
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDELFI: a computer oracle for recommending density functionals for excited states calculations†
Davide
Avagliano
 *ab,
Marta
Skreta
bc,
Sebastian
Arellano-Rubach
d and
Alán
Aspuru-Guzik
*abcefgh
*ab,
Marta
Skreta
bc,
Sebastian
Arellano-Rubach
d and
Alán
Aspuru-Guzik
*abcefgh
aDepartment of Chemistry, University of Toronto, 80 St. George Street, Toronto, ON M5S 3H6, Canada. E-mail: davide.avagliano@utoronto.ca; alan@aspuru.com
bDepartment of Computer Science, University of Toronto, 40 St. George Street, Toronto, ON M5S 2E4, Canada
cVector Institute for Artificial Intelligence, 661 University Ave. Suite 710, ON M5G 1M1, Toronto, Canada
dUniversity of Toronto Schools, 371 Bloor St W, Toronto, ON M5S 2R7, Canada
eDepartment of Materials Science & Engineering, University of Toronto, 184 College St, Toronto, M5S 3E4, Canada
fDepartment of Chemical Engineering & Applied Chemistry, University of Toronto, 200 College St, ON M5S 3E5, Toronto, Canada
gLebovic Fellow, Canadian Institute for Advanced Research (CIFAR), 66118 University Ave., M5G 1M1, Toronto, Canada
hAcceleration Consortium, 80 St George St, M5S 3H6, Toronto, Canada
First published on 13th February 2024
Abstract
Density functional theory (DFT) is the workhorse of computational quantum chemistry. One of its main limitations is that choosing the right functional is a non-trivial task left for human experts. The choice is particularly hard for excited state calculations when using its time-dependent formulation (TD-DFT). This is due to the approximations of the method, but also because the photophysical properties of a molecule are defined by a manifold of states that all need to be properly described. This includes not only the relative energy of the states, but also capturing the correct character, order, and intensity of the transitions. In this work, we developed a neural network to recommend functionals to be used on molecules for TD-DFT calculations, by simultaneously considering all these properties for a manifold of states. This was possible by developing a scoring system to define the accuracy of an excited state's calculation against a higher-accuracy reference. The scoring system is generalizable to any level of theory; we here applied it to evaluate the performance of common functionals of different rungs against a higher accuracy method on a large set of organic molecules. The results are collected in a database that we released and made open, providing four million data points to the community for future applications. The scoring system assigns a value between zero and one hundred to each functional for each molecule, transforming the complicated task of learning photophysical properties into a simpler regression task. We used the dataset to train a graph attention neural network to predict the scores for unseen molecules. We call this oracle DELFI (Data-driven EvaLuation of Functionals by Inference), which can be used to quickly screen and predict the ranking of functionals to calculate the optical properties of organic molecules. We validated DELFI in two in silico experiments: choosing a common functional for a series of spiropyran-merocyanine isomers and a unique functional to screen a large dataset of over 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 organic photovoltaic molecules, for which an extensive benchmark would be unfeasible. A corresponding web application allows DELFI to be easily run and the results to be analyzed, alleviating the hurdle of choosing the right functional for TD-DFT calculations.
000 organic photovoltaic molecules, for which an extensive benchmark would be unfeasible. A corresponding web application allows DELFI to be easily run and the results to be analyzed, alleviating the hurdle of choosing the right functional for TD-DFT calculations.
1 Introduction
Density functional theory (DFT)1 and its time-dependent formulation (TD-DFT)2 for calculations beyond the electronic ground state are the most widely used computational methods for the calculation of electronic energies, due to their hardly beatable accuracy/cost trade-off for medium-to-large size molecular systems. However, the main limitation of DFT lies in the impossibility of knowing the exact form of the electron exchange and correlation for molecules and the need to use an approximate functional.3 Although a plethora of exchange–correlation functionals have been developed,4 even reaching high accuracy for specific problems and universal recognition in the community,5 there is arguably a unique solution for the combination system/property/functional.6 This is particularly true for the calculation of electronically excited states, where several additional challenges arise due to the treating of highly correlated and multi-reference objects as excited states.7,8 TD-DFT, in its linear response formulation,9 the most popular one, and in the limit of the adiabatic approximation10 cannot describe double/multiple excitations11 and intersection seams between the ground and first excited state potential energy surfaces,12 and it problematically describes highly delocalized states as charge transfer or Rydberg states.13,14 While the first two problems cannot be overcome within the adiabatic approximation, for the latter several modifications have been introduced to mitigate the errors of ground state functionals and improve the performances of TD-DFT, leading to the development of long-range corrected functionals.15 However, a general recipe for a functional that can satisfactorily and universally describe optical molecular properties has not been found, and arguably cannot be found. As a consequence, since DFT and TD-DFT popularity is so high, one of the most common questions a computational chemist faces while approaching a problem to solve is: which exchange–correlation functional should I use?16 A generally accepted approach to finding an answer is to benchmark a bunch of functionals against the results obtained with a higher-level method or experimental results,17,18 if available. In this case, a one-by-one state comparison is needed, since it is missing a general quantitative way to estimate the quality of an excited state calculation. Additionally, this approach is prohibitive when studying large-size molecular systems, or when deciding on a unique functional to screen the optical properties of a large set of molecules, for example in the field of material discovery. In these cases, a single pick is made, based on literature research and chemical intuition. More generally, both approaches are limited to a small subset of choices and might easily leave out the outperforming functional. Alternatively, general qualitative indexes have been proposed, or specific types of excitations have been focused on, but always requiring actually running the calculations a priori.19–22 Machine learning could facilitate this task by learning patterns between molecular properties and functional performances, as promisingly shown very recently for ground state properties.23 However, in the case of optical properties like UV/Vis absorption, many electronic states need to be considered, and their energies are not the only quantities to consider when evaluating the performance of a functional, but also the character, intensity and order of the excited states need to be properly described by TD-DFT.24 Indeed, a wrong description of the order, character, and brightness of two states might lead to misinterpretation of photochemical pathways and reactivity. An easy yet important example can be the canonical nucleobase UV/Vis absorption, where a misinterpretation of order and character of La, Lb and nπ* states would lead to a wrong interpretation of their photophysical properties.25 To the best of our knowledge, there is not a systematic, universal, and transferable excited state evaluation scheme that is able to keep together all this information for a bunch of excited states that could be used for the assessment of the quality of a method or functional. In this paper, we aim to fill this large gap by introducing a general scheme to evaluate excited state calculations obtained with a lower accuracy electronic structure method with respect to a more accurate one. In particular, we developed a scoring system that considers at the same time differences in energy, character, and brightness of a certain number of states and then assigns a final score between 0 and 100. By applying this scoring system to the calculation obtained with 38 different functionals of different quality on a dataset of organic molecules, we will be able to transform the functional recommendation in a regression problem, train a graph attention neural network (GAT),26 and provide a transferable TD-DFT functional recommender. We called the recommender DELFI (Data-driven EvaLuation of Functionals by Inference) and we will show here its applicability to provide a set of functionals, and their relative score, to be tested and benchmarked as outperforming a given molecular system or database. Finally, we provide a web application that we developed to analyze the scoring system on which the data is trained and to directly run DELFI by just providing the SMILES of a molecule, including tools to analyze its results in an interactive and user-friendly way, providing a powerful tool for chemists of any level of programming experience approaching a TD-DFT calculation.2 Excited state scoring system
There is no unique metric to define the accuracy of an excited states calculation, since not only the relative energy of each state is relevant, but also the intensity and character of such excitations play a crucial role in the interpretation of UV/Vis absorption spectra, photochemical pathways and reactivity. Commonly, the evaluation is based on simply comparing the results with experiments or higher accuracy calculations. But how can we systematically and generally evaluate the performance of a given method/functional? Being able to correctly calculate the energy of the brightest state is definitely the first fundamental prerequisite, but there is not a systematic error for any TD-DFT functional, with a difference in energy that can vary from 0.3 to more than 1 eV. The ideal goal of a calculation would also be to identify the correct character of the states, i.e. the orbitals involved in the electronic transitions, as well as their relative intensity and order. We here propose an excited state scoring system that is based on weighing together the energy, character, and brightness of an excited state calculation with respect to a higher accuracy reference method. Given a set of calculated singlet states (but the same approach can be extended for states of other multiplicities), each state is evaluated singularly and the final score of the method/functional is given by their sum (Fig. 1). The score of a single state is in the range of 0–1 and it is the combination of a partial score due to the similarity of the character, the energy differences, and the brightness differences, with respect to the more accurate method, assigned in the following way: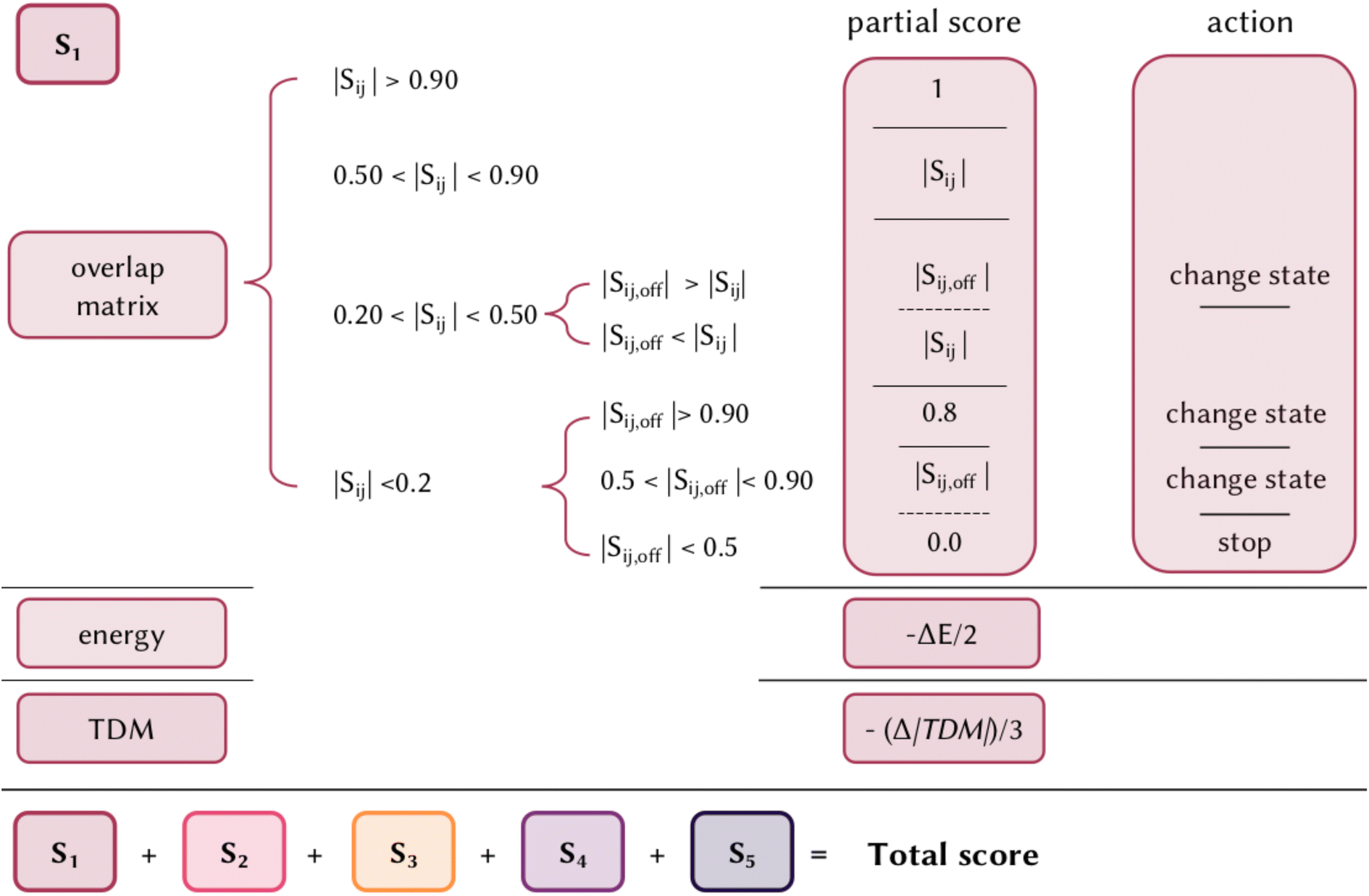 | ||
| Fig. 1 A summary of the DELFI excited state scoring system proposed in this work. First, the overlap matrix between the one-electron transition density matrix of the two calculations is calculated. According to the diagonal and/or the off-diagonal values, the matching states between the two calculations are chosen and a partial score is assigned for each state. This partial score is then refined by removing a penalty due to the difference in energy and TDM between the states of the two calculations. A score between 0 and 1 is assigned to each state, which indicates the similarity between the two. Finally, the scores for each state are summed up to give a final score to the excited state calculation. | ||
