DOI:
10.1039/D3SC06341K
(Edge Article)
Chem. Sci., 2024,
15, 4068-4074
Supramolecular copolymerization of hydrophobic and hydrophilic monomers in liquid crystalline media†
Received
25th November 2023
, Accepted 4th February 2024
First published on 5th February 2024
Abstract
In the case of covalent polymers, immiscible polymers can be integrated by covalently linking them together, but such a strategy is not possible in supramolecular polymers. Here we report the supramolecular copolymerization of two porphyrin-based monomers, C10P2H and TEGPCu with side chains bearing cyanobiphenyl (CB) groups at the ends of hydrophobic alkyl or hydrophilic tetraethylene glycol chains, respectively. These monomers undergo self-sorting supramolecular polymerization in highly diluted solutions ([monomer] = 3.4 × 10−9 mol% (2.0 × 10−8 mol L−1)) in nonpolar media due to the incompatibility of the side chains. Surprisingly, these monomers undergo supramolecular copolymerization under high concentration conditions ([monomer] = 7.7 mol%) in the medium of 4-cyano-4′-pentyloxybiphenyl (5OCB) to form a columnar liquid crystalline phase under thermodynamic conditions, where the individual columns are composed of supramolecular block copolymers. The combination of CB ends of both monomers and the 5OCB medium is essential for the two monomers to form an integrated structure in a condensed system without phase separation.
Introduction
Polymer mixtures may undergo macroscopic phase separation due to the small contribution of mixing entropy at high degrees of polymerization.1,2 When mixing enthalpy is positive and large, as in mixing hydrophobic and hydrophilic polymers, immiscibility is further increased. Covalent linkage between the ends of such polymers to make a block copolymer prevents macroscopic phase separation, resulting in integrated structures such as microphase-separated structures.3–6 In contrast to covalent polymers, the monomers in supramolecular polymers are connected noncovalently.7–14 Therefore, we cannot force the two immiscible supramolecular polymers to form a block copolymer because the non-covalent connection between the two is energetically disfavored.15,16 Currently, a guiding principle to mix two immiscible supramolecular polymers to form an integrated structure is missing.
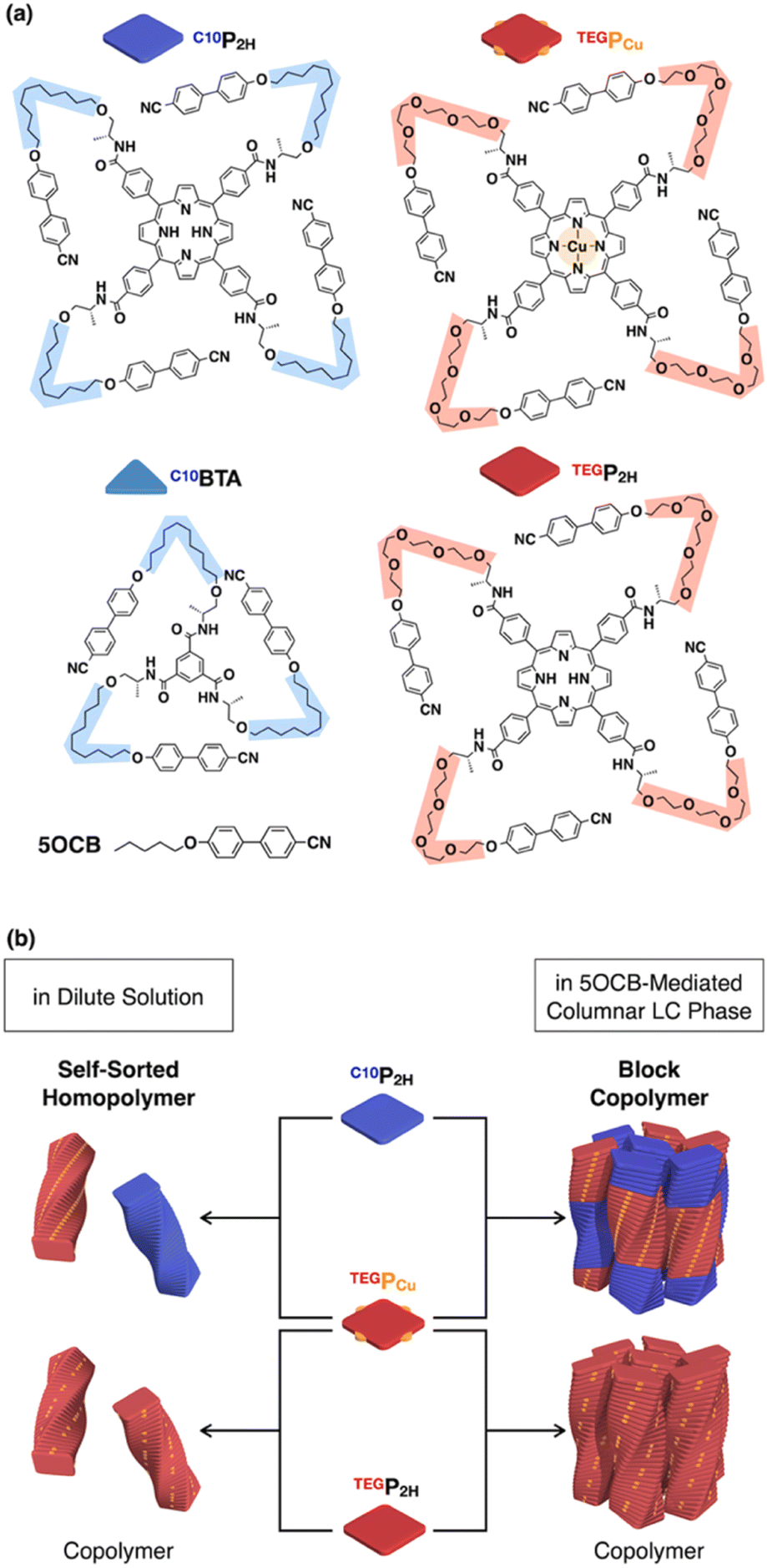
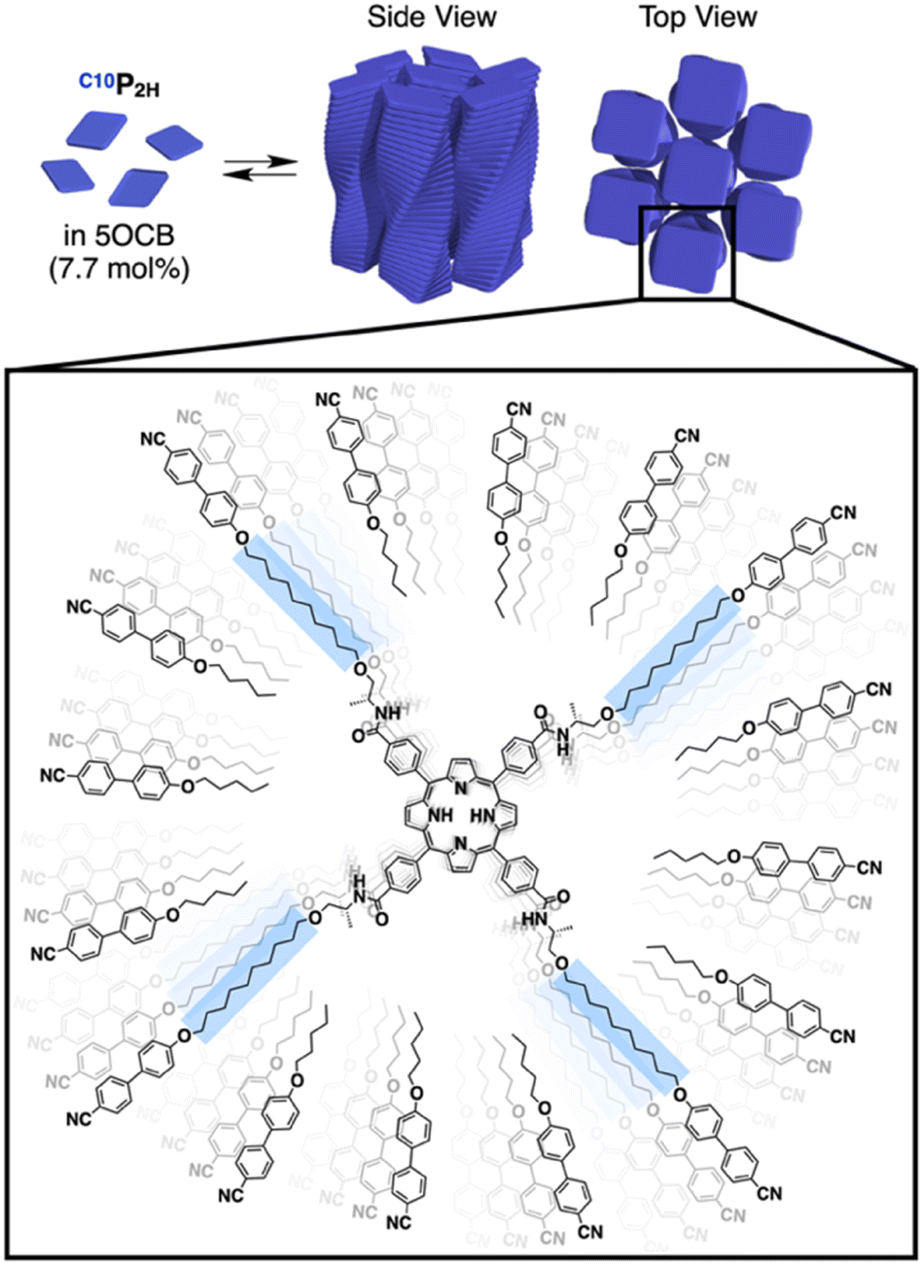
In this study, we report that monomers C10P2H and TEGPCu (Fig. 1a), which form hydrogen-bonded (H-bonded) supramolecular polymers and are not compatible with each other, can be copolymerized without macroscopic phase separation. This was achieved by employing nematic-forming 4-cyano-4′-pentyloxybiphenyl (5OCB) as a polymerization medium (Fig. 1b, middle). This liquid crystal (LC) system is designed based on our recent report on the formation of columnar LCs by supramolecular polymerization of