
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn artificial metalloenzyme that can oxidize water photocatalytically: design, synthesis, and characterization†
Ehider A.
Polanco‡
a,
Laura V.
Opdam‡
a,
Leonardo
Passerini
b,
Martina
Huber
 b,
Sylvestre
Bonnet
*a and
Anjali
Pandit
*a
b,
Sylvestre
Bonnet
*a and
Anjali
Pandit
*a
aLeiden Institute of Chemistry, Leiden University, Einsteinweg 55, 2333 CC Leiden, The Netherlands. E-mail: a.pandit@lic.leidenuniv.nl; bonnet@chem.leidenuniv.nl
bDepartment of Physics, Huygens-Kamerlingh Onnes Laboratory, Leiden University, Niels Bohrweg 2, 2333 CA, Leiden, The Netherlands
First published on 30th January 2024
Abstract
In nature, light-driven water oxidation (WO) catalysis is performed by photosystem II via the delicate interplay of different cofactors positioned in its protein scaffold. Artificial systems for homogeneous photocatalytic WO are based on small molecules that often have limited solubility in aqueous solutions. In this work, we alleviated this issue and present a cobalt-based WO-catalyst containing artificial metalloenzyme (ArM) that is active in light-driven, homogeneous WO catalysis in neutral-pH aqueous solutions. A haem-containing electron transfer protein, cytochrome B5 (CB5), served to host a first-row transition-metal-based WO catalyst, CoSalen (CoIISalen, where H2Salen = N,N′-bis(salicylidene)ethylenediamine), thus producing an ArM capable of driving photocatalytic WO. The CoSalen ArM formed a water-soluble pre-catalyst in the presence of [Ru(bpy)3](ClO4)2 as photosensitizer and Na2S2O8 as the sacrificial electron acceptor, with photocatalytic activity similar to that of free CoSalen. During photocatalysis, the CoSalen–protein interactions were destabilized, and the protein partially unfolded. Rather than forming tens of nanometer sized CoOx nanoparticles as free CoSalen does under photocatalytic WO conditions, the CB5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CoSalen ArM showed limited protein cross-linking and remained soluble. We conclude that a weak, dynamic interaction between a soluble cobalt species and apoCB5 was formed, which generated a catalytically active adduct during photocatalysis. A detailed analysis was performed on protein stability and decomposition processes during the harsh oxidizing reaction conditions of WO, which will serve for the future design of WO ArMs with improved activity and stability.
:CoSalen ArM showed limited protein cross-linking and remained soluble. We conclude that a weak, dynamic interaction between a soluble cobalt species and apoCB5 was formed, which generated a catalytically active adduct during photocatalysis. A detailed analysis was performed on protein stability and decomposition processes during the harsh oxidizing reaction conditions of WO, which will serve for the future design of WO ArMs with improved activity and stability.
1. Introduction
In nature, the challenging reaction of light-driven water oxidation (WO) catalysis is performed by photosystem II (PSII). In this system, light capture and charge separation processes are coupled with WO catalysis via different cofactors that are carefully positioned in a protein matrix.1,2 As water splitting is one of the most attractive ways to achieve sustainable H2 production as replacement for fossil fuels, the design of artificial photocatalysts for water oxidation has generated tremendous interest during the last two decades. Researchers have managed to develop photocatalytic water oxidation systems based on small molecules or macromolecules,3–6 but many of these compounds are based on precious metals.7–11 Recently, a significant variety of earth-abundant metal complexes have been proposed as more sustainable and accessible water oxidation catalysts (WOCs).4,12–16 Still, in homogeneous systems a major drawback of most of these catalysts, whether based on abundant or scarce metal, is their limited solubility in aqueous solutions.12,17In this work, we present, to our knowledge, one of the first studies of cobalt-based WO-catalyst (WOC) containing artificial metalloenzymes (ArMs) capable of homogeneous WO photocatalysis. During evolution countless enzymes have developed, approximately half of which are metalloproteins.18 The function of metalloenzymes is defined by the properties of their metal cofactors, which are carefully tuned by the protein environment to drive a specific chemical reaction. This tuning leads to high specificity and makes complex chemical reactions possible under extraordinarily mild conditions.19,20 Those potential advantages have led to the development of very active research on ArMs,21–24 particularly in the field of renewable energy.25–30 ArMs are being developed as potential greener alternatives to different reactions such as O2 reduction, cyclopropanation, or the Diels–Alder reaction, which can be obtained in much milder conditions compared to classical chemical synthesis, and in aqueous solvents.31–42 Of the two half-reactions of water splitting, i.e., water oxidation and proton reduction,3,43–46 the oxidation of water remains the most challenging, both kinetically and thermodynamically. Various photoactive ArMs based on modified hydrogenases, flavodoxin, or myoglobin, have been designed that can drive H2 evolution under light irradiation.25,47,48 However, examples of ArMs that can catalyse O2 evolution are scarce49–51 and only a few cases have been reported of ArMs capable of inducing chemical or photoelectrochemical WO catalysis.48,50,51 Extreme redox chemistry is required in PSII to photocatalyse WO.52 In fact, nature did not find a solution to prevent protein damage during this reaction and in plants, for example, the protein unit at the active site of PSII is replaced about every hour.53 Rational design of WOC ArMs calls for a systematic study of photostability and of possible side reactions occurring during photocatalytic WO with ArMs, as the harsh oxidative reaction conditions necessary to drive this reaction are expected to be detrimental to the protein scaffold.
Recently, we reported on the use of semi-native gel electrophoresis to study the interaction between haem proteins and small-molecule WOCs.54 Our study led to the selection of a protein–WOC couple comprising cytochrome B5 (CB5), an electron-transfer protein, and the earth-abundant WOC [CoIISalen], where H2Salen = N,N′-bis(salicylidene)ethylenediamine, to form an artificial protein (Fig. 1). Now, we report on the ability of CB5:CoSalen ArMs to drive homogeneous WO in photocatalytic conditions using [Ru(bpy)3](ClO4)2 as photosensitizer and Na2S2O8 as sacrificial electron acceptor. Moreover, we show that the ArMs prevent the formation of Co nanoparticles during photocatalysis and form a water-soluble protein–catalyst system. Finally, we present a detailed analysis of the stability and decomposition pathways of this ArM as a key step towards understanding side reactions that may occur when ArMs are exposed to severe oxidizing reaction conditions.
 | ||
| Fig. 1 General scheme for the production of photoactive CB5:CoSalen artificial proteins. HoloCB5 (left, PDB 1CYO), and apoCB5 binding CoSalen (right, model created using protein visualization software YASARA).55 | ||
2. Results
2.1 Preparation and binding stoichiometries
To prepare the CB5:CoSalen ArM, the haem cofactor of CB5 was removed using Teale's method (see ESI, Section B†). This method consists of dissociating haem by lowering the pH of the protein solution to 2 and removing it with 2-butanone. This step was followed by a buffer exchange to 20 mM NaPi (sodium phosphate) at pH 8.0, first via dialysis and then by a desalting column. Then, two different ArMs samples were prepared by reacting the resulting apo protein (apoCB5) with either 1 or 5 equivalents (eq.) of CoSalen at 4 °C. The effective stoichiometry of the isolated samples, i.e., the CB5:CoSalen molar ratio of the ArM, was determined experimentally using inductively coupled plasma mass spectrometry (ICP-MS) for measuring the metal concentration, and a BCA kit (bicinchoninic acid kit) for quantifying the protein concentration. For the sample prepared with 1 eq. of CoSalen, the final apoCB5:CoSalen ratio varied from 1:0.6 to 1:0.8, while for those prepared with 5 eq. CoSalen, the final apoCB5:CoSalen ratio was 1:4. Throughout the manuscript these ArMs will be referred to as ArM1 and ArM2, respectively. As a control, samples were prepared by reacting the holoCB5 protein (i.e., the protein still bearing its haem cofactor) with 5 eq. CoSalen; these samples will be referred to as ArM3. They had a CB5:CoSalen ratio of 1:2.7, indicating that up to 3 CoSalen could bind. To investigate whether the different ArM samples showed a dynamic equilibrium of bound and unbound CoSalen or static attachment of the metal complexes, ArM2 samples were passed three times over a microspin Bio-Rad P6 column, and the apoCB5:CoSalen ratio was measured after each passage. The ratio did not vary significantly between measurements (Table S1†), which was a clear indication that all CoSalen molecules were stably attached to the protein scaffold. Taken together, the results suggest that CoSalen may bind both to the haem binding site and to alternative, yet undefined, CB5 binding sites.
2.2 Spectroscopic characterization
The purified CB5:CoSalen samples were characterized by UV-vis spectroscopy (Fig. 2A–C). In each CB5:CoSalen sample spectral characteristics of both CB5 (π–π* bands) and CoSalen (d–d and MLCT bands) were observed. Upon interacting with apo- or holoCB5, small shifts compared to the free CoSalen complex from 368 nm to 377 nm in the d–d band were observed, as well as a red-shift of the MLCT band from 235 nm to 257 nm. Both shifts indicated a change in the coordination environment of the metal complex suggesting that CoSalen had indeed been coordinated to CB5 in the case of ArM1 and ArM2. In ArM3 the shift in the MLCT band was also observed, indicating that coordination of CoSalen has taken place, the d–d band was hidden under the haem Soret-peak. As seen in Fig. 2C, the binding of CoSalen to holoCB5 in ArM3 did not lead to loss of the coordinated haem, as the haem Soret band at 413 nm and Q bands at 532 nm and 560 nm were unaffected. The binding of CoSalen to both apo- and holoCB5 was further investigated by electron spray ionization mass spectrometry (ESI-MS, see Fig. S3†). As previously observed,54 a peak characteristic of the apoCB5 protein (mass = 10093 Da, calculated (calc) mass = 10093 Da), as well as a peak characteristic of apoCB5 binding 1 CoSalen (mass = 10417 Da, calc = 10418 Da), was observed for ArM1, while for ArM2, more peaks appeared, indicating that up to 3 CoSalen complexes were bound to apoCB5 (mass = 11066 Da, calc mass = 11068 Da). This number is lower than the binding ratio determined via ICP-MS, probably due to the denaturing conditions employed during ESI-MS that can result in the release of CoSalen from the protein. The ArM3 control samples prepared from holoCB5 and 5 eq. of CoSalen showed peaks corresponding to apoCB5 (mass = 10093 Da), apoCB5:CoSalen 1:1 (mass = 10417 Da), and apoCB5:CoSalen 1:2 (mass = 10741 Da, calc mass = 10743 Da). No binding of haem was observed, suggesting that the histidine–haem interactions did not survive the conditions of the ESI-MS analysis and that the apoCB5–CoSalen interaction is stronger. Release of haem under ESI-MS conditions was also observed in holoCB5 (data not shown).