• The first fundamental step is to assign the correct states for comparison since the simply adiabatic order might not correspond for two different calculations. For that, the overlap matrix between the one-electron transition density matrix of the method to be evaluated and the one of a reference calculation is considered. Starting from the diagonal element, i.e. starting from states at the corresponding adiabatic order, we identify three possible scenarios: (i) the overlap is larger than 0.5, (ii) smaller than 0.2, or (iii) in the range 0.2–0.5. If the overlap is larger than 0.5, the same character is assumed. If the overlap is between 0.5 and 0.9, the corresponding value is assigned as a partial score, if the overlap is larger than 0.9 a value of 1 is assigned. In case the overlap is <0.5, we cannot assume the same pure character of the two states, but we check if the order of the state might be inverted, by looking at the off-diagonal element of the overlap matrix. If any off-diagonal element is larger than the diagonal one, that value is assigned and a change of state will be considered in the next steps of the scoring assignment. If none of the off-diagonal elements is larger than the diagonal element, then the value of the latter is assigned. The same principle is followed if the diagonal element is <0.2: an off-diagonal state with a similar character is searched for and if found, the corresponding value is assigned as a partial score and the corresponding state will be considered in the next steps. Two exceptions are considered, (i) if the overlap is larger than 0.9, a maximum value of 0.8 is given to slightly penalize the error in the order of states, and (ii) if none of the off-diagonal elements are larger than 0.5, this state is considered missing in the reference calculation and a total score of 0 is assigned to that state.
• After the correct states to be compared have been identified, their energy difference (ΔE) in eV is calculated. After the first step, each state has an assigned partial score between 0 and 1. As the full difference in energy between the two calculations could set too frequently to zero the partial score of the state, and consequently limit the capability of discriminating the quality of different calculations, only half of this value is subtracted from the partial score.
• Last, the same thing is done for the difference in the magnitude of the transition dipole moment (Δ|TDM|) in Debye, which measures the probability of the transition. As the TDM is known to be highly overestimated in TD-DFT, for the purpose of this project only a third of this difference is subtracted from the partial score. For a dark state this penalization will be basically null, while it is useful to slightly differentiate the error in TDM for bright states.
In summary, the score for an excited state is given by the overlap with a corresponding state of the reference calculation, minus (ΔE)/2, minus (Δ|TDM|)/3. The scaling of ΔE and Δ|TDM| was empirically chosen to not penalize excessively due to energy and intensity differences, and to give greater weight to the first over the second. While the scoring system is straightforward to use in terms of relative accuracy between two or more methods or functionals, it is less trivial to identify a threshold for an excited state calculation to be considered overall generally accurate. Obviously, there is not a general answer and it depends on the method considered. The strengths of this scoring system are the inclusion of all the terms in one metric and being general and transferable to any electronic structure method; it can detect the absence of double excitation or the presence of spurious charge transfer states or misinterpretation of mixed and pure states; it can be extended to include any number of excited states of any multiplicity, or just focusing on a manifold of states; it is easy to interpret and can be applied to compare quickly different methods/functionals. For example, if five excited states are considered, each with a score between 0 and 1, by multiplying by 20, a score between 0 and 100 is given to a calculation, which can be interpreted as the percentage expressing the similarity to a higher method, and several functionals can be evaluated on the same molecule by comparing their scores. In the web application we developed, we added an interactive page to interpret the scoring system, as will be shown in Section 5.
3 Scoring applied to the QM8 dataset
We wanted to apply the scoring system to evaluate and compare the performances of several functionals on a large set of molecules. We chose the QM8 database, as it was specifically designed to model the electronic spectra of organic molecules.27,28 It collects excited state calculations on more than 20000 small organic molecules. These molecules contain up to 8 CONF atoms and the largest are 18 entries with 26 atoms. We chose 38 functionals, whose accuracy was recently reviewed again for TD-DFT calculations,29 of different rungs,30 from local density approximation (LDA) to generalized-gradient approximation, including meta (mGGA) and hybrid and several long-range corrected functionals, namely:
• LDA: SPW92 (ref. 31).
• GGA: B97-D,32 MPW91,33 PBE,34 BLYP,35 N12 (ref. 36).
• mGGA: B97M-V,37 mBEEF,38 M06-L,39 revM06 L,40 MN15-L,41 revTPSS,42 TPSS.43
• Hybrid GGA: ωB97X-D,44 CAM-B3LYP,45 ωB97X-V,46 SOGGA11-X,47 LRC-wPBE,48 LRC-wPBEh,49 MPW1K,50 PBE0,51 HSEHJS,52,53 rcamB3LYP,54 MPW1PW91,33 BHHLYP,55,56 PBE50,57 B3LYP,58,59 HFLYP.56
• Hybrid mGGA: BMK,60 M06-SX,61 M06-2X,62 ωB97M-V,63 wM05-D,64 MN15,65 PW6B95,66 SCAN0,67 M11,68 revTPSSh,69 TPSSh,70 MN12-SX.71
The very large number of molecules and calculations required (21238 molecules and 828282 single points) forced us to some notable, but necessary, approximations. We used as a reference method the second-order Algebraic Diagrammatic Construction (ADC(2))72 scheme of the polarization propagator and a triple zeta basis set (def2-TZVP),73 calculating the first five singlet excitations. Although not as accurate as higher-quality methods, the quality of ADC(2) has been extensively benchmarked and validated in the literature.24,74 The reliability of this method is well assessed in the community and many works use ADC(2) as a method to benchmark TD-DFT75 or assume its goodness for vertical excitations, as well as for dynamics, without benchmarking it over more accurate methods.76–79 The overall error on vertical excitations for organic molecules is known to be around 0.2 eV with respect to experimental data, and the method is also a good choice, as done in this work, to spot the most common pitfalls of TD-DFT mentioned above, since ADC(2) manages to describe for example CT or double excitations.80,81 However, more accurate methods, such as the golden standard CCSD(T), are either too computationally expensive to calculate more than a hundred thousand vertical excitations in total, or they are inapplicable in an automatic fashion, as complete active space methods, since it would require ad hoc active space selection for each of the more than 20000 molecules. Calculating five singlets ensures that the first absorption band and eventual dark states present in the UV/Vis region are covered. However, our scoring system is not intended to provide the closest energy value to the experimental results, but only the strongest similarity between the TD-DFT and the reference calculations. The scoring for each functional in this section indicates the similarity only with respect to the five lowest singlet states, at one specific geometry, obtained with the more accurate method ADC(2), which still carries its own limitations being a single reference method, in the gas phase. All the details of the quantum chemical calculations are reported in Section 7. We can analyze the distribution of the scores for each functional along the dataset, as well as how many times a functional is the top performer, and identify trends in the performances (Fig. 2). A group of functionals seems to be more frequently the top performer, namely rcamB3LYP, ωB97X-V, PBE50, and BHHLYP. Together they perform the best on over 60% of the molecules. These functionals are also the more frequent ones with scores above 80 and 75, and in almost half of the dataset (total number of molecules considered is 21228) they have a score larger than 60, a lower bound value that we recommend as a threshold for a functional to be considered for benchmarking since it would be derived by an average error of 0.3 on the overlap of the states and a 0.2 eV with a comparable TDM (see Section 5 for details in the interpretation of the scoring system). Despite the approximations used, the scores and the trends obtained are in line with the recommendations obtained with the latest and more accurate functional benchmarks.29 Some of these functionals can perform similarly or very differently for different molecules and these data only show the general trend of the scoring along the dataset. Combining distribution and similarity matrix (Fig. S1†) can provide more information, for example showing how rcamB3LYP, ωB97X-V, PBE50, and BHHLYP show a similar distribution of scores, while being totally different from the one of HFLYP, which on the other hand has very few scores higher than 60, but still performs the best in more than a thousand cases, showing how 100% of Hartree–Fock (HF) exact exchange can perform better on the subset of molecules where long-range corrected and partial HF differ the most from ADC(2). Comparing the pairwise similarity of the scores of the functional along the dataset also shows that performances are in line with Jacob's Ladder and that families of functionals perform similarly (Fig. S1†). The similarity matrix can be extremely useful to extrapolate information beyond the set of functionals tested here, where the computational chemistry intuition combined with the performances of a set of functionals, can drive the choice of a functional out of this list, but with parameters that would fit with the best-performing ones or their family. For this reason we make this analysis available in the web application (Section 5). Indeed, we here reported only a few possible examples of analysis, but we generated a large amount of data that we made available with a set of analysis and visualization tools on a web application, which is described in detail in Section 5, that allow a customized and more in-depth interpretation of these data. To validate the choices of the parameters we used, we tried some different combinations of the scoring system. First of all, we didn't include the TDM in the calculation. In this case, for 97% of the molecules the top functional is matching with the one obtained with the full scoring system. This dropped to 75% when we included entirely the difference in energy, but this is due to an over-penalization of the score of a state that set to zero the associated partial score. Finally, we analyzed the effect of not penalizing states according to the overlap element, and this led to the opposite effect of high scores by not penalizing states that are similar in energy but notably with different characters. In conclusion, the definition of our system is reasonable for the scope of this work, generating a balanced score, that reflects literature benchmarks and provides a range of values that can be learnt by our GAT.