disk-shaped monomers in a nematic LC medium composed of rod-shaped molecules.17–19 The introduction of a mesogenic moiety at the termini of the disk-shaped monomer promotes the order-increasing mesophase transition from 1D to 2D to form a columnar LC ordering (Fig. 2). We have previously reported disk-shaped monomers having benzenetricarboxamide (BTA)17,18 and porphyrin cores.19 In the present work, we newly synthesized porphyrin-centered TEGPCu, bearing hydrophilic side chains. C10P2H and TEGPCu are inherently immiscible due to the incompatible combination of the side chain units (conflicting pair). Thus, these two monomers do not mix homogeneously in bulk, and undergo self-sorting polymerization when mixed in a diluted solution of a nonpolar medium. Surprisingly, when these monomers are polymerized together in 5OCB, the entire material formed a columnar oblique (Colob) LC phase, where the individual column is composed of a block copolymer of the two monomers. The presence of 5OCB may suppress the macroscopic phase separation of two incompatible monomers and instead results in an integrated structure composed of supramolecular block copolymers. Considering that the supramolecular block copolymer is usually formed under kinetic conditions,20–29 our system is one of the rare examples of the formation of a block sequence under thermodynamic conditions.30–34 The present study may serve as a guiding principle for the control of the block sequence in supramolecular copolymers, that can lead to controlled microphase-separated structures as in conventional covalent copolymers.
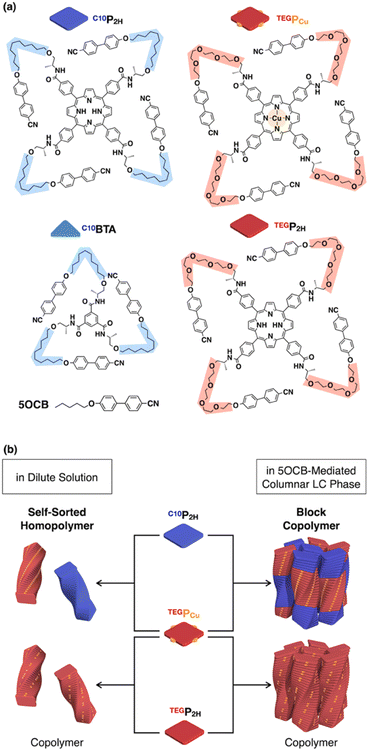 |
| | Fig. 1 (a) Molecular structures of H-bonding monomers TEGP2H, TEGPCu, C10P2H, and C10BTA, and nematic LC molecule 5OCB. (b) Schematic representations of the supramolecular copolymerization of three different monomer pairs in 5OCB. | |
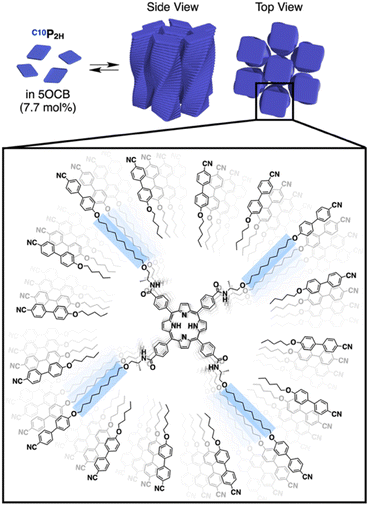 |
| | Fig. 2 Supramolecular polymerization of C10P2H in a nematic-forming 5OCB medium, resulting in an order-increasing mesophase transition into a columnar LC structure. | |
Results and discussion
Monomer design and formation of columnar LCs
We have previously reported benzene-centered C10BTA17 and porphyrin-centered C10P2H,19 both having oxycyanobiphenyl (OCB) terminated side chains appended with a chiral aliphatic chain (C10) that is connected to the core with an amide linkage. In this study, we newly synthesized two porphyrin-centered monomers, TEGPCu and TEGP2H with chiral tetraethylene glycol (TEG) chains (Fig. 1a). The center of the monomer is either a free-base (P2H) or copper(II) (PCu) porphyrin unit. It is known that the fluorescence of P2H is quenched by electron exchange with PCu upon stacking (Dexter mechanism).35–38 This allows us to evaluate whether the two monomers were copolymerized or not by using the fluorescence profile of the mixture.
