
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA water-soluble Pd4 molecular tweezer for selective encapsulation of isomeric quinones and their recyclable extraction†
Dharmraj
Prajapati
a,
Pallab
Bhandari
 a,
Ennio
Zangrando
b and
Partha Sarathi
Mukherjee
*a
a,
Ennio
Zangrando
b and
Partha Sarathi
Mukherjee
*a
aDepartment of Inorganic and Physical Chemistry, Indian Institute of Science, Bangalore-560012, India. E-mail: psm@iisc.ac.in
bDepartment of Chemical and Pharmaceuticals Sciences, University of Trieste, Trieste 34127, Italy
First published on 27th January 2024
Abstract
Quinones (QN) are one of the main components of diesel exhaust particulates that have significant detrimental effects on human health. Their extraction and purification have been challenging tasks because these atmospheric particulates exist as complex matrices consisting of inorganic and organic compounds. In this report, we introduce a new water soluble Pd4L2 molecular architecture (MT) with an unusual tweezer-shaped structure obtained by self-assembly of a newly designed phenothiazine-based tetra-imidazole donor (L) with the acceptor cis-[(tmeda)Pd(NO3)2] (M) [ tmeda = N,N,N′,N′-tetramethylethane-1,2-diamine]. The molecular tweezer encapsulates some quinones existing in diesel exhaust particulates (DEPs) leading to the formation of host–guest complexes in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio. Moreover, MT binds phenanthrenequinone (PQ) more strongly than its isomer anthraquinone (AQ), an aspect that enables extraction of PQ with a purity of 91% from an equimolar mixture of the two isomers. Therefore, MT represents an excellent example of supramolecular receptor capable of selective aqueous extraction of PQ from PQ/AQ with many cycles of reusability.
:1 molar ratio. Moreover, MT binds phenanthrenequinone (PQ) more strongly than its isomer anthraquinone (AQ), an aspect that enables extraction of PQ with a purity of 91% from an equimolar mixture of the two isomers. Therefore, MT represents an excellent example of supramolecular receptor capable of selective aqueous extraction of PQ from PQ/AQ with many cycles of reusability.
Introduction
Nature has developed numerous intricately designed nanoscale structures with low symmetry for selective recognition of substrates with no symmetry and their catalytic transformations.1 These natural biological systems take advantage of folded peptide chains as scaffolds to specifically position active sites and catalytic centers within-well defined cavities.2 Taking inspiration from these biological systems, synthetic chemists have evolved supramolecular systems in order to resemble the natural archetypes. These include both metal mediated assemblies3–5 and purely organic analogues.6 Like biological systems, these artificial receptors employing noncovalent interactions have promised to find application in encapsulation,7 sustainable synthesis,8 light harvesting,9 separation,10 drug delivery,11 and stabilization of reactive species.12Among these supramolecular systems, metallosupramolecular assemblies have gained special interest because in these systems more functionalities can be achieved by using suitable ligands.13 However, many reported metallosupramolecular architectures are insoluble or poorly soluble in water, which restricts their use in organic solvents. Design of water-soluble large architectures with confined hydrophobic pockets is always a challenging task. In this regard, metal–ligand coordination driven self-assembly has become an important tool to construct water-soluble self-assembled architectures.13 Owing to its gentle reaction conditions and the ability to predesign the resulting assembly, coordination driven self-assembly is considered a superior approach14 to other synthetic approaches involving covalent or weak non-covalent interactions, for the construction of large discrete architectures. In recent times various water-soluble supramolecular complexes have been synthesized using this methodology for the stabilization of unstable conformers15 and application in catalysis16 and separation.17
Quinones are highly redox active and toxicologically important components of diesel exhaust particulates (DEPs) responsible for environmental pollution.18 They are known to initiate many reactions associated with several toxicological events. Due to their strong electrophilicity and redox activity, they can be involved in the redox cycle with their semi quinone radical anions that ultimately lead to the production of reactive oxygen species (ROS), including superoxide, hydrogen peroxide, and hydroxyl radicals.19 Formation of these ROS in biological systems causes oxidative stress, which may entail lipid peroxidation, enzyme inactivation, and DNA damage with consequent gene mutation and cancer.20 Among the detected polluting quinones, 9-10 phenanthrenequinone (PQ) is highly toxic and was found to have a relatively higher concentration in the environment.21
Exposure to quinone pollutants is an increasing public concern and inhaling PQ from polluted air can cause diseases such as lung cancer, asthma, and allergic inflammation.18 Notably, PQ is recognized for its potent inhibitory effects on numerous enzymes, including nitric oxide synthases, protein kinases, and cyclooxygenase-2.21
Despite their highly toxic nature, quinones are useful chemicals in chemical industries and may serve as substrates for a variety of flavoenzymes and as fluorescent probes due to their high electron accepting ability. For example, PQ is used for fabricating organic electroluminescence devices, supercapacitors, and electrochemical energy storage and conversion devices.22 It is also used in dyes, as a hardener for dental restorage and for organic synthesis. Furthermore, the chemical framework of naphthoquinone serves as the core structure for numerous natural compounds, with K vitamin being one of the most prominent examples.23 Therefore, not only for their industrial applications but also for the issues caused by their particulates in the environment, their extraction and purification are both important.24 This is a challenging task because atmospheric particulates are found to be complex matrices made up of both inorganic compounds such as metals, salts, elemental carbon and different organic compounds such as alkanes, alcohols, fatty acids and polycyclic aromatic hydrocarbons (PAHs).25 Traditional methods such as microwave assisted extraction, solid phase extraction, and pressurized liquid extraction have been applied for extraction of organic compounds in diesel exhaust particulates. However, these methods are expensive, tedious, time consuming and usually require large volumes of solvents. Thus, alternative practical methods for their extraction and separation in simpler ways are required.
Herein, we report the design and synthesis of a water soluble Pd4 molecular tweezer (MT) as a potential host for the encapsulation of aromatic quinones. MT has an unusual shape formed by coordination-driven self-assembly of a new phenothiazine-based tetra-imidazole ligand L (L = 1,3,7,9-tetra(1H-imidazole-1-yl)-10-methyl-10H-phenothiazine) with a 90° cis-blocked Pd(II) acceptor (M) (Scheme 1). MT was characterized by various spectroscopic techniques, and the structure was elucidated by single crystal X-ray diffraction study. This newly engineered water-soluble molecular tweezer (MT), characterized by its hydrophobic inner cavity surrounded by the aromatic backbone of L, has demonstrated its compatibility for encapsulating quinones in water.
 | ||
| Scheme 1 Self-assembly of a M4L2 molecular tweezer (MT) and its selective host–guest complexation with phenanthrenequinone (PQ) over anthraquinone (AQ) in water. | ||
Experiments involving host–guest complexation and guest extraction, conducted with individual quinones as well as a mixture of quinones, strongly indicated the creation of a 1:1 inclusion complex with MT. Furthermore, it is capable of extracting phenanthrenequinone (PQ) with a selectivity of 91%, even in the presence of an equimolar amount of isomeric AQ and PQ in an aqueous medium (Scheme 1). Thus, MT represents a distinctive illustration of a discrete, water-soluble coordination architecture with the potential for effortless and selective extraction of environmentally abundant quinone PQ.
