
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceControlling product selectivity during dioxygen reduction with Mn complexes using pendent proton donor relays and added base†
Emma N.
Cook
 ,
Ian M.
Courter
,
Diane A.
Dickie
and
Charles W.
Machan
*
,
Ian M.
Courter
,
Diane A.
Dickie
and
Charles W.
Machan
*
Department of Chemistry University of Virginia, PO Box 400319 McCormick Rd, Charlottesville, VA 22904-4319, USA. E-mail: machan@virginia.edu
First published on 13th February 2024
Abstract
The catalytic reduction of dioxygen (O2) is important in biological energy conversion and alternative energy applications. In comparison to Fe- and Co-based systems, examples of catalytic O2 reduction by homogeneous Mn-based systems is relatively sparse. Motivated by this lack of knowledge, two Mn-based catalysts for the oxygen reduction reaction (ORR) containing a bipyridine-based non-porphyrinic ligand framework have been developed to evaluate how pendent proton donor relays alter activity and selectivity for the ORR, where Mn(p-tbudhbpy)Cl (1) was used as a control complex and Mn(nPrdhbpy)Cl (2) contains a pendent –OMe group in the secondary coordination sphere. Using an ammonium-based proton source, N,N′-diisopropylethylammonium hexafluorophosphate, we analyzed catalytic activity for the ORR: 1 was found to be 64% selective for H2O2 and 2 is quantitative for H2O2, with O2 binding to the reduced Mn(II) center being the rate-determining step. Upon addition of the conjugate base, N,N′-diisopropylethylamine, the observed catalytic selectivity of both 1 and 2 shifted to H2O as the primary product. Interestingly, while the shift in selectivity suggests a change in mechanism for both 1 and 2, the catalytic activity of 2 is substantially enhanced in the presence of base and the rate-determining step becomes the bimetallic cleavage of the O–O bond in a Mn-hydroperoxo species. These data suggest that the introduction of pendent relay moieties can improve selectivity for H2O2 at the expense of diminished reaction rates from strong hydrogen bonding interactions. Further, although catalytic rate enhancements are observed with a change in product selectivity when base is added to buffer proton activity, the pendent relays stabilize dimer intermediates, limiting the maximum rate.
Introduction
Increasing atmospheric CO2 concentrations has led to global warming and climate change, creating an undeniable need for renewable energy processes. Activation and reduction of dioxygen (O2) is an attractive route for the development of alternative energy and industrial-scale oxidation reactions due to its abundance and oxidizing power.1–9 The oxygen reduction reaction (ORR) to the 4H+/4e− product, H2O, is an ideal cathodic half reaction in hydrogen fuel cells and selectivity for the 2H+/2e− product, H2O2, is also attractive because of the large-scale use of H2O2 as an oxidant in a number of industrial processes.2,3,7,9–11 Molecular complexes offer a compelling way to study and optimize these electrocatalytic reactions because of the fidelity with which the active sites can be characterized relative to heterogeneous systems that may contain a distribution of active sites, as well as the possibility of using iterative synthetic modifications to test mechanistic hypotheses.Interest in bioinspired and biomimetic systems has spurred the development of a significant number of Fe- and Co-based porphyrinic systems.3,9,12,13 By comparison, Mn has been less widely studied, despite its reactivity toward O2 and prevalence in dioxygen-dependent biological systems.1,2,9 Because of the relatively high spin-pairing energy of Mn ions, open-shell configurations with side-on coordination modes of O2 are often observed to be relatively stable on the potential energy surface of catalytic reactions, limiting activity.2,14–16 This issue arises, in part, as a result of the low basicity of O atoms in side-on Mn–O2 species, preventing effective protonation and cleavage of the O–O bond.2,14 This remains a challenge in the development of Mn-based catalysts for ORR, where destabilization of side-on Mn–O2 intermediates (and other stable reactive oxygen species, ROSs) is crucial to achieve enhanced rates of catalytic turnover (Fig. 1).2,14
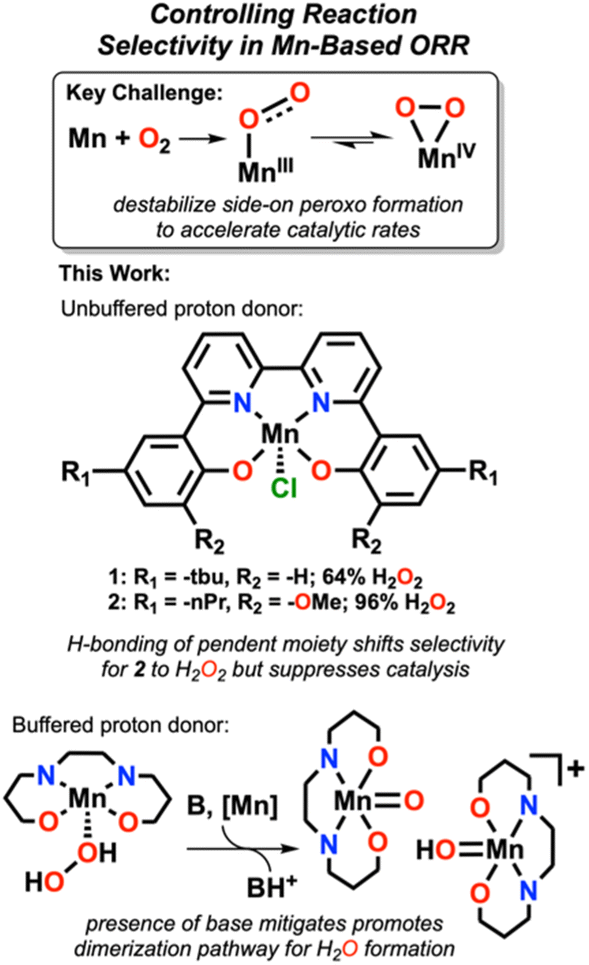 | ||
| Fig. 1 Summary of the work described here. | ||
There are a number of strategies that can be employed to avoid stable Mn ROS intermediates, including synthetically modifying the supporting ligand framework to tune the electronic structure at the metal center, the introduction of hydrogen-bonding interactions that alter the potential energy surface for Mn–O2 intermediates, or the use of steric hindrance at the active site.2 For instance, to explore how secondary-sphere ligand modification influenced ORR activity and selectivity, Nocera and coworkers introduced a xanthene-based pendent hangman moiety in a Mn tetraphenylporphyrin complex for ORR.17 They found that the hangman group promoted intramolecular proton transfer during catalysis and that hydrogen-bonding interactions between Mn-bound O2 and proton donors helped to favor end-on coordination modes.17 A firm fundamental understanding of how synthetic control over ORR at Mn-based active sites can avoid these potential thermodynamic limitations during catalysis remains an important question. Among the reports on Mn-based ORR catalysts, there are only a handful of non-porphyrinic based systems, despite the possibilities for alternative synthetic approaches for reaction control.2,18–23
Previous studies on the electrochemical ORR mediated by Co(N2O2) molecular complexes with 2,2′-bipyridine based ligand backbone24 found that the introduction of a –OMe pendent relay in the secondary coordination sphere resulted in a shift in selectivity from H2O to H2O2, as well as a shift in the observed rate-determining step (RDS) based on acid strength.25 It was hypothesized that directed protonation of the proximal oxygen of a Co–OOH intermediate was mediated by the pendent –OMe group.25 Reasoning that pendent relays could favor end-on O2 coordination and accelerate proton transfer in analogous Mn-based systems,22,23,26 two new Mn-based non-porphyrinic electrocatalysts for ORR have been prepared. Using an alkyl ammonium-based proton source, it was found that a secondary sphere –OMe pendent relay shifts selectivity to quantitative H2O2 formation under unbuffered conditions (proton donor is present without its conjugate base), although the rate of catalysis is suppressed. However, under buffered conditions (the proton donor and its conjugate base are present in equivalent amounts), hydrogen bonding between the ligand framework and the added acid is mitigated, resulting in a dramatic increase in rate and a shift in selectivity to H2O production. Interestingly, the shift in selectivity occurs in complexes with and without the pendent relay, with systematic variation of added base concentration implying that the change in mechanism is related to the deprotonation of a Mn(III)–OOH intermediate. Further, the complex with the pendent relay experiences a shift in RDS for catalysis under buffered conditions, which is proposed to originate from the pendent relay stabilizing Mn dimer formation relative to O2 binding and activation.
Overall, the proposed mechanism suggests that the 4H+/4e− product H2O is accessed by deprotonating an intermediate Mn(III) hydrogen peroxide complex species, implying that proton activity buffering at a pKa which favors an intermediate metal hydroperoxo is a potential point of reaction control over the selectivity of product formation, as well as over hydrogen-bonding interactions. These observations are representative of the complexity of ORR with Mn active sites: proton transfer is required to facilitate the reduction of superoxide to hydroperoxide (which we attribute to the competition between end-on and side-on coordination modes), but O–O bond scission occurs most efficiently if a second proton transfer to the hydroperoxide does not occur.