 | ||
| Fig. 2 UV-vis (A–C) and CD (D–F) spectra from left to right, ArM1 (A and D), ArM2 (B and E) and ArM3 (C and F). In each spectrum, ArM1 (orange), ArM2 (red), ArM3 (pink), holoCB5 (blue), apoCB5 (black), and CoSalen (grey). In the UV-vis spectra, the spectra of apoCB5 and holoCB5 were normalized to match the protein concentration determined for the CB5:CoSalen sample in the same panel, and the spectra of the CoSalen cofactor alone were normalized to the cobalt concentration in the CB5:CoSalen sample. | ||
Circular dichroism (CD) was used to determine the influence of CoSalen binding on protein folding (Fig. 2D–F). To visualise the protein structure, homology models of both proteins apo- and holoCB5 were generated using the Swiss Model software (Fig. 3).56–60HoloCB5 contains a beta-sheet core and 5 alpha helices, 4 of which frame the haem binding pocket. Haem is coordinated by two histidines, H44 and H68, coordinated in an axial position to iron. According to the homology models, upon haem removal, the alpha-helical character around the binding site was lost,61 while the beta-sheet core remained mostly intact.62
 | ||
| Fig. 3 Homology model of holoCB5 (A) and apoCB5 (B) showing each histidine explicitly (the homology models were prepared as detailed in ESI Section A†). | ||
The CD spectrum of apoCB5 contained a mixture of beta-sheet, unfolded strand, and alpha-helix character (Fig. 2) as expected from the computed structure shown in Fig. 3.63 In ArM1 a slight loss of alpha-helical character occurred with respect to apoCB5, while the alpha-helical contribution was further reduced for ArM2, which binds more CoSalen per protein than ArM1 (Fig. 2D). The CD spectrum of ArM3 was found to be very similar to that of ArM1 (Fig. 2F) indicating that a significant loss of alpha-helical fold also occurs upon binding of CoSalen to holoCB5. The changes in the protein secondary structures compared to apoCB5, observed for all ArMs, indicate that CoSalen binding has a destabilizing effect on the protein structure.
As the protein scaffold contains a single, fluorescent tryptophan residue (W27) located near one of the coordinating histidines (H20, see Fig. S4†), steady-state fluorescence spectroscopy was also used to analyse the interaction between CoSalen and apoCB5 in the ArM samples. The fluorescence emission of tryptophan varies from 330 nm to 350 nm, depending on the hydrophobicity of its local environment.64 Inside redox proteins for example, tryptophan can be involved in electron transfer reactions, and its fluorescence fingerprint has been used to study excitation energy transfer and redox reactions in biological systems.65–67 In addition, some transition metals have been reported to quench fluorescence emission in proteins.68–74 The effect of CoSalen binding on the tryptophan fluorescence of CB5 is shown in Fig. 4. The maximum emission of W27 from apoCB5 was found at 340 nm. The fluorescence in holoCB5 was quenched with respect to apoCB5, which is a known consequence of the paramagnetic iron centre of the haem porphyrin acting as an emission quencher.75,76 Similarly, the emission of ArM2 was less intense than that of ArM1, which was less intense than that of apoCB5 at 340 nm. We consider this observation to be a consequence of W27 fluorescence quenching by the bound cobalt complex, which was confirmed by a titration experiment, following the peak at 415 nm, which originates from the presence of CoSalen (Fig. S5†). We further noted a blueshift in the fluorescence maxima of both ArM1 and ArM2, compared to that of apoCB5 with maxima at 336 and 327 nm respectively. The fluorescence characteristics spectra of ArM1 and ArM2 resembled the characteristics of holoCB5 with maxima at 337 nm more closely than those for apoCB5, in terms of low emission because of quenching and in terms of the blue-shift of the maximum (Fig. 4). The observed blueshift reports an increase in the hydrophobicity of the tryptophan environment, especially for ArM2. The observations indicate that the fluorescence quenching of W27 is due to the presence of CoSalen and suggest that binding of the complex provides W27 with a more hydrophobic environment, which might be provided by coordination of CoSalen to H20 located close to W27 (see Fig. S4†).
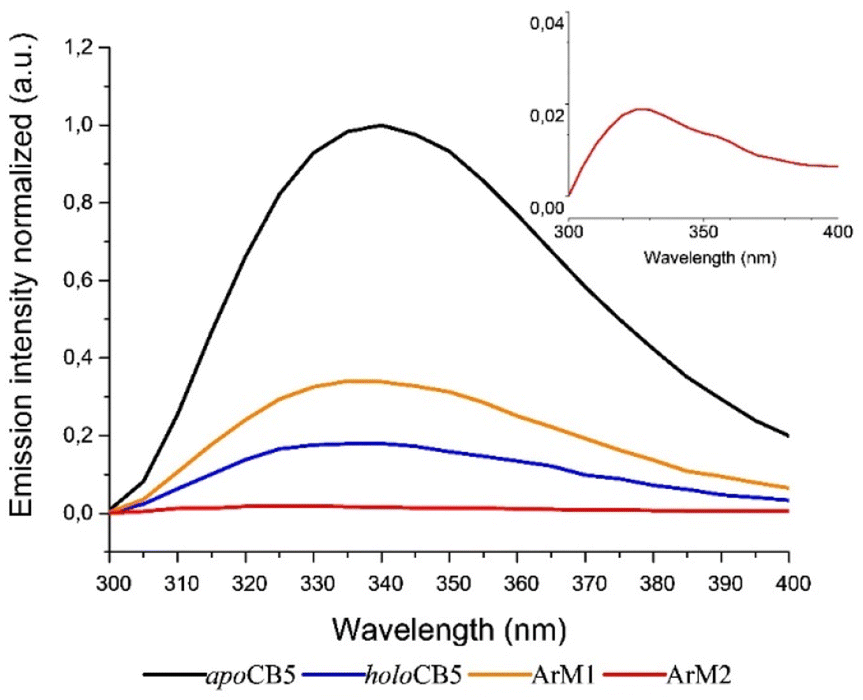 | ||
| Fig. 4 Plot of steady-state fluorescence emission of the W27 residue in apoCB5 (black), holoCB5 (blue), ArM1 (orange) and ArM2 (red) when excited at 280 nm. Inlet: zoom of ArM2 adduct emission from 300 to 400 nm. | ||
Size exclusion chromatography multi-angle light scattering (SEC-MALS) was used to analyse potential protein–protein interactions in the cobalt ArMs (Fig. 5). Apo- and holoCB5 were both found to be primarily monomeric. ArM1 was essentially monomeric, and minor fractions of dimers and octamers were observed. For ArM2 the monomeric peak was still the major species, and in addition, a significant population of multimers was found. Overall, reaction of 1 eq. of the CoSalen complex with apoCB5 (producing ArM1) appears to alter to a minor extent the aggregation state of the protein, while the addition of 5 equivalents of CoSalen (producing ArM2) led to significant multimerization, which may result from the coordination of two exterior histidine residues of two different proteins to the same CoSalen complex.
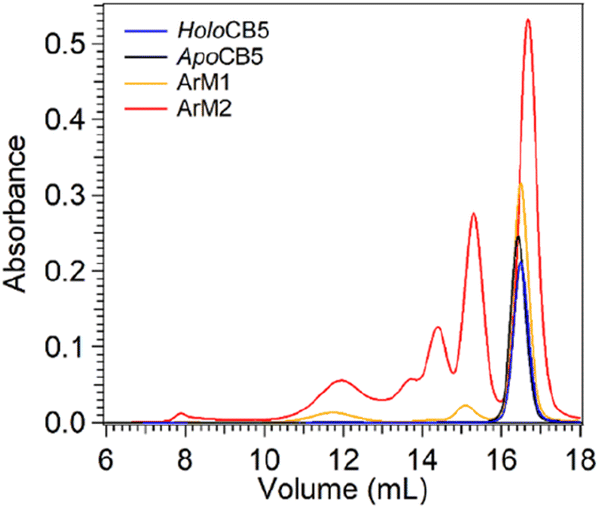 | ||
| Fig. 5 SEC-MALS analysis of holoCB5 (blue), apoCB5 (black), ArM2 (red) and ArM1 (orange). The assignment of the aggregation state of each peak, based on MALS data (not shown), is indicated in the figure. | ||
2.3 Binding location and oxidation state of CoSalen bound to CB5
Haem metalloproteins typically bind metal-based cofactors via histidine residues.77 To better characterize the binding site of CoSalen to CB5, histidine side chain-focused 15N–1H heteronuclear single quantum coherence spectroscopy (HSQC) NMR with a large spectral window (LSW HSQC) was performed on apoCB5, ArM1 and ArM2. As shown in the homology models in Fig. 3, CB5 contains 5 histidine residues: two in the haem binding pocket, H44 and H68, and three on the protein exterior, H20, H31 and H85. Site-directed mutagenesis of the outer histidine residues of apoCB5 was first used for their assignment (Tables S2 and S3†). In this manner, the “outer residues” H31 and H85, as well as binding pocket residues H44 and H68 could be assigned in the LSW HSQC spectrum. H20 was not observed in the LSW HSQC spectrum. The pKa value of each histidine residue in apoCB5 was determined using a 5-point pH titration from pH 6 to 8 (Fig. S6†). The chemical shifts obtained were fitted using eqn (1)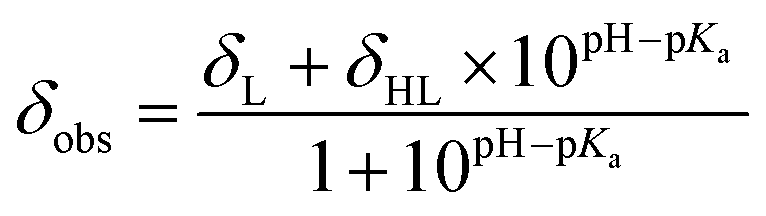 | (1) |
 | ||
| Fig. 6 LSW 15N–1H HSQC NMR of apoCB5 (black), ArM1 ((A) orange) and ArM2 ((B) red). Peak assignments are shown in the figure and in Tables S2 and S3,† with the labelling of the histidine ring as in C. | ||
The NMR spectra of the CB5:CoSalen ArMs suggest a diamagnetic Co(III) oxidation state, since for a paramagnetic state NMR line broadening or strong downfield shifts upon binding of His to cobalt are expected, which was not observed (Fig. 6 and S7†). Continuous wave EPR was performed to determine the oxidation state of CoSalen in ArM1 and ArM2. EPR of CoSalen in solution was also measured, both in aqueous buffer and in chloroform (Fig. 7A and B). In an aqueous environment, CoSalen was found to be high spin Co(II)78,79 (S = 3/2), while in chloroform the complex was found to be in the low spin Co(II) state (S = 1/2).80 This difference may be related to the presence of some Cl− in chloroform, which may coordinate CoSalen axially, introducing a stronger ligand field. The spectra acquired for ArM2 (Fig. 7C and D), with cobalt concentrations comparable to those used for the free CoSalen, did not give any EPR signal attributable to cobalt species (Fig. 7C and D, red spectrum), despite the use of signal enhancing conditions (e.g. high power, high modulation amplitude, low temperature). We, therefore, speculate that CoSalen is in the EPR silent Co(III) state when bound to CB5. To test this theory ArM2 was subjected to reduction by ascorbic acid before re-measuring an EPR spectrum (Fig. 7D, black spectrum). In this case, the spectrum resembled the one obtained for the CoSalen dissolved in the NaPi buffer of a high spin Co(II) state (Fig. 7A). After a chromatography step that would remove any unbound CoSalen, ICP-MS showed cobalt concentrations compatible with CB5 being fully complexed by CoSalen, proving that the Co-EPR signal observed after reduction derives from CoSalen bound to CB5. In conclusion, the EPR results are fully consistent with the picture that CoSalen is bound to CB5 in the diamagnetic Co(III) oxidation state, in agreement with NMR data.
