
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDe novo synthesis of inherently chiral heteracalix[4]aromatics from enantioselective macrocyclization enabled by chiral phosphoric acid-catalyzed intramolecular SNAr reaction†
Xing-Chi
Li
a,
Ying
Cheng
 *a,
Xu-Dong
Wang
b,
Shuo
Tong
*c and
Mei-Xiang
Wang
*c
*a,
Xu-Dong
Wang
b,
Shuo
Tong
*c and
Mei-Xiang
Wang
*c
aCollege of Chemistry, Beijing Normal University, Beijing 100875, China. E-mail: ycheng@bnu.edu.cn
bLaboratory of Molecular Recognition and Function, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China
cMOE Key Laboratory of Bioorganic Phosphorous Chemistry and Chemical Biology, Department of Chemistry, Tsinghua University, Beijing 100084, China. E-mail: tongshuo@mail.tsinghua.edu.cn; wangmx@mail.tsinghua.edu.cn
First published on 26th January 2024
Abstract
We report herein the synthesis of highly enantiopure inherently chiral N3,O-calix[2]arene[2]triazines from enantioselective macrocyclization enabled by chiral phosphoric acid-catalyzed intramolecular nucleophilic aromatic substitution reaction. In contrast to documented examples, the inherent chirality of the acquired compounds arises from one heteroatom difference in the linking positions of heteracalix[4](het)arenes.
Inherent chirality was proposed by Böhmer1,2 in 1994 to describe the handedness of calix[4]arene3–5 derivatives devoid of any symmetry elements except for a C1 rotation axis. As a typical example, introduction of four different substituents into either upper or lower rims of a conventional cone-configured calix[4]arene can lead to a pair of isomers which are non-superimposable mirror images of each other. The inherent chirality has now been widely used6 by macrocyclic and supramolecular chemists as a specific term to differentiate curved chiral macrocycles different from well-known central, axial, planar and helical molecules.
Despite unique stereochemical structures, tantalizing properties and potential applications in chemistry, materials and life science, inherently chiral compounds have remained largely underexplored.6,7 Research progress has been very slow since the concept of inherent chirality was introduced nearly thirty years ago. One of the main obstacles impeding and delaying the development of the field is the difficulty in accessing necessary optically active inherently chiral materials. Indeed, most reported methods to obtain enantiopure inherently chiral macrocycles, for instance, rely on the tedious and inefficient resolution8 of racemic samples by means of analytical HPLC using expensive columns coated with various chiral stationary phases and multistep asymmetric synthesis employing chiral auxiliary groups.9 The only attempts to synthesize inherently chiral calixarenes through lipase-catalyzed enantioselective acetylation of tris(O-2-hydroxyethylated)calixarenes10 and to construct tetraazacalix[4]arene from intramolecular C–N bond formation under the catalysis of Pd(dba)2/(R)-SEGPHOS11 suffer from very low chemical yield and enantioselectivity, respectively.
The breakthroughs in the catalytic enantioselective synthesis of inherently chiral compounds have not been reported until very recently12–14 (Scheme 1). In 2020, we12 showed that linear precursors A undergo transition metal catalyzed enantioselective macrocyclization reaction to afford ABCD-type chiral tetraazacalix[4]aromatics B with an ee of >99%. It represents the extraordinary example of excellent transfer and expression of chirality from catalyst Pd/(R,Sp)-JOSIPHOS to inherently chiral macrocycles in the step of palladium-catalyzed C–N cross coupling reaction. Catalytic enantioselective desymmetrization reactions of calix[4]arene derivatives featuring C–Br/C–H bond coupling were developed successfully last year.13,14 In the presence of PdBr2 and chiral BINOL-derived phosphoramidite as a catalyst, calix[4]arene substrates C are transformed into 9H-fluorene-embedded inherently chiral calixarenes Dvia a transannular C–H arylation in which an unusual enantioselective 1,5-palladium migration step is probably involved.13 Interestingly, the palladium-catalyzed reaction of the same reactants C using a chiral bifunctional phosphine-carboxylate ligand proceeds through an alternative C–H arylation pathway to deliver fluorenone-bearing inherent chiral calix[4]arene products E.14 The catalyst-steering divergent reactions of readily available starting materials provide attractive methods to generate structurally diversified inherently chiral calixarenes.