Results and discussion
Synthesis and characterization
The p-tbudhbpy[H]2 and nPrdhbpy[H]2 ligands were synthesized according to previously reported procedures.25 Metalation of p-tbudhbpy[H]2 to generate Mn(p-tbudhbpy)Cl 1 was achieved via a modified literature procedure,22 where a stoichiometric amount of manganese(II) acetate tetrahydrate was sparged with compressed air, allowed to reflux with p-tbudhbpy[H]2 for 3 hours in MeOH solution, and induced to precipitate by the addition of a saturated NaCl solution. Metalation of nPrdhbpy[H]2 was achieved by an analogous synthetic procedure.UV-vis, ESI-MS, and microanalyses are consistent with the proposed formulation of the Mn complexes shown in Fig. 1. Evans' method measurements exhibited μeff = 4.78 ± 0.11 and μeff = 4.59 ± 0.10 for 1 and 2, respectively; both values are consistent with high-spin d4 Mn(III) complexes (Tables S1 and S2†).27,28 Single crystals suitable for X-ray diffraction studies of complex 2 were grown by slow cooling of a saturated, boiling acetonitrile (MeCN) solution layered with diethyl ether. The solid-state structure of 2 is a dimeric species where a single O atom from each atom is coordinated in the axial position of a second equivalent of complex 2 to create a six-coordinate environment for each Mn center, analogous to our previous studies on a comparable Fe complex (Fig. 2C and D).29
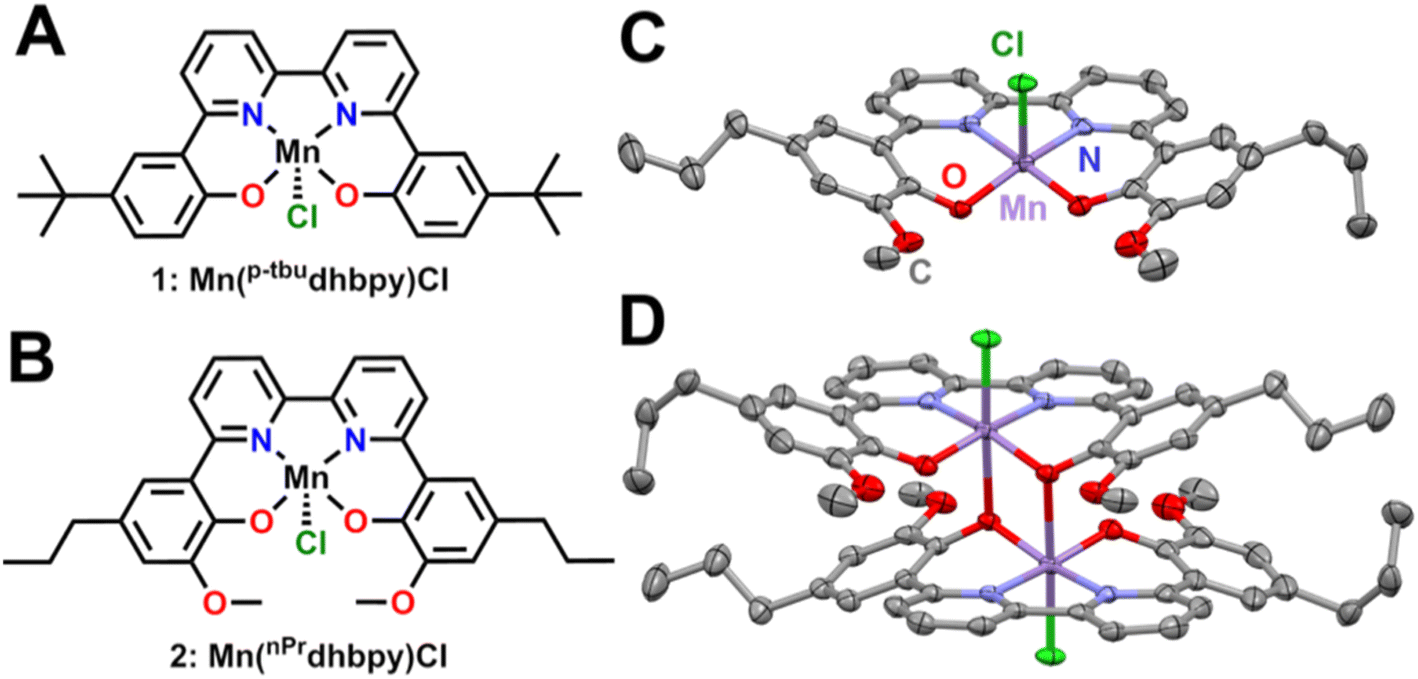 | ||
| Fig. 2 (A) Structure of 1 Mn(p-tbudhbpy)Cl, (B) structure of 2 Mn(nPrdhbpy)Cl, (C) molecular structure of Mn(nPrdhbpy)Cl 2 obtained from single crystal X-ray diffraction studies by omitting one Mn complex from the (D) dimeric solid-state species. Purple = Mn, red = O, green = Cl, gray = C; thermal ellipsoids 50%, H atoms and disordered atoms omitted for clarity. CCDC 2255849.† | ||
N,N′-Diisopropylethylammonium hexafluorophosphate (DIPEAHPF6) was synthesized according to a previously reported procedure.30 Toluene was brought to reflux in the presence of ammonium hexafluorophosphate (PF6) and N,N′-diisopropylethylamine (DIPEA), resulting in the precipitation of a solid product after 48 hours. The suspension was cooled to 0 °C on an ice bath before being filtered and washed with dichloromethane (DCM). Rotary evaporation of the DCM solvent yielded a spectroscopically pure product: NMR spectroscopic characterization and microanalysis were consistent with a N,N′-diisopropylethylammonium salt with a PF6− counteranion. Single crystals suitable for X-ray diffraction studies were grown via slow evaporation from a concentrated DCM solution (Fig. S3†). 1H NMR spectroscopy was used to estimate a pKa of 18.7 for DIPEAHPF6 in MeCN via titration and competition experiments (see ESI†).31
Electrochemical analysis of 1 and 2
Cyclic voltammetry studies were performed on 1 and 2 in MeCN solution with 0.1 M tetrabutylammonium hexafluorophosphate (TBAPF6) supporting electrolyte (Fig. 3). Under an inert Ar atmosphere, 1 (Fig. S9†) and 2 (Fig. S16†) show reversible features attributed to a Mn(III/II) redox event at −0.58 and −0.55 V vs. Fc+/Fc, respectively. Variable scan-rate studies of both 1 and 2 under Ar gas saturation showed a diffusion-limited response, consistent with homogenous species (Fig. S8 and S15,† respectively). Upon O2 saturation, there is an observed slight increase in current density at the Mn(III/II) redox event as well as the partial loss of the return oxidation feature for both 1 (Fig. S9†) and 2 (Fig. S16†), indicative of a reaction involving O2 and the reduced Mn(II) metal center. Conversely, there is a quasi-reversible redox feature observed at more negative potentials for 2 under O2 saturation, suggestive of a Mn superoxide/peroxide reduction following O2 binding and activation.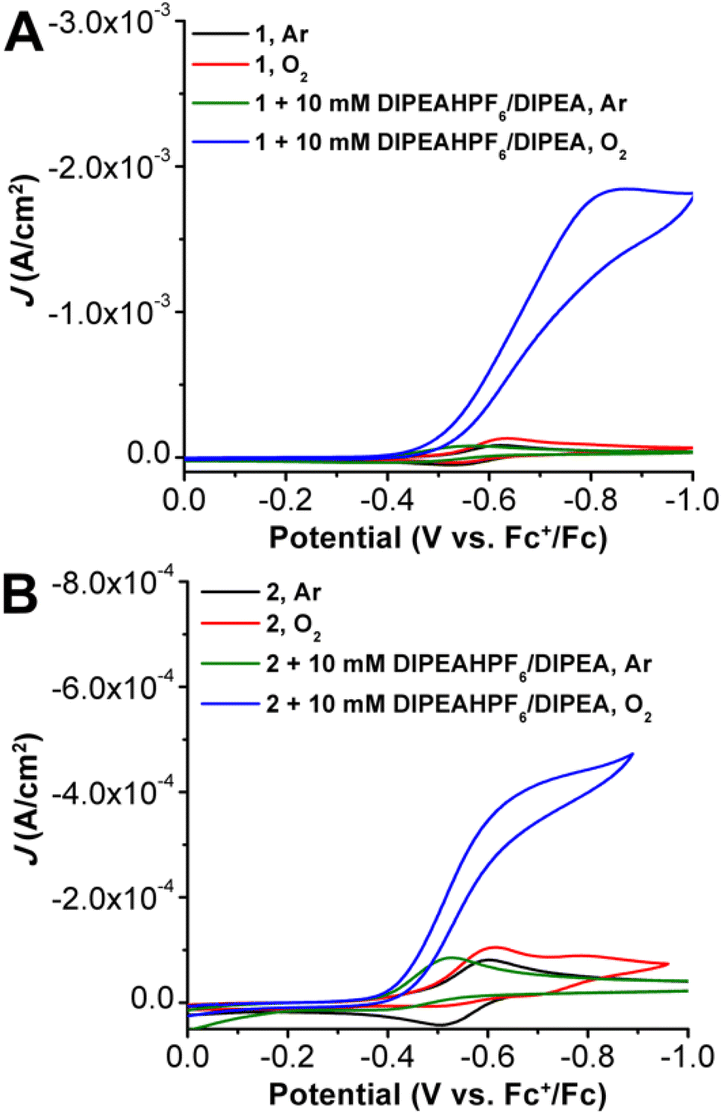 | ||
| Fig. 3 (A) CVs of Mn(p-tbudhbpy)Cl 1 under Ar and O2 with and without 10 mM DIPEAHPF6/DIPEA present. (B) CVs of Mn(nPrdhbpy)Cl 2 under Ar and O2 with and without 10 mM DIPEAHPF6/DIPEA present. Conditions: 0.5 mM Mn, 0.1 M TBAPF6/MeCN; glassy carbon working electrode, glassy carbon rod counter electrode, Ag/AgCl pseudoreference electrode; referenced to Fc+/Fc internal standard; 100 mV s−1 scan rate. | ||
Initial addition of 10 mM of the DIPEAHPF6 proton donor to both 1 and 2 under inert conditions resulted in the loss of reversibility of the Mn(III/II) redox couple as well as a shift to more positive potentials, Ep,a = −0.56 V vs. Fc+/Fc for 1 (Fig. S9†) and −0.53 V vs. Fc+/Fc for 2 (Fig. S16†). This is consistent with a contribution from an EC mechanism (reversible electron transfer followed by an irreversible chemical reaction), which is proposed to be the strong hydrogen-bonding interactions and protonation of an O atom in the inner-coordination sphere following formal reduction to Mn(II), consistent with our previous studies on acids with similar pKa values.22,32 Although titration of increasing amounts of DIPEAHPF6 beyond 10 mM did not cause a further potential shift for 1, the maximum positive potential shift for 2 was reached after the addition of 20 mM. Similarly, titration of increasing amounts of the DIPEAHPF6 proton donor in the presence of 10 mM DIPEA revealed that the Mn(III) reduction potential observed for 1 and 2 shifted to more positive potentials, however this shift was not dependent on the concentration of DIPEAHPF6 (Fig. S11 and S17†).