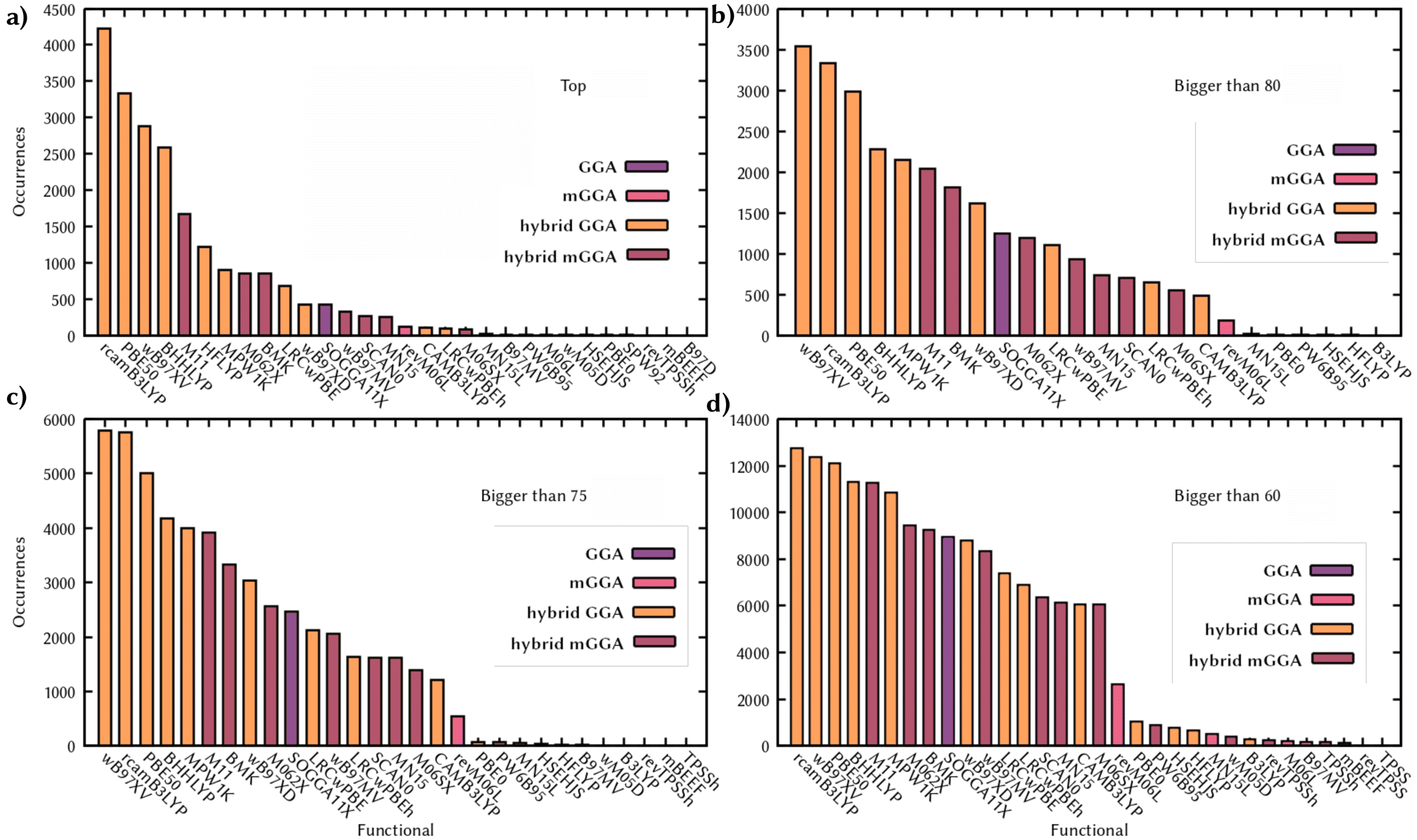 | ||
| Fig. 2 Scores distributions along the QM8 dataset. Occurrences of the functional with resulting highest score (a) and times for each functional with a score higher than 80 (b), 75 (c), and 60 (d). Histograms are colored according to the rung of the functional. | ||
However, we collected all the vertical excitations we ran on QM8, as well as all the overlap matrices calculated, in a database that we released and it is open and available at https://figshare.com/projects/DELFI/185308. This extended-QM8 database contains more than 4 million data points of singlet vertical excitations and oscillator strengths (5 excited states for each molecule), at ADC(2) and TD-DFT with the 38 functionals used. We believe that the dataset will be extremely useful for the community for computational chemistry and machine learning research on the optical properties of organic molecules. Additionally, we provided the overlap matrices for each functional of each molecule and we offer the possibility to the community of trying different combinations of the scoring system and have a complete overview on the huge amount of data we generated and provided.
4 Training and validation of the functional predictor
4.1 Training the scores predictor
Having defined a numerical evaluation of each functional and collected a large amount of data on the QM8 dataset, we trained a neural network to predict the score for each functional given a certain molecule. The goal of such a model is to quickly screen density functionals and cheaply provide a recommendation about a set of functionals within a certain threshold of score that should be properly benchmarked, potentially with a larger basis set and against experimental values, to find the most accurate functional to compute vertical excitations and UV/Vis spectrum of a molecule. We represented the molecules of the QM8 dataset using a 2D graph and trained a GAT for multitask regression, where a final message passing layer gives a set of 38 scores (one for each functional) for a given input molecule (Fig. 3a). Details on the implementation are described in Section 7. The test set used to analyze the performances of DELFI is composed of 2101 molecules (10% of the size of the dataset). We obtained an R-square of 0.7 and a minimum absolute error (MAE) of 6.5 (scores in the range 0–100) on all the 79838 scores predicted on the test set. We previously highlighted how it might be worth properly benchmarking a functional with a score above 60, so when we consider the predicted scores that are higher than this threshold, we have a true positive and negative accuracy of 76% and 95%, respectively. On average, 6 functionals per molecule are predicted above this threshold and, in 84% of the cases, the top performing functional is rightfully predicted in these six and 34% of the time it is correctly predicted with the highest score. When we considered the matching between which are the top five functionals calculated and predicted, 10% of the time 5/5 functionals match, reaching 78% considering when at least one match is found. However, performances and errors are not uniform for each of the 38 single regression tasks, as the distributions of the scores are also not uniform along the dataset for each of the functionals. We report in Section S2 of the ESI† performances and errors on the predicted score for the test set for each of the functionals individually. We also report, in the same section of the ESI,† the calculated uncertainty for each of the functionals, following the procedure described in Section 7. The uncertainty on the predicted scores for the molecules in the test set turns out to be extremely low for each of the functionals and smaller than the overall MAE (6.5). Before applying DELFI on test cases, it is important to underline the domain of applicability and when it should be used, remembering that the top functional does not necessarily reflect the one with the closest energies to experimental values, but only predicts the functional that matches the most the reference method at the level of theory used for the dataset, which carries its own limitation and experimental error.74 If possible, the top recommendation should be benchmarked. The score of the functionals is relative to a higher accuracy calculation, which still carries its own limitations. The dataset used to train the model contains relatively small organic molecules, and does not include, for example, transition metal complexes. The scores are obtained considering only five singlet states at a single geometry in the gas phase. As such, the indication provided by DELFI is meaningful for the calculation of a manifold of singlet vertical excitations and might yield inaccurate recommendations when including solvent effects, considering triplet state or excited state dynamics. Only 38 functionals are tested, but ranging among different types and different contributions of hybrid parameters and long-range corrections. The best functional might not be in this list, but the recommendation of DELFI could be useful to identify the family of functionals that perform the best.