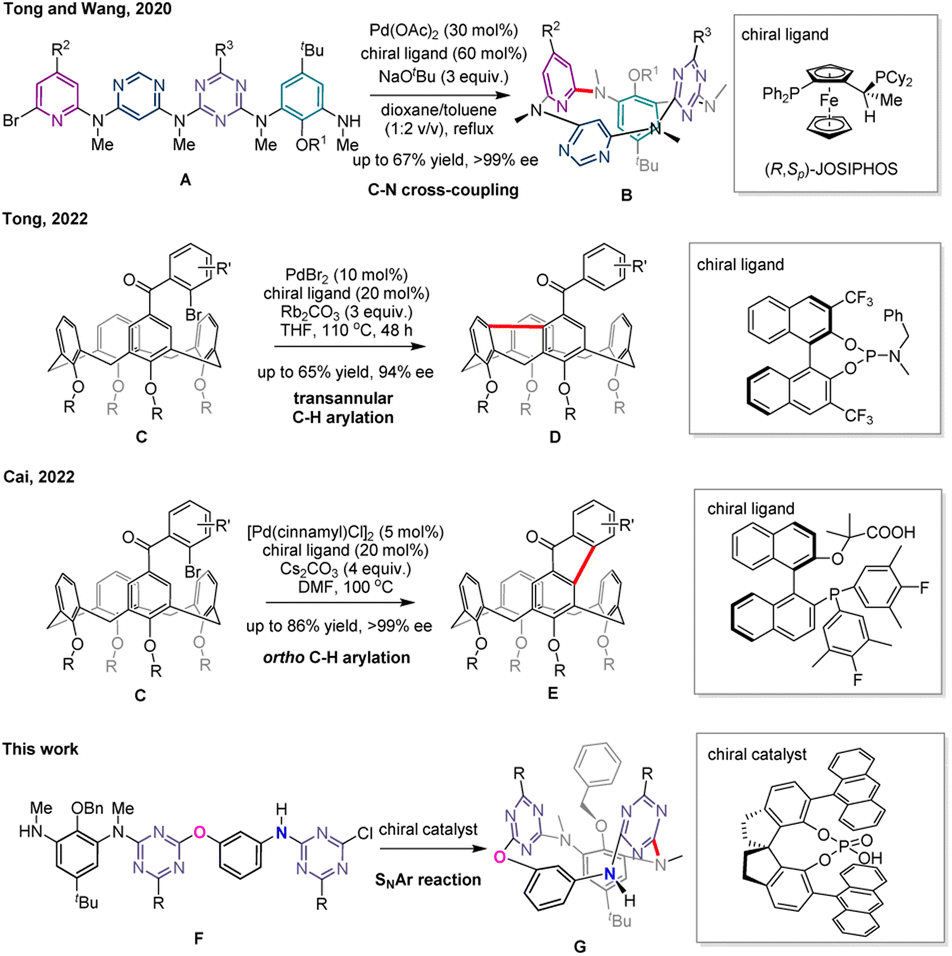 | ||
| Scheme 1 Previous and current studies on catalytic enantioselective synthesis of inherently chiral calixarene derivatives. | ||
Almost all inherently chiral calix[4](het)arenes reported to date are composed of differently substituted m-(het)arylene units or varied heteroaromatic rings.6–10,12–14 For instance, enantiomers D and E contain fluorene and fluorenone segments, respectively, in addition to m-phenylene rings.13,14 As an exciting example, the nitrogen-bridged ABCD-type calix[4]arenes B are assembled from benzene, pyridine, pyrimidine and triazine rings with the same methylamino linkages.12 Heteracalix[n]aromatics, or heteroatom-bridged calix(n)(het)arenes, are an important class of synthetic macrocyclic host compounds in (supra)molecular chemistry.15–19 They are easily prepared and functionalized by a number of methods. Fine self-tuning of the macrocyclic conformation and cavity owing to the interplay between heteroatom linkages and their adjacent (hetero)aromatic rings endows heteracalix[n]aromatics with powerful and versatile properties15–20 including selective molecular recognition and self-assembly, fabrication of sophisticated (supra)molecular architectures and functional materials, and formation and stabilization of high-valent arylcopper species.21 In contrast to conventional calixarenes, heteracalixaromatics enjoy a very rich diversity of molecular structures which results from the various combinations of heteroatoms and aromatic components. One of the most unique and advantageous structural features is the generation of inherent chirality by merely varying heteroatoms in the linking positions of three-dimensional calix[4](het)arene skeletons consisting of the same aromatic moieties.11,22 In other words, calix[4](het)arenes lose their molecular symmetries simply due to the variation of connection atoms in between aromatic units. We report herein a distinct type of inherently chiral N3,O-calix[2]arene[2]triazine in which symmetrically aligned aromatic rings are bridged by one oxygen and three nitrogen atoms. We show that these unprecedented chiral macrocycles G were synthesized with excellent enantioselectivity from the chiral phosphoric acid-catalyzed intramolecular nucleophilic aromatic substitution (SNAr) reaction of reactants F (Scheme 1). Further development of the molecular diversity of inherently chiral N3,O-calix[2]arene[2]triazines is demonstrated by post-macrocyclization functionalization reactions on the NH bridge without decrease of enantiopurity. To the best of our knowledge, inherently chiral calix[4](het)arenes obtained from the variation of bridging atoms are extremely rare, and successful synthesis of highly enantiopure isomers has not yet been achieved under asymmetric catalysis.11,22 There is also no precedent that chiral phosphoric acid acts as an organic catalyst to promote the intramolecular SNAr reaction inducing significantly the inherent chirality of macrocyclic molecules.
We commenced our study with de novo synthesis of the racemic form of inherently chiral N3,O-calix[2]arene[2]triazines by means of a fragment coupling strategy15 (Scheme 2). In the presence of diisopropylethylamine (DIPEA) or K2CO3 as an acid scavenger, 3-aminophenol and its derivatives 1a–e underwent two directional nucleophilic aromatic substitution reactions with 1,3-dichlorotriazine derivatives 2a–f at room temperature or refluxing in tetrahydrofuran (THF) to afford compounds 3a–j in 52–80% yields (Scheme S3†). Further treatment of 3a–j with 1,3-phenylenediamine derivatives 4a–c at ambient temperature under basic conditions in acetone or THF led to the formation of intermediates 5a–l in good to excellent yields (Scheme S5† summarizes chemical yields of each individual compound 5a–l). It should be addressed that the SNAr reaction of 3a–j with 4a–c took place preferentially and selectively on the aryloxy-substituted chlorotriazine ring rather than the arylamino-substituted one because the former is more electron-deficient than the latter (Scheme S5†). Macrocyclization of linear precursors 5a–l appeared not trivial. Following a conventional protocol to promote the intramolecular SNAr reaction of 5a in refluxing acetonitrile using Cs2CO3 as a base, desired macrocyclization product 6a was obtained in a yield not exceeding 15%. Employment of other inorganic and organic bases, and different solvents did not improve the formation of 6a at all (entries 1–8, Table S1†). Pleasingly, the intramolecular SNAr reaction of 5a proceeded efficiently under acidic conditions. For example, in the presence of 0.5 to 1 equivalent of HCl, CH3CO2H, p-TsOH and CF3CO2H (TFA), compound 5a underwent cyclization reaction to yield 6a as the sole heteracalixaromatic product in chemical yields increasing from 66% to 68%, 71% and 73% (entries 9–13, Table S1†). The target product 6a was obtained in 76% yield when 2 equivalents of TFA were applied (entry 14, Table S1†). The TFA-mediated intramolecular SNAr reaction was very general, and all intermediates 5 tested were converted into the corresponding macrocycles 6 in the yields ranging from 69% to 92% (Scheme 2).