The observation of these shifts in the presence of the proton donor and its conjugate base, as well as the absence of a Nernstian relationship suggested that the axial chloride ligand was participating in related chemical steps during the EC mechanism. To better understand the role of DIPEA and DIPEAHPF6 in axial Cl− ligand solvation, CVs of 1 (Fig. S13 and S14,† respectively) were taken in the presence of 0.1 M TBACl. The addition of increasing amounts of both DIPEA by itself and DIPEAHPF6 with a fixed concentration of DIPEA (buffered) in the presence of excess Cl− under Ar saturation conditions suppressed positive potential shifts of the Mn(III/II) redox feature in 1. The suppression of the potential shift in the presence of excess Cl− suggests that the shift observed originally is due to axial ligand solvation. Since it was observed that increasing concentrations of DIPEAHPF6 led to a loss of reversibility for the Mn(III/II) feature, these data taken in aggregate imply that axial Cl− ligand loss enables hydrogen-bonding interactions and protonation of the O atom in the inner coordination sphere upon reduction.
Similar studies were conducted on 2, where the addition of 0.1 M TBACl to a solution of 2 resulted in a negative potential shift of a quasireversible Mn(III/II) feature to −0.76 V vs. Fc+/Fc (Fig. S22†). Addition of 10 mM DIPEA caused a positive potential shift of this Mn(III/II) redox feature back to −0.56 V vs. Fc+/Fc (Fig. S22†) and the titration of increasing amounts of DIPEAHPF6 under buffered conditions suppressed the observed positive potential shift (Fig. S23†). As was the case with complex 1, it is hypothesized that the presence of both DIPEA and DIPEAHPF6 contribute to axial Cl− ligand loss for 2, with irreversibility again suggesting strong hydrogen-bonding interactions and protonation involving a ligand O atom bound to Mn. Consistent with this, UV-vis spectroscopic analysis of 1 and 2 in the presence of DIPEA and DIPEAHPF6 showed no evidence of interaction with Mn(III) (Fig. S58–S61†).
Under O2 saturation in the presence of 10 mM DIPEAHPF6, there is an increase in current at the Mn(III/II) reduction feature for both 1 (Fig. S9†) and 2 (Fig. S16†), indicative of electrocatalytic activity for the ORR. Likewise, under O2 saturation in the presence of buffered DIPEAHPF6 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of ammonium to its conjugate base), there is an increase in current density at the Mn(III/II) redox couple for both 1 and 2, consistent with catalytic O2 reduction (Fig. 3). Effective overpotentials (η) for the ORR by 1 and 2 calculated under buffered conditions were determined to be 0.58 and 0.56 V, respectively (see ESI†). Notably, the catalytic current density observed under buffered conditions with complex 2 is much higher than unbuffered conditions (Fig. S19†), suggesting that the presence of conjugate base enhances electrocatalytic ORR by 2, vide infra. Oxidative current due to amine oxidation precludes the use of rotating ring-disk methods for H2O2 detection under these conditions for both 1 and 2 (Fig. S24†).
:1 ratio of ammonium to its conjugate base), there is an increase in current density at the Mn(III/II) redox couple for both 1 and 2, consistent with catalytic O2 reduction (Fig. 3). Effective overpotentials (η) for the ORR by 1 and 2 calculated under buffered conditions were determined to be 0.58 and 0.56 V, respectively (see ESI†). Notably, the catalytic current density observed under buffered conditions with complex 2 is much higher than unbuffered conditions (Fig. S19†), suggesting that the presence of conjugate base enhances electrocatalytic ORR by 2, vide infra. Oxidative current due to amine oxidation precludes the use of rotating ring-disk methods for H2O2 detection under these conditions for both 1 and 2 (Fig. S24†).
Spectrochemical studies with 1 and 2
Catalytic ORR experiments with 1 and 2 were then run under spectrochemical conditions using decamethylferrocene (Cp*2Fe) as a chemical reductant in solution with the DIPEAHPF6 proton donor under unbuffered and buffered (1:1 ratio of DIPEAHPF6 to DIPEA) conditions. UV-vis stopped-flow spectroscopy was used to determine the kinetic parameters of the ORR by monitoring the accumulation of oxidized [Cp*2Fe]+ at 780 nm (Fig. 4). Consistent with the electrochemical data, ORR by 1 showed minimal differences in activity under catalytic conditions with buffered and unbuffered proton donor (Fig. 4A). However, the catalytic response mediated by 2 showed a significant enhancement under buffered conditions compared to unbuffered (Fig. 4B), similar to what was observed under electrochemical conditions, vide supra.