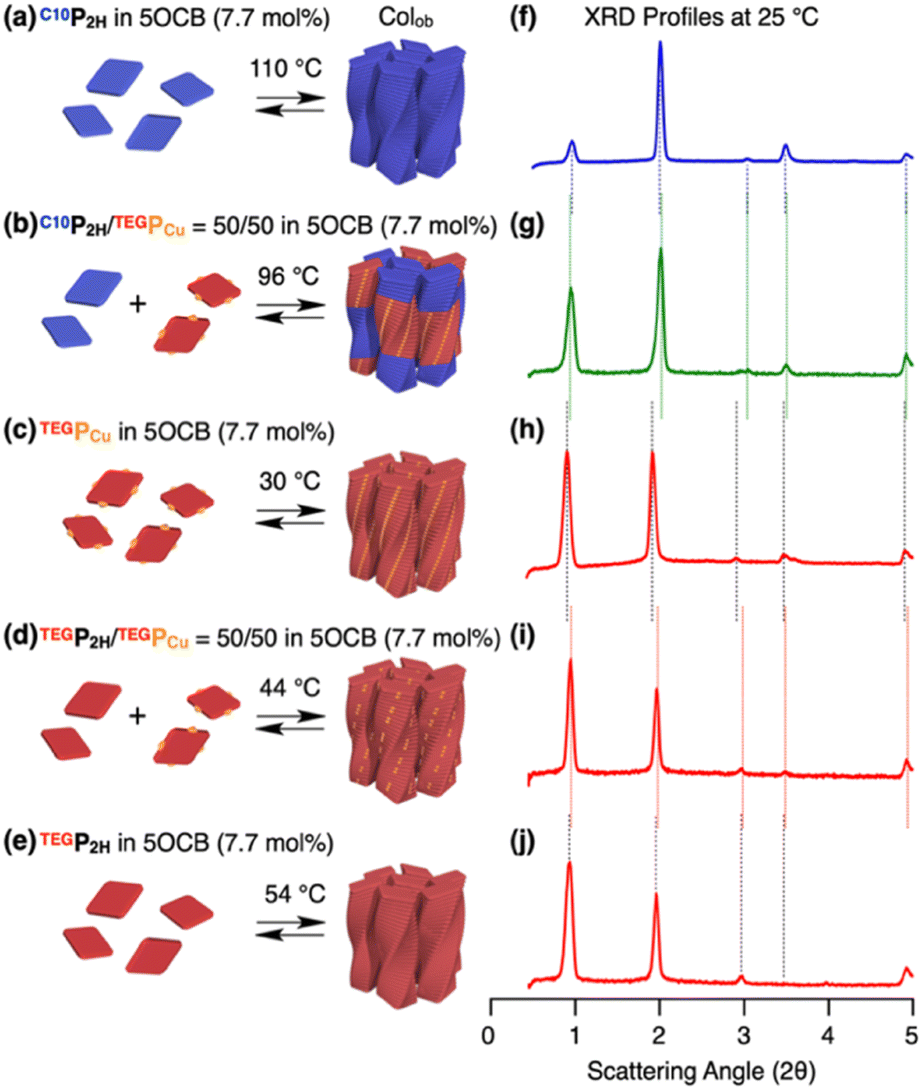
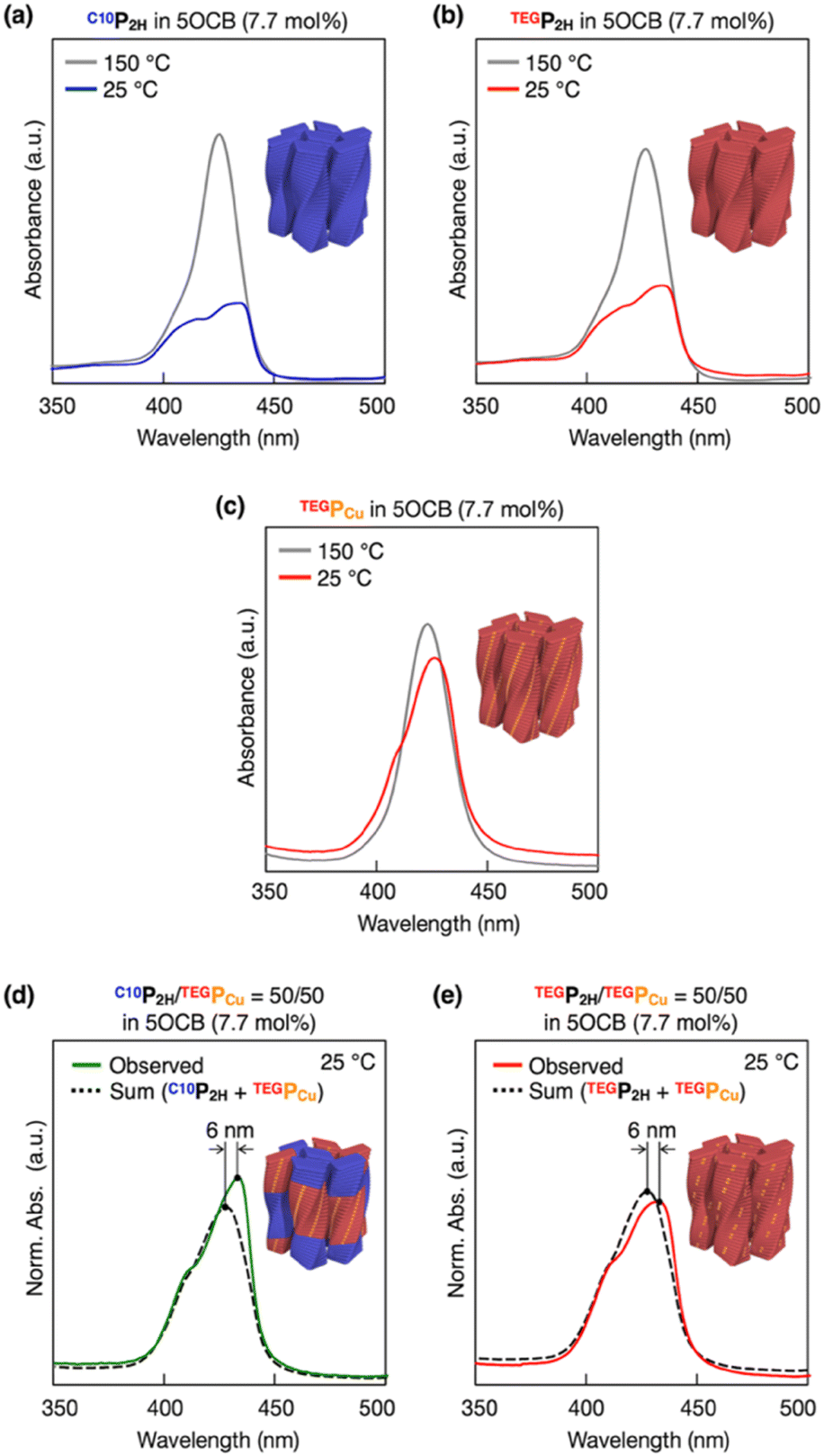
Based on previous studies, C10P2H was mixed with 5OCB at a concentration of 7.7 mol% and heated up to 200 °C to obtain the isotropic melt. Upon cooling, this mixture was found to undergo phase transition at 110 °C to give a Colob LC phase by H-bonding mediated supramolecular polymerization, which was confirmed by differential scanning calorimetry (DSC), Fourier-transform infrared (FT-IR) spectroscopy, polarized optical microscopy (POM), and X-ray diffraction (XRD) measurements (Fig. 3a, f, S3a, S6a, S8a, and S13a, b†). The mixture exhibited a mesophase in a wide temperature range between −20 and 110 °C including room temperature (a = 98.3 Å; b = 27.0 Å; γ = 70.7° at 25 °C) (Fig. S3a and S8a†). Electronic absorption spectra showed a sharp Soret band absorption at λmax = 426 nm in the isotropic phase at 150 °C (Fig. 4a, gray). This indicates that the porphyrin monomers are not stacked in the isotropic phase. On the other hand, in the LC phase at 25 °C, Soret band absorption exhibited both red-shifted (434 nm) and blue-shifted (418 nm) bands (Fig. 4a, blue), which is typical for the formation of porphyrin J-aggregates.39–43 Similarly, the newly synthesized TEGPCu and TEGP2H with TEG chains were also confirmed to form a Colob LC phase by supramolecular polymerization in 5OCB ([monomer] = 7.7 mol%) (TEGPCu: a = 99.6 Å; b = 25.7 Å; γ = 78.6°, TEGP2H: a = 105.7 Å; b = 28.2 Å; γ = 71.0° at 25 °C, Fig. 3c, e, h, j, S3b, c, S6b, c, S8b, c and S13c–f†). Electronic absorption spectra showed Soret band absorption at λmax = 423 nm for TEGPCu and λmax = 426 nm for TEGP2H in the isotropic phase at 150 °C (Fig. 4b and c, gray). In the LC phase at 25 °C, these bands exhibited both a red-shift (426 nm for TEGPCu and 434 nm for TEGP2H) and blue-shift (410 nm for TEGPCu and 418 nm for TEGP2H) (Fig. 4b and c, red). This is similar to that observed in the case of C10P2H (Fig. 4a) and suggests the formation of porphyrin J-aggregates for both TEGPCu and TEGP2H in their Col LC phase.