Results and discussion
Synthesis and characterization
A new tetra-imidazole building block 1,3,7,9-tetra(1H-imidazole-1-yl)-10-methyl-10H-phenothiazine (L) was synthesized in good yield from tetrabromo-N-methylphenothiazine (2) by a Cu(I) catalyzed C–N coupling reaction (Scheme S1, ESI†). Ligand L was totally characterized by 1H, 13C, 1H–1H COSY, 1H–1H NOESY-NMR and mass spectrometry (ESI-MS) analyses (Fig. S3–S9†). In order to synthesize the molecular tweezer (MT), a yellow solution of the 90° acceptor cis-(tmeda)Pd(NO3)2 (M) [tmeda = N,N,N′N′-tetramethylethylene-1,2-diamine] in DMSO was combined with a DMSO solution containing L in a 2:1 molar ratio. Subsequently, the mixture was heated at 60 °C for 12 h under stirring. After completion, the reaction mixture turned into a clear pale-yellow colored solution. Finally, the resulting mixture was mixed with an excess amount of ethyl acetate to obtain a whitish precipitate which was thoroughly washed using acetone and diethyl ether and dried under reduced pressure overnight to obtain the molecular tweezer as white powder in high yield (96%). Interestingly, self-assembled molecular tweezer MT is highly soluble in the aqueous medium contrary to the precursor L which is insoluble in water.
Fourteen distinct peaks in the aromatic region (δ = 8.74 to 7.44 ppm) and two methyl peaks in the aliphatic region (δ = 2.38 and 2.29 ppm) were observed in the proton NMR spectrum of the MT at room temperature in D2O solvent. Contrary to this, ligand L displayed six peaks in the aromatic region and one for the methyl protons in the aliphatic region (Fig. 1 and S5†). The aromatic region contains one broad peak with an integration of six protons and five sharp peaks, each with an integral ratio of 2 protons (Fig. S5†). The NMR signals corresponding to three protons (e, f, and i) merged with each other and formed a broad signal instead of being three different peaks for these protons. The observation of fourteen peaks in the 1H NMR spectrum of MT could be attributed to different orientations of the imidazole units around the metal centres, such that imidazole rings face different electronic environments depending on their proximity to other imidazole units within MT. In the diffusion ordered NMR (1H-DOSY, in D2O) spectrum of the product, a single horizontal band confirmed the existence of a single coordination architecture (Fig. 1 and S11†). Moreover, as a result of the coordination between the metal and ligand, significant downfield shifts were detected in the 1H NMR spectrum of the molecular tweezer, specifically in the proton peaks associated with imidazole. The assignment of these peaks was successfully accomplished through the utilization of 1H–1H COSY and 1H–1H NOESY NMR experiments (Fig. S12–S14†).
 | ||
| Fig. 1 Stacked partial 1H NMR spectra of the (a) MT in D2O and (b) ligand L in CDCl3 and (c) diffusion-ordered 1H NMR of MT in D2O. | ||
Although the NMR study provided clear evidence of single product formation, the precise composition of ligand L and M within the final product remained unknown. To address this, the water-soluble molecular tweezer MT was transformed into its PF6− analogue by subjecting its aqueous solution to an excess of KPF6 to facilitate easy fragmentation in ESI-MS analysis. The ESI-MS spectrum of the PF6− analogue of MT was recorded by dissolving it in acetonitrile solvent. The appearance of multiple peaks at m/z = 1358.1048, 856.4044, 606.0565 and 455.8466 with isotopic distribution patterns corresponding to [M4L2(PF6)6]2+, [M4L2(PF6)5]3+, [M4L2(PF6)4]4+ and [M4L2(PF6)3]5+ charge fragments, respectively (Fig. 2 and S15–S16†) definitively established the presence of an M4L2 assembly. While the NMR and ESI-MS analyses hinted at the formation of a single M4L2 self-assembled molecule, obtaining a precise solid-state structure of the resulting assembly was required to detail the nature and dimension of the nanocavity. To achieve this, we successfully cultivated suitable single crystals of MT through gradual acetone vapour diffusion into its aqueous solution. Subsequently, we collected single crystal X-ray diffraction data by employing synchrotron radiation. The XRD analysis of MT disclosed the formation of a new [4 + 2] self-assembled architecture having a molecular tweezer like topology (Fig. 3). MT crystallizes in triclinic space group P ![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with two whole molecular cations in the asymmetric unit of pseudo C2v symmetry and comparable geometry.
with two whole molecular cations in the asymmetric unit of pseudo C2v symmetry and comparable geometry.
 | ||
| Fig. 2 ESI-MS spectrum of the PF6− analogue of MT in acetonitrile. (Inset) Experimental isotopic distribution pattern of the [M4L2(PF6)4]4+ fragment. | ||
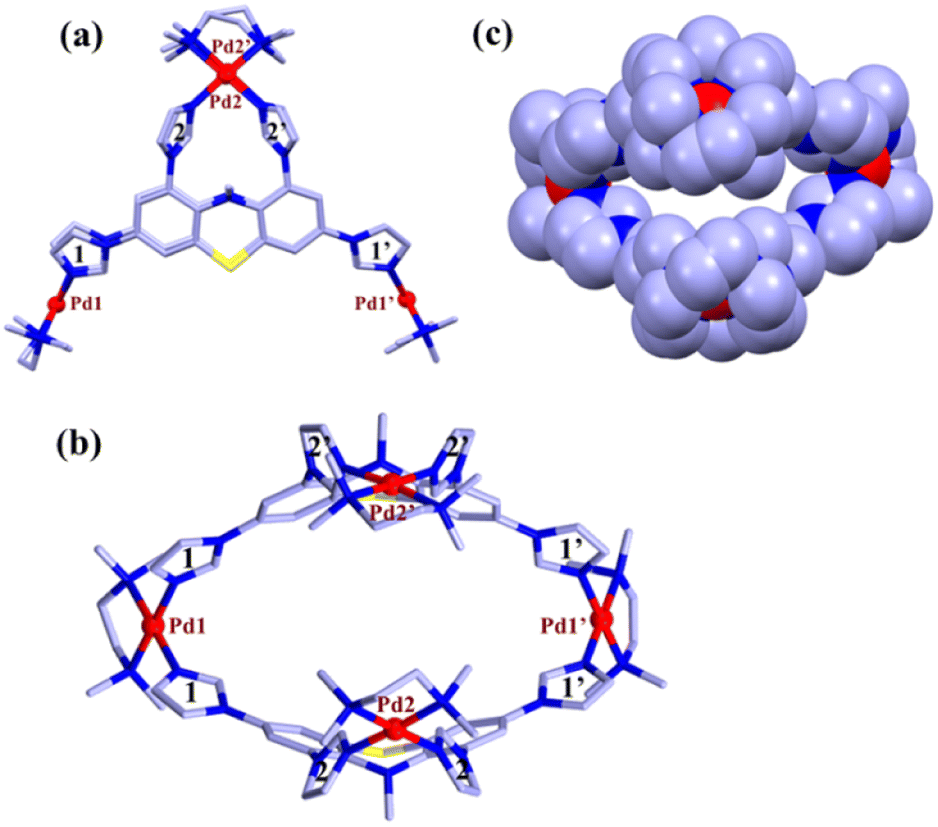 | ||