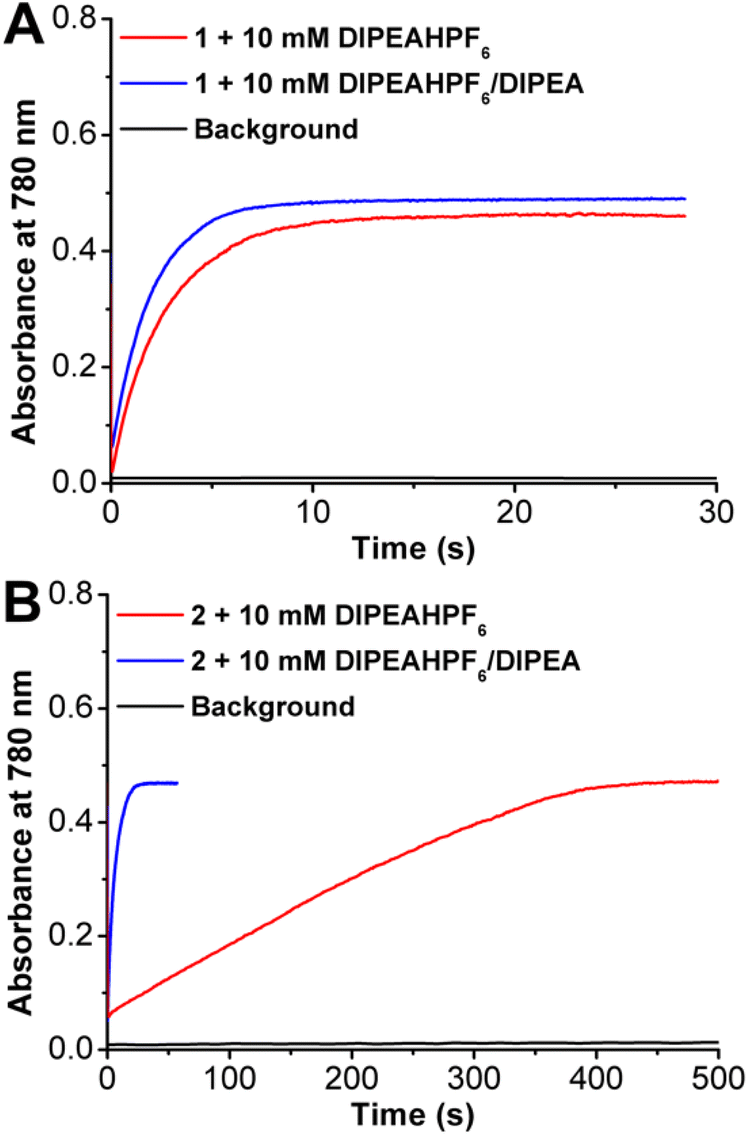 | ||
| Fig. 4 Change in absorbance at 780 nm over time as a result of the formation of [Cp*2Fe]+ by ORR catalyzed by Mn(p-tbudhbpy)Cl 1 (A) with DIPEAHPF6 (red) and DIPEAHPF6/DIPEA (blue) and Mn(nPrdhbpy)Cl 2 (B) with DIPEAHPF6 (red) and DIPEAHPF6/DIPEA (blue). Black: buffered control. Conditions: 40 μM [Mn], 4.05 mM [O2], 1 mM [Cp*2Fe], 10 mM [DIPEAHPF6/DIPEA]; control: 4.05 mM O2, 1 mM Cp*2Fe, 12.5 mM DIPEAHPF6/DIPEA. | ||
Variable concentration studies with 1 and DIPEAHPF6 under unbuffered conditions revealed a rate law for the ORR with first-order dependencies on [1] and [O2] and zero-order dependencies on [DIPEAHPF6] and [Cp*2Fe] (Fig. S36–S39†), corresponding to the rate law shown in eqn (1).
| Rateunbuffered = kcat[Mnp-tbu]1[O2]1 | (1) |
A Ti(O)SO4 colorimetric assay was used to determine the selectivity of the ORR as previously described.23,33 Aliquots were taken after the reaction was allowed to reach completion and the amount of H2O2 produced was quantified. Selectivity testing under unbuffered conditions revealed that 1 is 64.2 ± 6.9% selective for H2O2 corresponding to an ncat of 2.72 (Fig. S26† and Table 1). Under these unbuffered conditions (only proton donor present), control studies showed no degradation of H2O2via disproportionation, with quantitative recovery of H2O2 (Fig. S28 and Table S6†).
| Unbuffered | Buffered (15 s) | |||
|---|---|---|---|---|
| % H2O2 | % H2O | % H2O2 | % H2O | |
| 1 | 64.2 ± 6.9 | 35.8 ± 6.9 | 18.1 ± 4.1 | 81.9 ± 4.1 |
| 2 | 96.2 ± 4.1 | 3.8 ± 4.1 | 37.9 ± 6.7 | 62.1 ± 6.7 |
Similarly, variable concentration studies of ORR by 2 with unbuffered DIPEAHPF6 showed first-order dependencies on [2] and [O2] (Fig. S45–S49†). Selectivity studies showed 96.2 ± 4.1% H2O2 selectivity corresponding to an ncat = 2.08 (Fig. S27 and Table S5†), with control studies showing that 93.6 ± 4.6% H2O2 was recovered after 20 minutes (Fig. S29 and Table S7†), consistent with very slight activity for H2O2 disproportionation. Notably, the observation of an increased amount of the H2O2 product for complex 2 – which contains pendent proton donor relays – mirrors similar observations made with Co-based analogues of these compounds previously.25 Thus, eqn (2) is proposed as the rate law for ORR mediated by 2 under unbuffered conditions.
| Rateunbuffered = kcat[MnnPr]1[O2]1 | (2) |
Subsequent re-examination of 1 under buffered conditions (equal amounts of ammonium proton donor and its conjugate base) showed first-order dependencies on [1] and [O2] and zero-order dependencies on [1:1 DIPEAHPF6:DIPEA] and [Cp*2Fe] (Fig. S40–S44†). Systematically varying the concentration of either [DIPEAHPF6] or [DIPEA] against a fixed concentration of the other also exhibited no concentration dependence on the catalytic reaction (Fig. S42†). Selectivity studies revealed that after 15 s, 1 showed 81.9 ± 4.1% selectivity for H2O as the product (Fig. S30† and Table 1). H2O2 disproportionation was observed under catalytic conditions: only 10.8 ± 6.6% H2O2 was recovered after 105 s with 1:1 DIPEAHPF6:DIPEA present (Fig. S32 and Table S10†). Interestingly, control studies under buffered catalytic conditions without the presence of O2 revealed minimal degradation of H2O2 by the Mn(II) form of the complex (Fig. S34 and Table S12†), suggesting that the observed reaction selectivity difference is not due to disproportionation alone. These results are summarized in the following rate law, eqn (3), for ORR mediated by 1 under buffered conditions:
| Ratebuffered = kcat[Mnp-tbu]1[O2]1 | (3) |
Conversely, variable concentration studies of ORR catalyzed by 2 under buffered conditions revealed mechanistic differences in comparison to the data obtained for 1 and 2 under unbuffered conditions, as well as a change in selectivity. The ORR mediated by 2 showed a second-order dependence on [2], a first-order dependence on [1:1 DIPEAHPF6:DIPEA], an inverse first-order dependence on [Cp*2Fe] and a zero-order dependence on [O2] (Fig. S49–S53†). Unlike 1, when [DIPEA] was varied against a fixed concentration of [DIPEAHPF6] with 2, a first-order dependence on rate was observed; experiments where [DIPEAHPF6] was varied against a fixed [DIPEA] showed a zero-order dependence. These results lead to the proposed rate law shown in eqn (4). The observed first-order dependence on [DIPEA] implies that the dependence observed on [1:1 DIPEAHPF6:DIPEA] is a result of varying DIPEA concentration. This interpretation is validated by the zero-order dependence on [DIPEAHPF6] with fixed [DIPEA], ruling out proton activity or proton donor concentration as influencing the observed reaction rate.
| Rate = kcat[MnnPr]2[DIPEA]1[Cp*2Fe]−1 | (4) |
Selectivity studies of ORR under buffered conditions showed that after 15 s, 2 is 62.1 ± 6.7% selective for H2O (Fig. S33† and Table 1). Control studies showed H2O2 degradation consistent with disproportionation: after 150 s only 3.81 ± 0.83% of H2O2 was recovered under buffered conditions with complex 2. Again, H2O2 reduction was not observed under the buffered catalytic conditions when placed under an inert N2 atmosphere, suggesting that the change in selectivity is not exclusively due to disproportionation (Fig. S35 and Table S13†).
Computational studies on complex 2
To evaluate the initial steps of the reaction, computational studies on the thermodynamic positioning of likely intermediates for complex 2 during O2 reduction to H2O2 were subsequently undertaken (see ESI† for details). Geometry optimization was done with the Gaussian 16 package34 at B3LYP-D3(BJ)/def2-SVP level35–42 with a complete structural model and single point calculations for refining energy differences were completed with Orca 5.043 at the ωB97M-D4/def2-TZVPP level.39,40,44–48 At this level of theory, the calculated reduction potential of −0.53 V vs. Fc+/0 showed satisfactory agreement with the experimental potential of −0.55 V. Further, the anticipated ground states of S = 2 for Mn(III) and S = 5/2 for Mn(II) in this coordination environment were accurately reproduced, lending confidence to the chosen method.22 Electron transfer-proton transfer (ET-PT), proton transfer-electron transfer (PT-ET), and concerted electron-proton transfer (CEPT) pathways for reaction steps where a net hydrogen atom transfer occurred were considered; in all cases CEPT was found to be the most favorable and produced results consistent with experimental observation.
