenes upon polymerization indicates that the Hg(II) is playing a significant role in altering the redox properties of polymers and stabilizing the reduced species (Table 5). Moreover, PN6P-Ph showed relatively better redox peaks than the corresponding monomer and other two hexaazapentacene polymers (PN6P-Br and PN6P-Bu). Surprisingly, the reduction potential dropped only by 0.2 eV for tetraazaanthracene upon polymerization. In order to understand for such a significant drop in reduction potentials, simulations are performed on the dimers containing the Hg(II) metal at the center. HOMO of the dimers has no significant electron density on the metal center. On the other hand, LUMO of the hexaazapentacene dimers has electron density on the metal center as well as on the acene. Whereas the LUMO of the tetrazaanthracene dimer has no significant electron density on the metal center. This is also evident in the change in frontier orbital energy levels of dimers compared to that of monomers (Fig. 6, S13 and S14†). Mostly, the LUMOs of hexaazaapentacene dimers are affected due to the incorporation of metal center. This indicates that metal center facilitates the conjugation of hexaazapentancene LUMOs along the polymer backbone but not for the tetrazaanthracene. As a result, the LUMO of PN6Ps is extended along the polymer backbone through the metal centers. The extension of the electron density through the Hg(II) bridge in the LUMOs helped PN6Ps to significantly lower the reduction potential by ca. 2 eV. And the absence of such extension through Hg(II) bridge in PN4A lowered the reduction potential by only ca. 0.2 eV. Mulliken population analysis showed that the Hg(II) does not contribute to the LUMO orbitals of the Hg(II) bridged N4A dimer and indeed the spatial distribution of the LUMO orbital showed no electron density on the Hg(II) bridge. Whereas the Mulliken population analysis of Hg(II) bridged hexazapentacenes indicated 5%, 6% and 4% electron density on the Hg(II) center for N6P-Br, N6P-Bu, and N6P-Ph dimers respectively (Fig. S12†). Within the hexaazapentacenes the phenyl acetylene substituents stabilize the reduced species through extended conjugation giving rise to relatively reversible reduction waves. Thus, the pπ orbital interactions of the metal and hexaazapentacene provides a pathway for the delocalization of electron density along the polymer backbone and facilitate electrochemical reduction at lower negative potential. The electrochemical studies have unveiled a pivotal molecular design principle, the presence of dihydropyrazine (reduced pyrazines) reduces the extension of π-conjugation within the pyrazinacene but propitiously this allows the extension of electron density between the repeat units through the Hg(II) metal bridge and lowers the LUMO energy level. Simulations of the singly occupied molecular orbitals (SOMO) of dimers indicated that the odd electron is fully delocalized throughout the dimer including the metal center (Fig. 7). Since the monomer-precursors and polymers did not exhibit any oxidation potentials, the HOMO values were calculated using the optical band gap obtained from thin film UV-Vis absorption band red-edge (see Fig. S8†). Electrochemical studies of the control molecules (diphenylethynylmercury, and HgCl2) confirm that the observed redox peaks in polyazaacenes are arising from the polymer backbone as a result of conjugation between azaacene and Hg(II) and not from the Hg(II) center alone (see ESI Fig. S19 and S20† for the control molecules cyclic voltammetry data).
Table 5 Electrochemical properties of polymers and monomer-precursors
| Polymer/TIPS monomer |
E
optga (eV) |
LUMOCVb (eV) |
HOMOUVc (eV) |
|
From onset of thinfilm UV-Vis absorption spectra.
From reduction peak onset in CV.
HOMOUV calculated from Eoptg − LUMOCV values.