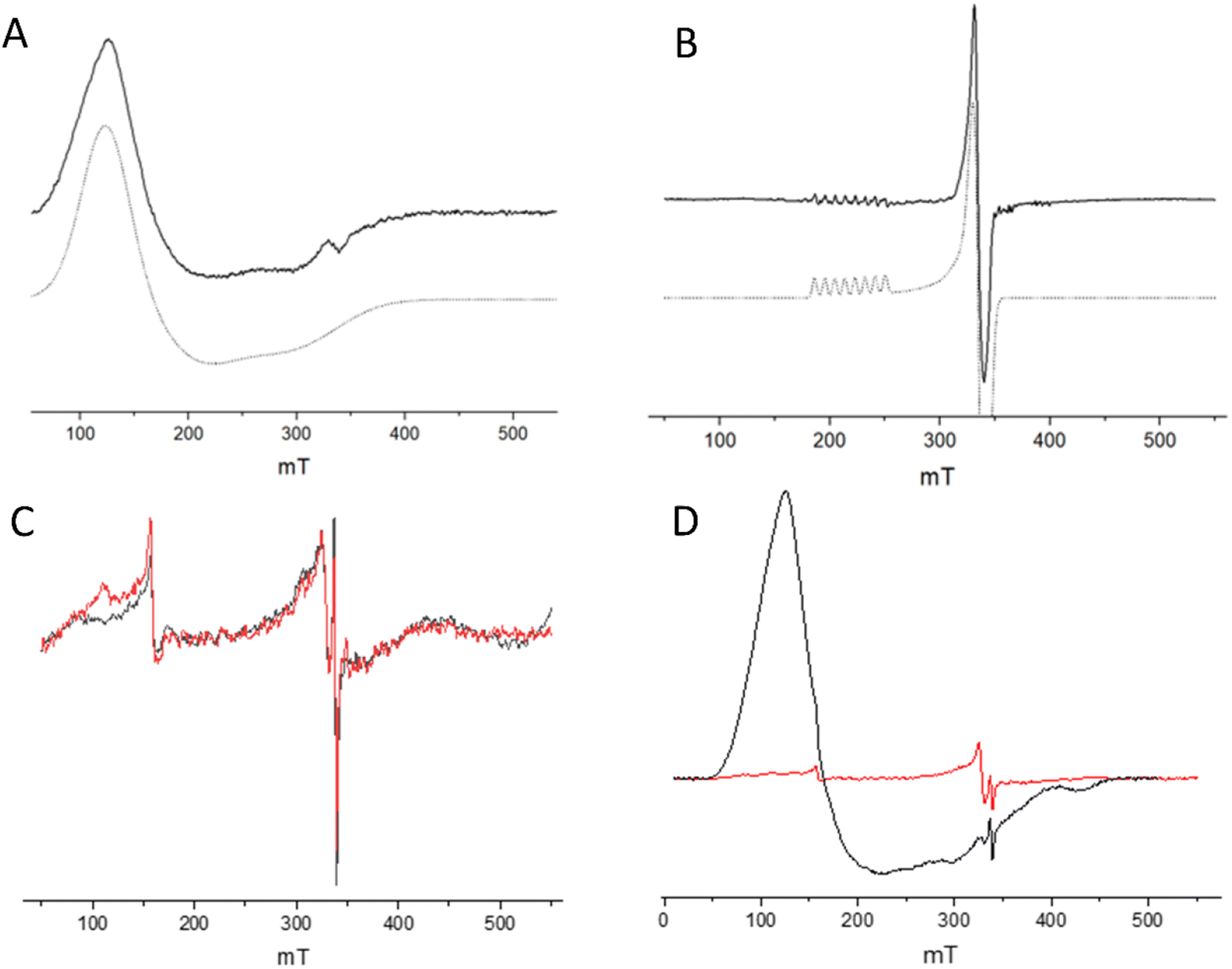 | ||
| Fig. 7 Low-temperature EPR spectra of CoSalen complex in solution and complexed with apoCB5. (A) CoSalen in aqueous buffer (solid line, black) and simulation as Co(II) S = 3/2 (dashed line, grey). (B) CoSalen in chloroform (solid line) and simulation as Co(II) S = 1/2 (dashed line). (C) CoSalen complexed with apoCB5, ArM2 ratio (red) and apoCB5 (black). (D) CoSalen complexed with apoCB5, ArM2 ratio (red) and after reduction with ascorbic acid (black). Signals in (C) are attributed to cavity background and spurious paramagnetic impurities. For details on experimental parameters and simulations, see ESI.† | ||
2.4 Water oxidation photoactivity of CB5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :CoSalen samples
:CoSalen samples
Once characterized, the capacity of the ArMs to oxidize water photocatalytically was tested in the presence of [Ru(bpy)3](ClO4)2 as photosensitizer (PS) and Na2S2O8 as sacrificial electron acceptor (SA) in a sodium phosphate buffer (80 mM) at pH 7.5 (for full experimental setup, see ESI†). Fig. 8 represents the O2 evolution of free CoSalen, apoCB5 (control), ArM1, ArM2, and ArM3, a control without catalyst is shown in Fig. S8.† As a reference, the O2 evolution activity of free CoSalen was also recorded under identical photocatalytic conditions. Free CoSalen produced 5.57 ± 0.85 μmol of O2 over 120 min irradiation (grey trace, Fig. 8A). Afterwards, the photoactivity of apoCB5 was tested. With this sample no oxygen production was observed, indicating that this protein was not capable of catalysing such a reaction (black trace, Fig. 8A). For the sample containing ArM1 as a catalyst, no O2 production was detectable either (orange trace, Fig. 8A). In contrast, the sample containing ArM2 was found to be catalytically active producing 4.24 ± 0.24 μmol of oxygen with a turnover number (TON) of 24 (red trace, Fig. 8A, Table 1). This performance was comparable, in terms of stability, with that of free CoSalen (TON 31, Table 1), but the maximal turnover frequency (TOFmax) of ArM2 (0.63 min−1) was approximately half compared to that of CoSalen (1.36 min−1) (Table 1). As will be discussed below, the combined results suggest that ArM2 carries one, inactive, CoSalen in the haem binding pocket and up to three additional CoSalens bound to the exterior His, implying that maximal 3 out of 4 CoSalen per protein are active. ArM3 generated less O2 (pink trace, Fig. 8A), as characterized by a twice lower TON of 16, but only a slightly lower TOFmax of 0.55 min−1 (Table 1). To our knowledge, these results are the first demonstrations that artificial proteins can perform photocatalytic water oxidation. However, the mechanism of such a reaction is far from obvious, and the question of whether the CoSalen–protein complex stayed intact under photocatalytic conditions, remained fully open at this stage. Loss of the cobalt could lead to catalysis performed by cobalt nanoparticles or some form of cobalt-bound protein species.