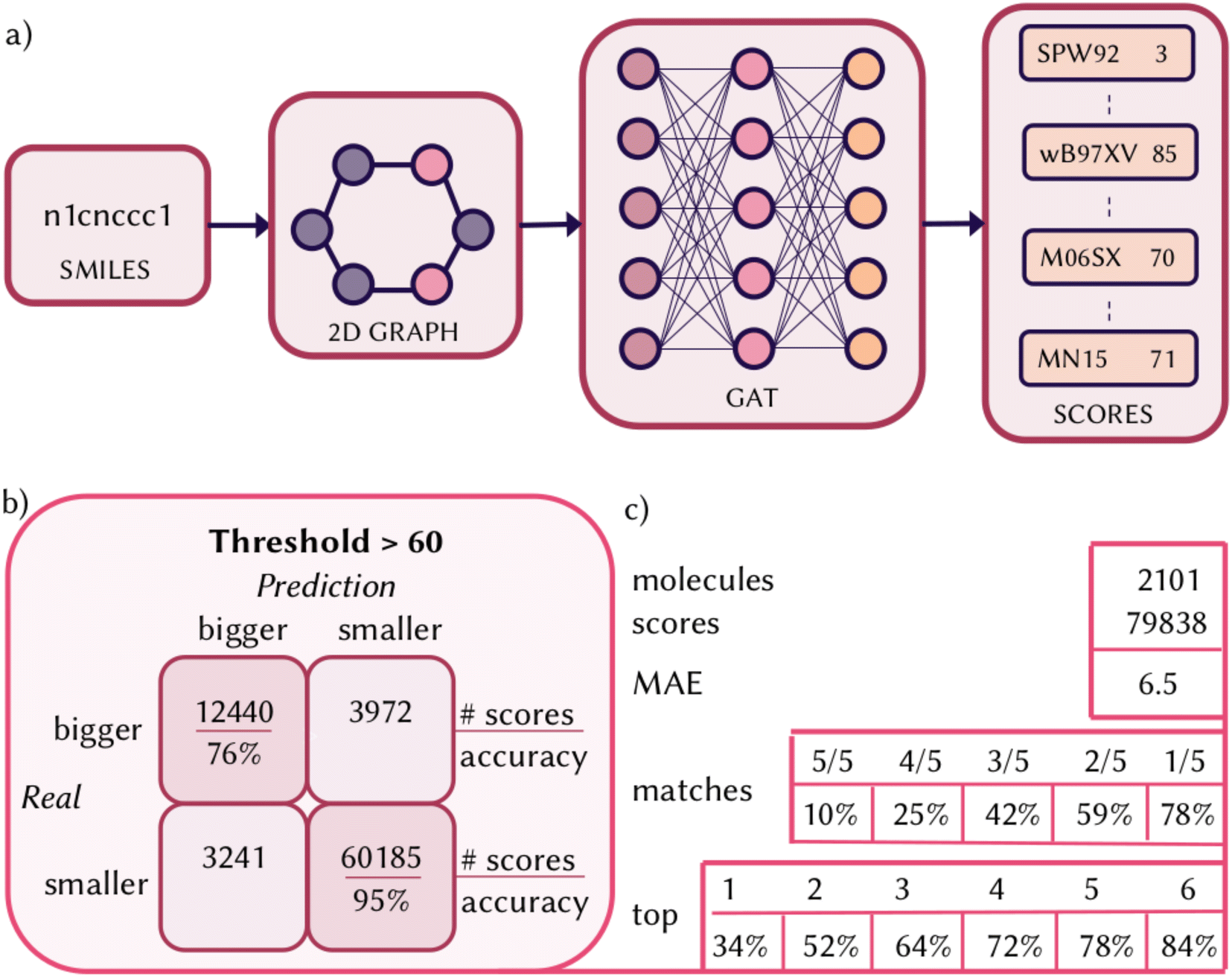 | ||
| Fig. 3 (a) Schematic representation of the model trained. A 2D graph is built starting from the SMILES of the molecules and a graph attention network (GAT) is trained for multitask regression to predict a score for each functional. (b and c) Performances of DELFI on QM8: confusion matrices for scores above the threshold of 60 (b); number of sample and mean absolute error (MAE), percentages of matches between test and prediction and top-k accuracy (c). | ||
4.2 Validation in real in silico experiments
| Molecule | R1 | R2 | Functional (score) | ||
|---|---|---|---|---|---|
| #1 | #2 | #3 | |||
| SP1 | H | H | ωB97X-V (80) | ωB97X-D (77) | LRC-wPBE (76) |
| MC1 | ωB97X-V (74) | rcamB3LYP (71) | M11 (71) | ||
| SP2 | H | NO2 | LRC-wPBE (59) | M11 (58) | ωB97X-V (57) |
| MC2 | ωB97X-V (64) | M11 (63) | LRC-wPBE (62) | ||
| SP3 | CH3 | NO2 | LRC-wPBE (60) | M11 (59) | ωB97X-V (58) |
| MC3 | ωB97M-V (63) | M11 (62) | ωB97X-V (62) | ||
| SP4 | H | OCH3 | ωB97X-V (81) | LRC-wPBE (76) | ωB97X-D (75) |
| MC4 | ωB97X-V (74) | rcamB3LYP (72) | M11 (71) | ||
| SP5 | NO2 | NO2 | ωB97M-V (61) | M11 (61) | ωB97X-V (60) |
| MC5 | ωB97M-V(66) | M11 (65) | ωB97X-V (64) | ||
To further validate the recommendations of DELFI, we chose two of these molecules, namely SP1 and SP2, and ran ADC(2) and TD-DFT calculations at the same level of theory as when the scores were obtained for the training set. All the functionals perform similarly and reproduce the same errors. In the case of SP1 (Fig. 4, left), for all three functionals the state order matches, and a systematic error in the energies is found. The similarity in the performances is reflected by the very similar calculated scores (Fig. 4, in parentheses). S1 and S5 are calculated to be less dark than in the reference, with ωB97X-V that mitigates this error more than the other two, and results have both the top predicted and calculated scores. In contrast, for SP2 (Fig. 4, right), the order of S1 and S2 is inverted with respect to the reference ADC(2). The calculated scores are lower than the one of SP1, and this important trend is successfully reproduced by the prediction of DELFI, which is able to capture the different performances of TD-DFT due to chemical changes in the molecules due to the presence of the nitro group. However, if one functional needs to be chosen to compare the photophysical properties of the whole set of the molecules considered, accounting for relative energies, brightness, and chemical intuition, ωB97X-V seems to actually be the best performer on these molecules with respect to the reference calculations, following the recommendation of DELFI that predicts to be always in the top three for the ten molecules considered and in particular the best performer in half of them. To confirm this, we calculated the first five excited states with all 38 functionals for SP1 and SP2 (Sections S4 and S5 of the ESI†), as well as the scores following our scoring system. The suggested functionals by DELFI reproduce the closest results with respect to the reference ADC(2) and are among the ones with the highest calculated scores, considering all 38 functionals, with ωB97X-V being, together with ωB97M-V, the top choice by both comparing energies with ADC(2) and according to the calculated scores.
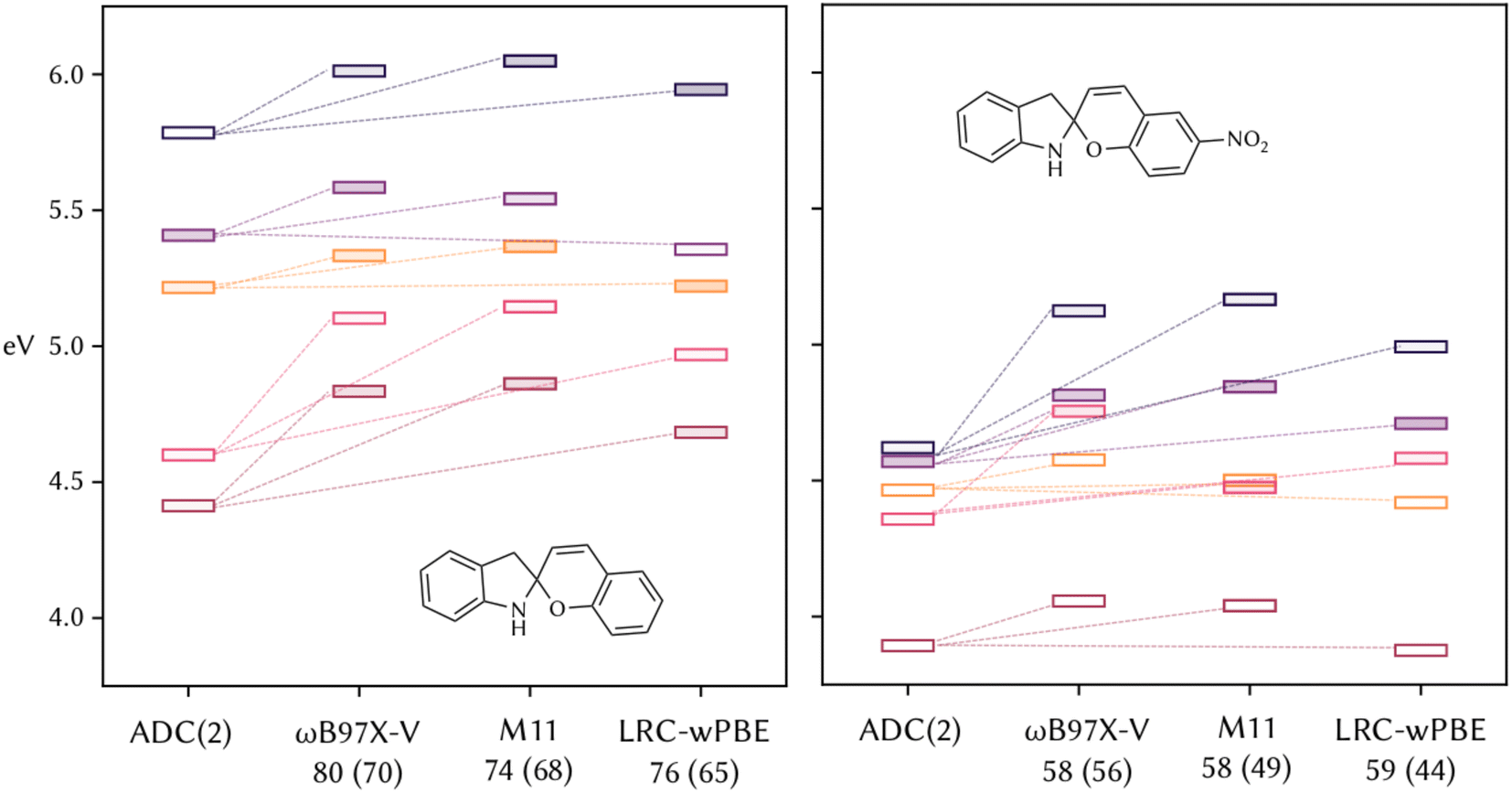 | ||
| Fig. 4 The first 5 singlet states calculated at ADC(2)/def2-TZVP and with the top three selected functionals for spiropyran bearing no substituent (left) and a nitro group on the chromene moiety (right). The color of the boxes represents the states with the same character; the intensity of the filling of the boxes is proportional to the intensity of the transitions; the dashed lines connect the matching state to the reference ADC(2). Predicted and calculated (in parentheses) scores are reported for each functional. | ||
000 entries, and we wanted to select a functional to screen the optical properties of these molecules to be used, for example, as molecular descriptors in a generative machine learning model. This dataset represents an additional test for the transferability of DELFI, as it includes molecules made of several fused rings and sulfur atoms, not present in the training set, but still photochemically relevant. Usually, what would drive the functional choice would be chemical intuition and a literature survey, trying to find a functional that is supposed to perform well on that family of molecules. DELFI can quickly predict the scores for each of the molecules and analyzing them can drastically facilitate the decision. We ran DELFI on the dataset and we obtained in a few seconds a clear indication of the functional to be used; indeed ωB97M-V turned out to be the top performer on 38747 out of 51281 (75%) molecules, with the second one being M11 in only 5% of the time (2578 molecules), giving a statistically robust indication of the functional to be used. Interestingly, this functional was only around 2% of the time the top performer on the training set of the model, showing how the graph representation allowed transferability of the prediction and not simply replicating the distribution of the scores of the learning. We wanted to validate the quality of the prediction by comparing the results of the calculations on a small ensemble of a few molecules contained in the dataset. We considered the first five entries, which are shown in Fig. 5. These molecules are larger than the one contained in QM8 and include several rings and sulfur atoms, among other moieties. We calculated the first five singlet states at ADC(2)/def2-TZVP and ωB97M-V/def2-TZVP at the same geometry, optimized at the B3LYP level (the same workflow followed for the generation of the training set). We report the results of these calculations in Table 2. ωB97M-V, the functional selected by DELFI, reproduces almost perfectly the results of the reference calculation in terms of energy and relative brightness of the first five excitations, for the first five molecules indexed in the dataset. On a large dataset, DELFI is able to recommend a clear winner which, on a small subset of exemplary molecules, perfectly matches with the reference. This was the case even though the molecules are more complex and contain atoms that are not present in the training set. However, it should be noted that for ωB97M-V, even if with the overall highest value, a value higher than 50 is predicted in only 69% of the molecules, with the percentage decreasing to 32 for scores higher than 60, alerting on how generally TD-DFT might perform poorly on some of these molecules and suggesting further analysis on single samples to have a more confident understanding of the optical properties of this class of compounds, beyond a few considered examples.
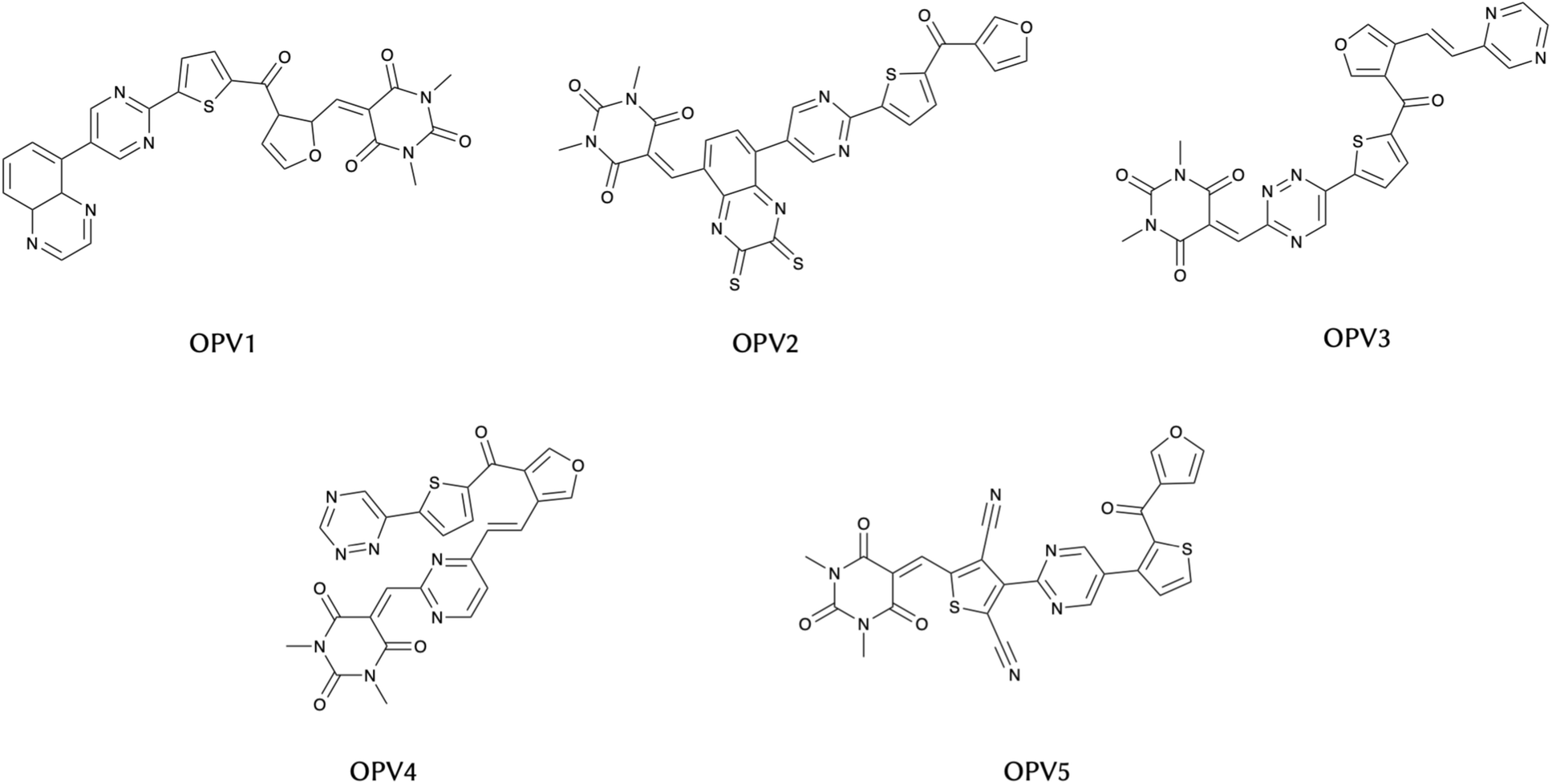 | ||
| Fig. 5 First five entries of the OPV dataset used in this work. The first five singlet states are calculated for each of these molecules at ADC(2) and ωB97M-V levels to test the performance of DELFI. | ||
| Molecule | Method | S1 | S2 | S3 | S4 | S5 |
|---|---|---|---|---|---|---|
| OPV1 | ADC(2) | 3.65 (0.21) | 3.79 (1.27) | 3.92 (0.00) | 4.04 (0.55) | 4.28 (0.03) |
| ωB97M-V | 3.73 (0.38) | 3.81 (0.75) | 4.07 (0.00) | 4.08 (0.02) | 4.31 (0.07) | |