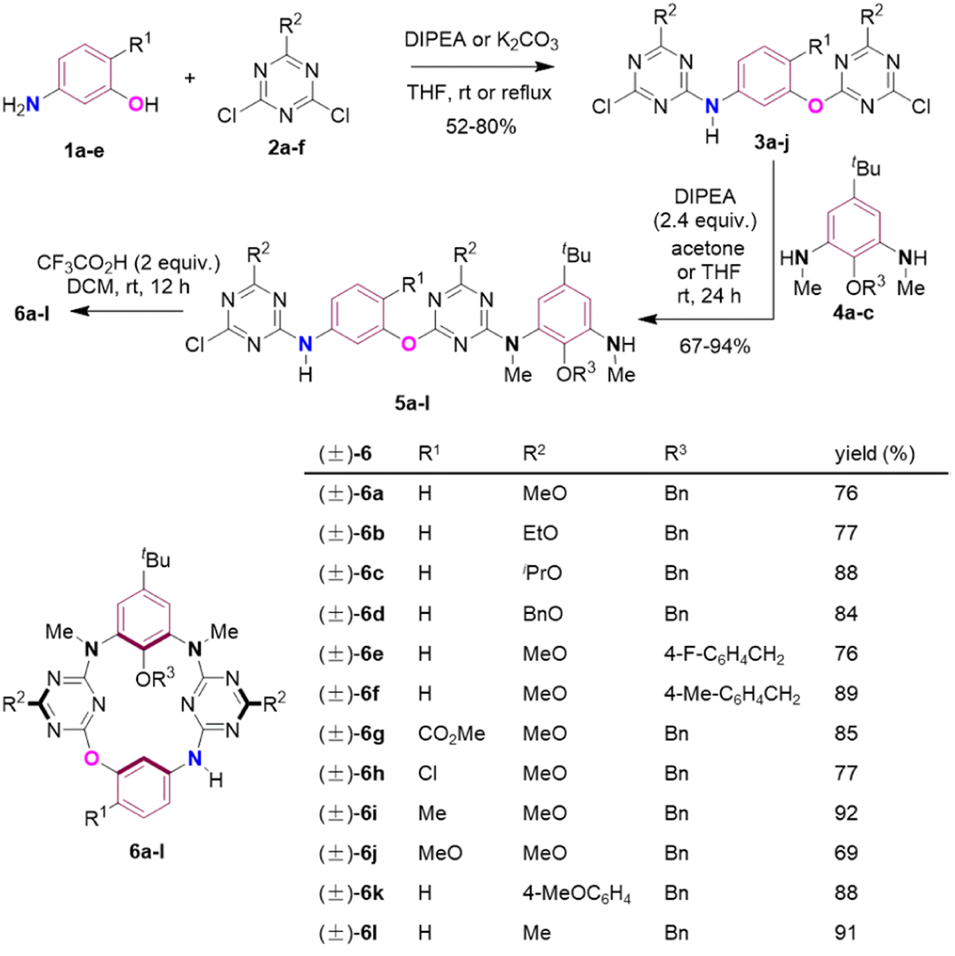 | ||
| Scheme 2 Synthesis of racemic N3,O-calix[2]arene[2]triazines 6. | ||
The structures of products were determined by mass spectrometry, NMR and X-ray crystallography. As revealed unambiguously by single crystal X-ray diffraction, the resulting N3,O-calix[2]arene[2]triazines such as 6a, 6d and 6j adopt the 1,3-alternate conformation in the solid state (Fig. 1). Two face-to-face paralleled benzene rings are at perpendicular positions relative to the plane defined by four bridging heteroatoms while two distal triazine rings are inclined to the plane yielding a V-shaped cavity which is occupied by the benzyl group attached to the phenol ring. Both nitrogen and oxygen atoms in linking positions form conjugations with their adjacent triazine rings. We were also delighted to find that all racemic samples of compounds 6a–l were resolved into enantiomers by means of HPLC using columns coated with chiral stationary phases (ESI†). It suggested that enantiomers of 1,3-alternate conformers of inherently chiral N3,O-calix[2]arene[2]triazines 6 are stable and do not racemize, rendering them attractive targets of asymmetric synthesis.