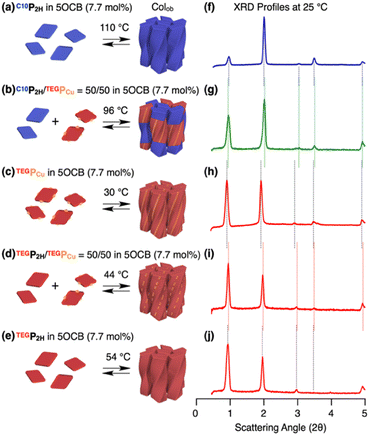 |
| | Fig. 3 (a–e) Schematic representations of the formation of columnar LCs and (f–j) XRD profiles at 25 °C of (a and f) C10P2H, (b and g) C10P2H/TEGPCu = 50/50, (c and h) TEGPCu, (d and i) TEGP2H/TEGPCu = 50/50 and (e and i) TEGP2H in 5OCB (7.7 mol%). The temperatures shown above the arrows in (a–e) are the LC phase transition temperatures obtained by DSC on cooling (5 °C min−1). | |
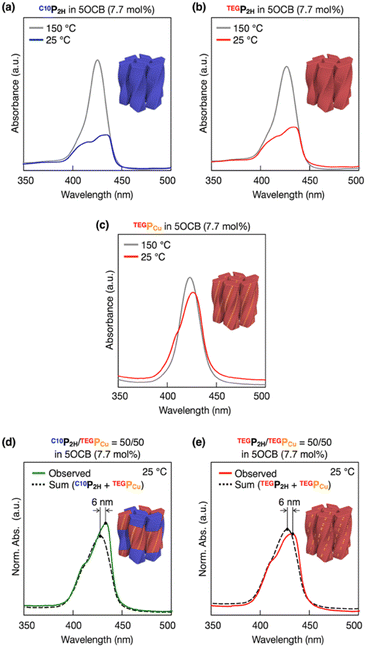 |
| | Fig. 4 Electronic absorption spectra of (a) C10P2H, (b) TEGP2H, (c) TEGPCu, (d) C10P2H/TEGPCu = 50/50 and (e) TEGP2H/TEGPCu = 50/50 in 5OCB (7.7 mol%) at 25 °C. For (a) C10P2H, (b) TEGP2H, and (c) TEGPCu in 5OCB (7.7 mol%), electronic absorption spectra at 150 °C are also shown (gray). The summation of the electronic absorption spectra of C10P2H in 5OCB (7.7 mol%) and TEGPCu in 5OCB (7.7 mol%) (d), and TEGP2H in 5OCB (7.7 mol%) and TEGPCu in 5OCB (7.7 mol%) (e) is also shown (black dashed lines). | |
Mixing of porphyrin monomers in columnar LC
We then performed a supramolecular copolymerization of a conflicting pair, C10P2H and TEGPCu, in 5OCB. The mixed LC of C10P2H/TEGPCu = 50/50 in 5OCB ([monomer] = 7.7 mol%) underwent a phase transition to the LC phase at 96 °C upon cooling (5 °C min−1) from its isotropic melt at 200 °C. XRD measurement at 25 °C confirmed the formation of the Colob LC phase (Fig. 3b, g S3d, S6d, S8d, and S14a, b†). Mixed LCs of C10P2H/TEGPCu = 75/25 and 25/75 in 5OCB ([monomer] = 7.7 mol%) also formed a Colob LC phase (Fig. S4c, e, and S9c, e†). These results indicate that the two monomers can be mixed and form an integrated structure in 5OCB at arbitrary mixing ratios without macroscopic phase separation. It is noted that C10P2H and TEGPCu have incompatible C10 and TEG side chains and undergo macroscopic phase separation in the bulk state (Fig. S27 and S28†) or in solution without 5OCB (Fig. S15 and S17†) (vide infra).44 Therefore, it is surprising that these two porphyrin derivatives form an integrated structure in the LC phase. The same mixing experiments were performed for the affinitive pair, TEGP2H and TEGPCu, both having common TEG side chains. The results indicate that they also form a single Colob LC phase without macroscopic phase separation (Fig. 3d, i, S3e, S6e, S8e, and S14c, d†).