| OPV2 | ADC(2) | 3.40 (1.51) | 3.80 (0.00) | 4.01 (0.01) | 4.19 (0.01) | 4.24 (0.27) |
| ωB97M-V | 3.37 (0.90) | 3.90 (0.00) | 4.22 (0.06) | 4.25 (0.06) | 4.30 (0.15) | |
| OPV3 | ADC(2) | 3.49 (0.00) | 3.79 (0.01) | 3.87 (0.05) | 4.01 (0.66) | 4.11 (0.55) |
| ωB97M-V | 3.42 (0.03) | 3.71 (0.01) | 3.97 (0.16) | 4.01 (0.69) | 4.19 (0.13) | |
| OPV4 | ADC(2) | 3.45 (0.01) | 3.85 (0.00) | 3.97 (0.00) | 4.03 (0.00) | 4.18 (0.00) |
| ωB97M-V | 3.38 (0.02) | 3.94 (0.00) | 4.02 (0.00) | 4.03 (0.00) | 4.21 (0.04) | |
| OPV5 | ADC(2) | 3.76 (0.01) | 3.80 (0.10) | 4.03 (0.23) | 4.11 (0.10) | 4.29 (0.14) |
| ωB97M-V | 3.80 (0.08) | 3.85 (0.00) | 4.10 (0.06) | 4.14 (0.04) | 4.41 (0.31) |
5 Analysis and predictions through a web application
Besides the theoretical definition of the scoring system and its application to QM8 to train a transferable functional predictor, we want to provide an easy-to-use and accessible web interface that could facilitate the choice of choosing the best functionals for a molecule by directly running DELFI and analyzing the results on a web application. The website, available at https://delfi-functional-predictor.streamlit.app, consists of four pages (Fig. 6). The first page includes a summary of the definition of the scoring system, and an interactive score calculator is available to insert customized values, helping the user to understand how the score is calculated and the weight of overlap between states, the difference in energy, and oscillator strength. The second page is dedicated to the analysis of the calculated scores for QM8, giving an overview of the distribution of scores on the training set of the model. The third page contains a plugin to run the prediction directly in the web application. There are two possibilities for the user: the first one is to enter a single SMILES manually and the 38 scores will be printed, or, alternatively, a list of SMILES can be provided and a data frame (in .csv) format will be generated and ready to be downloaded and/or analyzed. A disclaimer reminds the user of the nature of the molecules used in the training set and how the prediction of DELFI should be benchmarked if the molecules given with valid SMILES strings significantly differ from the one contained in the training set. The analysis can be performed on the last page, where a customized dataset can be uploaded. Histograms with the occurrences of each functional above a certain threshold, filtered histograms showing the recurrences of the best performing functionals, and correlation matrices can be easily visualized directly on that page. We strongly believe that this user-friendly application will increase notably the applicability of DELFI and be of support to chemists of all ranges of computational expertise. | ||
| Fig. 6 Screenshot of the four pages available in the DELFI web application. | ||
6 Conclusions and outlook
In this work, we have faced the challenging decision task of picking the right functional for TD-DFT excited state calculations. We first had to develop a general excited state scoring system that can quantify the quality of an excited state calculation with respect to a more accurate one, by simultaneously considering and proportionally weighing the character of the states, order of the states, and difference in energy and brightness for a set of excited states. We applied the scoring system to the calculation of 38 TD-DFT functionals with respect to an ADC(2) calculation for five singlet states for 21238 molecules, resulting in 828282 single-point calculations. The results of the calculations are open and available for the community and released in a large database that can be downloaded for future research and projects, together with the data necessary to try different combinations of scoring system and analysis. We used the scores we obtained to train a graph attention network. Based on this, we have developed a machine learning tool that can quickly screen the quality of density functionals for a single or set of molecules, and recommend a subset of functionals to be benchmarked on a specific molecule or chosen and used for a large set of molecules. This model, which we called DELFI, can be used to select a set of functionals to be tested on unseen molecules, as we demonstrated the applicability and transferability of DELFI on two in silico experiments, namely choosing a common functional for the calculation of the UV/Vis spectrum of a set of molecules with different degrees of functionalization and identifying a single functional to screen the photophysical descriptors of a very large dataset of organic molecules. DELFI gives system-specific recommendations, not learning score patterns in the test set, but discriminating chemical differences in the evaluation of the functionals. In these experiments we showed how DELFI obviates the need for benchmarks against a reference method like ADC(2), and as long as this method is sufficiently correct. Although the scoring system we developed is generalizable to any level of theory and number and multiplicity of states, a current limitation of DELFI lies in the generation of the training data, which are obtained using as a reference ADC(2), including only a manifold of singlet states, the exclusion of solvent effects and diffuse basis function, the use of only 38 functionals and the limited region of chemical space considered. We hope we made it clear throughout the text the range of application of DELFI and the goal of it: quickly screening density functionals for TD-DFT and producing a recommendation on which ones should be properly benchmarked or providing a clear indication when choosing a functional to screen a large number of molecules of prohibitive size, without the need for computationally unfeasible benchmarks. However, it is worth noticing how the results of the scoring system applied to QM8, and consequently, the recommendations of DELFI, are in line with extensive literature benchmarks. As all the data generated is freely available, as well as the code for the scoring system and the parameters of the model, we plan and encourage future efforts to improve the generalizability and applicability of the model, improving the quality of the training data and adding new functionals. Nonetheless, we strongly believe in the potential of DELFI to help the community with the task for which it was developed, so we wanted to provide an intuitive and user-friendly way to run and analyze DELFI. For this goal, we released a web application, freely available, to obtain predictions on unseen molecules, and analyze already available and new calculated/predicted results. Overall, we believe that all the work developed will drastically facilitate solving one of the most common hamletic doubts in computational chemistry.
7 Methods
7.1 Quantum mechanical calculations