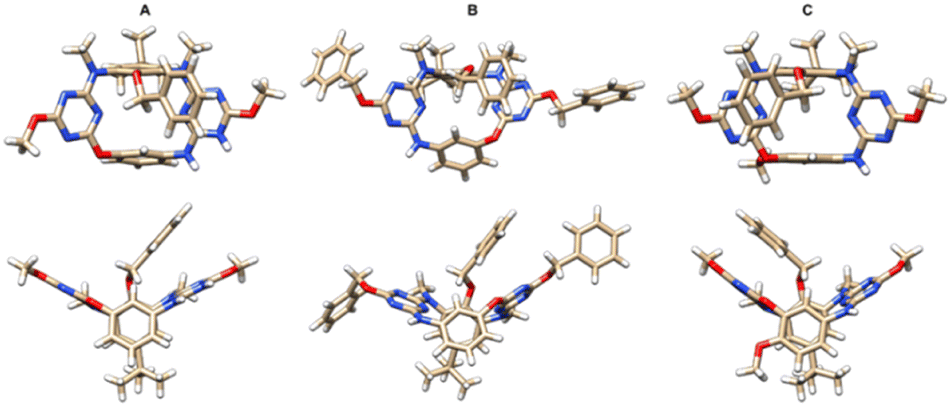 | ||
| Fig. 1 X-ray crystal and molecular structures of (±)-6a (A), (±)-6d (B) and (±)-6j (C) with top (top) and side (bottom) views. | ||
Given the facile formation of macrocycles 6 under acidic conditions, we set to investigate the asymmetric reaction of 5a as a model reaction using chiral phosphoric acids23,24 and N-triflyl phosphoramide24Cat-1 to Cat-18 as organic catalysts (Fig. 2). After first round screening both for activity and enantioselectivity (Table S2†), Cat-16, which is derived from (S)-6,6′-(9-anthryl)-2,2′,3,3′-tetrahydro-1,1′-spirobi[indene]-7,7′-diol, stood as a promising catalyst (entry 16, Table S2†). Further extensive examination of reaction conditions showed that the Cat-16 catalyzed reaction at 0 °C for 24 h in CCl4 gave product Sic-6a in 23% yield with 86% ee (entry 11, Table S3†). Chemical yields increased when the reaction was extended from 36 to 72 h (entries 12 and 13, Table S2†). Decreased enantioselectivity was observed with the increase of reaction temperatures (entries 15–17, Table S3†). Other solvents including xylenes, toluene, benzene, acetone and acetonitrile led to either low conversion or diminished enantioselectivity (entries 1–10, Table S3†). It should also be pointed out that a catalyst loading of 30 mol% is necessary as chemical yield dropped with the decrease of catalyst loading to 20 mol% (entry 18, Table S3†). Change of concentration of the substrate did not affect the enantioselectivity though the conversion was slightly affected (entries 19 and 20, Table S3†). To improve enantioselectivity, additives such as molecular sieves (3–5 Å), Ag2CO3, AgOAc and K2CO3 were tested in order to remove HCl released from the SNAr reaction in question, on the basis that HCl can promote non-enantioselective macrocyclization of 5a (vide supra). In all cases, additives appeared beneficial to increase conversion while attaining the same level of enantioselectivity (entries 21–30, Table S3†). For example, addition of K2CO3 (0.6 equiv.) to the Cat-16 (30 mol%) catalyzed reaction system led to the formation of product Sic-6a (87% ee) in a yield of 38% (entry 29, Table S3†). Employment of equimolar K2CO3 led to a diminished yield (entry 30, Table S3†). Finally, and gratifyingly, excellent enantioselective catalysis was achieved at 0 °C in a mixture of CCl4 and cyclohexane when K2CO3 (0.6 equiv.) was used as an additive, affording highly enantiopure Sic-6a in 36% yield and 93% ee (entry 40, Table S3†). Switch of the solvent system back to 1,2-dichloroethane resulted in a drastic decrease of the ee value of the product to 41% (entry 42, Table S3†).
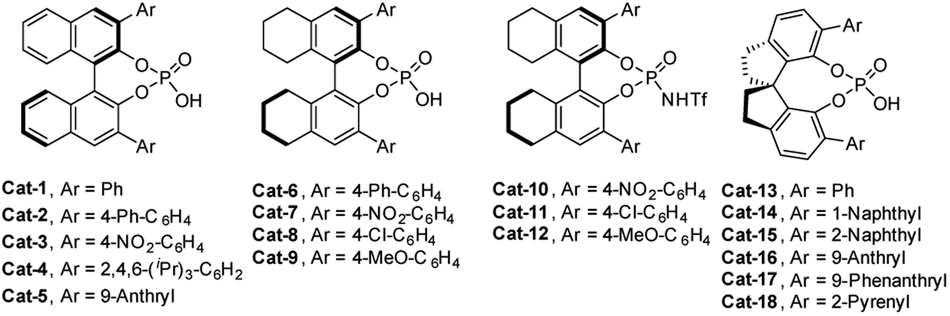 | ||
| Fig. 2 Chiral phosphoric acid catalysts selected to catalyze the macrocyclization of 5a. | ||
Under the optimized conditions, the scope of the chiral phosphoric acid Cat-16-catalyzed synthesis of inherently chiral O3,N-calix[4]aromatics 6a–l from the cyclization of 5a–l was surveyed. As illustrated in Scheme 3, reactants which bear an alkoxy group such as methoxy (5a), ethoxy (5b) and isopropoxy (5c) on the triazine moiety were transformed into the corresponding Sic-(+)-6a–c with an ee of 91.4–94.2%. A slight decrease of the ee value was observed when a larger benzyloxy (5d) is attached on triazine. The enantiopurity of product Sic-6d was nevertheless improved readily to 98.4% ee by recrystallization. Good enantioselectivity was attained when an electron-donating (Sic-6f) or electron-withdrawing (Sic-6e) group is present on the benzene ring of the aryloxy group at the lower rim. Functional groups such as ester (5g) were tolerated. More remarkably, irrespective of the nature of the substituent on the benzene ring embedded in the linear structure, macrocyclization of substrates 5g–j proceeded with good to excellent enantiocontrol, furnishing products Sic-(+)-6g–j with high enantiopurity. It should be noted that the marginal difference of enantioselection was observed when alkoxy was replaced with methyl (5l) on the triazine ring. Noticeably, however, substitution of alkoxy by an aryl group (5k) on the triazine subunit led to a considerable decrease of the enantioselectivity of asymmetric catalysis. The distinct substituent effect between aryl, alkyl and alkoxy on enantioselectivity implied the role of triazine played in catalysis (vide infra). As expected, macrocyclization of 5a and 5b applying the enantiomer of Cat-16 gave rise to Ric-(−)-6a and Ric-(−)-6b (Scheme 3).