Analysis by electronic absorption and fluorescence spectroscopy
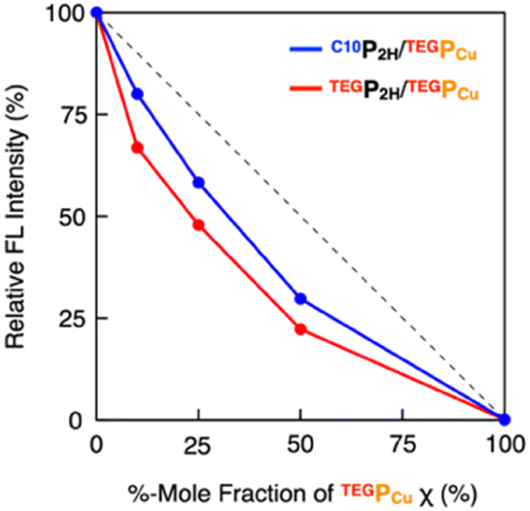
To investigate how C10P2H and TEGPCu are integrated in the LC phase, we performed electronic absorption and fluorescence spectroscopy of the mixed LC (Fig. 4 and 5). When the mixed LC of the conflicting pair (C10P2H/TEGPCu = 50/50 in 5OCB ([monomer] = 7.7 mol%)) was cooled from its isotropic melt at 150 °C to 25 °C at a ratio of 5 °C min−1, it showed a Soret band absorption peak at 433 nm (Fig. 4d, green solid line). Compared to a simple summation of the absorption spectra of C10P2H in 5OCB ([monomer] = 7.7 mol%) and TEGPCu in 5OCB ([monomer] = 7.7 mol%) (Fig. 4d, black dashed line), the absorption peak of the mixed LC exhibited a red-shift of 6 nm. Interestingly, the absorption spectral profile of the mixed LC of the affinitive pair (TEGP2H/TEGPCu = 50/50 in 5OCB ([monomer] = 7.7 mol%)) exhibited the same trend (Fig. 4e). This suggests that two monomers, no matter whether the monomer pair is conflicting (C10P2H/TEGPCu) or affinitive (TEGP2H/TEGPCu), are mixed in a single column in both mixed LCs. Then, the fluorescence spectra of LC samples were measured. LC samples containing only C10P2H or TEGP2H as a monomer showed characteristic fluorescence at λem = 650 nm and 715 nm upon excitation at λex = 590 nm (Fig. S16†). When TEGPCu was added to these LCs, the intensity of the fluorescence spectra decreased without a change in its shape (Fig. S16†), likely due to the electron exchange between P2H and PCu which happens only when they are stacked.35 Then, the fluorescence spectra of the mixed LCs at different %-mole fractions χ of PCu (χ = PCu/(PCu + P2H) = 0, 10, 25, 50, 100%) were recorded (Fig. S16†), and the intensity of the spectral peak at λem = 650 nm relative to that at χ = 0 (Fig. 5) was plotted. In both conflicting C10P2H/TEGPCu and affinitive TEGP2H/TEGPCu pairs, fluorescence quenching of P2H was observed at χ = 10 indicating the face-to-face stacking of P2H and PCu (i.e. copolymerization) in either of the mixed LCs. This is consistent with the results obtained from the absorption spectra (Fig. 4). In addition, the degree of fluorescence quenching for conflicting C10P2H/TEGPCu LC is smaller than that for affinitive TEGP2H/TEGPCu LC. This trend was also observed in relative fluorescence lifetimes of the same mixed LCs (Fig. S18 and Table S1†). This may suggest that although C10P2H and TEGPCu form a single columnar LC phase without macroscopic phase separation, the incompatibility of the side chains may favor homo-interaction over hetero-interaction at the microscopic level.
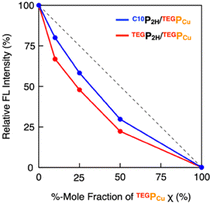 |
| | Fig. 5 Plots of relative fluorescence intensity (at λem = 650 nm and λex = 590 nm) at different %-mole fractions χ of TEGPCu for the mixed LC of C10P2H/TEGPCu (blue) and TEGP2H/TEGPCu (red) in 5OCB (7.7 mol%). | |
In sharp contrast, the supramolecular polymerization of the mixed samples in diluted solution ([monomer] = 3.4 × 10−9 mol% (2.0 × 10−8 mol L−1)) in dodecane/chlorocyclohexane (54![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :46) was distinct from that in the LC state. In the case of the conflicting C10P2H/TEGPCu pair, the electronic absorption spectrum of the mixed solution matched the summation of the absorption spectra of the individual polymers (Fig. S15a, c, d†). This indicates that the conflicting C10P2H/TEGPCu pair underwent self-sorting polymerization in a diluted solution. On the other hand, the absorption peak of the mixed solution of the affinitive TEGP2H/TEGPCu pair exhibited a red-shift of 4 nm compared to a simple summation of the absorption spectrum of TEGP2H and TEGPCu solutions (Fig. S15b, c, e†). This indicates that the affinitive TEGP2H/TEGPCu pair underwent copolymerization. These results were also supported by the fluorescence quenching experiment, where the fluorescence quenching was only obvious for the affinitive TEGP2H/TEGPCu pair (Fig. S17†). These results indicate that the copolymerization of C10P2H and TEGPCu occurs only in a medium of 5OCB under highly concentrated conditions.
:46) was distinct from that in the LC state. In the case of the conflicting C10P2H/TEGPCu pair, the electronic absorption spectrum of the mixed solution matched the summation of the absorption spectra of the individual polymers (Fig. S15a, c, d†). This indicates that the conflicting C10P2H/TEGPCu pair underwent self-sorting polymerization in a diluted solution. On the other hand, the absorption peak of the mixed solution of the affinitive TEGP2H/TEGPCu pair exhibited a red-shift of 4 nm compared to a simple summation of the absorption spectrum of TEGP2H and TEGPCu solutions (Fig. S15b, c, e†). This indicates that the affinitive TEGP2H/TEGPCu pair underwent copolymerization. These results were also supported by the fluorescence quenching experiment, where the fluorescence quenching was only obvious for the affinitive TEGP2H/TEGPCu pair (Fig. S17†). These results indicate that the copolymerization of C10P2H and TEGPCu occurs only in a medium of 5OCB under highly concentrated conditions.