| Fig. 3 (a) Capped stick representation of one of the two independent molecular tweezers MT (side view). (b) Top view of MT showing hydrophobic cavity. (c) Space filled model [colour codes: C (light blue), N (blue), S (yellow), and Pd (red)]. Hydrogen atoms, counter anions, and solvent molecules are omitted for the sake of clarity. | ||
The crystal structure showed (Fig. 3) that two ligands are encompassing along the two faces of the molecular tweezer in similar coordination mode. Two imidazole units (labelled as 2 and 2′) of each ligand chelate one metal center (Pd2 or Pd2′), whereas the other two imidazole units (1 and 1′) bridge atoms Pd1 and Pd1′ (Fig. 3). This binding mode of ligands leads to the formation of a molecular tweezer like structure with a hydrophobic cavity fenced by the aromatic wall of ligands L. The distance between Pd1 and Pd1′ in the two complexes averages to 13.75 whereas that between Pd2 and Pd2′ is 7.49 Å (Fig. 3 and S1†). In each ligand the imidazole units 2 and 2′ are twisted inward, and their mean planes form a dihedral angle of ca. 66.7°. On the other hand, the imidazole rings connected to each metal center Pd1 and Pd1′ form a dihedral angle of 74.2° (mean value for the two complexes). The geometries of the square planar Pd(II) centers are found to be quite regular with Pd–N bond distances in the range 2.017(3)-2.064(3) Å and cis and trans bond angles within 86.12(12)–94.74(13)° and 172.01(15)–179.55(13)°, respectively, for all the Pd centers. Indeed, the necessary steric strain required to close the structure seems to primarily originate from the ligands. This is favored by the flexible phenothiazine core of L where side phenyl rings form a dihedral angle of about 33°.
Host–guest studies
As previously discussed, both the solution-based and solid-state characterization studies revealed an unusual molecular tweezer-like structure of MT. The water-soluble molecular tweezer has an inner capacious hydrophobic pocket fenced by two ligands L having multiple aromatic rings. The water solubility, inherent hydrophobic cavity, and molecular tweezer-like structural features inspired us to explore MT as a molecular host to encapsulate various types of water insoluble aromatic guests in water. Among organic compounds to be considered as molecular guests, polyaromatic hydrocarbons (PAHs) have been well explored so far.17 However, there are a few reports dealing with highly polar PAHs i.e., oxygenated PAHs (such as quinones) as molecular guests.26 Herein, we explore the binding of quinones within the cavity of the molecular tweezer, and the scope of MT to act as a host was examined by treating a solution of MT in D2O (4.3 mM) with excess amounts of different quinones such as 1,4-naphthoquinone (NQ), 2-methyl-1,4-naphthoquinone (2-MeNQ), 1,2-acenaphthoquinone (AceNQ), 9,10-phenanthrenequinone (PQ), 9,10-anthraquinone (AQ), and 9-fluorenone (9-FN) at room temperature for 12 hours. The resulting suspensions were centrifuged to remove unbound guests and the clear solutions were used for further characterization. 1H NMR and UV-vis absorption studies were carried out for the characterization of the host–guest complexes (Fig. 4b–g and S17–S30†). These characterizations revealed the effective encapsulation of the guest molecules within MT.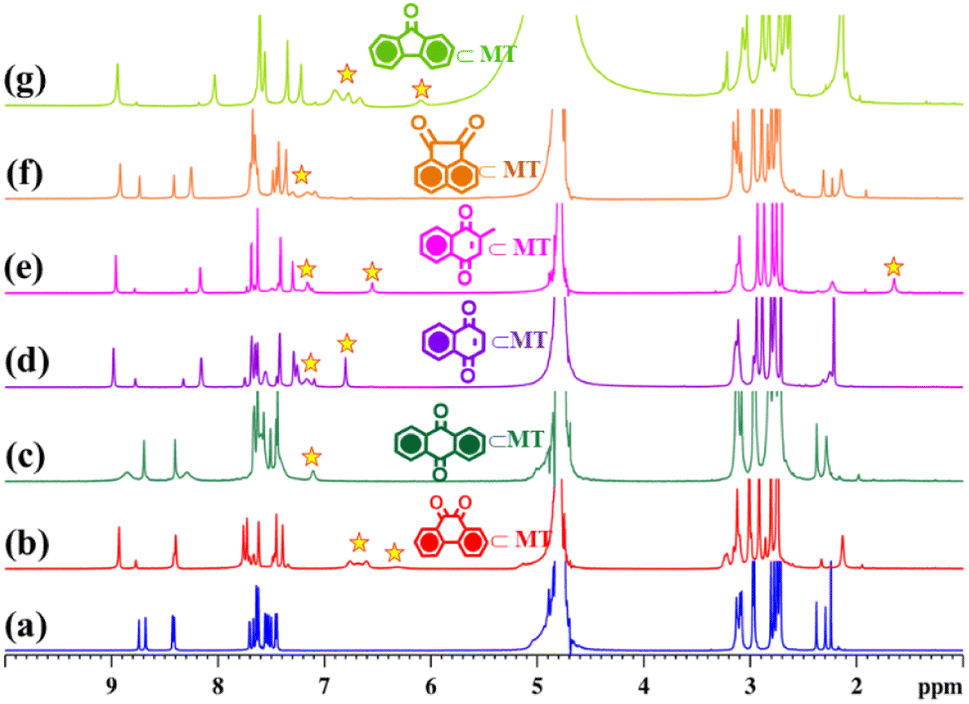 | ||
| Fig. 4 Partial 1H NMR stack plot of (a) MT, (b) PQ⊂MT, (c) AQ⊂MT, (d) NQ⊂MT, (e) 2-MeNQ⊂MT, (f) AceNQ⊂MT, and (g) 9-FN⊂MT in D2O showing the change in NMR spectra upon guest encapsulation by MT. Star marked peaks correspond to the encapsulated guests. | ||
Interestingly, when a colorless solution of MT in D2O was treated with an excess amount of water insoluble solid phenanthrenequinone (PQ), the color of the solution gradually turned orange within 2 h and it turned dark orange after a period of 12 h. The colour change upon host–guest complexation is presumably due to the charge-transfer interaction between the host and quinone guest. The 1H NMR spectrum of the inclusion complex PQ⊂MT showed distinct peaks within the aromatic region, accompanied by the appearance of four new peaks between 6.24–6.76 ppm (Fig. 4b and S17†). Downfield shifts of the Hd1 protons (0.2 ppm) and Hd2 protons (0.1 ppm) were also observed upon PQ encapsulation. This gradual shifting in the proton peaks of MT is attributed to the strong CH⋯π and π⋯π interactions occurring between the guest molecule and the aromatic wall of the MT. Surprisingly, we did not notice any shift in the peaks of the Hg protons as these pertain to the imidazole units which are coordinated to the same Pd center and hence are very close to each other (Fig. 3). Therefore, these imidazole rings could not interact with the guest molecule and no shift was observed. Furthermore, the new set of peaks associated with the encapsulated guest displayed a noticeable up field shift due to the aromatic shielding effect of the aromatic wall of the MT.