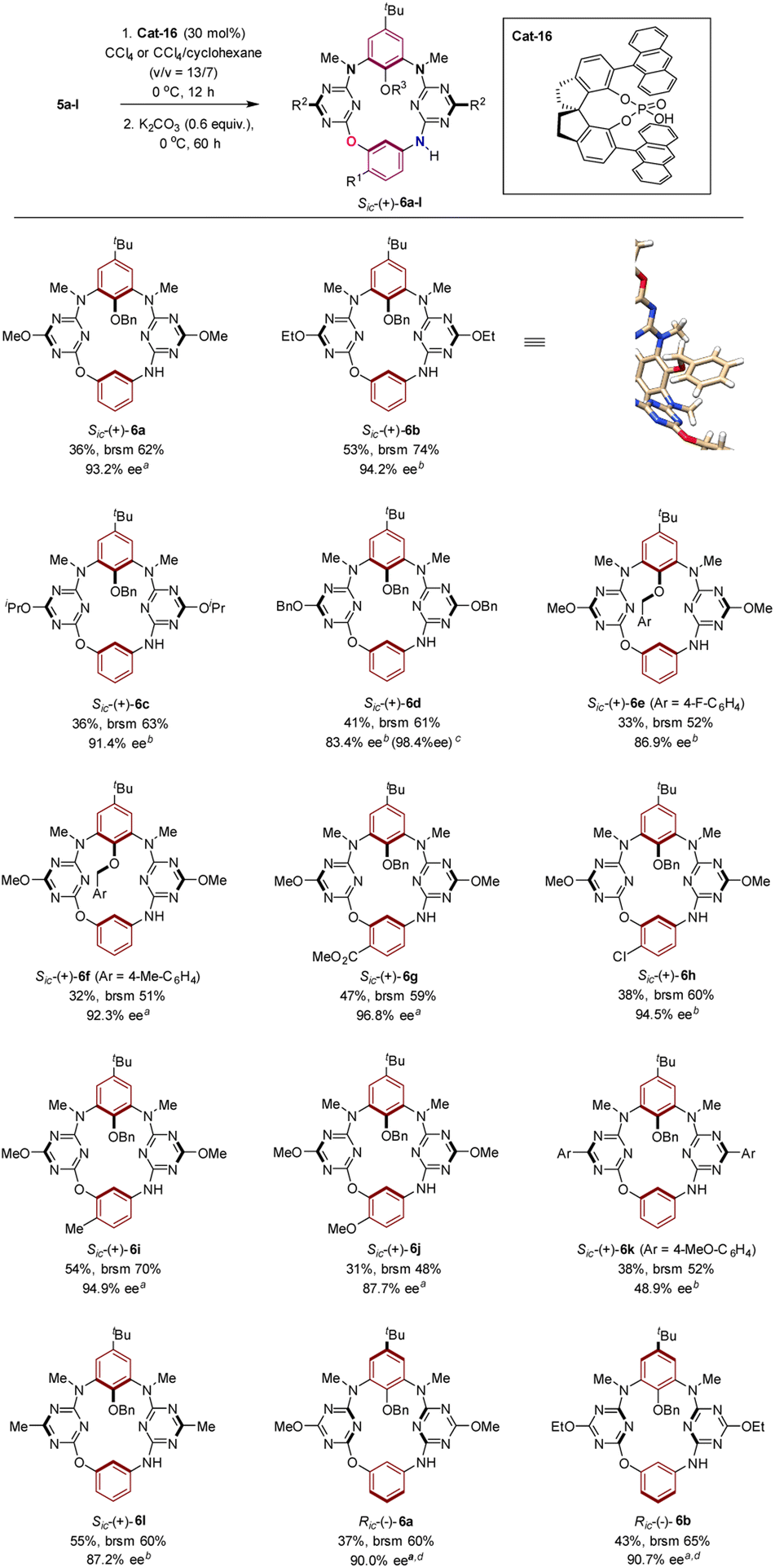 | ||
| Scheme 3 Catalytic enantioselective synthesis of inherently chiral N3,O-calix[2]arene[2]triazines 6a–l and the X-ray molecular structure of Sic-6b. aReaction was performed in a mixture of CCl4 and cyclohexane (v/v = 13/7). bReaction was carried out in pure CCl4. cThe ee value was obtained after recrystallization. dEnantiomer of Cat-16 was used as the chiral catalyst. | ||
The absolute configuration of inherently chiral products was assigned based on the single crystal X-ray molecular structure of Sic-6b,25 which is the predominant enantiomer from the Cat-16 catalyzed reaction of 5b (Scheme 3, Fig. S4 and Table S7†), and the comparison of circular dichroism spectra between chiral products (Fig. S7–S18†). Based on the outcomes of asymmetric induction and substituent effects aforementioned, we proposed a catalytic reaction process in which chiral phosphoric acid Cat-16 forms a pair of hydrogen bonds with the secondary amino-s-triazine moiety of 5 (Fig. 3, left). The catalytic mode was supported by theoretical calculations. According to DFT calculations (Fig. S28†), two optimized pre-organized complexes between the chiral catalyst Cat-16 and substrate 5a were obtained. The complex (Fig. 3, right) leading to Sic-(+)-6a has a lower binding energy by 5 kcal mol−1 in comparison to the complex leading to the enantiomer (Fig. S28†). Revealed by the computed structure (Fig. 3, right), the spiro phosphoric acid forms a pocket in which phosphoric acid binds secondary amino-s-triazine nicely through a pair of hydrogen bonds. One of the anthracene substituents of the catalyst forms evidently non-covalent π–π interactions with the benzene while the other forms C–H–π interaction with t-butyl of the substrate. There is also a weak interaction between phosphoric acid and the terminal amino group (dO–N = 3.885 Å) that brings SNAr reaction sites in close proximity to each other (dC–N = 2.801 Å). It is therefore the favourable multiple non-covalent bond interactions between the chiral catalyst and substrate that lead to enantioselective macrocyclization through the intramolecular SNAr reaction (Fig. 3). To verify the computed mode of asymmetric catalysis, substrate 5m was designed and synthesized (ESI†). Without the N–H binding site, the Cat-16 catalysed reaction of 5m under identical conditions gave the corresponding product Sic-8a merely in 7% yield and 42% ee (Scheme 4). It may be worth noting that activation of amino-substituted heteroaromatic rings using a chiral phosphoric acid to facilitate the SNAr reaction would open a new route to construct axial chiral bi(hetero)aryls.