From the QM8 dataset, only the molecules larger than 10 atoms were extracted. The reason for excluding the smallest molecules was to reduce the computational burden by not considering very small molecules with limited relevance from a photochemical point of view. The geometries reported in the dataset are optimized at the B3LYP level with the 6-31G(2df,p) basis set. We did not re-optimize the geometries because our goal was anyway to compare the performances of the functionals at the same given geometry. For each of these molecules, 39 QM calculations were run, 38 TD-DFT with the functional listed in Section 3, employing Tamm-Dancoff approximation89 and one ADC(2), using spin opposite scaling with a default parameter of 1.3. The exchange–correlation functionals were either directly loaded from Turbomole libraries or, if not available, from libxc.90 We used the def2-TZVP73 basis set and calculated five singlet excited states. Resolution of identity approximation was used in all the calculations.91,92 Solvent effects were never included in the calculations. All the QM calculations were run with Turbomole v. 7.3 (ref. 93 ) on the resources provided by the Digital Research Alliance of Canada. The transition density matrices and overlap among them were calculated using TheoDORE 3.0.94The one-electron transition density matrix for a transition from the ground (g) to an excited state (i) can be expressed as
| γg,i(re, rh) = n∫…∫Ψg(rh, r2,…, rn)Ψi(re, r2,…, rn)dr2drn | (1) |
7.2 Training and architecture of the GAT model
Starting from the SMILES strings, two-dimensional graphs were obtained using the MolGraphConvFeaturizer from DeepChem,96 which represents nodes using 30 atom features and edges using 11 bond features. These are the input features for a Graph Attention Network (GAT),26 also based on the DeepChem implementation. The architecture consists of two graph attention layers followed by a multilayer perceptron prediction layer.26 We used graph attention layers with a width of 128 dimensions and passed their aggregated outputs through a sigmoid activation function. We used dropout with 0.1 probability for the graph attention layers and 0.2 probability for the predictor layer, parameters chosen after tuning. The output represented score predictions for each of the 38 functionals. We used an L1 loss (mean average error) to minimize the distance between the predictions and ground truth functional scores for a given molecule. Before computing the loss, we normalized the functional scores to be between 0 and 1. We divided the dataset randomly into a train/validation/test split (80/10/10). We used the validation set to select the learning rate (0.001), batch size (128) and number of graph attention layers (2). A single multitask predictor was trained for the 38 scores. Other hyperparameters were left as the default implementation, as any deviation from the default optimized values worsened the performance of the model during any attempt at their optimization. The model was trained for 800 epochs until convergence was reached. The training was monitored by following the validation loss and early stopping was implemented to interrupt the training with a patience of 200 iterations. Uncertainty on the predictions on the test set was added with Monte Carlo dropout.97 We did 500 forward passes through the network, with dropout activated using the same dropout probability, and predicted the score for each of the molecules in the test. The variance of the 500 measurements was considered as a measure of uncertainty. In Section S2 of the ESI† we reported the average value of the variance for each of the molecules and for each of the functionals. The model can be downloaded on GitHub at https://github.com/aspuru-guzik-group/DELFI/tree/main.7.3 Validation
DELFI was run starting from the SMILES string of spiropyran and merocyanine structures obtained from the geometries used in ref. 87. Only a NO2 group for the degree of substitution was chosen for the QM calculations. The geometries were taken from the same work, using the first three top-performer recommended functionals. The so-called CCT isomer was used for the merocyanine. TD-DFT and ADC(2) QM calculations were run following the same protocol used for QM8. For the prediction of the best functional to be used to screen the OPVs, the dataset we used was collected in the framework of the Harvard Clean Energy project and downloaded at https://github.com/aspuru-guzik-group/ORGANIC. DELFI was run starting from the SMILES included in the dataset. The dataset used and the scores predicted are available at https://github.com/aspuru-guzik-group/DELFI/tree/main.7.4 Web application
The web application was created using Streamlit, an open-source Python library, and is hosted on Streamlit Cloud at Delfi Functional Predictor. It is split into four sections, used for visualizing the dataset, running predictions on your own dataset, visualizing your own dataset and explaining how the scoring system works.Data availability
Extended-QM8 is open and available at https://figshare.com/projects/DELFI/185308. It collects all the geometries, the vertical excitations calculated at ADC(2) and TD-DFT level with the 38 functionals, as well as all the overlap matrices between the different calculations. The scores, the model and its parameters are available at https://github.com/aspuru-guzik-group/DELFI/tree/main.Author contributions
DA: conceptualization, data curation, formal analysis, investigation, methodology, validation, visualization, writing – original draft, writing – review and editing; MS: methodology, writing – review and editing; SAR: software; AAG: conceptualization, funding acquisition, project administration, resources, writing – review and editing.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This research was enabled in part by support provided by SciNet (https://www.scinethpc.ca/) and the Digital Research Alliance of Canada (https://www.alliancecan.ca). Computations were performed on the Niagara supercomputer at the SciNet HPC Consortium. SciNet is funded by the Canada Foundation for Innovation; the Government of Ontario; the Ontario Research Fund – Research Excellence; and the University of Toronto. AAG thanks Anders G. Frøseth for his generous support. AAG also acknowledges the generous support of Natural Resources Canada and the Canada 150 Research Chairs program. This research was undertaken thanks in part to funding provided to the University of Toronto's Acceleration Consortium from the Canada First Research Excellence Fund (CFREF).References
- P. Hohenberg and W. Kohn, Inhomogeneous electron gas, Phys. Rev., 1964, 136, B864–B871 CrossRef.
- E. Runge and E. K. U. Gross, Density-functional theory for time-dependent systems, Phys. Rev. Lett., 1984, 52, 997–1000 CrossRef CAS.
- A. J. Cohen, P. Mori-Sánchez and W. Yang, Challenges for density functional theory, Chem. Rev., 2012, 112(1), 289–320 CrossRef CAS PubMed.
- N. Mardirossian and M. Head-Gordon, Thirty years of density functional theory in computational chemistry: an overview and extensive assessment of 200 density functionals, Mol. Phys., 2017, 115(19), 2315–2372 CrossRef CAS.
- D. Jacquemin, V. Wathelet, E. A. Perpète and C. Adamo, Extensive td-dft benchmark: Singlet-excited states of organic molecules, J. Chem. Theory Comput., 2009, 5(9), 2420–2435 CrossRef CAS PubMed.
- M. Bursch, J.-M. Mewes, A. Hansen and S. Grimme, Best-practice dft protocols for basic molecular computational chemistry, Angew. Chem., Int. Ed., 2022, 61(42), e202205735 CrossRef CAS PubMed.
- C. Adamo and D. Jacquemin, The calculations of excited-state properties with time-dependent density functional theory, Chem. Soc. Rev., 2013, 42, 845–856 RSC.