The most probable thermodynamic step from the starting complex [Mn(nPrdhbpy)(Cl)]0 under protic conditions is reduction followed by a hydrogen–bonding interaction between the ligand framework and an equivalent of [DIPEAH]+ to generate [Mn(nPrdhbpy[AH])(Cl)]0 (Fig. 5). Note that the [AH] notation indicates the hydrogen-bonding interaction involving a Mn-bound O atom from the ligand framework. Although formal proton transfer to the ligand framework with loss of DIPEA is exergonic by −7.2 kcal mol−1, the binding of O2 with accompanying Cl− release to generate the cationic species [Mn(nPrdhbpy[H])(η2-O2)]+ is uphill by 34.2 kcal mol−1, precluding its involvement in the catalytic cycle. Instead, O2 binding with loss of [DIPEAH][Cl] is endergonic by 20.2 kcal mol−1, which is consistent with experimental observations, generating [Mn(nPrdhbpy)(η2-O2)]0. In this structure, the bound O2 is in a side-on coordination mode with a bond length of 1.309 Å, consistent with reduction to superoxide, O2˙−. Reduction and protonation of this species had a favorable CEPT pathway (−0.01 V vs. Fc+/0), where proton transfer has occurred from [DIPEAH]+ to generate [Mn(nPrdhbpy)(η1-O2H)]0. The shift in coordination mode of the O2 fragment from side-on to end-on reflects additional reduction: the bond lengthens to 1.451 Å, consistent with a peroxide. Protonation of the proximal O atom to generate [Mn(nPrdhbpy)(η1-O2H2)]+ by an equivalent of [DIPEAH]+ is endergonic by 9.9 kcal mol−1 and displacement by Cl− to facilitate H2O2 release is favorable by −23.6 kcal mol−1. These reaction steps align with experimental observations on the catalytic cycle which produces H2O2 under unbuffered conditions.
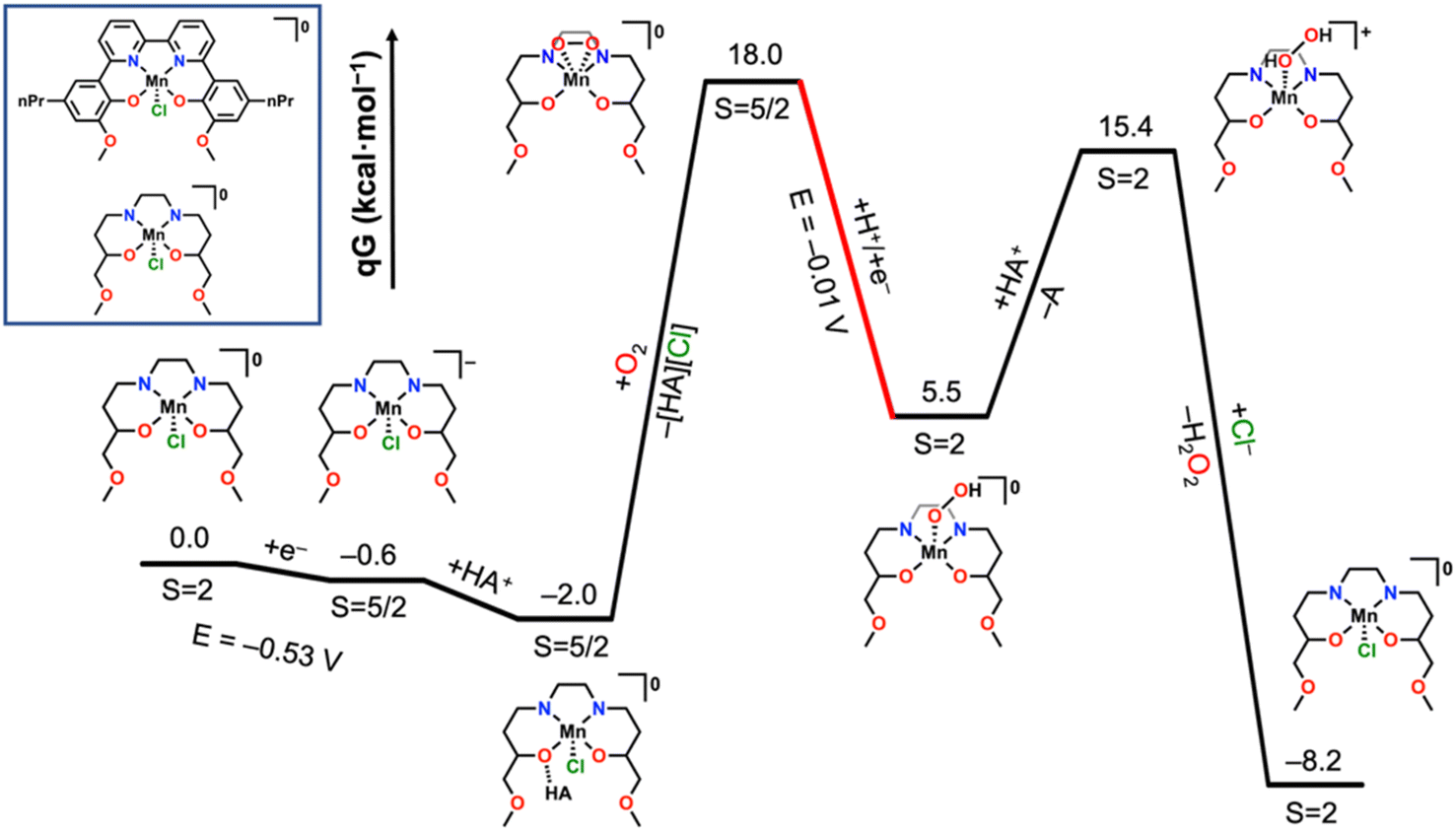 | ||
| Fig. 5 Free energy diagram from DFT methods showing key thermodynamic intermediates in the production of H2O2 by complex 2. Diagram considers the operating potential of catalysis to be −0.55 V vs. Fc+/0 and red indicates CEPT steps. DIPEA = A and DIPEAH+ = HA+. | ||
Next the favorability of dimerization from the hydroperoxide was assessed to explore the implied dimeric pathway to H2O production (Fig. S77†). The experimental studies described above established the viability of H2O2 as an intermediate species, therefore the [Mn(nPrdhbpy)(η1-O2H2)]+ adduct was considered as the starting point. Deprotonation of Mn-bound H2O2 by DIPEA to generate [Mn(nPrdhbpy)(η1-O2H)]0 is favorable by 9.9 kcal mol−1. Subsequent dimerization of [Mn(nPrdhbpy)(η2-O2H)]0 with an equivalent of [Mn(nPrdhbpy)(Cl)]0 is endergonic by 19.7 kcal mol−1 and generates [Mn(nPrdhbpy)(O)]0 (S = 3/2), [Mn(nPrdhbpy)(OH)]+ (S = 3/2), and Cl− as the products. Attempts to examine a stabilized bridging hydroperoxo dimer were unsuccessful, homolytic O–O bond scission occurred spontaneously in all cases. From the terminal manganese oxo [Mn(nPrdhbpy)(O)]0, a calculated CEPT potential of +1.00 V vs. Fc+/Fc was obtained for the production of a Mn(III) hydroxide [Mn(nPrdhbpy)(OH)]0, while the convergent pathway via the reduction of [Mn(nPrdhbpy)(OH)]+ was estimated to be +0.62 V; both processes are expected to be facile at the considered operating potential of −0.55 V vs. Fc+/Fc. Protonation of the neutral Mn–OH species to make the corresponding aquo species is uphill by 1.2 kcal mol−1 and subsequent H2O loss with Cl− coordination to Mn to regenerate the starting species and close the H2O2 reduction cycle is downhill by −20.2 kcal mol−1.