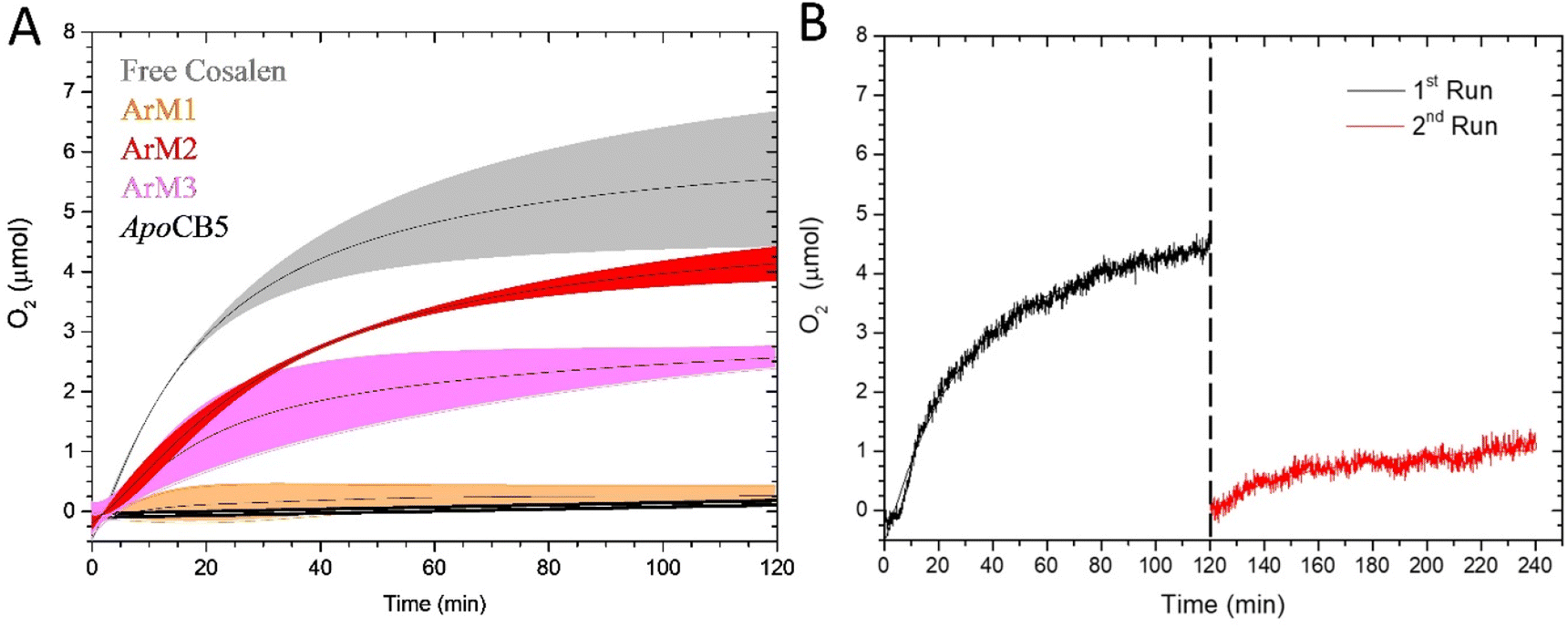 | ||
| Fig. 8 Photocatalytic O2 evolution by ArM1 (orange), ArM2 (red), ArM3 (pink), apoCB5 (black) and free CoSalen (grey). Conditions: [catalyst] 50 μM in Co, [Ru(bpy)3](ClO4)2 0.3 mM, Na2S2O8 5 mM, 80 mM NaPi pH 7.5, 25 °C, 450 nm LED (18–19 mW). For apoCB5, the reported concentration is the protein concentration (50 μM) (A). O2 evolution activity of ArM2 (B). The sample was irradiated for a total of 240 min. Irradiation started at t = 0 min. For the first run (black) the sample was irradiated with 450 nm LED (18–19 mW) in photocatalytic conditions: [catalyst] 50 μM in Co, [Ru(bpy)3](ClO4)2 0.3 mM and Na2S2O8 5 mM, 80 mM NaPi pH 7.5, 25 °C. After 120 min, irradiation was stopped and more [Ru(bpy)3](ClO4)2 and Na2S2O8 were added (same concentrations, dotted line). After the addition of another identical quantity of PS and SA, a second run (red) was started and the sample was irradiated for another 120 min. All experiments were performed in duplo. Data were fitted using OriginPro software. For raw data of panel (A), see Fig. S9 in ESI.† | ||
| Sample | O2 producedb (μmol) | TONb | TOFmaxc | QYO2d |
|---|---|---|---|---|
| a Conditions: PS = 0.3 mM [Ru(bpy)3](ClO4)2, SA = 5 mM Na2S2O8, [cat.] = 50 μM in Co, LED = 450 nm, 18–19 mW, 25 °C, 3.5 mL, 80 mM NaPi pH 7.5. b Value calculated as in eqn (S1) at 120 min using the [Co] from ICP-MS as catalyst concentration. c TOFmax = max value of TOF during photocatalysis. d Value obtained according to eqn (2) (Page S7). | ||||
| apoCB5 | — | — | — | — |
| Free CoSalen | 5.57 ± 0.85 | 31.80 ± 4.83 | 1.36 ± 0.03 | 0.073 ± 0.001 |
| ArM1 | — | — | — | — |
| ArM2 | 4.24 ± 0.24 | 24.25 ± 1.35 | 0.63 ± 0.07 | 0.036 ± 0.004 |
| ArM3 | 2.72 ± 0.15 | 15.54 ± 0.84 | 0.55 ± 0.09 | 0.032 ± 0.004 |
Finally, to test whether the TON of ArM2 was limited by the lifetime of the ArM or that of the photosensitizer or sacrificial agent, the photocatalysis was continued by the addition of fresh PS and SA after the activity of a first photocatalytic run had ceased (Fig. 8B). The addition of fresh PS and SA did lead to a continuation of the oxygen production, though the amount generated was significantly lower than that of the initial run. No precipitation was observed in the solution during the second run. Overall, this experiment proved that the activity of ArM2 was limited essentially by the PS and/or SA as is the case for free CoSalen,81 but suggested that the ArM was affected in such conditions.
2.5 Nanoparticle formation and ligand release
To test the stability of ArM2 under photocatalytic conditions, ICP-MS analysis was performed for samples at different irradiation time points, i.e., before, and after 0.5 min, 3 min, and 120 min irradiation in the presence of sacrificial agent and photosensitizer (Fig. S10†). Each sample was run over a size exclusion microspin Bio-Rad P6 column to remove any released or loosely attached small-molecule CoSalen. The initial apoCB5:CoSalen stoichiometry of ArM2 was 1:4.0 (see above), yet after 3 min of irradiation in the presence of PS and SA, this number dropped to 1:1.2, indicating that only one cobalt per protein remained firmly attached, i.e. withstanding the washing step during column purification. After 120 min of irradiation, the average number of CoSalen per CB5 had dropped to 0.2, suggesting that little cobalt was firmly attached to the protein scaffold. Overall, the ICP-MS data revealed that most CoSalen–protein interactions were destabilized within 10 min of blue light irradiation. Since water oxidation activity continued for approximately 120 min, we suspect that the photo catalytically active compound is not the original apoCB5:CoSalen adduct (see Discussion).
For photocatalytic water oxidation using free CoSalen as catalyst, [Ru(bpy)3](ClO4)2 as photosensitizer, and Na2S2O8 as sacrificial electron acceptor, it is known that the molecular complex CoSalen is a pre-catalyst of the catalytically active species and that the activity of this system can be ascribed to a mixture of cobalt oxide and cobalt hydroxide nanoparticles.16,81 To investigate if such nanoparticles were also formed in our artificial protein system during photocatalysis, dynamic light scattering (DLS) analysis was performed on photocatalytic mixtures containing ArM1, ArM2, ArM3, or free CoSalen as catalyst, [Ru(bpy)3](ClO4)2 and Na2S2O8 in a phosphate buffer solution at pH 7.5. Samples were measured three times: (i) at t = 0, (ii) after keeping the mixture in the dark for 120 min, and (iii) after blue light irradiation for 120 min (Fig. 9 and S18†).
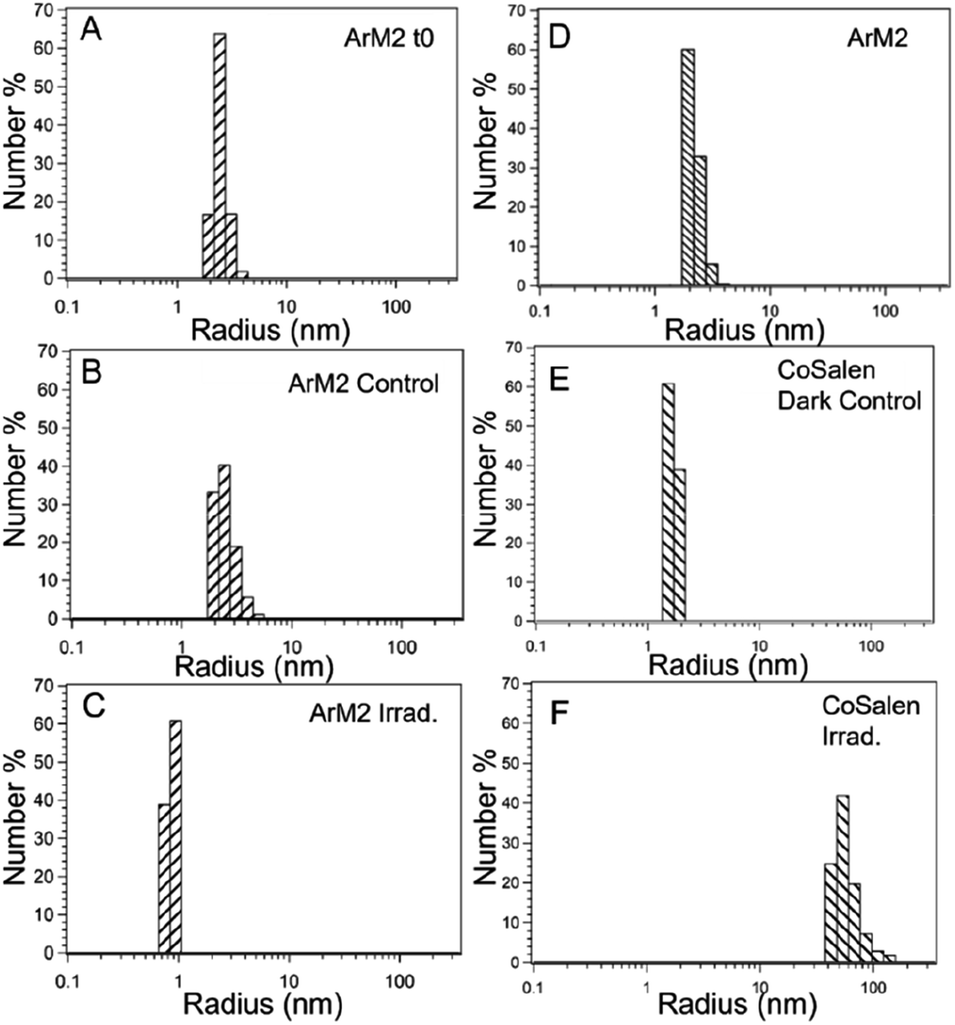 | ||
| Fig. 9 Dynamic light scattering (DLS) analysis for ArM2 and free CoSalen samples, kept in the dark or irradiated with blue light. Y-Axis indicates the percentage of the number of particles. All samples contained 50 μM CoSalen, either free or bound to CB5, in 80 mM NaPi pH 7.5 buffer. The following distributions can be observed: ArM2 directly after the addition of PS and SA (A), ArM2 + PS + SA after 120 min incubation in the dark (B), ArM2 + PS + SA after 120 min irradiation (C), ArM2 alone (D), free CoSalen + PS + SA kept in the dark (E), and free CoSalen + PS + SA after 120 min irradiation (F). Conditions: [Ru(bpy)3](ClO4)2 (PS); 0.3 mM, Na2S2O8 (SA):5 mM, T = 25 °C, light source 450 nm, 18–19 mV. | ||
The sample containing free CoSalen, PS and SA kept in the dark formed small nanoparticles of around 1–2 nm. After light irradiation for 120 min, the formation of much larger nanoparticles was detected, characterized by an average diameter of 47 nm. The behaviour of the ArM samples containing PS and SA was rather different. Before and after irradiation, the particle size was around 2 nm, which is the size of the CB5 protein determined by DLS, indicating that contrary to the free molecular catalyst, for the ArMs no large nanoparticles had formed upon irradiation. To test if intact H2Salen ligands may be part of the soluble species present in solutions after photocatalysis, a high-resolution mass spectrum (HR-MS) was measured in the positive mode to detect small molecules. Two solutions in the presence of [Ru(bpy)3](ClO4)2 and Na2S2O8 after 120 min irradiation were analysed: (a) ArM2 and (b) free CoSalen (Fig. S11 and S12†). Unbound H2Salen ligand was observed (m/z = 268) in the solutions for each case upon light irradiation in the presence of ruthenium photosensitizer and electron acceptor, indicating that in both cases of free CoSalen, and of the active ArM2 catalyst, the H2Salen ligand had been released within 120 min irradiation.