Analysis by electron spin resonance (ESR) spectroscopy
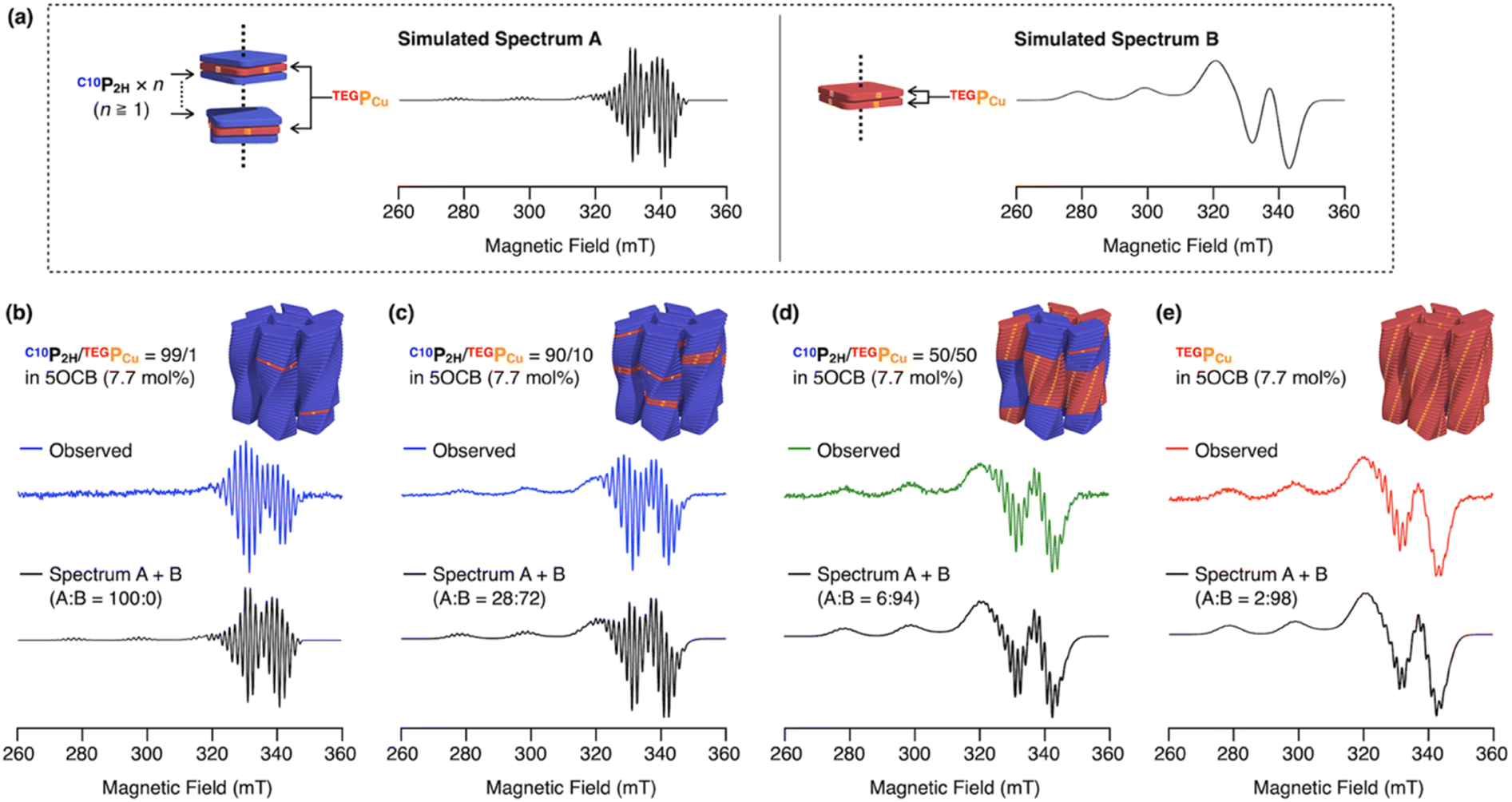
We investigated how the conflicting C10P2H/TEGPCu pair was mixed in a single column by ESR spectroscopy. In general, ESR spectra of Cu(II) change drastically when those spins interact with each other.45–48 It is known that dispersed PCu gives a sharp spectrum with a narrow line width (Fig. 6a, simulated spectrum A).45–47 On the other hand, when PCu are close to each other so that interactions between the Cu(II) spins due to orbital overlap take place, it is known to give a broad spectrum (Fig. 6a, simulated spectrum B).46–48 In the case where one (or more) P2H is intercalated between PCu, the interaction between the Cu(II) spins is prohibited thus giving a sharp spectrum like spectrum A (Fig. 6a). We expected that this characteristic spectral feature could be used to evaluate the internal structure of an individual column, that is a copolymer composed of C10P2H and TEGPCu. Thus, we measured the ESR spectra of four LC samples, C10P2H/TEGPCu = 99/1, 90/10, 50/50, and 0/100 in 5OCB ([monomer] = 7.7 mol%).44 Interestingly, we found that the ESR spectra of all four samples can be represented as either spectrum A or B, or their summation (Fig. 6b–e). For example, the mixed LC of C10P2H/TEGPCu = 99/1 in 5OCB ([monomer] = 7.7 mol%), forming a single Colob LC phase, gave a sharp ESR spectrum with a narrow line width identical to that of spectrum A (Fig. 6b). This indicates that TEGPCu monomers, which are present in only 1% of the total monomers, are separated from each other. On the other hand, the Colob LC phase of the single-monomer LC of TEGPCu in 5OCB ([monomer] = 7.7 mol%) gave an ESR spectrum that could be represented as a summation of a broad spectrum B (98%) and a sharp spectrum A (2%) (Fig. 6e). This shows that the TEGPCu monomers were mostly stacked with each other and only 2% formed a non-stacked disordered structure. When we performed ESR spectroscopy of the mixed LC of C10P2H/TEGPCu = 50/50 in 5OCB ([monomer] = 7.7 mol%), the spectrum could be represented as a summation of spectrum A (6%) and spectrum B (94%) (Fig. 6d). This result is surprising considering that an ESR spectrum of a statistically random co-stacking of TEGPCu and C10P2H would be the sum of 25% A and 75% B. The experimental result indicates that as much as 94% of the total TEGPCu monomers in the mixture are stacked with each other. The ESR spectrum of the mixed LC of C10P2H/TEGPCu = 90/10 in 5OCB ([monomer] = 7.7 mol%) was represented as a sum of 28% A and 72% B (Fig. 6c). Considering that a statistically random copolymer should be the sum of 81% A and 19% B, this result also showed that TEGPCu prefers the homo-interaction. Together with the fluorescence spectral profiles, the C10P2H/TEGPCu-based integrated Colob LCs are likely composed of supramolecular block copolymers of the two monomers.44 In the previous examples, the formation of supramolecular block copolymers is governed by the balance between the association energies between the monomers (homo- and hetero-interactions).30–34 In the present case, compatibilizing 5OCB seems to contribute to controlling this energy balance to realize a block sequence with the two monomers, which otherwise undergo self-sorting supramolecular polymerization.
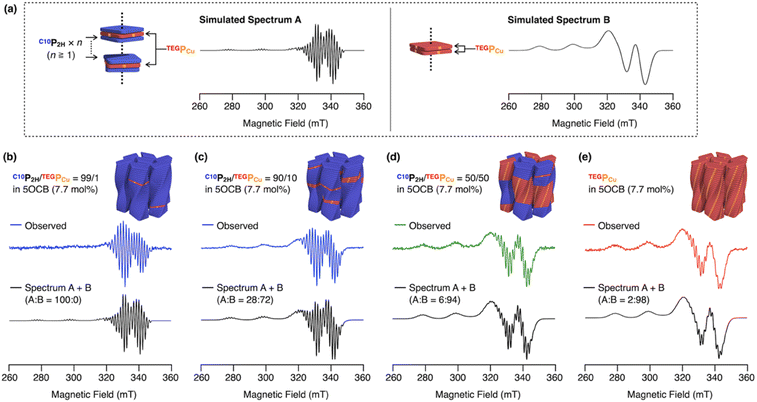 |
| | Fig. 6 (a) ESR spectra typical of PCu without Cu(II) spin–spin interactions (spectrum A) and with spin–spin interactions (spectrum B), generated by using the MATLAB® package of EasySpin®. (b–e) Obtained ESR spectra of (b) C10P2H/TEGPCu = 99/1, (c) C10P2H/TEGPCu = 90/10, (d) C10P2H/TEGPCu = 50/50 and (e) TEGPCu in 5OCB (7.7 mol%). The summation of spectrum A and spectrum B is also shown (black). | |
Molecular simulation of the LC structures
The above results are also supported by the coarse-grained molecular simulations. First, the columnar LC structure was constructed using C10P2H and 5OCB (Fig. S23†), and equilibrated using constant-temperature simulation. The columnar LC structure turned out to be stable for more than 100 ns without structural collapse (Fig. S24†). When half of the C10P2H molecules in the model were randomly replaced with TEGPCu and the same constant-temperature simulation was performed, the integrated columnar LC structure was stable as well (Fig. S25†). Finally, a simulation was performed in which all 5OCB molecules were changed to virtual solvent particles from the initial configuration of the columnar LC containing both C10P2H and TEGPCu. After 200 ns, the columnar structure collapsed and a single cluster composed of separated C10P2H and TEGPCu was formed at equilibrium (Fig. S26†). These results are consistent with our experimental results that both monomers do not mix homogeneously in the bulk state44 but can be integrated into a columnar structure in the presence of 5OCB.