- T. Neepa, Maitra. Perspective: Fundamental aspects of time-dependent density functional theory, J. Chem. Phys., 2016, 144(22), 220901 CrossRef PubMed.
- M. E. Casida and M. Huix-Rotllant, Progress in time-dependent density-functional theory, Annu. Rev. Phys. Chem., 2012, 63(1), 287–323 CrossRef CAS PubMed.
- K. Yair and R. Baer, Time-dependent exchange-correlation current density functionals with memory, J. Chem. Phys., 2004, 121(18), 8731–8741 CrossRef PubMed.
- N. T. Maitra, F. Zhang, R. J. Cave and K. Burke, Double excitations within time-dependent density functional theory linear response, J. Chem. Phys., 2004, 120(13), 5932–5937 CrossRef CAS PubMed.
- B. G. Levine, C. Ko, J. Quenneville and T. J. MartÍnez, Conical intersections and double excitations in time-dependent density functional theory, Mol. Phys., 2006, 104(5–7), 1039–1051 CrossRef CAS.
- A. Dreuw and M. Head-Gordon, Failure of Time-Dependent Density Functional Theory for Long-Range Charge-Transfer Excited States: The Zincbacteriochlorin Bacteriochlorin and Bacteriochlorophyll Spheroidene Complexes, J. Am. Chem. Soc., 2004, 126(12), 4007–4016 CrossRef CAS PubMed.
- N. T. Maitra, Charge transfer in time-dependent density functional theory, J. Phys.: Condens. Matter, 2017, 29(42), 423001 CrossRef PubMed.
- T. Takao and K. Hirao, Long-range correction for density functional theory, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4(4), 375–390 Search PubMed.
- A. Charaf-Eddin, A. Planchat, B. Mennucci, C. Adamo and D. Jacquemin, Choosing a functional for computing absorption and fluorescence band shapes with td-dft, J. Chem. Theory Comput., 2013, 9(6), 2749–2760 CrossRef CAS PubMed.
- D. Adèle, Laurent and Denis Jacquemin. Td-dft benchmarks: A review, Int. J. Quantum Chem., 2013, 113(17), 2019–2039 CrossRef.
- C. Suellen, R. Garcia Freitas, P.-F. Loos and D. Jacquemin, Cross-comparisons between experiment, td-dft, cc, and adc for transition energies, J. Chem. Theory Comput., 2019, 15(8), 4581–4590 CrossRef CAS PubMed.
- C. A. Guido, P. Cortona, B. Mennucci and C. Adamo, On the metric of charge transfer molecular excitations: A simple chemical descriptor, J. Chem. Theory Comput., 2013, 9(7), 3118–3126 CrossRef CAS PubMed.
- M. J. G. Peach, P. Benfield, T. Helgaker and D. J. Tozer, Excitation energies in density functional theory: An evaluation and a diagnostic test, J. Chem. Phys., 2008, 128(4), 044118 CrossRef PubMed.
- T. Le Bahers, C. Adamo and I. Ciofini, A qualitative index of spatial extent in charge-transfer excitations, J. Chem. Theory Comput., 2011, 7(8), 2498–2506 CrossRef CAS PubMed.
- H. Nitta and I. Kawata, A close inspection of the charge-transfer excitation by tddft with various functionals: An application of orbital- and density-based analyses, Chem. Phys., 2012, 405, 93–99 CrossRef CAS.
- C. Duan, A. Nandy, R. Meyer, N. Arunachalam and H. J. Kulik, A transferable recommender approach for selecting the best density functional approximations in chemical discovery, Nat. Comput. Sci., 2023, 3(1), 38–47 CrossRef PubMed.
- R. Sarkar, M. Boggio-Pasqua, P.-F. Loos and D. Jacquemin, Benchmarking td-dft and wave function methods for oscillator strengths and excited-state dipole moments, J. Chem. Theory Comput., 2021, 17(2), 1117–1132 CrossRef CAS PubMed.
- R. Improta, F. Santoro and L. Blancafort, Quantum mechanical studies on the photophysics and the photochemistry of nucleic acids and nucleobases, Chem. Rev., 2016, 116(6), 3540–3593 CrossRef CAS PubMed.
- P. Veličković, G. Cucurull, A. Casanova, A. Romero, P. Liò and Y. Bengio, Graph Attention Networks, 2018 Search PubMed.
- L. Ruddigkeit, R. van Deursen, L. C. Blum and J.-L. Reymond, Enumeration of 166 billion organic small molecules in the chemical universe database gdb-17, J. Chem. Inf. Model., 2012, 52(11), 2864–2875 CrossRef CAS PubMed.
- R. Ramakrishnan, M. Hartmann, E. Tapavicza and O. Anatole von Lilienfeld, Electronic spectra from TDDFT and machine learning in chemical space, J. Chem. Phys., 2015, 143(8), 084111 CrossRef PubMed.
- J. Liang, X. Feng, D. Hait and M. Head-Gordon, Revisiting the performance of time-dependent density functional theory for electronic excitations: Assessment of 43 popular and recently developed functionals from rungs one to four, J. Chem. Theory Comput., 2022, 18(6), 3460–3473 CrossRef CAS PubMed.
- P. John, Perdew and Karla Schmidt. Jacob’s ladder of density functional approximations for the exchange-correlation energy, AIP Conf. Proc., 2001, 577(1), 1–20 Search PubMed.
- J. P. Perdew and Y. Wang, Accurate and simple analytic representation of the electron-gas correlation energy, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45, 13244–13249 CrossRef PubMed.
- S. Grimme, Semiempirical gga-type density functional constructed with a long-range dispersion correction, J. Comput. Chem., 2006, 27(15), 1787–1799 CrossRef CAS PubMed.
- C. Adamo and V. Barone, Exchange functionals with improved long-range behavior and adiabatic connection methods without adjustable parameters: The mPW and mPW1PW models, J. Chem. Phys., 1998, 108(2), 664–675 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- B. Miehlich, A. Savin, H. Stoll and H. Preuss, Results obtained with the correlation energy density functionals of becke and lee, yang and parr, Chem. Phys. Lett., 1989, 157, 200 CrossRef CAS.
- R. Peverati and D. G. Truhlar, An improved and broadly accurate local approximation to the exchange–correlation density functional: The mn12-l functional for electronic structure calculations in chemistry and physics, Phys. Chem. Chem. Phys., 2012, 14, 13171 RSC.
- N. Mardirossian and M. Head-Gordon, Mapping the genome of meta-generalized gradient approximation density functionals: The search for b97m-v, J. Chem. Phys., 2015, 142, 074111 CrossRef PubMed.
- J. Wellendorff, K. T. Lundgaard, K. W. Jacobsen and T. Bligaard, mbeef: An accurate semi-local bayesian error estimation density functional, J. Chem. Phys., 2014, 140, 144107 CrossRef PubMed.
- Y. Zhao and D. G. Truhlar, A new local density functional for main-group thermochemistry, transition metal bonding, thermochemical kinetics, and noncovalent interactions, J. Chem. Phys., 2006, 125, 194101 CrossRef PubMed.
- Y. Wang, X. Jin, H. S. Yu, D. G. Truhlar and X. He, Revised m06-l functional for improved accuracy on chemical reaction barrier heights, noncovalent interactions, and solid-state physics, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 8487 CrossRef CAS PubMed.
- H. S. Yu, X. He and D. G. Truhlar, Mn15-l: Anew local exchange-correlation functional for kohn–sham density functional theory with broad accuracy for atoms, molecules, and solids, J. Chem. Theory Comput., 2016, 12, 1280 CrossRef CAS PubMed.
- J. P. Perdew, A. Ruzsinszky, G. I. Csonka, L. A. Constantin and J. Sun, Workhorse semilocal density functional for condensed matter physics and quantum chemistry, Phys. Rev. Lett., 2009, 103, 026403 CrossRef PubMed.
- J. Tao, J. P. Perdew, V. N. Staroverov and G. E. Scuseria, Climbing the density functional ladder: Nonempirical meta–generalized gradient approximation designed for molecules and solids, Phys. Rev. Lett., 2003, 91, 146401 CrossRef PubMed.
- J.-D. Chai and M. Head-Gordon, Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections, Phys. Chem. Chem. Phys., 2008, 10, 6615 RSC.
- T. Yanai, D. P. Tew and N. C. Handy, A new hybrid exchange–correlation functional using the coulomb-attenuating method (cam-b3lyp), Chem. Phys. Lett., 2004, 393, 51 CrossRef CAS.
- N. Mardirossian and M. Head-Gordon, ωb97x-v: A 10-parameter, range-separated hybrid, generalized gradient approximation density functional with nonlocal correlation, designed by a survival-of-the-fittest strategy, Phys. Chem. Chem. Phys., 2014, 16, 9904 RSC.
- R. Peverati and D. G. Truhlar, Communication: A global hybrid generalized gradient approximation to the exchange-correlation functional that satisfies the second-order density-gradient constraint and has broad applicability in chemistry, J. Chem. Phys., 2011, 135, 191102 CrossRef PubMed.
- M. A. Rohrdanz and J. M. Herbert, Simultaneous benchmarking of ground- and excited-state properties with long-range-corrected density functional theory, J. Chem. Phys., 2008, 129, 034107 CrossRef PubMed.
- M. A. Rohrdanz, K. M. Martins and J. M. Herbert, A long-range-corrected density functional that performs well for both ground-state properties and time-dependent density functional theory excitation energies, including charge-transfer excited states, J. Chem. Phys., 2009, 130, 054112 CrossRef PubMed.
- B. J. Lynch, P. L. Fast, M. Harris and D. G. Truhlar, Adiabatic connection for kinetics, J. Phys. Chem. A, 2000, 104, 4811 CrossRef CAS.
- C. Adamo and V. Barone, Toward reliable density functional methods without adjustable parameters: The pbe0 model, J. Chem. Phys., 1999, 110, 6158 CrossRef CAS.
- T. M. Henderson, B. G. Janesko and G. E. Scuseria, Generalized gradient approximation model exchange holes for range-separated hybrids, J. Chem. Phys., 2008, 128, 194105 CrossRef PubMed.