2.6 Stability of the CB5:CoSalen in the presence of a photosensitizer (PS) and sacrificial electron acceptor (SA)
The photostability of the ArM samples was first studied in the absence of PS and SA. The UV-vis spectrum of a solution containing 50 μM of either ArM1 or ArM2 in 80 mM phosphate buffer at pH 7.5 was recorded vs. time under constant irradiation with a 450 nm light source (Fig. S13A and B†). After 120 min of continuous irradiation, the spectra of both samples did not show any visible changes. In addition, high-resolution mass spectra of the above samples, measured after irradiation, showed the presence of CoSalen (m/z = 325.04) and the complex:protein adduct (i.e. m/z = 869 for ArM1 sample and m/z = 1230 for ArM2) and no trace of the free Salen ligand could be identified (Fig. S14 and S15†). In addition, semi-native and denaturing gel electrophoresis analysis of the irradiated ArM1 and ArM2 samples showed the stability of the protein under irradiation (Fig. S13C†). Overall, we concluded that when no PS and no SA was present the ArM1 and ArM2 samples were stable under blue light irradiation. The next step consisted of studying the protein–cobalt adduct integrity in the presence of the PS and SA. Fluorescence emission spectroscopy was employed to determine if ArM1 and ArM2 were also stable upon irradiation in the presence of the PS and SA in 80 mM NaPi pH 7.5 buffer (Fig. 10). The irradiated samples, containing ArM1 or ArM2 in the presence of PS and SA, were compared with the samples before irradiation. Upon irradiation, after one photocatalytic run, the tryptophan fluorescence emission of either the ArM1 or the ArM2 sample was no longer visible. This result suggested that under the oxidizing conditions of photocatalysis, chemical/structural changes did occur at the W27 site. ESI-MS spectra of ArM1, ArM2, ArM3 and apoCB5 were measured in the presence of PS and SA, directly after sample preparation or following 120 min incubation in the dark. The spectra of the samples before and after incubation in the dark were found to be quite similar (Fig. S16†). To study the effect of PS and SA on the protein in the dark, gel electrophoresis was employed.
 | ||
| Fig. 10 Tryptophan fluorescence emission spectrum of ArM samples before and after photocatalysis under irradiation by a 450 nm LED at 18–19 mW. ApoCB5 (black), ArM1 + PS + SA before (orange) and after photocatalysis (grey-dashed) (A). ApoCB5 (black), ArM2 + PS + SA before (red) and after photocatalysis (grey-dashed) (B). Inset: detailed view of ArM2 adduct emission from 300 to 400 nm before and after catalysis. Excitation: 280 nm. Conditions: PS = 0.3 mM [Ru(bpy)3](ClO4)2, SA = 5 mM Na2S2O8, [cat.] = 50 μM in Co, 25 °C, 3.5 mL, 80 mM NaPi pH 7.5. | ||
On SDS PAGE (Fig. S17†) ArM1 alone runs at the same height as apoCB5. However, on semi-native gel (Fig. S17†) two populations could be distinguished, one running at the height of apoCB5 and one running below, the latter being assigned as reacted CB5:CoSalen.54 The mass spectrum and SDS PAGE of the solution containing ArM3 looked very similar to ArM1. The sample containing ArM2, on the other hand, showed a ladder of multimers on semi-native gel in accordance with SEC-MALS (Fig. 5). The multimer bands observed in the semi-native gel of ArM2 (Fig. S17B,† lane 2, left image) did not show on the denaturing gel (Fig. S17B,† lane 2, right), as they did not withstand the denaturing conditions in the presence of SDS and β-mercaptoethanol. In contrast, SDS PAGE of ArM2 samples taken directly after the addition of PS and SA to the solution (Fig. S17B,† lane 3), also showed bands of dimers and trimers on denaturing gel, and further aggregation, seen as fading of the monomer band, was observed after 120 min dark incubation with PS and SA (Fig. S17B,† lane 4, right). This observation suggests a limited cross-linking effect of PS and SA on the CB5 protein observed after dark incubation (see Discussion section below). With the ArM1 sample, multimers were only observed on denaturing gel after 120 min incubation with PS and SA in the dark. For ArM3, the cross-linking effect was not observed.
To investigate the photostability of our most active artificial system, ArM2 was studied after irradiation in the presence of PS and SA. Both mass spectroscopy and gel electrophoresis were performed (Fig. S18†), the samples were tested after 0 to 2.5 min of irradiation. In the chromatogram of the C4 column attached to the mass spectrometer, ArM2 was observed, though the peak became broader and less intense as irradiation time increased. However, in the deconvoluted spectrum, signals for the complex:protein adduct were no longer observed, because the sample did not ionize anymore even after only 10 s of irradiation (data not shown), indicating that irradiation in the presence of PS and SA had a significant effect on the protein. Semi-native gel electrophoresis (Fig. S18B†) showed protein monomer, dimer, and trimer bands prior to irradiation. After only 5 seconds of irradiation, the bands faded, indicating an effect on the protein stability and a change in its charge upon irradiation in the presence of PS and SA, which is in line with the mass spectrometry results. On denaturing SDS PAGE (Fig. S18C†) the protein before irradiation showed a monomer band, while after the addition of PS and SA and 5 s irradiation, a ladder of multimers could be observed, revealing a cross-linking effect as was observed after dark incubation. In other words, 450 nm light irradiation in the presence of PS and SA affected the protein charge and stability even after just 5 seconds of irradiation.
The visibility of the protein signals in mass spectrometry and gel electrophoresis decreased after light irradiation in the presence of PS and SA. To gain a better view of the photocatalytic multimerization process, SEC-MALS was employed on ArM2 alone and in the presence of PS and SA, either kept in the dark or irradiated for 0.5 min and 120 min (Fig. 11). Compared to ArM2 alone, the sample kept in the dark with PS and SA contained a much smaller monomeric population and the population size of aggregates was increased.
 | ||
| Fig. 11 UV-vis absorbance at 280 nm during SEC-MALS analysis of samples containing ArM2 (50 μM) alone (red) and after combination with [Ru(bpy)3](ClO4)2 (0.3 mM) and Na2S8O4 (5 mM) and either kept in the dark (purple) or irradiated for 0.5 min (blue) or 120 min (yellow) at 450 nm (18–90 mV) in the photoreactor setup (see ESI† for the detailed setup). Full chromatograms are shown in panel (A). An enlarged view of the protein elution range with the aggregation state of each peak determined from MALS (data not shown) is shown in panel (B). | ||
After irradiation of ArM2 in the presence of PS and SA for 0.5 min or 120 min, the band of monomeric protein eluted slower, because of an increase in the protein mass (see Section 2.7). Furthermore, both in samples after 0.5 min and 120 min of irradiation, the major multimer populations corresponded to dimers and trimers, while in samples kept in the dark a much more uniform distribution of population sizes was observed. Finally, CD was used to observe the secondary structure of the protein after photocatalysis (Fig. 12). PS and SA were removed from the sample before the CD measurements using a Corning concentrator with a 5 kDa molecular weight cut-off (MWCO). The observed CD spectrum after photocatalysis showed a loss of alpha-helical character, and an increase in unfolded character, while some beta-sheet character remains. The data shows that while the exposure to light in the presence of PS and SA had a significant and rapid effect on the protein, it nonetheless remained intact, soluble, and partially folded.