|
| PN4A |
2.31 |
−3.27 |
−5.40 |
|
18 (N4A TIPS) |
2.85 |
−3.02 |
−5.87 |
| PN6P-Br |
2.17 |
−4.68 |
−6.85 |
|
11 (N6P-Br TIPS) |
2.54 |
−2.70 |
−5.24 |
| PN6P-Bu |
2.13 |
−4.50 |
−6.63 |
|
13 (N6P-Bu TIPS) |
2.50 |
−2.62 |
−5.12 |
| PN6P-Ph |
2.21 |
−4.42 |
−6.63 |
|
14 (N6P-Ph TIPS) |
2.45 |
−2.70 |
−5.15 |
 |
| | Fig. 7 DFT-B3LYP simulated SOMOs of radical anions of dimers. | |
3. Conclusions
In summary, we successfully overcame the synthetic challenges associated with the polymerization of pyrazinacenes, resulting in the generation of a series of highly sought-after and elusive pyrazinacene polymers, nearly a century after their initial discovery. The key strategies behind this successful synthesis included the design of pyrazinacene monomers that would yield solution-soluble conjugated polymers and the identification of a Pd and Cu-free dehydrohalogenation polymerization method. Achieving these milestones required the optimization and establishment of several synthetic steps within the 12-step synthetic pathway and screening the polymerization of pyrazinacene monomers using various Pd and Cu-catalyzed methods. Gratifyingly, pyrazinacenes undergo Pd and Cu-free dehydrohalogenation polymerization with HgCl2, resulting in high molecular weight organometallic conjugated pyrazinacene polymers within just a few minutes at room temperature. The dual role played by Hg(II) during the polymerization, coupled with the self-coupling of the RHgCl intermediates, lies at the core of this successful polymerization process. Notably, the self-coupling of intermediates challenges the strict stoichiometric balance typically required for step-growth polymerization. Thus, HgCl2, unlike other popular transition metal halides (such as PdCl2L2 and PtCl2L2), offers a novel step-growth polymerization method to generate high molecular weight organometallic conjugated polymers without the need for strict monomer stoichiometry. A combination of electrochemical studies and DFT-B3LYP simulations revealed that the presence of reduced pyrazine rings (i.e., NH protons) promotes interacene π-conjugation through the metal center, in contrast to completely oxidized tetrazaazaanthracene. In terms of molecular design principles, although dihydropyrazine reduces the extension of π-conjugation within the pyrazinacene, it extends the conjugation along the polymer backbone and lowers the LUMO energy level. This extended conjugation results in an approximately 2 eV lower reduction potential for the polymers compared to the monomeric unit, placing the LUMO energy levels within the range of −4.68 to −4.42 eV for these polymers, which is comparable to some of the best-known n-type polymers.64–67 Therefore, pyrazinacene conjugated polymers offer a straightforward strategy for generating n-type polymers with low-lying LUMOs capable of undergoing multi-electron reduction. These pyrazinacene polymers hold great promise across a wide range of applications, including organic electronics, energy conversion and storage, electrochemical transistors, catalysis, and sensors.68–75 Additionally, they help to narrow the gap between number of p-type and n-type polymers.
Data availability
Synthesis and characterization details of small molecules and polymers are included in the experimental ESI.† Simulation details for dimers and small molecules are provided in the simulations ESI.†
Author contributions
F. H., A. M. and V. S. F. performed the synthesis of monomers and polymers. F. H. performed the structural and optical characterizations, simulations and was involved in the draft writing. N. G. conceived the idea, guided the experimental work, involved in the data analysis, and prepared the manuscript.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
This work was supported by a National Science Foundation CAREER Grant (NSF-1944184) and Georgetown University. V. S. F. acknowledges the Georgetown University-Institute for Soft Matter Synthesis and Metrology Graduate Student Fellowship. We would like to thank Prof. Miklos Kertesz and Dr Rameswar Bhattacharjee for help pertaining to computations with density functional theory and Dr Jeffery A. Bertke for performing the single crystal X-ray Diffraction and analysis.
References
- T. Dallos, D. Beckmann, G. Brunklaus and M. Baumgarten, Thiadiazoloquinoxaline–Acetylene Containing Polymers as Semiconductors in Ambipolar Field Effect Transistors, J. Am. Chem. Soc., 2011, 133(35), 13898–13901 CrossRef CAS PubMed.
- S. Wang, M. Kappl, I. Liebewirth, M. Müller, K. Kirchhoff, W. Pisula and K. Müllen, Organic field-effect transistors based on highly ordered single polymer fibers, Adv. Mater., 2012, 24(3), 417–420 CrossRef CAS PubMed.
- P.-L. T. Boudreault, J.-F. Morin and M. Leclerc, Synthesis of poly (2, 7-carbazole) s and derivatives, Des. Synth. Conjugated Polym., 2010, 205–226 CAS.