Discussion
From the data summarized above, we are able to propose the following mechanisms for the ORR catalyzed by 1 and 2 under unbuffered and buffered conditions, described in Scheme 1. Starting at i, the reduction of Mn(III) to Mn(II) induces a hydrogen-bonding interaction between [DIPEAH]+ and the O atom in the inner coordination sphere to form ii, the resting state of the catalyst in solution. It is likely that this species exists in equilibrium with formal proton transfer, based on DFT results, UV-vis studies on chemically reduced species (Fig. S64 and S71†), and the observed irreversibility of the Mn(III)/(II) reduction. Control studies showed that neither DIPEAHPF6 or DIPEA interacted substantially with complexes in the Mn(III) oxidation state (Fig. S58–S61†). However, formal proton transfer renders O2 binding to Mn less favorable by 14 kcal mol−1 according to DFT methods, indicating that this pathway isn't viable catalytically.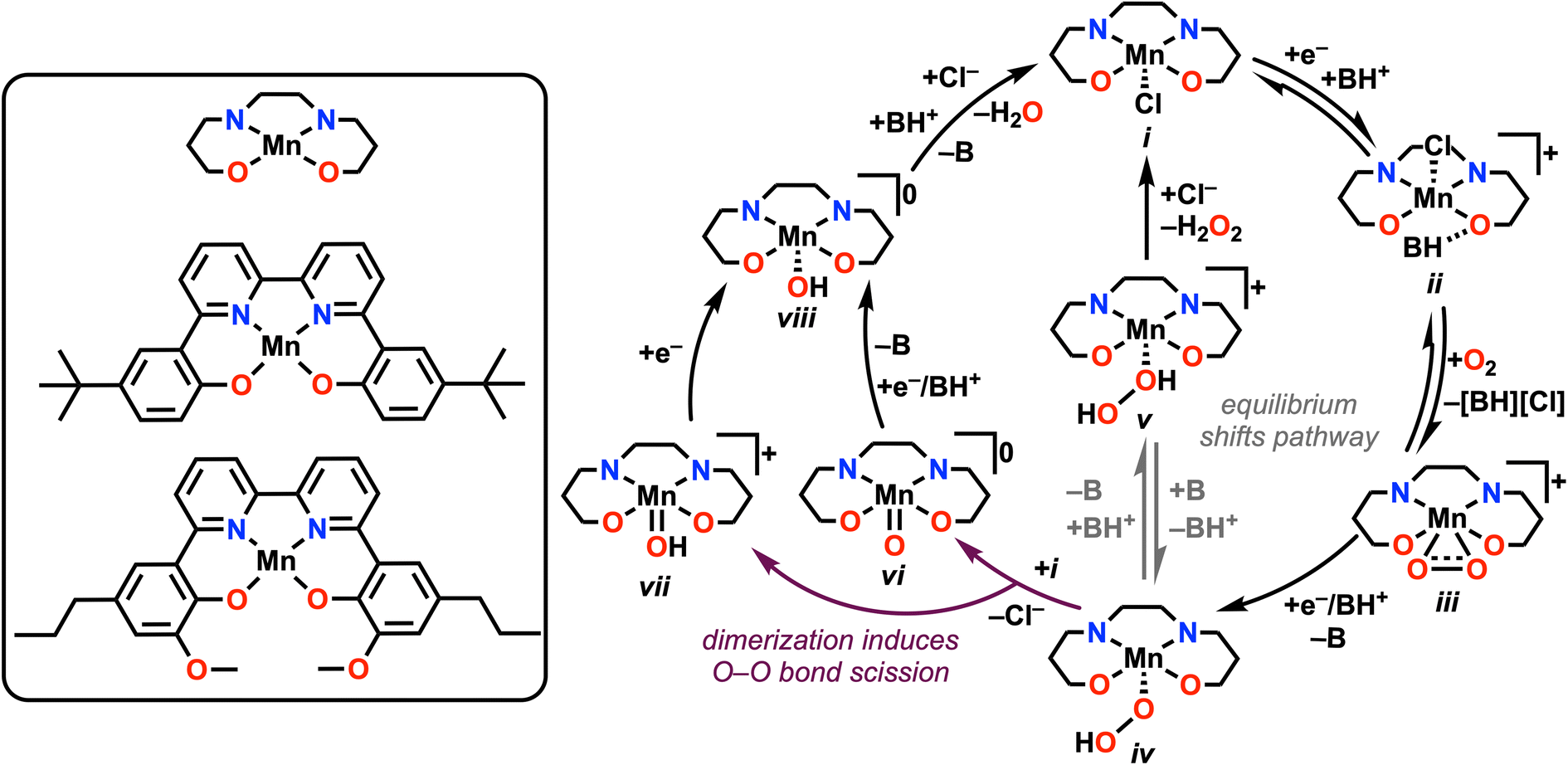 | ||
| Scheme 1 Proposed catalytic cycle for ORR, with the key equilibrium for switching between the two reaction pathways indicated in gray. | ||
Following this activation of the catalyst, rate-limiting O2 binding to Mn(II) to form a Mn(III)-superoxide species, iii, occurs with loss of [DIPEAH][Cl]. Based on this assignment, species ii is considered to be the resting state of the catalytic cycle under these conditions. Reduction and protonation by a CEPT pathway results in the formation of a Mn-hydroperoxo species, iv, whose protonation leads to the formation of a Mn–H2O2 intermediate, v. This is the primary reaction pathway for 1 (64.2 ± 6.9% selectivity) and 2 (96.2 ± 4.1% selectivity) under unbuffered conditions, as suggested by selectivity studies and the relative absence of activity for H2O2 disproportionation in separate testing. The observed selectivity enhancement for H2O2 during ORR mediated by 2 is also aligned with the participation of the pendent methoxy group in hydrogen bonding interactions with added acid, as we have observed previously.25 This mechanistic proposal is consistent with the general thermodynamic landscape obtained by DFT methods (Fig. 5). It is worth noting that H2O2 disproportionation is observed by the parent Mn(III) species of both complexes in the presence of DIPEA. Further, control studies with Mn(II) revealed minimal interaction with H2O2 (Fig. S34†), suggesting that disproportionation is reliant on the availability of formally Mn(III) complexes.
Under buffered conditions, complex 2 demonstrated a substantial increase in the observed catalytic rate in comparison to unbuffered conditions with an accompanying mechanistic divergence involving dimerization. While we do not observe a change in the rate-determining step or infer a change in the resting state for 1, it is proposed that the shift in selectivity from H2O2 under unbuffered conditions to H2O under buffered conditions is similarly due to the accessibility of a dimerization pathway in the presence of added base. Based on the experimental and computational data presented, it is likely that the observed mechanistic divergence arises from differences in equilibrium control over the speciation of complexes iv and v, as depicted in eqn (5).
Mn(O2H) + BH+ ![[left over right harpoons]](https://www.rsc.org/images/entities/char_21cb.gif) [Mn(H2O2)]+ + B [Mn(H2O2)]+ + B | (5) |
Therefore, under buffered conditions, an alternative reaction pathway dominates the observed selectivity. With suitable concentrations of base present, eqn (5) shifts to the left, favoring the hydroperoxo species, iv, which becomes the resting state for 2 under buffered conditions. Species iv dimerizes with an equiv of the Mn(III) complex i leading to the formation of species vi and vii from spontaneous O–O bond cleavage, with accompanying chloride loss. Following one-electron reduction or a one-electron CPET process, these products converge at a Mn(III) hydroxide species viii. Following this, formal protonation and displacement of H2O by Cl− complete the catalytic cycle. For complex 2, spontaneous O–O bond cleavage from dimerization becomes the RDS of the reaction, which is attributed to the pendent relay groups slowing dimer formation through intermolecular coordination and hydrogen-bonding. This proposal is consistent with the second-order rate dependence on [Mn] and first-order rate dependence on [DIPEA] under buffered conditions. Further, the inverse first-order dependence observed for [Cp*2Fe] implies that Mn(III) is required for dimerization, rather than Mn(II). This reaction pathway is in good agreement with that computed using DFT methods (Fig. S77†).
The differences in ORR catalyzed by 2 under unbuffered and buffered conditions in comparison to 1 suggest an important role for the pendent –OMe moiety in the secondary coordination sphere, as well as for the added base. Under unbuffered conditions, catalytic activity of 2 is suppressed in comparison to 1, which is observed under both electrochemical and spectrochemical conditions. This is attributed to strong hydrogen-bonding interactions between the –OMe groups and DIPEAHPF6 kinetically inhibiting O2 binding, the rate-determining step of the catalytic cycle. In addition to catalytic suppression, we observe a shift in selectivity from approximately 64% for H2O2 by 1 to 96% for H2O2 by 2 under unbuffered catalytic conditions. As introduced above, we propose that the observed shift in product selectivity is due to the hydrogen-bonding interaction of the –OMe group assisting in proton transfer to the proximal Mn–OOH oxygen in species iv, as we have previously suggested in similar Co systems.25
Interestingly, upon the addition of the DIPEA conjugate base to catalytic conditions with 2, catalysis is significantly enhanced under electro- and spectrochemical conditions. There is an accompanying change in mechanism, where the equilibrium responsible for H2O2 formation becomes unfavorable, allowing dimerization with accompanying O–O bond scission to become rate-determining. It is also likely that DIPEA mitigates the effects of the strong H-bonding interaction between DIPEAHPF6 and the –OMe moiety, allowing an increased rate of hydroperoxo intermediate generation. Indeed, in control studies with 2 chemically reduced to Mn(II) by CoCp2in situ, the addition of DIPEAHPF6 inhibited reactivity with O2: under unbuffered conditions the reaction between 2 and O2 took approximately 100 min to go to completion (Fig. S72†), compared with approximately 25 m for complex 1 (Fig. S65†). It is also worth noting the difference in selectivity under buffered conditions between 1 and 2, where 1 is 81.9% selective for H2O (Table 1) and 2 is only 62.7% selective for H2O. We attribute this difference to the H-bonding ability of the –OMe groups of 2 promoting the formation of species v during catalysis by directing the proton donor. However, the dimer pathway to water formation is the primary pathway under buffered conditions, as evidenced by the shift in observed rate law and the shift in product selectivity to H2O.