- A. V. Krukau, O. A. Vydrov, A. F. Izmaylov and G. E. Scuseria, Influence of the exchange screening parameter on the performance of screened hybrid functionals, J. Chem. Phys., 2006, 125, 224106 CrossRef PubMed.
- A. J. Cohen, P. Mori-Sánchez and W. Yang, Development of exchange-correlation functionals with minimal many-electron self-interaction error, J. Chem. Phys., 2007, 126, 191109 CrossRef PubMed.
- A. D. Becke, Density-functional exchange-energy approximation with correct asymptotic behavior, Phys. Rev. A, 1988, 38, 3098 CrossRef CAS PubMed.
- C. Lee, W. Yang and R. G. Parr, Development of the colle-salvetti correlation-energy formula into a functional of the electron density, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS PubMed.
- Y. A. Bernard, Y. Shao and A. I. Krylov, General formulation of spin-flip time-dependent density functional theory using non-collinear kernels: Theory, implementation, and benchmarks, J. Chem. Phys., 2012, 136, 204103 CrossRef PubMed.
- A. D. Becke, Density-functional thermochemistry. iii. the role of exact exchange, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields, J. Phys. Chem., 1994, 98, 11623 CrossRef CAS.
- A. D. Boese and J. M. L. Martin, Development of density functionals for thermochemical kinetics, J. Chem. Phys., 2004, 121, 3405 CrossRef CAS PubMed.
- Y. Wang, P. Verma, L. Zhang, Y. Li, Z. Liu, D. G. Truhlar and X. He, M06-sx screened-exchange density functional for chemistry and solid-state physics, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 2294 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, The m06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four m06-class functionals and 12 other functionals, Theor. Chem. Acc., 2008, 120, 215 Search PubMed.
- N. Mardirossian and M. Head-Gordon, ωb97m-v: A combinatorially optimized, range-separated hybrid, meta-gga density functional with vv10 nonlocal correlation, J. Chem. Phys., 2016, 144, 214110 CrossRef PubMed.
- Y.-S. Lin, C.-W. Tsai, G.-D. Li and J.-D. Chai, Long-range corrected hybrid meta-generalized-gradient approximations with dispersion corrections, J. Chem. Phys., 2012, 136, 154109 CrossRef PubMed.
- H. S. Yu, X. He, S. L. Li and D. G. Truhlar, Mn15: A kohn–sham global-hybrid exchange–correlation density functional with broad accuracy for multi-reference and single-reference systems and noncovalent interactions, Chem. Sci., 2016, 7, 5032 RSC.
- Y. Zhao and D. G. Truhlar, Design of density functionals that are broadly accurate for thermochemistry, thermochemical kinetics, and nonbonded interactions, J. Phys. Chem. A, 2005, 109, 5656 CrossRef CAS PubMed.
- K. Hui and J.-D. Chai, Scan-based hybrid and double-hybrid density functionals from models without fitted parameters, J. Chem. Phys., 2016, 144, 044114 CrossRef PubMed.
- R. Peverati and D. G. Truhlar, Improving the accuracy of hybrid meta-gga density functionals by range separation, J. Phys. Chem. Lett., 2011, 2, 2810 CrossRef CAS.
- G. I. Csonka, J. P. Perdew and A. Ruzsinszky, Global hybrid functionals: A look at the engine under the hood, J. Chem. Theory Comput., 2010, 6, 3688 CrossRef CAS.
- V. N. Staroverov, G. E. Scuseria, J. Tao and J. P. Perdew, Comparative assessment of a new nonempirical density functional: Molecules and hydrogen-bonded complexes, J. Chem. Phys., 2003, 119, 12129 CrossRef CAS.
- R. Peverati and D. G. Truhlar, Screened-exchange density functionals with broad accuracy for chemistry and solid-state physics, Phys. Chem. Chem. Phys., 2012, 14, 16187 RSC.
- A. Dreuw and M. Wormit, The algebraic diagrammatic construction scheme for the polarization propagator for the calculation of excited states, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2015, 5(1), 82–95 CAS.
- F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for h to rn: Design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- P.-F. Loos, F. Lipparini, M. Boggio-Pasqua, S. Anthony and D. Jacquemin, A mountaineering strategy to excited states: Highly accurate energies and benchmarks for medium sized molecules, J. Chem. Theory Comput., 2020, 16(3), 1711–1741 CrossRef CAS PubMed , PMID: 31986042..
- R. Pollice, P. Friederich, C. Lavigne, G. P. Gomes and A. Aspuru-Guzik, Organic molecules with inverted gaps between first excited singlet and triplet states and appreciable fluorescence rates, Matter, 2021, 4(5), 1654–1682 CrossRef CAS.
- R. Szabla, H. Kruse, P. Stadlbauer, J. Šponer and A. L. Sobolewski, Sequential electron transfer governs the uv-induced self-repair of dna photolesions, Chem. Sci., 2018, 9, 3131–3140 RSC.
- J. Novak, A. Prlj, N. Basarić, C. Corminboeuf and N. Došlić, Photochemistry of 1- and 2-naphthols and their water clusters: The role of 1ππ*(la) mediated hydrogen transfer to carbon atoms, Chem.–Eur. J., 2017, 23(34), 8244–8251 CrossRef CAS PubMed.
- M. A. Kochman, A. Tajti, C. A. Morrison and R. J. Dwayne Miller, Early events in the nonadiabatic relaxation dynamics of 4-(n,n-dimethylamino)benzonitrile, J. Chem. Theory Comput., 2015, 11(3), 1118–1128 CrossRef CAS PubMed.
- M. Elena Castellani, D. Avagliano, L. González and J. R. R. Verlet, Site-specific photo-oxidation of the isolated adenosine-5’-triphosphate dianion determined by photoelectron imaging, J. Phys. Chem. Lett., 2020, 11(19), 8195–8201 CrossRef PubMed.
- N. O. C. Winter, N. K. Graf, S. Leutwyler and C. Hättig, Benchmarks for 0–0 transitions of aromatic organic molecules: Dft/b3lyp, adc(2), cc2, sos-cc2 and scs-cc2 compared to high-resolution gas-phase data, Phys. Chem. Chem. Phys., 2013, 15, 6623–6630 RSC.
- H. Li, N. Reed, A. J. A. Aquino, H. Lischka and S. Tretiak, Comparison of lc-tddft and adc(2) methods in computations of bright and charge transfer states in stacked oligothiophenes, J. Chem. Theory Comput., 2014, 10(8), 3280–3289 CrossRef CAS PubMed.
- L. Kortekaas and W. R. Browne, The evolution of spiropyran: fundamentals and progress of an extraordinarily versatile photochrome, Chem. Soc. Rev., 2019, 48, 3406–3424 RSC.
- R. Klajn, Spiropyran-based dynamic materials, Chem. Soc. Rev., 2014, 43, 148–184 RSC.
- B. S. Lukyanov and M. B. Lukyanova, Spiropyrans: Synthesis, properties, and application, Journal Chemistry of Heterocyclic Compounds, 2005, 41(3), 281–311 CrossRef CAS.
- W. Szymański, J. M. Beierle, H. A. V. Kistemaker, W. A. Velema and B. L. Feringa, Reversible photocontrol of biological systems by the incorporation of molecular photoswitches, Chem. Rev., 2013, 113(8), 6114–6178 CrossRef PubMed.
- D. Avagliano, P. A. Sánchez-Murcia and L. González, Spiropyran meets guanine quadruplexes: Isomerization mechanism and dna binding modes of quinolizidine-substituted spiropyran probes, Chem.–Eur. J., 2020, 26(57), 13039–13045 CrossRef CAS PubMed.
- Y. Sheng, J. Leszczynski, A. A. Garcia, R. Rosario, D. Gust and J. Springer, Comprehensive theoretical study of the conversion reactions of spiropyrans: Substituent and solvent effects, J. Phys. Chem. B, 2004, 108(41), 16233–16243 CrossRef CAS.
- B. Sanchez-Lengeling, C. Outeiral, G. L. Guimaraes, and A. Aspuru-Guzik. Optimizing distributions over molecular space. an objective-reinforced generative adversarial network for inverse-design chemistry (organic). 2017 Search PubMed.
- S. Hirata and M. Head-Gordon, Time-dependent density functional theory within the tamm–dancoff approximation, Chem. Phys. Lett., 1999, 314, 291 CrossRef CAS.
- S. Lehtola, S. Conrad, M. J. T. Oliveira and M. A. L. Marques, Recent developments in libxc — a comprehensive library of functionals for density functional theory, SoftwareX, 2018, 7, 1–5 CrossRef.
- C. Hättig and F. Weigend, CC2 excitation energy calculations on large molecules using the resolution of the identity approximation, J. Chem. Phys., 2000, 113(13), 5154–5161 CrossRef.
- R. Bauernschmitt, M. Häser, T. Oliver and R. Ahlrichs, Calculation of excitation energies within time-dependent density functional theory using auxiliary basis set expansions, Chem. Phys. Lett., 1997, 264(6), 573–578 CrossRef CAS.
- TURBOMOLE V7.3 2017, a development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989-2007, TURBOMOLE GmbH, since 2007; available from http://www.turbomole.com.
- F. Plasser, TheoDORE: A toolbox for a detailed and automated analysis of electronic excited state computations, J. Chem. Phys., 2020, 152(8), 084108 CrossRef CAS PubMed.
- F. Plasser and H. Lischka, Analysis of excitonic and charge transfer interactions from quantum chemical calculations, J. Chem. Theory Comput., 2012, 8(8), 2777–2789 CrossRef CAS PubMed.
- S. Kearnes, K. McCloskey, M. Berndl, V. Pande and P. Riley, Molecular graph convolutions: moving beyond fingerprints, J. Comput.-Aided Mol. Des., 2016, 30, 595–608 CrossRef CAS PubMed.
- Y. Gal and Z. Ghahramani, Dropout as a bayesian approximation: Representing model uncertainty in deep learning, 2016 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc06440a |
| This journal is © The Royal Society of Chemistry 2024 |