- F. Garnier, R. Hajlaoui, A. Yassar and P. Srivastava, All-polymer field-effect transistor realized by printing techniques, Science, 1994, 265(5179), 1684–1686 CrossRef CAS PubMed.
- M. He, J. Li, M. L. Sorensen, F. Zhang, R. R. Hancock, H. H. Fong, V. A. Pozdin, D.-M. Smilgies and G. G. Malliaras, Alkylsubstituted thienothiophene semiconducting materials: structure− property relationships, J. Am. Chem. Soc., 2009, 131(33), 11930–11938 CrossRef CAS PubMed.
- R. J. Kline, M. D. McGehee, E. N. Kadnikova, J. Liu and J. M. Frechet, Controlling the field-effect mobility of regioregular polythiophene by changing the molecular weight, Adv. Mater., 2003, 15(18), 1519–1522 CrossRef CAS.
-
M. Leclerc and J.-F. Morin, Design and synthesis of conjugated polymers, John Wiley & Sons, 2010 Search PubMed.
- D. Lehnherr, J. Gao, F. A. Hegmann and R. R. Tykwinski, Pentacene-based dendrimers: synthesis and thin film photoconductivity measurements of branched pentacene oligomers, J. Org. Chem., 2009, 74(14), 5017–5024 CrossRef CAS PubMed.
- H. Sirringhaus, Device physics of solution-processed organic field-effect transistors, Adv. Mater., 2005, 17(20), 2411–2425 CrossRef CAS.
- S. Sivaramakrishnan, P.-J. Chia, Y.-C. Yeo, L.-L. Chua and P. K.-H. Ho, Controlled insulator-to-metal transformation in printable polymer composites with nanometal clusters, Nat. Mater., 2007, 6(2), 149–155 CrossRef CAS PubMed.
- H. N. Tsao, D. Cho, J. W. Andreasen, A. Rouhanipour, D. W. Breiby, W. Pisula and K. Müllen, The influence of morphology on high-performance polymer field-effect transistors, Adv. Mater., 2009, 21(2), 209–212 CrossRef CAS.
- H. Yan, Z. Chen, Y. Zheng, C. Newman, J. R. Quinn, F. Dötz, M. Kastler and A. J. N. Facchetti, A high-mobility electron-transporting polymer for printed transistors, Nature, 2009, 457(7230), 679–686 CrossRef CAS PubMed.
- M. Zhang, H. N. Tsao, W. Pisula, C. Yang, A. K. Mishra and K. Müllen, Field-effect transistors based on a benzothiadiazole− cyclopentadithiophene copolymer, J. Am. Chem. Soc., 2007, 129(12), 3472–3473 CrossRef CAS PubMed.
- B. Purushothaman, S. R. Parkin and J. E. Anthony, Synthesis and stability of soluble hexacenes, Org. Lett., 2010, 12(9), 2060–2063 CrossRef CAS PubMed.
- X.-Z. Bo, N. Tassi, C. Lee, M. Strano, C. Nuckolls and G. B. Blanchet, Pentacene-carbon nanotubes: Semiconducting assemblies for thin-film transistor applications, Appl. Phys. Lett., 2005, 87(20), 203510 CrossRef.
- T. Okamoto and Z. Bao, Synthesis of solution-soluble pentacene-containing conjugated copolymers, J. Am. Chem. Soc., 2007, 129(34), 10308–10309 CrossRef CAS PubMed.
- T. Okamoto, Y. Jiang, F. Qu, A. C. Mayer, J. E. Parmer, M. D. McGehee and Z. Bao, Synthesis and Characterization of Pentacene-
and Anthradithiophene- Fluorene Conjugated Copolymers Synthesized by Suzuki Reactions, Macromolecules, 2008, 41(19), 6977–6980 CrossRef CAS.
- S. K. Park, T. N. Jackson, J. E. Anthony and D. A. J. A. P. L. Mourey, High mobility solution processed 6, 13-bis (triisopropyl-silylethynyl) pentacene organic thin film transistors, J. Appl. Phys., 2007, 91(6), 063514 Search PubMed.
- W. Fudickar and T. Linker, Why triple bonds protect acenes from oxidation and decomposition, J. Am. Chem. Soc., 2012, 134(36), 15071–15082 CrossRef CAS PubMed.