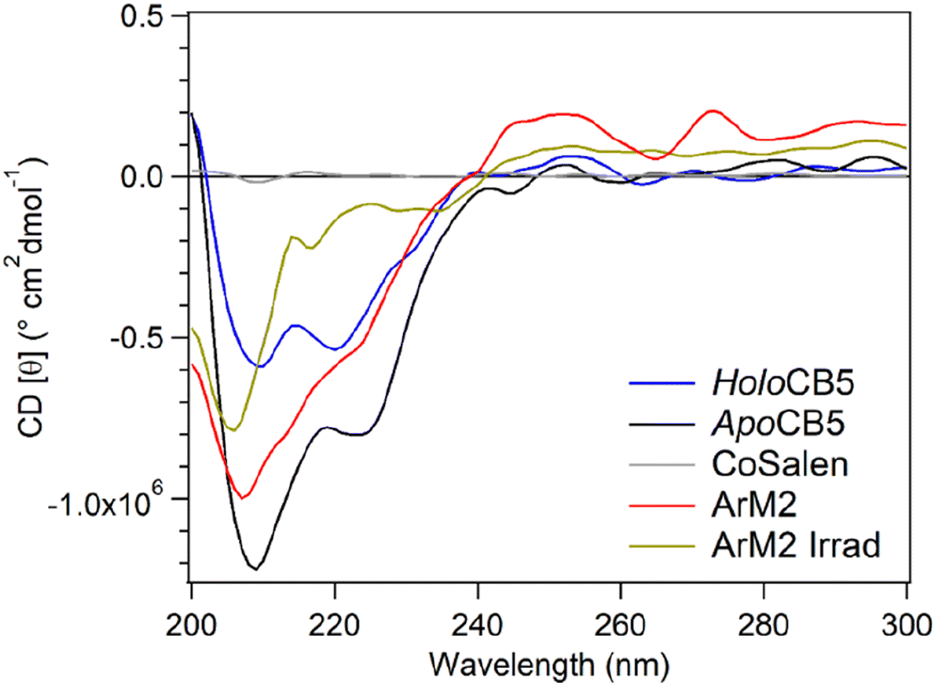 | ||
| Fig. 12 Circular dichroism (CD) analysis of ArM2 (red), holoCB5 (blue), apoCB5 (black), free CoSalen (grey) and ArM2 after irradiation in the presence of 0.3 mM [Ru(bpy)3](ClO4)2 and 5 mM Na2S2O8 after purification to remove small molecules (yellow). | ||
2.7 Protein association with a breakdown product of the photosensitizer
Samples of ArM2 + PS + SA that were either kept in the dark or had been irradiated, were purified over a Bio-Rad P6 spin column to remove small molecules. The samples that were kept dark were observed to be virtually colourless, while the samples that had been irradiated were orange in colour. We, therefore, suspected either aggregation of the orange, ruthenium containing, PS, or an association of the PS or a breakdown product thereof with ArM2. To investigate this phenomenon, ruthenium and cobalt concentration measurements by ICP-MS were recorded after purification of the sample over a microspin Bio-Rad P6 column with a 6 kDa MWCO that removed all small molecules, i.e., excess of PS and SA. These measurements revealed that while the concentration of cobalt decreased in the ArM2 sample during photocatalysis, the concentration of ruthenium increased considerably, giving a CB5:ruthenium ratio of 1:1 after 0.5 min of light irradiation. The presence of ruthenium in the purified samples indicated that either a ruthenium-containing aggregate with a molecular weight larger than 6 kDa was formed during irradiation of ArM2 in the presence of PS and SA, or that a ruthenium species got firmly attached to apoCB5.
A UV-vis spectrum was measured for the microspin Bio-Rad P6-purified sample to determine the spectral characteristics of this orange material. As shown in Fig. 13, a characteristic absorbance maximum around 450 nm was observed, which is characteristic for [Ru(bpy)3]2+. Since the orange material was observed after purification after irradiation of the sample in the presence of PS and SA, while ArM2 exposed to PS and SA in the dark was essentially colourless after purification, the bound ruthenium was hypothesized to be a breakdown product of the PS. To determine if the observed ruthenium species was an aggregate of photosensitizer-derived breakdown product, or if a breakdown product of the photosensitizer had indeed attached to the protein, SEC-MALS was used to monitor the co-elution of the protein and the ruthenium species. This was done by monitoring the UV-vis absorbance of the sample during elution of the SEC column at 280 nm, where both the protein and the ruthenium species absorb light, and at 450 nm, where the ruthenium species absorbs. The absorbance ratio A280/A450 was determined for a solution of ArM2, PS, and SA, at different time points during a photocatalytic oxygen evolution experiment (t = 0, 0.5, 120 min, or dark control, Table S4†). The dark ArM2 sample containing PS and SA showed similar ratios compared to ArM2 without PS and SA, showing that no interaction of the protein and the ruthenium photosensitizer took place prior to irradiation. After 0.5 min of irradiation, the A280/A450 ratio decreased, which, in combination with the before-mentioned ICP-MS results, indicated an increase in photosensitizer breakdown product concentration with respect to CB5. This spectral signature of the ruthenium compound was present with each multimer of the protein as it was eluted from the SEC column, suggesting that the ruthenium compound was indeed attached to the protein.
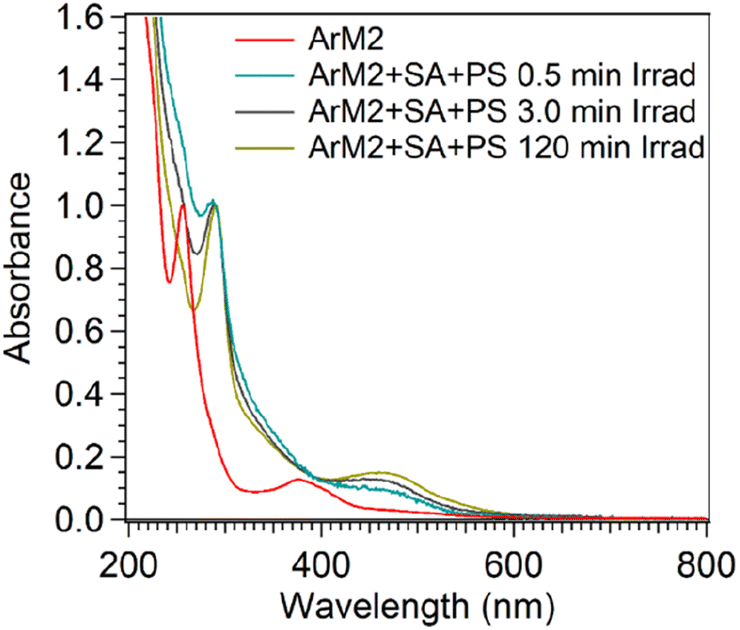 | ||
| Fig. 13 The stability of ArM2 under photocatalytic conditions. Normalized (to the lambda max) UV-vis spectra measured after purification over a P6 column of ArM2 samples kept in the dark (red), irradiated for 0.5 min (blue), 3 min (black), or 120 min (yellow) with a 450 nm LED (18–19 mW) in the photoreactor in the presence 0.3 mM [Ru(bpy)3](ClO4)2 and 5 mM Na2S2O8. | ||
3. Discussion
In this study, we analysed one of the rare, reported cases of light-induced generation of O2 using an artificial metalloenzyme bearing an abundant metal cofactor. For most reported photoactive water oxidation systems using small-molecule cobalt catalysts, the instability or decomposition of the complex leads to the formation of cobalt nanoparticles or deposits.81–86 By contrast, with ArM2 we present a system in which nanoparticle formation is avoided.For ArM1, photocatalytic activity was not observed in the presence of PS, SA, and blue light. NMR data revealed that CoSalen was likely coordinated in the binding pocket of apoCB5 between two axial histidines, H44 and H68. This bi-axial coordination in the pocket of CB5 is different to what can be found in the literature for artificial haem proteins, where most of the cofactors are coordinated only by one residue in the pocket of the protein such that one coordination site on the metal is left open.87,88 Bi-axial coordination leaves no free position on cobalt to coordinate water, and while in some cases redox reactions can take place on the ligands rather than at the metal centre,89–94 the coordination is likely the reason why ArM1 is catalytically inactive. Other potential explanations for the inactivity are that, while CB5 is a relatively small and open protein, either water or the excited photosensitizer could be restricted in its ability to approach the cobalt compound. In any case, the environment provided by the protein stabilized the complex in such a way that in our photocatalytic conditions no active cobalt species capable of water oxidation could be formed. By contrast, ArM2 was found to be catalytically active. Hence, the results suggest that the exterior bound CoSalen molecules determine photocatalytic activity. If ArM2 carries one, inactive, CoSalen in the haem binding pocket and up to three additional CoSalens bound to the exterior His, maximal 3 out of 4 CoSalen per protein are active. Indeed, the productivity of this ArM, producing 4.24 μmol O2 with a TON per CoSalen of 24.3, is ∼75% of the productivity of free CoSalen, 5.57 μmol O2, TON of 31.8 per CoSalen, under the same conditions. Furthermore, ArM3, binding ∼2.7 CoSalen per protein as well as haem, was photocatalytically active under the same conditions as ArM2 but generated less O2. This suggests that the presence of haem negatively influences the O2 production. It has been reported that Fe3+ from haem bound to proteins can be oxidized by the triplet excited state of [Ru(bpy)3]2+ generated during photocatalysis and that such a redox process is reversible and might lead to charge recombination, thereby lowering the efficacy of photocatalysis.95,96
Further, side-reactions involving protein oxidation could potentially lead to ArM oligomerization and reduction of the ArM activity. Loss of tryptophan fluorescence was observed after irradiation in the presence of PS and SA. This can be ascribed to: (a) the reaction of singlet oxygen generated by the direct reaction of the triplet forming photosensitizer;97,98 or (b) the sulphate radical (SO4˙−) generated by 1-electron transfer from the excited state of the photosensitizer to sodium persulfate. SO4˙− is a strong oxidant that might induce an electron transfer (ET) from the indole ring, leading to an oxidized, non-emissive aromatic moiety. The presence of oxidizing species could affect the protein fold and result in oligomerization. Oxidation of tyrosine leading to cross-linking after the addition of PS and SA, in the dark has been previously observed in the literature.99,100 We observed that multimers or small aggregates present in the PS- and SA-free ArM2 sample became resistant to denaturation upon exposure to PS and SA in the dark as well as under irradiation (Fig. S16 and S17†) and to a lesser degree upon irradiation of ArM2 in the absence of PS and SA (Fig. S10†). Finally, a surprising result of irradiation of ArM2 in the presence of PS and SA was an interaction between the PS and the protein scaffold (Section 2.7). The PS would bind to the proteins in a 1:1 fashion, despite the large (6-fold) access of PS, indicating specificity of the protein binding. The interaction may be electrostatic in nature, due to the positive charge of the PS, since apoCB5 has an overall negative charge, or dative, due to the exchange of a bipyridine ligand under oxidative conditions with an amino acid, e.g. histidine.12
Purification of the ArM2 sample after photocatalysis followed by ICP-MS revealed that CoSalen did not remain bound to the protein in its original form. Within the first minutes of the start of the reaction, 3 out of 4 CoSalen had weakened protein interactions as the compounds could be separated from the protein through column purification. The ArM2 system, however, remained soluble and did not show significant changes in particle size, which corresponded to the size of a protein monomer or dimer, whereas free CoSalen formed particles of ∼47 nm during photocatalysis (Fig. 10). Hence, our results suggest that upon exposure to light, in the presence of the PS and SA, the protein–cobalt interactions were destabilized but in contrast to the free CoSalen system, no large, cobalt-containing particles had formed. Thus, the artificial cobalt–protein complex is a pre-catalyst, like CoSalen, but unlike free CoSalen it forms a non-aggregated, protein-interacting, catalytically active species (Fig. 14). The proposed hypothesized reaction scheme is summarized in Fig. 14: upon light activation of the CoSalen ArMs in the presence of a PS and SA, dynamic interaction between cobalt and the protein generates an active, soluble, adduct with photocatalytic water-oxidation activity.