Mixing of porphyrin monomers and BTA monomers in columnar LC
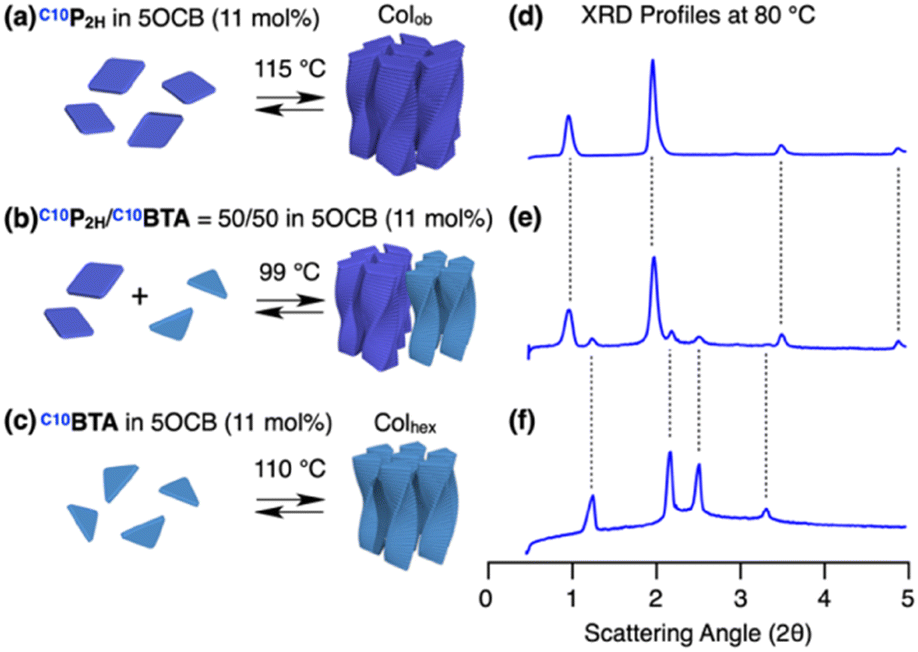
The above experiments showed that two porphyrin-centered monomers having incompatible side chains, C10P2H and TEGPCu can be integrated in the presence of compatibilizing 5OCB. We then considered mixing C10P2H and C10BTA (Fig. 1). Although both monomers have the same side chains, their different core sizes and number of amide groups make H-bonding-based copolymerization difficult. In such cases, is it possible for 5OCB to compatibilize C10P2H and C10BTA?. When C10BTA was mixed in 5OCB at a concentration of 11 mol%, a single columnar hexagonal LC phase (a = 41.6 Å at 80 °C) was formed (Fig. 7c, f, S5e, S7b and S11b†). In the case of C10P2H, it formed a Colob LC phase (a = 100.0 Å; b = 27.8 Å; γ = 71.6° at 80 °C) at 11 mol% concentration in 5OCB (Fig. 7a, d, S5a, S7a and S11a†). The mixed LC of C10P2H/C10BTA = 50/50 in 5OCB ([monomer] = 11 mol%) underwent a phase transition to a LC phase at 99 °C upon cooling (5 °C min−1) from its isotropic melt at 200 °C (Fig. 7b and S5c†). The XRD profile obtained at 80 °C was represented as a summation of the diffraction patterns of the columnar LCs of C10P2H in 5OCB ([monomer] = 11 mol%) and C10BTA in 5OCB ([monomer] = 11 mol%) (Fig. 7e). This indicates that both monomers formed their own columnar LC phases separately (Fig. 7d–f). In the POM images of the mixed LC, two different textures were observed, which were similar to that observed for the single monomer LC of C10P2H and C10BTA, indicating macroscopic phase separation (Fig. S7†). The samples with C10P2H/C10BTA = 25/75 and 75/25 also showed similar phase separation (Fig. S12†). These results suggest that 5OCB cannot compatibilize a pair of monomers having different preferences in H-bonding pattern and/or LC ordering.
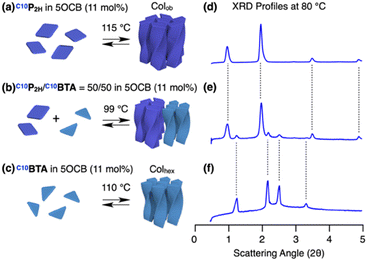 |
| | Fig. 7 (a–c) Schematic representations of the formation of columnar LC and (d–f) XRD profiles at 80 °C of (a and d) C10P2H, (b and e) C10P2H/C10BTA = 50/50 and (c and f) C10BTA in 5OCB (11 mol%). The temperatures shown above the arrows in (a–c) are the LC phase transition temperatures obtained by DSC on cooling (5 °C min−1). | |
Conclusions
In this study, we found that C10P2H and TEGPCu, which have mutually incompatible side chains, form an integrated structure consisting of supramolecular block copolymers in the columnar LC phase in the presence of 5OCB. The key for this achievement is the compatibilization by 5OCB, which forms a columnar structure together with supramolecular polymers, suppressing the macroscopic phase separation of both monomers. This strategy of mixing incompatible polymers by using a mediator is expected to lead to the realization of microphase-separated structures in supramolecular polymers.
Data availability
All data are available in the manuscript or in the ESI.†
Author contributions
D. M., Y. I. and T. A. designed the research. D. M. and X.-J. Z. prepared and characterized the materials. K. F. designed and conducted ESR measurements. N. A. carried out MD simulation. D. M. and Y. I. wrote the manuscript, with contributions from all authors.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The synchrotron radiation experiments were performed on BL44B2 at the Super Photon Ring (SPring-8) with the approval of RIKEN (Proposal No. 20200031, 20210015, and 20220053). We acknowledge Dr K. Kato and Dr T. Hoshino from RIKEN SPring-8 center for their generous support for the synchrotron radiation experiments. This work was financially supported by a JSPS Grant-in-Aid for Scientific Research (S) (18H05260) on “Innovative Functional Materials based on Multi-Scale Interfacial Molecular Science” and Specially Promoted Research (23H05408) on “Solid-State Materials Science of Supramolecular Polymers and Their Applications” for T. A. Y. I. is grateful for a JSPS Grant-in-Aid for Young Scientist (A) (16H06035), Scientific Research (B) (21H01903) and the JST PRESTO grant JPMJPR21Q1. D. M. thanks the University Fellowship Founding Project for Innovation Creation in Science and Technology, “Fellowship for Integrated Materials Science and Career Development”.
Notes and references
- P. J. Flory, J. Chem. Phys., 1941, 9, 660 CrossRef CAS.
- M. L. Huggins, J. Phys. Chem., 1942, 46, 151–158 CrossRef CAS.
- C. M. Bates and F. S. Bates, Macromolecules, 2017, 50, 3–22 CrossRef CAS.
- L. Leibler, Macromolecules, 1980, 13, 1602–1617 CrossRef CAS.
- F. S. Bates and G. H. Fredrickson, Annu. Rev. Phys. Chem., 1990, 41, 525–557 CrossRef CAS PubMed.
- M. W. Matsen and F. S. Bates, Macromolecules, 1996, 29, 1091–1098 CrossRef CAS.
- J. A. A. W. Elemans, A. E. Rowan and R. J. M. Nolte, J. Mater. Chem., 2003, 13, 2661–2670 RSC.
- T. F. A. De Greef, M. M. J. Smulders, M. Wolffs, A. P. H. J. Schenning, R. P. Sijbesma and E. W. Meijer, Chem. Rev., 2009, 109, 5687–5754 CrossRef CAS PubMed.
- T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335, 813–817 CrossRef CAS PubMed.
- K. Liu, Y. Kang, Z. Wang and X. Zhang, Adv. Mater., 2013, 25, 5530–5548 CrossRef CAS PubMed.