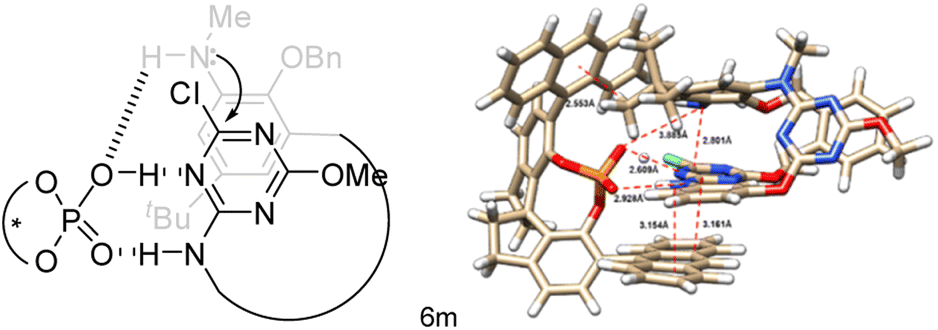 | ||
| Fig. 3 Proposed (left) and computed (right) catalytic mode for enantioselective macrocyclization. | ||
 | ||
| Scheme 4 Control reaction of 5m under identical catalytic conditions. | ||
The secondary amino moiety in the bridging position and the ester on the benzene ring would provide useful handles for derivatization of the acquired products. To demonstrate the synthetic value of the method, and also to develop the diversity of inherently chiral N3,O-calix[2]arene[2]triazines, functionalization reactions on the bridging amino group were conducted (Scheme 5). Thus, assisted by NaH, enantioenriched compounds Sic-6a (90.0–92.0% ee) and Sic-6g (96.8% ee) underwent N-alkylation with methyl iodide (7a), allylic bromide (7b), propargyl bromide (7c) and 4-bromobenzyl bromide (7d) efficiently at 0 °C in DMF. The corresponding N-functionalized products Sic-8a–h (90.4–96.7% ee)25 were isolated almost quantitatively. N-Arylation product Sic-10 (89.9% ee) was also prepared straightforwardly from the reaction between Sic-6a (90.0% ee) and 9,9′-(6-chloro-1,3,5-triazine-2,4-diyl)bis(9H-carbazole) 9 in refluxing acetone when Cs2CO3 was present. The conceivable versatile reactivities of unsaturated carbon–carbon bonds, Caryl–Br bonds introduced and of the ester group installed on the benzene ring would now render compounds Sic-8a–h a unique and invaluable platform for further fabrication of a wide variety of unprecedented chiral chemical entities.
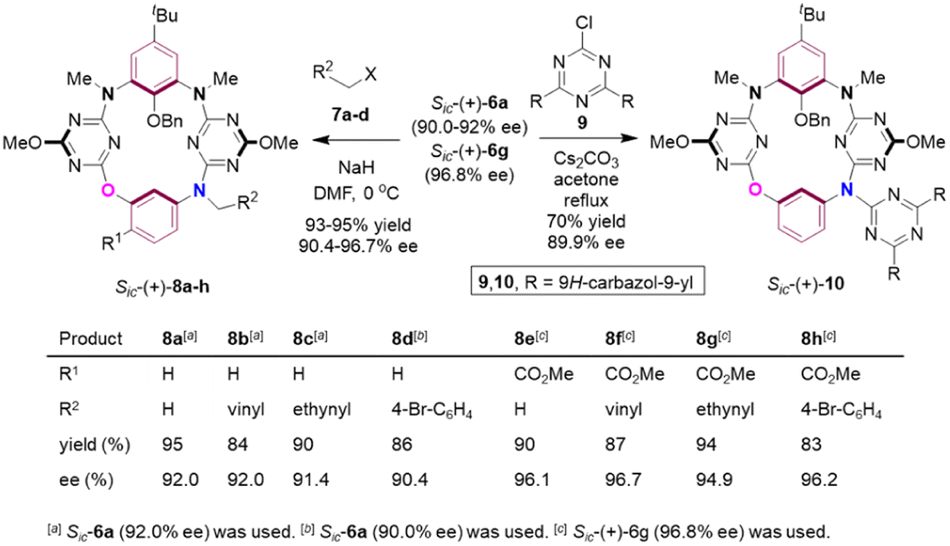 | ||
| Scheme 5 Synthetic applications. | ||
The inherent chirality of heteracalix[4](het)arenes was found very stable and no racemization took place after heating Sic-(+)-6a at 150 °C for 48 h in o-dichlorobenzene (Scheme S8†). The stability of inherent chirality was attributable to the presence of 2-(benzyloxy)-5-(tert-butyl)-1,3-phenylene in which bulky substituents on both upper and lower rims inhibit the inversion of the macrocyclic ring. It is especially worth addressing that all resulting N3,O-calix[2]arene[2]triazines are distinct inherently chiral calix[4](het)arene species. In contrast to well-documented inherently chiral calix[4](het)arenes which are composed of different (hetero)aromatic subrings and the same bridging units, the inherent chirality of all N3,O-calix[2]arene[2]triazines in question stems from non-superimposable mirror imaged macrocyclic scaffolds generated by the introduction of different heteroatom linkages. Compound Sic-(+)-8a25 represents, for instance, an extraordinary example as one oxygen atom difference in the bridging position causes desymmetrization of the molecular structure. The outcomes demonstrated an alternative approach to the design and synthesis of inherently chiral heteracalix[4](het)arenes simply by taking advantage of variable heteroatom linkages.