The observed rate constants (Rfit/ncat values) under identical buffered conditions for 1 and 2 were kobs = 1.23 ± 0.17 × 10−1 s−1 and kobs = 0.706 ± 0.25 × 10−1 s−1, respectively. While addition of one equivalent of conjugate base for every equivalent of proton donor mitigates some of the hydrogen-bonding induced suppression observed for 2 under unbuffered conditions, the accompanying inhibition of dimerization results in a slight decrease in the rate of catalysis relative to 1. Therefore, it has been demonstrated that the introduction of a pendent relay into homogeneous molecular Mn-based electrocatalysts plays an essential role in ORR through the hydrogen bond-assisted stabilization of key intermediates and by favoring the direct protonation of the proximal oxygen of the Mn–OOH intermediate. The relative hydrogen-bond donor ability of DIPEAHPF6 is such that inhibition of catalysis can also occur, which is mitigated through the introduction of the conjugate base. However, the mitigation of this effect results in a change in mechanism, where O–O bond scission occurs spontaenously upon dimerization. Thus, the conjugate base plays an important role during catalysis, allowing for an on-cycle dimer pathway that shifts the reaction pathway towards the formation of water.
It is worth briefly discussing the comparison between the data reported here for 1 and those reported previously with phenolic proton donors for a related complex with additional tert-butyl substituents.22,23 With buffered phenolic proton donors, the observed rate law for the ORR mediated by this more sterically hindered complex showed first-order dependencies on [catalyst] and [O2], but no dependence on proton donor activity. Similarly, the results obtained in the presence of a buffered ammonium proton donor with higher activity (pKa(MeCN) = 18.7) described here again demonstrate that proton activity is not relevant to the observed rate laws. The observed relevance of an off-cycle EC reaction involving the protonation of the ligand framework also aligns with the results of a potential-pKa diagram obtained during the previous study where acids with pKa < 20.11 were found to protonate the ligand framework of the tetra tert-butylated complex. The systems reported here show greater stability with respect to any H2O2 generated, which we had previously observed not to be the case in the presence of phenol and phenolate derivatives, consistent with the ease with which they can be oxidized as well as their competency as ligands relative to the sterically hindered ammonium/amine pairs used here.49–51 Indeed, control testing shows that the DIPEA does not react under experimental conditions (Fig. S58 and S59†) and is too sterically hindered to coordinate to Mn.
Conclusion
Here we report the synthesis and characterization of two new Mn(N2O2) homogeneous catalysts for ORR with an ammonium-based proton source in MeCN solution. Introduction of a pendent –OMe group in the secondary coordination sphere proved to play an important role during catalysis. Under unbuffered conditions, the pendent relay enabled quantitative selectivity for H2O2 due to directed protonation of the proximal O atom in a key Mn(III)–OOH intermediate. However, strong hydrogen-bonding interactions between the proton source and the pendent –OMe moiety suppressed catalytic activity. The addition of the proton donor's conjugate base enhanced the catalytic response and shifted the rate-determining step to O–O bond cleavage induced by dimerization. These results describe the importance of secondary sphere ligand modifications in the tunability of catalytic ORR mediated by Mn complexes, including the role of hydrogen-bond donor ability in directing product selectivity and the benefits of buffered conditions. Destabilization of certain Mn–O2 intermediates can be achieved through secondary-sphere modifications, however, later steps in the catalytic cycle must also be considered, since they can impact in negative ways. The continued challenge for improved Mn-based ORR catalysts is to drive the initial O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O reduction with a proton donor while retaining activity for subsequent O–O bond scission. Based on these conclusions, we are exploring the use of proton relays with variable basicity in ligand frameworks which disfavor dimerization, in order to improve the activity of monomeric catalyst pathways mediated by Mn complexes.
O reduction with a proton donor while retaining activity for subsequent O–O bond scission. Based on these conclusions, we are exploring the use of proton relays with variable basicity in ligand frameworks which disfavor dimerization, in order to improve the activity of monomeric catalyst pathways mediated by Mn complexes.
Data availability
The datasets supporting this article have been uploaded as part of the ESI.†Author contributions
ENC and CWM conceptualized the project; ENC, IMC, DAD, and CWM designed and conducted experiments; CWM supervised the project. The manuscript was written with analysis and contributions from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the University of Virginia for infrastructural support. E. N. C., I. M. C., and C. W. M. acknowledge N.S.F. CHE-2102156 for support. Single crystal X-ray diffraction experiments were performed on a diffractometer at the University of Virginia funded by the NSF-MRI program (CHE-2018870). E. N. C. acknowledges the Jefferson Scholars Foundation for support from a Jefferson Arts and Sciences Dissertation Year Fellowship.References
- E. N. Cook and C. W. Machan, Bioinspired mononuclear Mn complexes for O2 activation and biologically relevant reactions, Dalton Trans., 2021, 50(46), 16871–16886, 10.1039/D1DT03178C.
- E. N. Cook and C. W. Machan, Homogeneous catalysis of dioxygen reduction by molecular Mn complexes, Chem. Commun., 2022, 58(84), 11746–11761, 10.1039/D2CC04628H.
- M. L. Pegis, C. F. Wise, D. J. Martin and J. M. Mayer, Oxygen reduction by homogeneous molecular catalysts and electrocatalysts, Chem. Rev., 2018, 118(5), 2340–2391, DOI:10.1021/acs.chemrev.7b00542.
- C. W. Machan, Advances in the molecular catalysis of dioxygen reduction, ACS Catal., 2020, 10(4), 2640–2655, DOI:10.1021/acscatal.9b04477.
- E. I. Solomon, S. Goudarzi and K. D. Sutherlin, O2 activation by non-heme iron enzymes, Biochemistry, 2016, 55(46), 6363–6374, DOI:10.1021/acs.biochem.6b00635.
- E. I. Solomon and S. S. Stahl, Introduction: oxygen reduction and activation in catalysis, Chem. Rev., 2018, 118(5), 2299–2301, DOI:10.1021/acs.chemrev.8b00046.
- X. Huang and J. T. Groves, Oxygen activation and radical transformations in heme proteins and metalloporphyrins, Chem. Rev., 2018, 118(5), 2491–2553, DOI:10.1021/acs.chemrev.7b00373.
- S. Hong, Y.-M. Lee, K. Ray and W. Nam, Dioxygen activation chemistry by synthetic mononuclear nonheme iron, copper and chromium complexes, Coord. Chem. Rev., 2017, 334, 25–42, DOI:10.1016/j.ccr.2016.07.006.
- S. Bhunia, A. Ghatak and A. Dey, Second sphere effects on oxygen reduction and peroxide activation by mononuclear iron porphyrins and related systems, Chem. Rev., 2022, 122(14), 12370–12426, DOI:10.1021/acs.chemrev.1c01021.
- J. M. Campos-Martin, G. Blanco-Brieva and J. L. G. Fierro, Hydrogen peroxide synthesis: an outlook beyond the anthraquinone process, Angew. Chem., Int. Ed., 2006, 45(42), 6962–6984, DOI:10.1002/anie.200503779.
- M. Kiani, X. Q. Tian and W. Zhang, Non-precious metal electrocatalysts design for oxygen reduction reaction in polymer electrolyte membrane fuel cells: recent advances, challenges and future perspectives, Coord. Chem. Rev., 2021, 441, 213954, DOI:10.1016/j.ccr.2021.213954.
- M. L. Pegis, D. J. Martin, C. F. Wise, A. C. Brezny, S. I. Johnson, L. E. Johnson, N. Kumar, S. Raugei and J. M. Mayer, Mechanism of catalytic O2 Reduction by iron tetraphenylporphyrin, J. Am. Chem. Soc., 2019, 141(20), 8315–8326, DOI:10.1021/jacs.9b02640.
- R. Zhang and J. J. Warren, Recent developments in metalloporphyrin electrocatalysts for reduction of small molecules: strategies for managing electron and proton transfer reactions, ChemSusChem, 2021, 14(1), 293–302, DOI:10.1002/cssc.202001914.
- Q. M. Phung and K. Pierloot, The dioxygen adducts of iron and manganese porphyrins: electronic structure and binding energy, Phys. Chem. Chem. Phys., 2018, 20(25), 17009–17019, 10.1039/C8CP03078B.
- J. P. T. Zaragoza and D. P. Goldberg, in Dioxygen-Dependent Heme Enzymes, The Royal Society of Chemistry, 2019, pp. 1–36 Search PubMed.
- R. B. VanAtta, C. E. Strouse, L. K. Hanson and J. S. Valentine, Peroxo(tetraphenylporphinato)manganese(III) and chloro(tetraphenylporphinato)manganese(II) anions. Synthesis, crystal structures, and electronic structures, J. Am. Chem. Soc., 1987, 109(5), 1425–1434, DOI:10.1021/ja00239a024.
- G. Passard, D. K. Dogutan, M. Qiu, C. Costentin and D. G. Nocera, Oxygen reduction reaction promoted by manganese porphyrins, ACS Catal., 2018, 8(9), 8671–8679, DOI:10.1021/acscatal.8b01944.