 | ||
| Fig. 14 Proposed pathway of photocatalytic water oxidation using the ArM2 pre-catalyst compared to the free CoSalen pre-catalyst. | ||
4. Conclusions
Here, a novel artificial CB5:CoSalen metalloenzyme ArM2 active for water oxidation under photocatalytic conditions, was developed and characterized. The analogue ArM1, where CoSalen was bound via axial histidine coordination to the haem binding pocket of the protein, lacked catalytic activity. ArM2, in which CoSalen binds both in the binding pocket and to the protein exterior, showed water oxidation activity in photocatalytic conditions, like free CoSalen. Slightly lower activity was observed from the ArM3 analogue that still contained haem while CoSalen was bound to the protein exterior. In the dark, addition of [Ru(bpy)3](ClO4)2 and Na2S2O8 led to limited protein cross-linking. However, one equivalent of the ruthenium photosensitizer was found to interact strongly with ArM2 upon irradiation. During photocatalysis, the protein partially unfolded and protein–CoSalen interactions were destabilized. Strikingly, while the CoSalen catalyst formed tens of nm sized nanoparticles capable of performing photocatalysis, no such large nanoparticles were found for the CoSalen ArMs that were photocatalytically active while remaining soluble in aqueous, neutral-pH solution. This work, which is among the first studies of artificial proteins performing photocatalytic water oxidation, shows the potential and possible advantages of protein scaffolds as water-soluble support of a catalytically active molecule in the conditions of photocatalytic water oxidation.
Data availability
The data that supports this article are available in the ESI.†Author contributions
E. A. P., L. O., A. P. and S. B. designed the project concept. A. P. and S. B. guided the research. E. A. P. and L. O. performed all the experiments related to photocatalysis and protein characterization. M. H. and L. P. performed EPR measurements. E. A. P., L. O., L. P., M. H., A. P. and S. B. wrote the manuscript with contributions from all authors.Conflicts of interest
There are no conflicts of interests to declare.Acknowledgements
We thank Dr B. I. Florea for ESI-MS support, Ing. S. G. Romeijn for the training on the fluorimeter and MSc T. G. J. Knetsch for instruction on the DLS. Dr W. Elings, Dr R. Dasgupta and Dr K. B. Sai Sankar Gupta for instruction on the NMR spectrometer. Ing. H. van den Elst for HR-MS measurements, A. J. Cramer-Blok for recording the SEC-MALS spectra and Dr S. Zheng for the ICP-MS measurements.References
- D. J. Vinyard, G. M. Ananyev and G. C. Dismukes, Annu. Rev. Biochem., 2013, 82, 577–606 CrossRef CAS PubMed.
- W. Lubitz, E. J. Reijerse and J. Messinger, Energy Environ. Sci., 2008, 1, 15–31 RSC.
- M. D. Kärkäs, O. Verho, E. V. Johnston and B. Åkermark, Chem. Rev., 2014, 114, 11863–12001 CrossRef PubMed.
- C. Liu, D. van den Bos, B. den Hartog, D. van der Meij, A. Ramakrishnan and S. Bonnet, Angew. Chem., Int. Ed., 2021, 60, 13463–13469 CrossRef CAS.
- S. Ye, C. Ding, M. Liu, A. Wang, Q. Huang and C. Li, Adv. Mater., 2019, 31, 1902069 CrossRef CAS PubMed.
- R. Matheu, P. Garrido-Barros, M. Gil-Sepulcre, M. Z. Ertem, X. Sala, C. Gimbert-Suriñach and A. Llobet, Nat. Rev. Chem, 2019, 3, 331–341 CrossRef CAS.
- R. Matheu, M. Z. Ertem, C. Gimbert-Suriñach, X. Sala and A. Llobet, Chem. Rev., 2019, 119, 3453–3471 CrossRef CAS PubMed.
- S. W. Gersten, G. J. Samuels and T. J. Meyer, J. Am. Chem. Soc., 1982, 104, 4029–4030 CrossRef CAS.
- F. Liu, J. J. Concepcion, J. W. Jurss, T. Cardolaccia, J. L. Templeton and T. J. Meyer, Inorg. Chem., 2008, 47, 1727–1752 CrossRef CAS.
- J. K. Hurst, J. L. Cape, A. E. Clark, S. Das and C. Qin, Inorg. Chem., 2008, 47, 1753–1764 CrossRef CAS PubMed.
- U. Hintermair, S. M. Hashmi, M. Elimelech and R. H. Crabtree, J. Am. Chem. Soc., 2012, 134, 9785–9795 CrossRef CAS PubMed.
- B. Limburg, E. Bouwman and S. Bonnet, Coord. Chem. Rev., 2012, 256, 1451–1467 CrossRef CAS.
- T. Liu, B. Zhang and L. Sun, Chem.–Asian J., 2019, 14, 31–43 CrossRef CAS PubMed.
- S. C. Silver, J. Niklas, P. Du, O. G. Poluektov, D. M. Tiede and L. M. Utschig, J. Am. Chem. Soc., 2013, 135, 13246–13249 CrossRef CAS PubMed.
- K. G. Kottrup, S. D'Agostini, P. H. van Langevelde, M. A. Siegler and D. G. H. Hetterscheid, ACS Catal., 2018, 8, 1052–1061 CrossRef CAS PubMed.
- D. Nesterov and O. Nesterova, Catalysts, 2018, 8, 602 CrossRef.
- B. Limburg, E. Bouwman and S. Bonnet, ACS Catal., 2016, 6, 5273–5284 CrossRef CAS.
- K. Degtyarenko, Encyclopedia of Genetics, Genomics, Proteomics and Bioinformatics, 2005 Search PubMed.
- K. Vong, I. Nasibullin and K. Tanaka, Bull. Chem. Soc. Jpn., 2021, 94, 382–396 CrossRef CAS.
- K. Faber, Biotransformations in organic chemistry: a textbook, Springer, 2011 Search PubMed.
- K. Yamamura and E. T. Kaiser, J. Chem. Soc. Chem. Commun., 1976, 830–831 RSC.
- M. E. Wilson and G. M. Whitesides, J. Am. Chem. Soc., 1978, 100, 306–307 CrossRef CAS.
- F. Schwizer, Y. Okamoto, T. Heinisch, Y. Gu, M. M. Pellizzoni, V. Lebrun, R. Reuter, V. Köhler, J. C. Lewis and T. R. Ward, Chem. Rev., 2018, 118, 142–231 CrossRef CAS PubMed.
- S. Lopez, L. Rondot, C. Leprêtre, C. Marchi-Delapierre, S. Ménage and C. Cavazza, J. Am. Chem. Soc., 2017, 139, 17994–18002 CrossRef CAS.
- C. Esmieu, P. Raleiras and G. Berggren, Sustain. Energy Fuels, 2018, 2, 724–750 RSC.
- M. R. Cerón, M. Izquierdo, N. Alegret, J. A. Valdez, A. Rodríguez-Fortea, M. M. Olmstead, A. L. Balch, J. M. Poblet and L. Echegoyen, Chem. Commun., 2016, 52, 64–67 RSC.
- A. Pinnola, J. Exp. Bot., 2019, 70, 5527–5535 CrossRef CAS PubMed.
- R. E. Blankenship, D. M. Tiede, J. Barber, G. W. Brudvig, G. Fleming, M. Ghirardi, M. R. Gunner, W. Junge, D. M. Kramer, A. Melis, T. A. Moore, C. C. Moser, D. G. Nocera, A. J. Nozik, D. R. Ort, W. W. Parson, R. C. Prince and R. T. Sayre, Science, 2011, 332, 805–809 CrossRef CAS PubMed.
- P. Hosseinzadeh and Y. Lu, Biochim. Biophys. Acta, Bioenerg., 2016, 1857, 557–581 CrossRef CAS PubMed.
- P. K. Wawrzyniak, A. Alia, R. G. Schaap, M. M. Heemskerk, H. J. M. de Groot and F. Buda, Phys. Chem. Chem. Phys., 2008, 10, 6971 RSC.
- Y. Yu, C. Hu, L. Xia and J. Wang, ACS Catal., 2018, 8, 1851–1863 CrossRef CAS.
- H. M. Key, P. Dydio, D. S. Clark and J. F. Hartwig, Nature, 2016, 534, 534–537 CrossRef CAS.
- J. Bos and G. Roelfes, Curr. Opin. Chem. Biol., 2014, 19, 135–143 CrossRef CAS.
- K. Oohora, Y. Kihira, E. Mizohata, T. Inoue and T. Hayashi, J. Am. Chem. Soc., 2013, 135, 17282–17285 CrossRef CAS PubMed.
- S. Lopez, L. Rondot, C. Cavazza, M. Iannello, E. Boeri-Erba, N. Burzlaff, F. Strinitz, A. Jorge-Robin, C. Marchi-Delapierre and S. Ménage, Chem. Commun., 2017, 53, 3579–3582 RSC.
- I. Hamachi, S. Tanaka and S. Shinkai, J. Am. Chem. Soc., 1993, 115, 10458–10459 CrossRef CAS.
- J. Liu, S. Chakraborty, P. Hosseinzadeh, Y. Yu, S. Tian, I. Petrik, A. Bhagi and Y. Lu, Chem. Rev., 2014, 114, 4366–4469 CrossRef CAS PubMed.
- S. N. Natoli and J. F. Hartwig, Acc. Chem. Res., 2019, 52, 326–335 CrossRef CAS.
- A. D. Liang, J. Serrano-Plana, R. L. Peterson and T. R. Ward, Chem. Res., 2019, 52, 585–595 CrossRef CAS.
- N. J. Porter, E. Danelius, T. Gonen and F. H. Arnold, J. Am. Chem. Soc., 2022, 144, 8892–8896 CrossRef CAS PubMed.