Conclusions
In conclusion, we have shown the synthesis of highly enantiopure inherently chiral N3,O-calix[2]arene[2]triazines from enantioselective macrocyclization of linear precursors by means of chiral phosphoric acid-catalyzed intramolecular nucleophilic aromatic substitution reaction. We have also demonstrated efficient functionalization of the resulting macrocycles based on the reactions of bridging the secondary amino moiety. The synthetic method developed and the inherent chirality induced by heteroatom-dictated desymmetrization would open a new opportunity to design and construct a wide variety of diverse chiral macrocycles of unique structural features and potential applications. Study on the functionalization and molecular recognition of inherently chiral heteracalixaromatics26 is being actively pursued in this laboratory and the results will be reported in due course.Data availability
All experimental and computational data are available in ESI.†Author contributions
Ying Cheng, Shuo Tong and Mei-Xiang Wang conceived the project and supervised the study. Xing-Chi Li conducted experiments and Xu-Dong Wang performed computational calculations. All authors contributed to discussions and wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Natural Science Foundation of China (21920102001, 22050005, 21821001, and 22371161) and Tsinghua University Dushi Program for financial support.Notes and references
- V. Böhmer, D. Kraft and M. Tabatabai, J. Inclusion Phenom. Mol. Recognit. Chem., 1994, 19, 17 CrossRef.
- A. Dalla Cort, L. Mandolini, C. Pasquini and L. Schiaffino, New J. Chem., 2004, 28, 1198 RSC.
- C. D. Gutsche, Calixarenes: An Introduction, Royal Society of Chemistry, Cambridge, U.K, 2nd edn, 1989 Search PubMed.
- Calixarenes 2001, ed. Z. Asfari, V. Böhmer, J. Harrowfield, J. Vicens and M. Saadioui, Kluwer Academic, The Netherlands, 2001 Search PubMed.
- Calixarenes and Beyond, ed. P. Neri, J. L. Sessler and M.-X. Wang, Springer, Switzerland, 2016 Search PubMed.
- A. Szumna, Chem. Soc. Rev., 2010, 39, 4274 RSC , and references cited therein.
- (a) G. E. Arnott, Chem.–Eur. J., 2018, 24, 1744 CrossRef CAS PubMed , and references cited therein; (b) M. Tang and X. Yang, Eur. J. Org Chem., 2023, 26, e202300738 CrossRef CAS.
- For selected examples, see: (a) V. Böhmer, L. Merkel and U. Kunz, J. Chem. Soc., Chem. Commun., 1987, 896 RSC; (b) L. Zetta, A. Wolff, W. Vogt, K.-L. Platt and V. Böhmer, Tetrahedron, 1991, 47, 1911 CrossRef CAS; (c) S.-Y. Li, Y.-W. Xu, J.-M. Liu and C.-Y. Su, Int. J. Mol. Sci., 2011, 12, 429 CrossRef CAS PubMed; (d) J.-T. Li, L.-X. Wang, D.-X. Wang, L. Zhao and M.-X. Wang, J. Org. Chem., 2014, 79, 2178 CrossRef CAS PubMed; (e) S. J. Nemat, H. Jedrzejewska, A. Prescimone, A. Szumna and K. Tiefenbacher, Org. Lett., 2020, 22, 5506 CrossRef CAS PubMed; (f) D. Sokolova, G. M. Piccina and K. Tiefenbacher, Angew. Chem., Int. Ed., 2022, 61, e202203384 CrossRef CAS PubMed; (g) P. Lhotak, Org. Biomol. Chem., 2022, 20, 7377 RSC; (h) S.-Y. Guo, Z.-A. Zhang, S. Tong, Q.-H. Guo, R. Hua and M.-X. Wang, Chem. Sci., 2023, 14, 8393 RSC; (i) W. Zhu, Y. Cheng, S. Tong and M.-X. Wang, Org. Lett., 2023, 25, 5105 CrossRef CAS PubMed.