- E. Moulin, J. J. I. Armao and N. Giuseppone, Acc. Chem. Res., 2019, 52, 975–983 CrossRef CAS PubMed.
- M. Wehner and F. Würthner, Nat. Rev. Chem, 2020, 4, 38–53 CrossRef CAS.
- T. Aida and E. w. Meijer, Isr. J. Chem., 2020, 60, 33–47 CrossRef CAS.
- P. K. Hashim, J. Bergueiro, E. W. Meijer and T. Aida, Prog. Polym. Sci., 2020, 105, 101250 CrossRef CAS.
- B. Adelizzi, N. J. Van Zee, L. N. J. de Windt, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2019, 141, 6110–6121 CrossRef CAS PubMed.
- H. M. M. ten Eikelder, B. Adelizzi, A. R. A. Palmans and A. J. Markvoort, J. Phys. Chem. B, 2019, 123, 6627–6642 CrossRef CAS PubMed.
- K. Yano, Y. Itoh, F. Araoka, G. Watanabe, T. Hikima and T. Aida, Science, 2019, 363, 161–165 CrossRef CAS PubMed.
- K. Yano, T. Hanebuchi, X.-J. Zhang, Y. Itoh, Y. Uchida, T. Sato, K. Matsuura, F. Kagawa, F. Araoka and T. Aida, J. Am. Chem. Soc., 2019, 141, 10033–10038 CrossRef CAS PubMed.
- X.-J. Zhang, D. Morishita, T. Aoki, Y. Itoh, K. Yano, F. Araoka and T. Aida, Chem.–Asian J., 2022, 17, e202200223 CrossRef CAS PubMed.
- W. Zhang, W. Jin, T. Fukushima, A. Saeki, S. Seki and T. Aida, Science, 2011, 334, 340–343 CrossRef CAS PubMed.
- Q. Wan, W.-P. To, X. Chang and C.-M. Che, Chem, 2020, 6, 945–967 CAS.
- W. Wagner, M. Wehner, V. Stepanenko and F. Würthner, CCS Chem., 2019, 1, 598–613 CrossRef CAS.
- W. Wagner, M. Wehner, V. Stepanenko and F. Würthner, J. Am. Chem. Soc., 2019, 141, 12044–12054 CrossRef CAS PubMed.
- A. Sarkar, R. Sasmal, C. Empereur-mot, D. Bochicchio, S. V. K. Kompella, K. Sharma, S. Dhiman, B. Sundaram, S. S. Agasti, G. M. Pavan and S. J. George, J. Am. Chem. Soc., 2020, 142, 7606–7617 CrossRef CAS PubMed.
- A. Sarkar, R. Sasmal, A. Das, S. S. Agasti and S. J. George, Chem. Commun., 2021, 57, 3937–3940 RSC.
- A. Sarkar, R. Sasmal, A. Das, A. Venugopal, S. S. Agasti and S. J. George, Angew. Chem., Int. Ed., 2021, 60, 18209–18216 CrossRef CAS PubMed.
- S. H. Jung, D. Bochicchio, G. M. Pavan, M. Takeuchi and K. Sugiyasu, J. Am. Chem. Soc., 2018, 140, 10570–10577 CrossRef CAS PubMed.
- C. Jarrett-Wilkins, X. He, H. E. Symons, R. L. Harniman, C. F. J. Faul and I. Manners, Chem.–Eur. J., 2018, 24, 15556–15565 CrossRef CAS PubMed.
- D. Görl, X. Zhang, V. Stepanenko and F. Würthner, Nat. Commun., 2015, 6, 7009 CrossRef PubMed.
- B. Adelizzi, A. Aloi, A. J. Markvoort, H. M. M. Ten Eikelder, I. K. Voets, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2018, 140, 7168–7175 CrossRef CAS PubMed.
- A. Sarkar, T. Behera, R. Sasmal, R. Capelli, C. Empereur-mot, J. Mahato, S. S. Agasti, G. M. Pavan, A. Chowdhury and S. J. George, J. Am. Chem. Soc., 2020, 142, 11528–11539 CrossRef CAS PubMed.
- L. López-Gandul, A. Morón-Blanco, F. García and L. L. Sánchez, Angew. Chem., Int. Ed., 2023, 62, e202308749 CrossRef PubMed.
- L. N. J. de Windt, Z. Fernández, M. Fernández-Míguez, F. Freire and A. R. A. Palmans, Chem.–Eur. J., 2022, 28, e202103691 CrossRef CAS PubMed.
- B. Adelizzi, P. Chidchob, N. Tanaka, B. A. G. Lamers, S. C. J. Meskers, S. Ogi, A. R. A. Palmans, S. Yamaguchi and E. W. Meijer, J. Am. Chem. Soc., 2020, 142, 16681–16689 CrossRef CAS PubMed.
- O. Ohno, Y. Ogasawara, M. Asano, Y. Kajii, Y. Kaizu, K. Obi and H. Kobayashi, J. Phys. Chem., 1987, 91, 4269–4273 CrossRef CAS.
- S. Prathapan, T. E. Johnson and J. S. Lindsey, J. Am. Chem. Soc., 1993, 115, 7519–7520 CrossRef CAS.
- M. S. Asano, K. Okamura, A. Jin-mon, S. Takahashi and Y. Kaizu, Chem. Phys., 2013, 419, 250–260 CrossRef CAS.
- G. A. Schick, I. C. Schreiman, R. W. Wagner, J. S. Lindsey and D. F. Bocian, J. Am. Chem. Soc., 1989, 111, 1344–1350 CrossRef CAS.
- J. H. van Esch, M. C. Feiters, A. M. Peters and R. J. M. Nolte, J. Phys. Chem., 1994, 98, 5541–5551 CrossRef CAS.
- R. van der Weegen, A. J. P. Teunissen and E. W. Meijer, Chem.–Eur. J., 2017, 23, 3773–3783 CrossRef CAS PubMed.
- N. Sasaki, M. F. J. Mabesoone, J. Kikkawa, T. Fukui, N. Shioya, T. Shimoaka, T. Hasegawa, H. Takagi, R. Haruki, N. Shimizu, S. Adachi, E. W. Meijer, M. Takeuchi and K. Sugiyasu, Nat. Commun., 2020, 11, 3578 CrossRef PubMed.
- T. Yamaguchi, T. Kimura, H. Matsuda and T. Aida, Angew. Chem., Int. Ed., 2004, 43, 6350–6355 CrossRef CAS PubMed.
- S. Okada and H. Segawa, J. Am. Chem. Soc., 2003, 125, 2792–2796 CrossRef CAS PubMed.
- See ESI†.
- K. Tanaka, A. Tengeiji, T. Kato, N. Toyama and M. Shionoya, Science, 2003, 299, 1212–1213 CrossRef CAS PubMed.
- S. Richert, I. Kuprov, M. D. Peeks, E. A. Suturina, J. Cremers, H. L. Anderson and C. R. Timmel, Phys. Chem. Chem. Phys., 2017, 19, 16057–16061 RSC.
- N. Toyama, M. Asano-Someda and Y. Kaizu, Mol. Phys., 2003, 101, 733–742 CrossRef CAS.
- F. Hajjaj, K. Tashiro, H. Nikawa, N. Mizorogi, T. Akasaka, S. Nagase, K. Furukawa, T. Kato and T. Aida, J. Am. Chem. Soc., 2011, 133, 9290–9292 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Yoshimitsu
Itoh
a,
Yoshimitsu
Itoh