- A. Maliakal, K. Raghavachari, H. Katz, E. Chandross and T. Siegrist, Photochemical stability of pentacene and a substituted pentacene in solution and in thin films, Chem. Mater., 2004, 16(24), 4980–4986 CrossRef CAS.
- E. Kumarasamy, S. N. Sanders, A. B. Pun, S. A. Vaselabadi, J. Z. Low, M. Y. Sfeir, M. L. Steigerwald, G. E. Stein and L. M. Campos, Properties of Poly- and Oligopentacenes Synthesized from Modular Building Blocks, Macromolecules, 2016, 49(4), 1279–1285 CrossRef CAS.
- C. L. Anderson, T. Zhang, M. Qi, Z. Chen, C. Yang, S. J. Teat, N. S. Settineri, E. A. Dailing, A. Garzon-Ruiz, A. Navarro, Y. Lv and Y. Liu, Exceptional Electron-Rich Heteroaromatic Pentacycle for Ultralow Band Gap Conjugated Polymers and Photothermal Therapy, J. Am. Chem. Soc., 2023, 145(9), 5474–5485 CrossRef CAS PubMed.
- J. Kim, A.-R. Han, J. H. Seo, J. H. Oh and C. J. C. o. M. Yang, β-Alkyl substituted Dithieno [2, 3-d; 2′, 3′-d′] benzo [1, 2-b; 4, 5-b′] dithiophene Semiconducting Materials and Their Application to Solution-Processed Organic Transistors, Chem. Mater., 2012, 24(17), 3464–3472 CrossRef CAS.
- B. Cirera, A. Sánchez-Grande, B. de la Torre, J. Santos, S. Edalatmanesh, E. Rodríguez-Sánchez, K. Lauwaet, B. Mallada, R. Zbořil and R. J. N. n. Miranda, Tailoring topological order and π-conjugation to engineer quasi-metallic polymers, Nat. Nanotechnol., 2020, 15(6), 437–443 CrossRef CAS PubMed.
- G. Li, Y. Wu, J. Gao, J. Li, Y. Zhao and Q. Zhang, Synthesis, Physical Properties, and Anion Recognition of Two Novel Larger Azaacenes: Benzannelated Hexazaheptacene and Benzannelated N, N′-Dihydrohexazaheptacene, Chem.–Asian J., 2013, 8(7), 1574–1578 CrossRef CAS PubMed.
- P. Anzenbacher, D. S. Tyson, K. Jursíková and F. N. Castellano, Luminescence lifetime-based sensor for cyanide and related anions, J. Am. Chem. Soc., 2002, 124(22), 6232–6233 CrossRef CAS PubMed.
- O. B. Berryman, V. S. Bryantsev, D. P. Stay, D. W. Johnson and B. P. Hay, Structural criteria for the design of anion receptors: the interaction of halides with electron-deficient arenes, J. Am. Chem. Soc., 2007, 129(1), 48–58 CrossRef CAS PubMed.
- H. T. Chifotides, B. L. Schottel and K. R. Dunbar, The π-accepting arene HAT (CN) 6 as a halide receptor through charge transfer: multisite anion interactions and self-assembly in solution and the solid state, Angew. Chem., 2010, 122(40), 7360–7365 CrossRef.
- P. Gamez, T. J. Mooibroek, S. J. Teat and J. Reedijk, Anion binding involving π-acidic heteroaromatic rings, Acc. Chem. Res., 2007, 40(6), 435–444 CrossRef CAS PubMed.
- B. P. Hay and V. S. Bryantsev, Anion–arene adducts: C–H hydrogen bonding, anion–π interaction, and carbon bonding motifs, Chem. Commun., 2008, 21, 2417–2428 RSC.
- R. E. Dawson, A. Hennig, D. P. Weimann, D. Emery, V. Ravikumar, J. Montenegro, T. Takeuchi, S. Gabutti, M. Mayor and J. Mareda, Experimental evidence for the functional relevance of anion–π interactions, Nat. Chem., 2010, 2(7), 533–538 CrossRef CAS PubMed.
- J. Mareda and S. J. C. A. E. J. Matile, Anion–π slides for transmembrane transport, Chem.–Eur. J., 2009, 15(1), 28–37 CrossRef CAS PubMed.