- T. Lazarides, I. V. Sazanovich, A. J. Simaan, M. C. Kafentzi, M. Delor, Y. Mekmouche, B. Faure, M. Réglier, J. A. Weinstein, A. G. Coutsolelos and T. Tron, J. Am. Chem. Soc., 2013, 135, 3095–3103 CrossRef CAS PubMed.
- V. Robert, E. Monza, L. Tarrago, F. Sancho, A. De Falco, L. Schneider, E. N. Ngoutane, Y. Mekmouche, P. R. Pailley, A. J. Simaan, V. Guallar and T. Tron, ChemPlusChem, 2017, 82, 607–614 CrossRef CAS PubMed.
- M. Orio and D. A. Pantazis, Chem. Commun., 2021, 57, 3952–3974 RSC.
- H. Ahmad, S. K. Kamarudin, L. J. Minggu and M. Kassim, Renew. Sustain. Energy Rev., 2015, 43, 599–610 CrossRef CAS.
- B. You and Y. Sun, Acc. Chem. Res., 2018, 51, 1571–1580 CrossRef CAS PubMed.
- J. Qi, W. Zhang and R. Cao, Adv. Energy Mater., 2018, 8, 1701620 CrossRef.
- D. J. Sommer, M. D. Vaughn and G. Ghirlanda, Chem. Commun., 2014, 50, 15852–15855 RSC.
- S. R. Soltau, P. D. Dahlberg, J. Niklas, O. G. Poluektov, K. L. Mulfort and L. M. Utschig, Chem. Sci., 2016, 7, 7068–7078 RSC.
- M.-C. Kim and S.-Y. Lee, Chem.–Asian J., 2018, 13, 334–341 CrossRef CAS PubMed.
- Z. Abdi, R. Bagheri, Z. Song and M. M. Najafpour, Sci. Rep., 2019, 9, 11499 CrossRef PubMed.
- C. Casadevall, H. Zhang, S. Chen, D. J. Sommer, D.-K. Seo and G. Ghirlanda, Catalysts, 2021, 11, 626 CrossRef CAS.
- J. F. Allen and J. Nield, Trends Plant Sci., 2017, 22, 97–99 CrossRef CAS PubMed.
- V. M. Johnson and H. B. Pakrasi, Microorganisms, 2022, 5, 10 Search PubMed.
- L. V. Opdam, E. A. Polanco, B. de Regt, N. Lambertina, C. Bakker, S. Bonnet and A. Pandit, Anal. Biochem., 2022, 653, 114788 CrossRef CAS PubMed.
- E. Krieger and G. Vriend, Bioinformatics, 2014, 30, 2981–2982 CrossRef CAS PubMed.
- S. Bienert, A. Waterhouse, T. A. De Beer, G. Tauriello, G. Studer, L. Bordoli and T. Schwede, Nucleic Acids Res., 2017, 45, D313–D319 CrossRef CAS PubMed.
- N. Guex, M. C. Peitsch and T. Schwede, Electrophoresis, 2009, 30, S162–S173 CrossRef PubMed.
- A. Waterhouse, M. Bertoni, S. Bienert, G. Studer, G. Tauriello, R. Gumienny, F. T. Heer, T. A. P. de Beer, C. Rempfer and L. Bordoli, Nucleic Acids Res., 2018, 46, W296–W303 CrossRef CAS PubMed.
- G. Studer, C. Rempfer, A. M. Waterhouse, R. Gumienny, J. Haas and T. Schwede, Bioinformatics, 2020, 36, 1765–1771 CrossRef CAS PubMed.
- M. Bertoni, F. Kiefer, M. Biasini, L. Bordoli and T. Schwede, Sci. Rep., 2017, 7, 1–15 CrossRef CAS PubMed.
- F. W. J. Teale, Biochim. Biophys. Acta, 1959, 35, 543 CrossRef CAS PubMed.
- C. J. Falzone, Y. Wang, B. C. Vu, N. L. Scott, S. Bhattacharya and J. T. J. Lecomte, Biochemistry, 2001, 40, 4879–4891 CrossRef CAS PubMed.
- N. J. Greenfield, Nat. Protoc., 2006, 1, 2876–2890 CrossRef CAS PubMed.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, 1983, pp. 341–381 Search PubMed.
- Y. Chen and M. D. Barkley, Biochemistry, 1998, 37, 9976–9982 CrossRef CAS PubMed.
- C. A. Royer, Chem. Rev., 2006, 106, 1769–1784 CrossRef CAS PubMed.
- M. M. Lee and B. R. Peterson, ACS Omega, 2016, 1, 1266–1276 CrossRef CAS PubMed.
- K. D. Bhatt, H. S. Gupte, B. A. Makwana, D. J. Vyas, D. Maity and V. K. Jain, J. Fluoresc., 2012, 22, 1493–1500 CrossRef CAS PubMed.
- L. Fabbrizzi, M. Licchelli, P. Pallavicini, D. Sacchi and A. Taglietti, Analyst, 1996, 121, 1763 RSC.
- Y. Li, Z. Csók, P. Szuroczki, L. Kollár, L. Kiss and S. Kunsági-Máté, Anal. Chim. Acta, 2013, 799, 51–56 CrossRef CAS PubMed.
- Y. Chang, Z. Zhang, H. Liu, N. Wang and J. Tang, Analyst, 2016, 141, 4719–4724 RSC.
- D. Kong, F. Yan, Z. Han, J. Xu, X. Guo and L. Chen, RSC Adv., 2016, 6, 67481–67487 RSC.
- E. B. Azimi, A. Badiei, M. Jafari, A. B. Dehkordi, J. B. Ghasemi and G. M. Ziarani, New J. Chem., 2019, 43, 12087–12093 RSC.
- W. Boonta, C. Talodthaisong, S. Sattayaporn, C. Chaicham, A. Chaicham, S. Sahasithiwat, L. Kangkaew and S. Kulchat, Mater. Chem. Front., 2020, 4, 507–516 RSC.
- L. D. Newton, S. I. Pascu, R. M. Tyrrell and I. M. Eggleston, Org. Biomol. Chem., 2019, 17, 467–471 RSC.
- S. Koga, S. Yoshihara, H. Bando, K. Yamasaki, Y. Higashimoto, M. Noguchi, S. Sueda, H. Komatsu and H. Sakamoto, Anal. Biochem., 2013, 433, 2–9 CrossRef CAS PubMed.
- A. L. J. Smith, A. Kahraman and J. M. Thornton, Proteins: Struct., Funct., Bioinf., 2010, 78, 2349–2368 CrossRef PubMed.
- B. Bennett, in Metals in Biology, Springer, 2010, pp. 345–370 Search PubMed.
- J. Zarembowitch and O. Kahn, Inorg. Chem., 1984, 23, 589–593 CrossRef CAS.
- M. C. R. Symons, T. Taiwo, A. M. Sargeson, M. M. Ali and A. S. Tabl, Inorg. Chim. Acta., 1996, 241, 5–8 CrossRef CAS.
- S. Fu, Y. Liu, Y. Ding, X. Du, F. Song, R. Xiang and B. Ma, Chem. Commun., 2014, 50, 2167–2169 RSC.
- D. den Boer, Q. Siberie, M. A. Siegler, T. H. Ferber, D. C. Moritz, J. P. Hofmann and D. G. H. Hetterscheid, ACS Catal., 2022, 12, 4597–4607 CrossRef PubMed.
- D. Hong, J. Jung, J. Park, Y. Yamada, T. Suenobu, Y.-M. Lee, W. Nam and S. Fukuzumi, Energy Environ. Sci., 2012, 5, 7606 RSC.
- M. Risch, D. Shevchenko, M. F. Anderlund, S. Styring, J. Heidkamp, K. M. Lange, A. Thapper and I. Zaharieva, Int. J. Hydrogen Energy, 2012, 37, 8878–8888 CrossRef CAS.
- M. W. Kanan and D. G. Nocera, Science, 2008, 321, 1072–1075 CrossRef CAS PubMed.
- D. Shevchenko, M. F. Anderlund, A. Thapper and S. Styring, Energy Environ. Sci., 2011, 4, 1284 RSC.
- K. Oohora and T. Hayashi, Dalton Trans., 2021, 50, 1940–1949 RSC.
- Y. Miyazaki, K. Oohora and T. Hayashi, J. Organomet. Chem., 2019, 901, 120945 CrossRef CAS.
- H. M. Castro-Cruz and N. A. Macías-Ruvalcaba, Coord. Chem. Rev., 2022, 458, 214430 CrossRef CAS.
- O. R. Luca and R. H. Crabtree, Chem. Soc. Rev., 2013, 42, 1440–1459 RSC.
- J. I. van der Vlugt, Eur. J. Inorg. Chem., 2012, 2012, 363–375 CrossRef CAS.
- V. Lyaskovskyy and B. de Bruin, ACS Catal., 2012, 2, 270–279 CrossRef CAS.
- D. J. Sommer, M. D. Vaughn, B. C. Clark, J. Tomlin, A. Roy and G. Ghirlanda, Biochim. Biophys. Acta, Bioenerg., 2016, 1857, 598–603 CrossRef CAS PubMed.
- S. Salzl, M. Ertl and G. Knör, Phys. Chem. Chem. Phys., 2017, 19, 8141–8147 RSC.
- J. Chen and W. R. Browne, Coord. Chem. Rev., 2018, 374, 15–35 CrossRef CAS.
- J. R. Scott, A. Willie, M. McLean, P. S. Stayton, S. G. Sligar, B. Durham and F. Millett, J. Am. Chem. Soc., 1993, 115, 6820–6824 CrossRef CAS.
- G. E. Ronsein, M. C. B. Oliveira, S. Miyamoto, M. H. G. Medeiros and P. Di Mascio, Chem. Res. Toxicol., 2008, 21, 1271–1283 Search PubMed.
- M. Ehrenshaft, L. J. Deterding and R. P. Mason, Free Radic. Biol. Med., 2015, 89, 220–228 CrossRef CAS PubMed.
- B. G. Soliman, G. C. Lindberg, T. Jungst, G. J. Hooper, J. Groll, T. B. Woodfield and K. S. Lim, Adv. Healthcare Mater., 2020, 9, 1901544 CrossRef CAS PubMed.
- J. W. Bjork, S. L. Johnson and R. T. Tranquillo, Biomaterials, 2011, 32, 2479–2488 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc05870k |
| ‡ Authors with equal contribution to the work. |
| This journal is © The Royal Society of Chemistry 2024 |