To further prove the encapsulation of PQ by MT 2D-NMR studies were carried out. The 1H–1H COSY NMR spectrum (Fig. S18a†) of the inclusion complex PQ⊂MT showed strong correlation between the extra peaks (the peaks of encapsulated PQ), which is due to the host–-guest interaction. In addition, a 2D NOESY NMR spectrum showed cross correlation between the encapsulated PQ peak at 6.76 ppm with Hd and Hf protons of the MT and the peak at 6.61 ppm with He and Hf protons of the MT (Fig. S18b†), which further confirms the encapsulation of PQ by MT in water. Moreover, appearance of a single diffusion coefficient for all the proton peaks (logD = −9.68) of the host and guest in the 1H DOSY NMR spectrum (Fig. S18c†) of PQ⊂MT further confirms the host–guest adduct formation. The host–guest stoichiometry was determined to be 1:1 by assessing the integration of 1H NMR peaks of PQ to the integration of Hg proton of the host (Fig. 4b and S17b†). Like PQ, other quinones upon complexation with colorless solution of MT showed a color change in their respective host–guest complexes. The formation of these host–guest adducts of the quinones with MT was studied by 1H NMR (Fig. 4 and S19–S28†). In all the cases, the host–guest ratio was estimated to be 1:1 by comparing the integration of 1H NMR guest peaks to the integration of the Hg proton of the host.
Complexation of quinones with MT was further explored by UV-visible study. The absorption spectra of the ligand L and MT were recorded in CHCl3 and H2O, respectively (Fig. 5 and S29†).
The absorption spectrum of L showed three absorption peaks at 244, 273 and 327 nm, which can be attributed to π–π* transitions. Molecular tweezer MT showed a strong absorption band at 202 nm, a broad band at 263 nm and a broad hump centered around 324 nm due to π–π* transitions originated from the L. Phenanthrenequinone (PQ) encapsulation resulted in an additional band at 257 nm and a peak at 260 nm (blue shifted by 3 nm), whereas no shift of bands at 202 and 324 nm peaks was observed (Fig. 5). In addition to this a shoulder at 192 nm and a red shifted broad hump at 327 nm with slightly increased intensity were observed. According to literature reports, PQ has strong absorption bands at 257, 263 and 319 nm,27,19b while PQ⊂MT showed a blue shifted peak at 260 nm, an additional absorption band at 257 and a red shifted band at 327 nm. On the other hand, for AQ⊂MT the peak at 263 was shifted to 260 nm (a blue shift), whereas peaks at 202 and 324 nm remain unchanged upon complexation with MT (Fig. 5).
 | ||
| Fig. 5 Normalized absorption spectra of MT, PQ⊂MT and AQ⊂MT at room temperature in water (10−5 M solution). | ||
Host–guest complex 9-FN⊂MT showed two new bands at 248 and 257 nm in addition to the bands due to MT, which originated from π–π* transitions of the guest 9-FN (Fig. S29†). Similar kinds of absorption spectral features were detected by the encapsulation of NQ (naphthoquinone) and 2-MeNQ (2-methyl naphthoquinone) by MT. 2-MeNQ⊂MT exhibited two extra bands at 250 and 262 nm due to π–π* transitions from 2-MeNQ. Likewise, NQ⊂MT showed new bands at 251 and 262 nm due to π–π* transitions of NQ (Fig. S29†).
We have observed that MT is a potential host for different quinones such as phenanthrenequinone, anthraquinone, naphthoquinone, 2-methylnaphthoquinone, acenaphthoquinone and 9-fluorenone in water. The guest uptake affinity of MT towards the interested quinones (PQ and AQ) was quantified using the association constant (Ka). To determine Ka, 1H NMR titration experiments were carried out. For this study a stock solution of MT was prepared in 0.5 mL D2O (5 mM). As the quinone guests tested are not soluble in water, stock solutions of AQ and PQ in DMSO-d6 (0.04 M) were prepared. The 1H NMR titration was carried out by adding aliquots of guest stock solution (4.2 μL) to the solution of the host MT and 1H NMR spectra were recorded immediately after each addition (Fig. S31 and S33†). The change in the chemical shift of the Hd1 proton of guest⊂MT, obtained from the 1H NMR titration experiments, was plotted vs. the equivalents of the guest added. The plots also support the formation of a host–guest complex with a stoichiometry of 1:1 for the guests tested. To calculate apparent association constants, the chemical shifts of the host–guest complexes corresponding to the protons Hd1 and Hd2 with the equivalents of guest added were fitted using the BindFit program (online software “supramolecular.org”) using the 1:1 (host:guest) binding model.28 The association constants were found to be 3.93 × 102 and 4.6 × 103 M−1 for AQ⊂MT and PQ⊂MT, respectively (Fig. S32 and S34†). The relatively high association constants of MT with guest PQ are presumably due to a more compatible fitting of it within the inner hydrophobic cavity of MT.
Unfortunately, several attempts to grow suitable single crystals of the host–guest complexes by conventional slow evaporation and the vapour diffusion technique for X-ray diffraction remained unsuccessful. Therefore, computational calculations were conducted to investigate the difference in binding affinity between the molecular tweezer (MT) and two typical isomeric guests, 9,10-phenanthrenequinone (PQ) and 9,10-anthraquinone (AQ). The computational optimization of both the host–guest inclusion complexes was carried out by the PM6 semiempirical method, with water modelled as solvent and by using the polarization continuum model (PCM) in the ground state. Initially, the free host structure (MT) was optimized, revealing a structural similarity to that obtained from crystallographic study (Fig. S35a†). The interatomic distances between the metal centers, Pd1⋯Pd1′ and Pd2⋯Pd2′, were determined to be 13.88 Å and 7.54 Å, respectively, in close agreement to those obtained from SC-XRD analysis of MT. In the energy-minimized structures of both the inclusion complexes, the guest molecules were found to align parallel to the inner wall cavity of MT (Fig. S35b and d†). This orientation led to the stabilization of host–guest adducts through multiple π⋯π and CH⋯π interactions of the guest molecule with the phenothiazine cores and imidazole units (coordinated with Pd2 and Pd2′) of MT, respectively. Furthermore, single-point energy calculations were conducted using the DFT method for all inclusion complexes (Table S1†). These calculations revealed that PQ⊂MT exhibited the lowest energy configuration, when diketone functional groups of the guest were oriented upwards (towards Pd2 and Pd2′), indicating superior size matching. Consequently, this computational study supports the conclusion that PQ exhibits a stronger binding affinity within the molecular tweezer (MT) in an aqueous medium.