- M. Mascal, I. Yakovlev, E. B. Nikitin and J. C. Fettinger, Fluoride-selective host based on anion–π interactions, ion pairing, and hydrogen bonding: synthesis and fluoride-ion sandwich complex, Angew. Chem., 2007, 119(46), 8938–8940 CrossRef.
- B. L. Schottel, H. T. Chifotides and K. R. Dunbar, Anion-π interactions, Chem. Soc. Rev., 2008, 37(1), 68–83 RSC.
- G. Gil-Ramírez, E. C. Escudero-Adán, J. Benet-Buchholz and P. Ballester, Quantitative evaluation of anion–π interactions in solution, Angew Chem. Int. Ed. Engl., 2008, 47(22), 4114–4118 CrossRef PubMed.
- D. Miklik, S. Fatemeh Mousavi, Z. Buresova, A. Middleton, Y. Matsushita, J. Labuta, A. Ahsan, L. Buimaga-Iarinca, P. A. Karr, F. Bures, G. J. Richards, P. Svec, T. Mori, K. Ariga, Y. Wakayama, C. Morari, F. D'Souza, T. A. Jung and J. P. Hill, Pyrazinacenes exhibit on-surface oxidation-state-dependent conformational and self-assembly behaviours, Commun. Chem., 2021, 4(1), 29 CrossRef CAS PubMed.
- G. J. Richards, A. Cador, S. Yamada, A. Middleton, W. A. Webre, J. Labuta, P. A. Karr, K. Ariga, F. D'Souza, S. Kahlal, J. F. Halet and J. P. Hill, Amphiprotism-Coupled Near-Infrared Emission in Extended Pyrazinacenes Containing Seven Linearly Fused Pyrazine Units, J. Am. Chem. Soc., 2019, 141(50), 19570–19574 CrossRef CAS PubMed.
- G. J. Richards and J. P. Hill, The Pyrazinacenes, Acc. Chem. Res., 2021, 54(16), 3228–3240 CrossRef CAS PubMed.
- G. J. Richards, J. P. Hill, N. K. Subbaiyan, F. D'Souza, P. A. Karr, M. R. Elsegood, S. J. Teat, T. Mori and K. Ariga, Pyrazinacenes: aza analogues of acenes, J. Org. Chem., 2009, 74(23), 8914–8923 CrossRef CAS PubMed.
- G. J. Richards, S. Ishihara, J. Labuta, D. Miklík, T. Mori, S. Yamada, K. Ariga and J. P. Hill, Fluorescent mesomorphic pyrazinacenes, J. Mater. Chem. C, 2016, 4(48), 11514–11523 RSC.
- L. Ahrens, J. Butscher, V. Brosius, F. Rominger, J. Freudenberg, Y. Vaynzof and U. H. F. Bunz, Azaacene Dimers: Acceptor Materials with a Twist, Chem.–Eur. J., 2020, 26(2), 412–418 CrossRef CAS PubMed.
- U. Bunz, A. Appleton, S. Barlow, S. Marder and K. Hardcastle, N,N-Dihydrotetraazaheptacene: A Synthetic Strategy towards Larger Acenes with Ambient Stability, Synlett, 2011, 2011(14), 1983–1986 CrossRef.
- U. H. Bunz, J. U. Engelhart, B. D. Lindner and M. Schaffroth, Large N-heteroacenes: new tricks for very old dogs?, Angew. Chem., Int. Ed., 2013, 52(14), 3810–3821 CrossRef CAS PubMed.
- Z. He, R. Mao, D. Liu and Q. Miao, Highly Electron-Deficient hexaazapentacene, Org. Lett., 2012, 14(16), 4190–4193 CrossRef CAS PubMed.
- C. Z. Karaman, S. Göker, S. O. Hacioğlu, T. Hacıefendioğlu, E. Yıldırım and L. Toppare, Altering electronic and optical properties of novel benzothiadiazole comprising homopolymers via π bridges, J. Electrochem. Soc., 2021, 168(3), 036514 CrossRef CAS.