- For selected examples, see: (a) P. C. B. Page, H. Heaney and E. P. Sampler, J. Am. Chem. Soc., 1991, 121, 6751 CrossRef; (b) K. Iwamoto, H. Shimizu, K. Araki and S. Shinkai, J. Am. Chem. Soc., 1993, 115, 3997 CrossRef CAS; (c) Y.-D. Cao, J. Luo, Q.-Y. Zheng, C.-F. Chen, M.-X. Wang and Z.-T. Huang, J. Org. Chem., 2004, 69, 206 CrossRef CAS PubMed; (d) Z.-X. Xu, C. Zhang, Z.-T. Huang and C.-F. Chen, Org. Lett., 2008, 10, 477 CrossRef CAS PubMed; (e) W.-Z. Zhang, K. Yang, S.-Z. Li, H. Ma, J. Luo, K.-P. Wang and W.-S. Chung, Eur. J. Org Chem., 2015, 765 CrossRef CAS; (f) M. Tlusty, H. Dvorakova, J. Cejka, M. Kohout and P. Lhotak, New J. Chem., 2020, 44, 6490 RSC; (g) L. Hodson, K. J. Visagie, M.-P. Smith, L. Loots, D. Kuter, T. M. Snayer and G. E. Arnott, Chem. Commun., 2021, 57, 11045 RSC; (h) F.-M. Yang, J. Luo, Y.-C. Chen and W.-S. Chung, Synthesis, 2023, 55, 2786 CrossRef CAS.
- J. K. Browne, M. A. McKervey, M. Pitarch, J. A. Russell and J. S. Millership, Tetrahedron Lett., 1998, 39, 1787 CrossRef CAS.
- K. Ishibashi, H. Tsue, H. Takahashi and R. Tamura, Tetrahedron: Asymmetry, 2009, 20, 375 CrossRef CAS.
- S. Tong, J.-T. Li, D.-D. Liang, Y.-E. Zhang, Q.-Y. Feng, X. Zhang, J. Zhu and M.-X. Wang, J. Am. Chem. Soc., 2020, 142, 14432 CrossRef CAS PubMed.
- X. Zhang, S. Tong, J. Zhu and M.-X. Wang, Chem. Sci., 2023, 14, 827 RSC.
- Y.-Z. Zhang, M.-M. Xu, X.-G. Si, J.-L. Hou and Q. Cai, J. Am. Chem. Soc., 2022, 144, 22858 CrossRef CAS PubMed.
- (a) M.-X. Wang, Chem. Commun., 2008, 4541 RSC; (b) M.-X. Wang, Acc. Chem. Res., 2012, 45, 182 CrossRef CAS PubMed.
- W. Maes and W. Dehaen, Chem. Soc. Rev., 2008, 37, 2393 RSC.
- H. Tuse, K. Ishibashi and R. Tamura, Top. Heterocycl. Chem., 2008, 17, 73 Search PubMed.
- D.-X. Wang and M.-X. Wang, Azacalixaromatics in Calixarenes and Beyond, ed. P. Neri, J. L. Sessler and M.-X. Wang, Springer, 2016, ch. 14, pp. 363–397 Search PubMed.
- R. Hudson and J. L. Katz, Oxacalixarenes in Calixarenes and Beyond, ed. P. Neri, J. L. Sessler and M.-X. Wang, Springer, 2016, ch. 15, pp. 399–420 Search PubMed.
- D.-X. Wang and M.-X. Wang, Acc. Chem. Res., 2020, 53, 1364 CrossRef CAS PubMed.
- Q. Zhang, S. Tong and M.-X. Wang, Acc. Chem. Res., 2022, 55, 2796 CrossRef CAS PubMed.
- M.-X. Wang and H.-B. Yang, J. Am. Chem. Soc., 2004, 126, 15412 CrossRef CAS PubMed.
- (a) D. Uraguchi and M. Terada, J. Am. Chem. Soc., 2004, 126, 5356 CrossRef CAS PubMed; (b) T. Akiyama, J. Itoh, K. Yokota and K. Fuchibe, Angew. Chem., 2004, 116, 1592 ( Angew. Chem., Int. Ed. , 2004 , 43 , 1566 ) CrossRef; (c) M. Terada, Chem. Commun., 2008, 4097 RSC; (d) T. Akiyama, J. Itoh and K. Fuchibe, Adv. Synth. Catal., 2006, 348, 999 CrossRef CAS.
- (a) D. Nakashima and H. Yamamoto, J. Am. Chem. Soc., 2006, 128, 9626 CrossRef CAS PubMed; (b) M. Rueping, W. Ieawsuwan, A. P. Antonchick and B. J. Nachtsheim, Angew. Chem., 2007, 119, 2143 ( Angew. Chem., Int. Ed. , 2007 , 46 , 2097 ) CrossRef; (c) M. Rueping, B. J. Nachtsheim, S. A. Moreth and M. Bolte, Angew. Chem., 2008, 120, 603 ( Angew. Chem., Int. Ed. , 2008 , 47 , 593 ) CrossRef; (d) D. Parmar, E. Sugiono, S. Raja and M. Rueping, Chem. Rev., 2014, 114, 9047 CrossRef CAS PubMed.
- Deposition Numbers 2303849 (racemic 6a), 2303851 (racemic 6d), 2303853 (racemic 6j), 2304018 (racemic 8d), 2304020 (enantiopure Sic-6b) and 234021 (enantiomer Sic-8a) contain the supplementary crystallographic data for this paper.†.
- Our preliminary study showed that Sic-(+)-6a and Ric-(−)-6a are able to form a 1
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complex with S-naproxen in CDCl3 with the association constant being 374 M−1 and 389 M−1, respectively.
:1 complex with S-naproxen in CDCl3 with the association constant being 374 M−1 and 389 M−1, respectively.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2303849, 2303851, 2303853, 2304018, 2304020 and 2304021. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc06436k |
| This journal is © The Royal Society of Chemistry 2024 |