Extraction of phenanthrenequinone from anthraquinone
Selective extraction of one quinone from complex diesel exhaust particulates present in the environment as toxic pollutants is a challenging task. MT has a suitable inner cavity size and stronger uptake affinity for phenanthrenequinone than its isomer anthraquinone. Such a finding was very interesting and led to the idea that MT could be used for the selective extraction of phenanthrenequinone from a mixture of anthraquinone (AQ) and phenanthrenequinone (PQ). To check this, first competitive encapsulations of PQ and AQ by the molecular tweezer were studied. An orange D2O solution of PQ⊂MT was reacted with 3 eq. of solid AQ, and in a reverse manner, a pale yellow D2O solution of AQ⊂MT was treated with PQ with continuous stirring at room temperature for 12 h. Interestingly, we noticed that the colour of the AQ⊂MT solution changed to orange after the phenanthrenequinone addition, whereas the colour of PQ⊂MT remained unchanged upon AQ addition. In both the cases, the resulting orange suspension was centrifuged to remove unbound solid guests and the clear orange solution was examined by 1H NMR analysis (Fig. 6d and e). The spectral pattern of AQ⊂MT solution changed to the characteristic pattern of PQ⊂MT after the addition of PQ (Fig. 6e). However, introduction of AQ to PQ⊂MT doesn't change the spectral pattern of PQ⊂MT (Fig. 6d). Thus, from the above-described competitive experiments, it could be further confirmed that the molecular tweezer has a higher binding affinity for phenanthrenequinone than its isomer anthraquinone. | ||
| Fig. 6 Partial 1H NMR stack plot of (a) MT, (b) PQ⊂MT, (c) AQ⊂MT, (d) PQ⊂MT after treatment with AQ, (e) AQ⊂MT after treatment with PQ, (f) MT after treatment with an equimolar mixture of AQ and PQ in D2O, and (g) CDCl3 extract of the aqueous solution formed by treating an equimolar mixture of AQ and PQ with MT. Blue sphere, red tringle and cyan square marked peaks correspond to AQ, PQ and internal standard 1,3,5-trimethoxybenzene respectively. | ||
The compelling difference in uptake ability of molecular tweezer towards PQ and AQ encouraged us to test its use in the extraction of PQ from an equimolar mixture of AQ and PQ. A colourless D2O solution of MT was treated with an equimolar mixture (3 eq. each) of AQ and PQ under continuous stirring at room temperature for 12 h. The resulting orange suspension was centrifuged to remove unbound solid guests and the clear orange solution was characterized by 1H NMR analysis. 1H NMR spectrum of the orange supernatant was almost identical to that of PQ⊂MT (Fig. 6f and Scheme 2). This result clearly demonstrates that MT preferentially encapsulates PQ over its isomer AQ in water to form predominately an inclusion complex PQ⊂MT. To isolate PQ in high purity, the resulting aqueous solution of the inclusion complex PQ⊂MT was extracted with chloroform (Scheme 2). After removing the CHCl3 solvent, the collected solid was examined by 1H NMR analysis to assess the composition of guest/s using 1,3,5-trimethoxybenzene as an internal standard (Fig. 6g). The 1H-NMR spectrum of the isolated solid gave signals consistent with that of phenanthrenequinone with a purity of 91%, clearly demonstrating that MT successfully extracts phenanthrenequinone from a mixture of its isomer anthraquinone in water with quite high purity. The D2O solution of MT obtained after guest extraction showed an almost identical 1H NMR spectrum (Fig. S37†) to that of the pure synthesized MT. Furthermore, ESI-MS analysis confirmed the presence of the intact host MT (Fig. S38†), which urged us to reuse MT for such separation. To do this, the extracted D2O solution of MT was reused for five cycles for phenanthrenequinone extraction without much noticeable reduction in efficiency (Fig. S36†).
 | ||
| Scheme 2 Schematic presentation of the extraction of PQ (phenanthrenequinone) from a mixture of AQ (anthraquinone) and PQ by MT. | ||
Conclusions
In conclusion, we report here synthesis of a water-soluble molecular tweezer (MT) with unusual structural topology by coordination driven self-assembly of a newly designed phenothiazine-based low symmetrical tetra-imidazole ligand (L) with a cis blocked 90° Pd(II) acceptor (M) in a 1:2 molar ratio. X-ray diffraction study of complex MT revealed the formation of an unusual tweezer-shaped architecture, and the NMR and ESI-MS results were consistent with this structure. The molecular tweezer (MT) could encapsulate some hydrophobic quinones of different sizes present in diesel exhaust particulates. The host–guest complexes were found to have a stoichiometry of 1:1 as revealed from the 1H NMR studies. The unique structural feature of the molecular tweezer with a hydrophobic cavity made it an efficient extracting agent for selective capture of phenanthrenequinone. This stronger binding ability of MT was used for the extraction of phenanthrenequinone with a purity of 91% from an equimolar mixture of isomeric phenanthrenequinone and anthraquinone by simple aqueous extraction. This procedure represents an important finding as extraction, enrichment, and identification of traces of quinones from the complex matrix of atmospheric particulates is a challenging task. Importantly, molecular tweezer was reused for five cycles without any noticeable loss in extraction efficiency. Thus, our present study represents a simple and reliable method for the separation of quinones by a simple aqueous extraction method by using a water-soluble complex species.
Data availability
All data are provided in the ESI† and additional data can be available upon request.Author contributions
P. S. M. and D. P. designed the project and devised the experiments. D. P. carried out the experimental work and analysed the data. E. Z. collected and solved the crystallographic data. P. B. optimized structures and carried out theoretical calculations. P. S. M. supervised the whole project. All the authors contributed to the writing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
P. S. M. thanks the SERB (New Delhi, India) for financial support with a research grant.References
- J. A. Kelly, A. R. Sielecki, B. D. Sykes, M. N. G. James and D. C. Phillips, Nature, 1979, 282, 875–878 CrossRef CAS PubMed.
- (a) K. Wu, E. Benchimol, A. Baksi and G. H. Clever, ChemRxiv, 2023, preprint, DOI:10.26434/chemrxiv-2023-5gb4q; (b) D. Ringe and G. A. Petsko, Science, 2008, 320, 1428–1429 CrossRef CAS PubMed.