- G. P. Kini, S. K. Lee, W. S. Shin, S.-J. Moon, C. E. Song and J.-C. Lee, Achieving a solar power conversion efficiency exceeding 9% by modifying the structure of a simple, inexpensive and highly scalable polymer, J. Mater. Chem. A, 2016, 4(47), 18585–18597 RSC.
- C.-S. Ke, C.-C. Fang, J.-Y. Yan, P.-J. Tseng, J. R. Pyle, C.-P. Chen, S.-Y. Lin, J. Chen, X. Zhang and Y.-H. Chan, Molecular engineering and design of semiconducting polymer dots with narrow-band, near-infrared emission for in vivo biological imaging, ACS Nano, 2017, 11(3), 3166–3177 CrossRef CAS PubMed.
- H. Reiss, F. Rominger, J. Freudenberg and U. H. Bunz, Peralkynylated Tetraazaacene Derivatives, Chem.–Eur. J., 2020, 26(5), 1013–1016 CrossRef CAS PubMed.
- S. Maier, N. Hippchen, F. Rominger, J. Freudenberg and U. H. Bunz, Cyclodimers and Cyclotrimers of 2, 3-Bisalkynylated Anthracenes, Phenazines and Diazatetracenes, Chem.–Eur. J., 2021, 27(66), 16320–16324 CrossRef CAS PubMed.
- G. Zhu and G. Zhang, Access to benzo-fused aza [7] helicene via unexpected indolization of alkyne-amine, Org. Chem. Front., 2021, 8(19), 5336–5344 RSC.
- V. Williams and T. Swager, An improved synthesis of poly (p-phenylenebutadiynylene)s, J. Polym. Sci., Part A: Polym. Chem., 2000, 38(S1), 4669–4676 CrossRef CAS.
- J.-H. Lee, M. D. Curtis and J. W. Kampf, Unusual Thermal Polymerization of 1, 4-Bis-5-(4,4′-dialkyl-2,2′-bithiazolyl)-1, 3-butadiynes: Soluble Polymers from Diacetylenes, Macromolecules, 2000, 33(6), 2136–2144 CrossRef CAS.
- J. Xu, B. Meng, J. Liu and L. Wang, A high molecular weight organometallic conjugated polymer incorporated with Hg(ii), Chem. Commun., 2020, 56(42), 5701–5704 RSC.
- L. Xu, J. Sun, T. Tang, H. Zhang, M. Sun, J. Zhang, J. Li, B. Huang, Z. Wang and Z. Xie, Metallated graphynes as a new class of photofunctional 2D organometallic nanosheets, Angew Chem. Int. Ed. Engl., 2021, 60(20), 11326–11334 CrossRef CAS PubMed.
- R. E. Dessy, W. L. Budde and C. Woodruff, The Formation of Carbon-Metal Bonds, J. Am. Chem. Soc., 1962, 84(7), 1172–1178 CrossRef CAS.
- R. Cross and C. Jenkins, Symmetrisation of organomercuric halides on alumina, silica and magnesium oxide, J. Organomet. Chem., 1973, 56, 125–129 CrossRef CAS.
-
E. Frankland, 3.3 Product Class 3: Organometallic Complexes of Mercury, 2004 Search PubMed.
- W.-Y. Wong, L. Liu and J.-X. Shi, Triplet Emission in Soluble Mercury(II) Polyyne Polymers, Angew. Chem., 2003, 115(34), 4198–4202 CrossRef.
- C. L. Ho, Z. Q. Yu and W. Y. Wong, Multifunctional polymetallaynes: properties, functions and applications, Chem. Soc. Rev., 2016, 45(19), 5264–5295 RSC.
- W.-Y. Wong and C.-L. Ho, Di-, oligo- and polymetallaynes: Syntheses, photophysics, structures and applications, Coord. Chem. Rev., 2006, 250(19–20), 2627–2690 CrossRef CAS.
- S. Saha, Anion-Induced Electron Transfer, Acc. Chem. Res., 2018, 51(9), 2225–2236 CrossRef CAS PubMed.
- T. Takeda, T. Ikemoto, S. Yamamoto, W. Matsuda, S. Seki, M. Mitsuishi and T. Akutagawa, Preparation, Electronic and Liquid Crystalline Properties of Electron-Accepting Azaacene Derivatives, ACS Omega, 2018, 3(10), 13694–13703 CrossRef CAS PubMed.