- M. Gennari, D. Brazzolotto, J. Pécaut, M. V. Cherrier, C. J. Pollock, S. Debeer, M. Retegan, D. A. Pantazis, F. Neese, M. Rouzières, R. Clérac and C. Duboc, Dioxygen activation and catalytic reduction to hydrogen peroxide by a thiolate-bridged dimanganese(II) complex with a pendant thiol, J. Am. Chem. Soc., 2015, 137(26), 8644–8653, DOI:10.1021/jacs.5b04917.
- G. A. Hamilton and A. Revesz, Oxidation by molecular oxygen. IV. A possible model reaction for some amine oxidases1-3, J. Am. Chem. Soc., 1966, 88(9), 2069–2070, DOI:10.1021/ja00961a054.
- D. F. Evans and T. S. Sheriff, The production of hydrogen peroxide from dioxygen and hydroxylamine catalysed by manganese complexes, J. Chem. Soc., Chem. Commun., 1985, 20, 1407–1408, 10.1039/C39850001407.
- R. L. Shook, S. M. Peterson, J. Greaves, C. Moore, A. L. Rheingold and A. S. Borovik, Catalytic reduction of dioxygen to water with a monomeric manganese complex at room temperature, J. Am. Chem. Soc., 2011, 133(15), 5810–5817, DOI:10.1021/ja106564a.
- S. L. Hooe, A. L. Rheingold and C. W. Machan, Electrocatalytic reduction of dioxygen to hydrogen peroxide by a molecular manganese complex with a bipyridine-containing schiff base ligand, J. Am. Chem. Soc., 2018, 140(9), 3232–3241, DOI:10.1021/jacs.7b09027.
- S. L. Hooe and C. W. Machan, Dioxygen Reduction to hydrogen peroxide by a molecular Mn complex: mechanistic divergence between homogeneous and heterogeneous reductants, J. Am. Chem. Soc., 2019, 141(10), 4379–4387, DOI:10.1021/jacs.8b13373.
- A. W. Nichols, J. S. Kuehner, B. L. Huffman, P. R. Miedaner, D. A. Dickie and C. W. Machan, Reduction of dioxygen to water by a Co(N2O2) complex with a 2,2′-bipyridine backbone, Chem. Commun., 2021, 57, 516–519, 10.1039/d0cc06763f.
- A. W. Nichols, E. N. Cook, Y. J. Gan, P. R. Miedaner, J. M. Dressel, D. A. Dickie, H. S. Shafaat and C. W. Machan, Pendent relay enhances H2O2 selectivity during dioxygen reduction mediated by bipyridine-based Co–N2O2 complexes, J. Am. Chem. Soc., 2021, 143(33), 13065–13073, DOI:10.1021/jacs.1c03381.
- S. L. Hooe, E. N. Cook, A. G. Reid and C. W. Machan, Non-covalent assembly of proton donors and p-benzoquinone anions for co-electrocatalytic reduction of dioxygen, Chem. Sci., 2021, 12(28), 9733–9741, 10.1039/d1sc01271a.
- P. Claude, Paramagnetic susceptibility by NMR: the “solvent correction” removed for large paramagnetic molecules, J. Chem. Educ., 1997, 74(7), 815–816, DOI:10.1021/ed074p815.
- G. A. Bain and J. F. Berry, Diamagnetic corrections and Pascal's constants, J. Chem. Educ., 2008, 85(4), 532–536, DOI:10.1021/ed085p532.
- A. W. Nichols, S. L. Hooe, J. S. Kuehner, D. A. Dickie and C. W. Machan, Electrocatalytic CO2 reduction to formate with molecular Fe(III) complexes containing pendent proton relays, Inorg. Chem., 2020, 59(9), 5854–5864, DOI:10.1021/acs.inorgchem.9b03341.
- S. Saba, R. Hernandez, C. C. Choy, K. Carta, Y. Bennett, S. Bondi, S. Kolaj and C. Bennett, A simple and efficient one-step protocol for the preparation of alkyl-substituted ammonium tetrafluoroborate and hexafluorophosphate salts, J. Fluorine Chem., 2013, 153, 168–171, DOI:10.1016/j.jfluchem.2013.05.007.
- R. Shivapurkar and D. Jeannerat, Determination of the relative pKa's of mixtures of organic acids using NMR titration experiments based on aliased 1H–13C HSQC spectra, Anal. Methods, 2011, 3(6), 1316–1322, 10.1039/C0AY00771D.
- P. Zanello, The Royal Society of Chemistry, Cambridge, UK, 2003, pp. 49–135.
- C. W. Anson and S. S. Stahl, Cooperative electrocatalytic O2 reduction involving Co(salophen) with p-hydroquinone as an electron–proton transfer mediator, J. Am. Chem. Soc., 2017, 139(51), 18472–18475, DOI:10.1021/jacs.7b11362.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision B.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- A. D. Becke, Density-functional thermochemistry. III. The role of exact exchange, J. Chem. Phys., 1993, 98(7), 5648–5652, DOI:10.1063/1.464913.
- C. Lee, W. Yang and R. G. Parr, Development of the colle-salvetti correlation-energy formula into a functional of the electron density, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37(2), 785–789 CrossRef CAS PubMed.
- S. H. Vosko, L. Wilk and M. Nusair, Accurate spin-dependent electron liquid correlation energies for local spin density calculations: a critical analysis, Can. J. Phys., 1980, 58(8), 1200–1211, DOI:10.1139/p80-159.
- P. J. Stephen, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields, J. Phys. Chem., 1994, 98(45), 11623–11627 CrossRef.
- F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7(18), 3297–3305, 10.1039/b508541a.
- F. Weigend, Accurate coulomb-fitting basis sets for H to Rn, Phys. Chem. Chem. Phys., 2006, 8(9), 1057–1065, 10.1039/b515623h.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132(15), 154104, DOI:10.1063/1.3382344.
- S. Grimme, S. Ehrlich and L. Goerigk, Effect of the damping function in dispersion corrected density functional theory, J. Comput. Chem., 2011, 32(7), 1456–1465, DOI:10.1002/jcc.21759.
- F. Neese, Software update: the ORCA program system—Version 5.0, WIREs Comput. Mol. Sci., 2022, 12(5), e1606, DOI:10.1002/wcms.1606.
- N. Mardirossian and M. Head-Gordon, ωB97M-V: a combinatorially optimized, range-separated hybrid, meta-GGA density functional with VV10 nonlocal correlation, J. Chem. Phys., 2016, 144(21), 214110, DOI:10.1063/1.4952647.
- A. Hellweg, C. Hättig, S. Höfener and W. Klopper, Optimized accurate auxiliary basis sets for RI-MP2 and RI-CC2 calculations for the atoms Rb to Rn, Theor. Chem. Acc., 2007, 117(4), 587–597, DOI:10.1007/s00214-007-0250-5.
- E. Caldeweyher, C. Bannwarth and S. Grimme, Extension of the D3 dispersion coefficient model, J. Chem. Phys., 2017, 147(3), 034112, DOI:10.1063/1.4993215.
- E. Caldeweyher, S. Ehlert, A. Hansen, H. Neugebauer, S. Spicher, C. Bannwarth and S. Grimme, A generally applicable atomic-charge dependent London dispersion correction, J. Chem. Phys., 2019, 150(15), 154122, DOI:10.1063/1.5090222.
- A. Najibi and L. Goerigk, DFT-D4 counterparts of leading meta-generalized-gradient approximation and hybrid density functionals for energetics and geometries, J. Comput. Chem., 2020, 41(30), 2562–2572, DOI:10.1002/jcc.26411.
- P. Chaau Yan Poon, M. A. Dedushko, X. Sun, G. Yang, S. Toledo, E. C. Hayes, A. Johansen, M. C. Piquette, J. A. Rees, S. Stoll, E. Rybak-akimova and J. A. Kovacs, How metal ion lewis acidity and steric properties influence the barrier to dioxygen binding, peroxo O–O bond cleavage, and reactivity, J. Am. Chem. Soc., 2019, 141, 15046–15057, DOI:10.1021/jacs.9b04729.
- J.-Z. Li, W. Hu, Y. Wang, J.-B. Zhong and S.-X. Li, A Kinetic study of phenol oxidation with H2O2 catalysed by crowned schiff base mn(III) complexes in micellar media, Prog. React. Kinet. Mech., 2012, 37(1), 30–41, DOI:10.3184/146867812X13242290723318.
- D. B. Broughton and R. L. Wentworth, Mechanism of decomposition of hydrogen peroxide solutions with manganese dioxide. I, J. Am. Chem. Soc., 1947, 69(4), 741–744, DOI:10.1021/ja01196a003.
Footnote |
| † Electronic supplementary information (ESI) available: Computational coordinates, experimental and computational details, as well as supplementary data from additional cyclic voltammetry, spectrochemical, spectroscopic experiments are available. Deposition numbers 2255849 and 2255850 contain the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre. CCDC 2255849 and 2255850. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc02611f |
| This journal is © The Royal Society of Chemistry 2024 |