- (a) B. Huang, L. Mao, X. Shi and H. B. Yang, Chem. Sci., 2021, 12, 13648–13663 RSC; (b) R. Saha, J. Sahoo, M. Venkateswarulu, M. De and P. S. Mukherjee, Inorg. Chem., 2022, 61, 17289–17298 CrossRef CAS PubMed; (c) W. W. Wang, Y. X. Yang and H. B. Yang, Chem. Soc. Rev., 2016, 45, 2656–2693 RSC; (d) J. D. Xiao and H. L. Jiang, Acc. Chem. Res., 2019, 52, 356–366 CrossRef CAS PubMed; (e) M. Zhao, S. Ou and C. D. Wu, Acc. Chem. Res., 2014, 47, 1199–1207 CrossRef CAS PubMed; (f) X. Fu, X. Lin, X. Ren, H. Cong, C. Liu and J. Huang, Chin. Chem. Lett., 2021, 32, 565–568 CrossRef CAS; (g) Z. Wang, H.-F. Su, Y.-Z. Tan, S. Schein, S.-C. Lin, W. Liu, S.-A. Wang, W.-G. Wang, C.-H. Tung, D. Sun and L.-S. Zheng, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 12132–12137 CrossRef CAS PubMed; (h) B.-L. Han, Z. Wang, R. K. Gupta, L. Feng, S. Wang, M. Kurmoo, Z.-Y. Gao, S. Schein, C.-H. Tung and D. Sun, ACS Nano, 2021, 15, 8733–8741 CrossRef CAS PubMed.
- (a) I. A. Bhat, E. Zangrando and P. S. Mukherjee, Inorg. Chem., 2019, 58, 11172–11179 CrossRef CAS PubMed; (b) T. R. Cook, Y.-R. Zheng and P. J. Stang, Chem. Rev., 2013, 113, 734–777 CrossRef CAS PubMed; (c) M. Fujita, D. Oguro, M. Miyazawa, H. Oka, K. Yamaguchi and K. Ogura, Nature, 1995, 378, 469–471 CrossRef CAS; (d) C. M. Hong, R. G. Bergman, K. N. Raymond and F. D. Toste, Acc. Chem. Res., 2018, 51, 2447–2455 CrossRef CAS PubMed; (e) P. Howlader, S. Ahmed, S. Mondal, E. Zangrando and P. S. Mukherjee, Inorg. Chem., 2022, 61, 8121–8125 CrossRef CAS PubMed; (f) R. Saha, B. Mondal and P. S. Mukherjee, Chem. Rev., 2022, 122, 12244–12307 CrossRef CAS PubMed.
- (a) A. A. Adeyemo and P. S. Mukherjee, Beilstein J. Org. Chem., 2018, 14, 2242–2249 CrossRef CAS PubMed; (b) R. Banerjee, D. Chakraborty and P. S. Mukherjee, J. Am. Chem. Soc., 2023, 145, 7692–7711 CrossRef CAS PubMed; (c) S. Bhattacharyya, A. Chowdhury, R. Saha and P. S. Mukherjee, Inorg. Chem., 2019, 58, 3968–3981 CrossRef CAS PubMed; (d) L. J. Jongkind, X. Caumes, A. P. T. Hartendorp and J. N. H. Reek, Acc. Chem. Res., 2018, 51, 2115–2128 CrossRef CAS PubMed; (e) D. M. Kaphan, M. D. Levin, R. G. Bergman, K. N. Raymond and F. D. Toste, Science, 2015, 350, 1235–1238 CrossRef CAS PubMed; (f) J. Liu, L. Chen, H. Cui, J. Zhang, L. Zhang and C.-Y. Su, Chem. Soc. Rev., 2014, 43, 6011–6061 RSC; (g) M. Morimoto, S. M. Bierschenk, K. T. Xia, R. G. Bergman, K. N. Raymond and F. D. Toste, Nat. Catal., 2020, 3, 969–984 CrossRef CAS; (h) P. C. Purba, S. Bhattacharyya, M. Maity, S. Mukhopadhyay, P. Howlader and P. S. Mukherjee, Chem. Commun., 2019, 55, 8309–8312 RSC; (i) I. Sinha and P. S. Mukherjee, Inorg. Chem., 2018, 57, 4205–4221 CrossRef CAS PubMed; (j) D. Zhang, T. K. Ronson and J. R. Nitschke, Acc. Chem. Res., 2018, 51, 2423–2436 CrossRef CAS PubMed.
- (a) M. Liu, L. Zhang, M. A. Little, V. Kapil, M. Ceriotti, S. Yang, L. Ding, D. L. Holden, R. Balderas-Xicohténcatl, D. He, R. Clowes, S. Y. Chong, G. Schütz, L. Chen, M. Hirscher and A. I. Cooper, Science, 2019, 366, 613–620 CrossRef CAS PubMed; (b) P.-F. Wei, M.-Z. Qi, Z.-P. Wang, S.-Y. Ding, W. Yu, Q. Liu, L.-K. Wang, H.-Z. Wang, W.-K. An and W. Wang, J. Am. Chem. Soc., 2018, 140, 4623–4631 CrossRef CAS PubMed; (c) H. Xu, J. Gao and D. Jiang, Nat. Chem., 2015, 7, 905–912 CrossRef CAS PubMed; (d) J. Zhang, X. Han, X. Wu, Y. Liu and Y. Cui, J. Am. Chem. Soc., 2017, 139, 8277–8285 CrossRef CAS PubMed.
- (a) V. Abdul Rinshad, J. Sahoo, M. Venkateswarulu, N. Hickey, M. De and P. Sarathi Mukherjee, Angew. Chem., Int. Ed., 2023, 62, e202218226 CrossRef CAS PubMed; (b) R. Banerjee, D. Chakraborty, W.-T. Jhang, Y.-T. Chan and P. S. Mukherjee, Angew. Chem., Int. Ed., 2023, e202305338 CAS; (c) K. G. Dutton, D. A. Rothschild, D. B. Pastore, T. J. Emge and M. C. Lipke, Inorg. Chem., 2020, 59, 12616–12624 CrossRef CAS PubMed; (d) B. Roy, A. K. Ghosh, S. Srivastava, P. D'Silva and P. S. Mukherjee, J. Am. Chem. Soc., 2015, 137, 11916–11919 CrossRef CAS PubMed; (e) H. Takezawa, T. Murase and M. Fujita, J. Am. Chem. Soc., 2012, 134, 17420–17423 CrossRef CAS PubMed.
- (a) A. K. Bar, R. Chakrabarty and P. S. Mukherjee, Inorg. Chem., 2009, 48, 10880–10882 CrossRef CAS PubMed; (b) C. J. Brown, F. D. Toste, R. G. Bergman and K. N. Raymond, Chem. Rev., 2015, 115, 3012–3035 CrossRef CAS PubMed; (c) J. Jiao, C. Tan, Z. Li, Y. Liu, X. Han and Y. Cui, J. Am. Chem. Soc., 2018, 140, 2251–2259 CrossRef CAS PubMed; (d) V. Martí-Centelles, A. L. Lawrence and P. J. Lusby, J. Am. Chem. Soc., 2018, 140, 2862–2868 CrossRef PubMed; (e) H. Takezawa, K. Shitozawa and M. Fujita, Nat. Chem., 2020, 12, 574–578 CrossRef CAS PubMed; (f) C. Zhao, Q.-F. Sun, W. M. Hart-Cooper, A. G. DiPasquale, F. D. Toste, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2013, 135, 18802–18805 CrossRef CAS PubMed.