- N. Jun-ichi Nishida, M. Shio, F. Eiichi, T. Hirokazu, T. Masaaki and Y. Yoshiro, Preparation, Characterization, and FET Properties of Novel Dicyanopyrazinoquinoxaline Derivatives, Org. Lett., 2004, 6(12), 2007–2010 CrossRef PubMed.
- T. Du, Y. Liu, C. Wang, Y. Deng and Y. Geng, n-Type Conjugated Polymers Based on an Indandione-Terminated
Quinoidal Building Block, Macromolecules, 2022, 55(14), 5975–5984 CrossRef CAS.
- H. Sun, X. Guo and A. Facchetti, High-performance n-type polymer semiconductors: applications, recent development, and challenges, Chem, 2020, 6(6), 1310–1326 CAS.
- Y. Zhang, Y. Wang, C. Gao, Z. Ni, X. Zhang, W. Hu and H. Dong, Recent advances in n-type and ambipolar organic semiconductors and their multi-functional applications, Chem. Soc. Rev., 2023, 52(4), 1331–1381 RSC.
- Y. Wang, E. Zeglio, L. Wang, S. Cong, G. Zhu, H. Liao, J. Duan, Y. Zhou, Z. Li and D. Mawad, Green Synthesis of Lactone-Based Conjugated Polymers for n-Type Organic Electrochemical Transistors, Adv. Funct. Mater., 2022, 32(16), 2111439 CrossRef CAS.
- S. R. Bheemireddy, M. P. Hautzinger, T. Li, B. Lee and K. N. Plunkett, Conjugated Ladder Polymers by a Cyclopentannulation Polymerization, J. Am. Chem. Soc., 2017, 139(16), 5801–5807 CrossRef CAS PubMed.
- H. Zhou, L. Yang, S. C. Price, K. J. Knight and W. You, Enhanced photovoltaic performance of low-bandgap polymers with deep LUMO levels, Angew. Chem., 2010, 122(43), 8164–8167 CrossRef.
- J. Han, Y. Jiang, E. Tiernan, C. Ganley, Y. Song, T. Lee, A. Chiu, P. McGuiggan, N. Adams and P. J. A. C. I. E. Clancy, Blended Conjugated Host and Unconjugated Dopant Polymers Towards N-type All-Polymer Conductors and High-ZT Thermoelectrics, Angew. Chem., Int. Ed., 2023, 62(23), e202219313 CrossRef CAS PubMed.
- D. Yoo, X. Luo, T. Hasegawa, M. Ashizawa, T. Kawamoto, H. Masunaga, N. Ohta, H. Matsumoto, J. Mei and T. Mori, n-Type Organic Field-Effect Transistors Based on Bisthienoisatin Derivatives, ACS Appl. Electron. Mater., 2019, 1(5), 764–771 CrossRef CAS.
-
Y. Zhou, J. Lee and L. Fang, n-Type Electron-Accepting Materials for Organic Solar Cells (OSC), in Organic and Hybrid Solar Cells, ed. Huang H. and Huang J., Springer International Publishing, Cham, 2014, pp. 97–119 Search PubMed.
- M. Ball, Y. Zhong, B. Fowler, B. Zhang, P. Li, G. Etkin, D. W. Paley, J. Decatur, A. K. Dalsania and H. Li, Macrocyclization in the design of organic n-type electronic materials, J. Am. Chem. Soc., 2016, 138(39), 12861–12867 CrossRef CAS PubMed.
- Y. Zhang, D. Hanifi, E. Lim, S. Chourou, S. Alvarez, A. Pun, A. Hexemer, B. Ma and Y. Liu, Enhancing the Performance of Solution-Processed n-Type Organic Field-Effect Transistors by Blending with Molecular “Aligners”, Adv. Mater., 2014, 26(8), 1223–1228 CrossRef CAS PubMed.
- Y. Yu, C. Dong, A. F. Alahmadi, B. Meng, J. Liu, F. Jäkle and L. Wang, A p-π* conjugated triarylborane as an alcohol-processable n-type semiconductor for organic optoelectronic devices, J. Mater. Chem. C, 2019, 7(24), 7427–7432 RSC.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab
*ab