- (a) K. Acharyya, S. Bhattacharyya, S. Lu, Y. Sun, P. S. Mukherjee and P. J. Stang, Angew. Chem., Int. Ed., 2022, 61, e202200715 CrossRef CAS PubMed; (b) K. Acharyya, S. Bhattacharyya, H. Sepehrpour, S. Chakraborty, S. Lu, B. Shi, X. Li, P. S. Mukherjee and P. J. Stang, J. Am. Chem. Soc., 2019, 141, 14565–14569 CrossRef CAS PubMed; (c) S. Chen, K. Li, F. Zhao, L. Zhang, M. Pan, Y.-Z. Fan, J. Guo, J. Shi and C.-Y. Su, Nat. Commun., 2016, 7, 13169 CrossRef CAS PubMed; (d) A. Kumar, R. Saha and P. S. Mukherjee, Chem. Sci., 2021, 12, 5319–5329 RSC; (e) Z. Zhang, Z. Zhao, Y. Hou, H. Wang, X. Li, G. He and M. Zhang, Angew. Chem., Int. Ed., 2019, 58, 8862–8866 CrossRef CAS PubMed.
- (a) W. Brenner, T. K. Ronson and J. R. Nitschke, J. Am. Chem. Soc., 2017, 139, 75–78 CrossRef CAS PubMed; (b) X. Chang, S. Lin, G. Wang, C. Shang, Z. Wang, K. Liu, Y. Fang and P. J. Stang, J. Am. Chem. Soc., 2020, 142, 15950–15960 CrossRef CAS PubMed; (c) Y. Liu, H. Wang, P. Liu, H. Zhu, B. Shi, X. Hong and F. Huang, Angew. Chem., Int. Ed., 2021, 60, 5766–5770 CrossRef CAS PubMed; (d) P. C. Purba, M. Maity, S. Bhattacharyya and P. S. Mukherjee, Angew. Chem., Int. Ed., 2021, 60, 14109–14116 CrossRef CAS PubMed; (e) D. Zhang, T. K. Ronson, R. Lavendomme and J. R. Nitschke, J. Am. Chem. Soc., 2019, 141, 18949–18953 CrossRef CAS PubMed; (f) D. Zhang, T. K. Ronson, Y.-Q. Zou and J. R. Nitschke, Nat. Rev. Chem., 2021, 5, 168–182 CrossRef CAS PubMed; (g) C. Zhu, H. Tang, K. Yang, Y. Fang, K.-Y. Wang, Z. Xiao, X. Wu, Y. Li, J. A. Powell and H.-C. Zhou, J. Am. Chem. Soc., 2021, 143, 12560–12566 CrossRef CAS PubMed.
- (a) J. E. M. Lewis, E. L. Gavey, S. A. Cameron and J. D. Crowley, Chem. Sci., 2012, 3, 778–784 RSC; (b) F. Schmitt, J. Freudenreich, N. P. E. Barry, L. Juillerat-Jeanneret, G. Süss-Fink and B. Therrien, J. Am. Chem. Soc., 2012, 134, 754–757 CrossRef CAS PubMed.
- (a) C. M. Hong, D. M. Kaphan, R. G. Bergman, K. N. Raymond and F. D. Toste, J. Am. Chem. Soc., 2017, 139, 8013–8021 CrossRef CAS PubMed; (b) Y. Inokuma, T. Arai and M. Fujita, Nat. Chem., 2010, 2, 780–783 CrossRef CAS PubMed; (c) R. Saha and P. S. Mukherjee, Dalton Trans., 2020, 49, 1716–1720 RSC; (d) J. H. Tang, Y. Li, Q. Wu, Z. Wang, S. Hou, K. Tang, Y. Sun, H. Wang, H. Wang, C. Lu, X. Wang, X. Li, D. Wang, J. Yao, C. J. Lambert, N. Tao, Y. W. Zhong and P. J. Stang, Nat. Commun., 2019, 10, 4599 CrossRef PubMed.
- (a) S. Bhattacharyya, M. Maity, A. Chowdhury, M. L. Saha, S. K. Panja, P. J. Stang and P. S. Mukherjee, Inorg. Chem., 2020, 59, 2083–2091 CrossRef CAS PubMed; (b) A. Brzechwa-Chodzyńska, W. Drożdż, J. Harrowfield and A. R. Stefankiewicz, Coord. Chem. Rev., 2021, 434, 213820 CrossRef; (c) L. J. Chen, H. B. Yang and M. Shionoya, Chem. Soc. Rev., 2017, 46, 2555–2576 RSC; (d) S. Goeb and M. Sallé, Acc. Chem. Res., 2021, 54, 1043–1055 CrossRef CAS PubMed; (e) R. J. Li, J. Tessarolo, H. Lee and G. H. Clever, J. Am. Chem. Soc., 2021, 143, 3865–3873 CrossRef CAS PubMed; (f) S. J. Wezenberg, Chem. Lett., 2020, 49, 609–615 CrossRef CAS; (g) K. Wu, K. Li, S. Chen, Y.-J. Hou, Y.-L. Lu, J.-S. Wang, M.-J. Wei, M. Pan and C.-Y. Su, Angew. Chem., Int. Ed., 2020, 59, 2639–2643 CrossRef CAS PubMed; (h) L. Xu, Y.-X. Wang, L.-J. Chen and H.-B. Yang, Chem. Soc. Rev., 2015, 44, 2148–2167 RSC.
- (a) J. L. Bolliger, A. M. Belenguer and J. R. Nitschke, Angew. Chem., Int. Ed., 2013, 52, 7958–7962 CrossRef CAS PubMed; (b) M. Fujita, M. Tominaga, A. Hori and B. Therrien, Acc. Chem. Res., 2005, 38, 369–378 CrossRef CAS PubMed; (c) A. B. Grommet, J. B. Hoffman, E. G. Percástegui, J. Mosquera, D. J. Howe, J. L. Bolliger and J. R. Nitschke, J. Am. Chem. Soc., 2018, 140, 14770–14776 CrossRef CAS PubMed; (d) R. Hayes, S. A. Bernard, S. Imberti, G. G. Warr and R. Atkin, J. Phys. Chem. C, 2014, 118, 21215–21225 CrossRef CAS; (e) P. Mal, D. Schultz, K. Beyeh, K. Rissanen and J. R. Nitschke, Angew. Chem., Int. Ed., 2008, 47, 8297–8301 CrossRef CAS PubMed; (f) W. Meng, J. K. Clegg, J. D. Thoburn and J. R. Nitschke, J. Am. Chem. Soc., 2011, 133, 13652–13660 CrossRef CAS PubMed; (g) E. G. Percástegui, J. Mosquera, T. K. Ronson, A. J. Plajer, M. Kieffer and J. R. Nitschke, Chem. Sci., 2019, 10, 2006–2018 RSC; (h) M. Yamashina, Y. Sei, M. Akita and M. Yoshizawa, Nat. Commun., 2014, 5, 4662 CrossRef CAS PubMed; (i) K. Yazaki, M. Akita, S. Prusty, D. K. Chand, T. Kikuchi, H. Sato and M. Yoshizawa, Nat. Commun., 2017, 8, 15914 CrossRef CAS PubMed.
- (a) P. Bhandari, R. Modak, S. Bhattacharyya, E. Zangrando and P. S. Mukherjee, JACS Au, 2021, 1, 2242–2248 CrossRef CAS PubMed; (b) P. Howlader, B. Mondal, P. C. Purba, E. Zangrando and P. S. Mukherjee, J. Am. Chem. Soc., 2018, 140, 7952–7960 CrossRef CAS PubMed; (c) R. Saha, A. Devaraj, S. Bhattacharyya, S. Das, E. Zangrando and P. S. Mukherjee, J. Am. Chem. Soc., 2019, 141, 8638–8645 CrossRef CAS PubMed.
- (a) P. Das, A. Kumar, P. Howlader and P. S. Mukherjee, Chem.–Eur. J., 2017, 23, 12565–12574 CrossRef CAS PubMed; (b) P. Howlader, P. Das, E. Zangrando and P. S. Mukherjee, J. Am. Chem. Soc., 2016, 138, 1668–1676 CrossRef CAS PubMed.
- (a) Y. Liu, H. Wang, L. Shangguan, P. Liu, B. Shi, X. Hong and F. Huang, J. Am. Chem. Soc., 2021, 143, 3081–3085 CrossRef CAS PubMed; (b) D. Chakraborty, R. Saha, J. K. Clegg and P. S. Mukherjee, Chem. Sci., 2022, 13, 11764–11771 RSC; (c) P. Howlader, S. Mondal, S. Ahmed and P. S. Mukherjee, J. Am. Chem. Soc., 2020, 142, 20968–20972 CrossRef CAS PubMed; (d) P. Howlader, E. Zangrando and P. S. Mukherjee, J. Am. Chem. Soc., 2020, 142, 9070–9078 CrossRef CAS PubMed; (e) D. Prajapati, P. Bhandari, N. Hickey and P. S. Mukherjee, Inorg. Chem., 2023, 62, 9230–9239 CrossRef CAS PubMed; (f) A. B. Sainaba, M. Venkateswarulu, P. Bhandari, K. S. A. Arachchige, J. K. Clegg and P. S. Mukherjee, J. Am. Chem. Soc., 2022, 144, 7504–7513 CrossRef CAS PubMed.
- N. Kishikawa, M. Wada, Y. Ohba, K. Nakashima and N. Kuroda, J. Chromatogr. A, 2004, 1057, 83–88 CrossRef CAS PubMed.
- (a) Y. Kumagai and N. Shimojo, Environ. Health Prev. Med., 2002, 7, 141–150 CrossRef CAS PubMed; (b) J. Zhang, S. Wang, J. Lalevée, F. Morlet-Savary, E. S. H. Lam, B. Graff, J. Liu, F. Xing and P. Xiao, J. Polym. Sci., 2020, 58, 792–802 CrossRef CAS.
- (a) T. P. Devasagayam, J. P. Kamat, H. Mohan and P. C. Kesavan, Biochim. Biophys. Acta, 1996, 1282, 63–70 CrossRef PubMed; (b) T. Hancock-Chen and J. C. Scaiano, J. Photochem. Photobiol., B, 2000, 57, 193–196 CrossRef CAS PubMed.
- (a) Y. Kumagai, T. Hayashi, T. Miyauchi, A. Endo, A. Iguchi, M. Kiriya-Sakai, S. Sakai, K. Yuki, M. Kikushima and N. Shimojo, Am. J. Physiol. Regul. Integr. Comp. Physiol., 2001, 281, R25–30 CrossRef CAS PubMed; (b) Y. Kumagai, H. Nakajima, K. Midorikawa, S. Homma-Takeda and N. Shimojo, Chem. Res. Toxicol., 1998, 11, 608–613 Search PubMed; (c) N. Rudra-Ganguly, S. T. Reddy, P. Korge and H. R. Herschman, J. Biol. Chem., 2002, 277, 39259–39265 CrossRef CAS PubMed; (d) K. Taguchi, Y. Kumagai, A. Endo, M. Kikushima, Y. Ishii and N. Shimojo, J. Health Sci., 2001, 47, 571–574 Search PubMed; (e) B. H. Wang, B. Ternai and G. M. Polya, Biol. Chem. Hoppe-Seyler, 1994, 375, 527–535 CrossRef CAS PubMed.
- (a) S. V. Venkatesan, A. Nandy, K. Karan, S. R. Larter and V. Thangadurai, Electrochem. Energy Rev., 2022, 5, 16 CrossRef CAS; (b) X. Wang, T. Ji, J. Zhao and Z. Ding, J. Mater. Sci.: Mater. Electron., 2023, 34, 1006 CrossRef CAS.
- (a) J. Gaultier and C. Hauw, Acta Crystallogr., 1965, 18, 179–183 CrossRef CAS; (b) J. E. Rhoads and M. T. Fliegelman, J. Am. Med.Assoc., 1940, 114, 400–401 CAS.
- C. Walgraeve, S. Chantara, K. Sopajaree, P. D. Wispelaere, K. Demeestere and H. R. V. Langenhove, Atmos. Environ., 2015, 107, 262–272 CrossRef CAS.
- Z. Jiao, Y. Bai, Y. Zeng, S. Lv, H. Fan and W. Huang, Anal. Methods, 2017, 9, 345–351 RSC.
- (a) P. Pananusorn, A. Ruengsuk, A. Docker, K. Khamphaijun, K. Sirivibulkovit, M. Sukwattanasinitt, J. Tantirungrotechai, P. Saetear, T. Limpanuparb and T. Bunchuay, ACS Appl. Mater. Interfaces, 2022, 14, 6810–6817 CrossRef CAS PubMed; (b) K. Yazaki, N. Kishi, M. Akita and M. Yoshizawa, Chem. Commun., 2013, 49, 1630–1632 RSC.
- A. Kuboyama, F. Kobayashi and S. Morokuma, Bull. Chem. Soc. Jpn., 1975, 48, 2145–2148 CrossRef CAS.
- (a) D. Brynn Hibbert and P. Thordarson, Chem. Commun., 2016, 52, 12792–12805 RSC; (b) P. Thordarson, Chem. Soc. Rev., 2011, 40, 1305–1323 RSC . http://supramolecular.org.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra, ESI-MS, binding constant calculations, optimized structures, and experimental details (PDF). CCDC 2259512. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc05093a |
| This journal is © The Royal Society of Chemistry 2024 |