
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInterplay of chloride levels and palladium(II)-catalyzed O-deallenylation bioorthogonal uncaging reactions†
Gean M.
Dal Forno
 a,
Eloah
Latocheski
a,
Claudio D.
Navo
b,
Brunno L.
Albuquerque
a,
Albert L.
St John
a,
Frédéric
Avenier
c,
Gonzalo
Jiménez-Osés
bd and
Josiel B.
Domingos
*a
a,
Eloah
Latocheski
a,
Claudio D.
Navo
b,
Brunno L.
Albuquerque
a,
Albert L.
St John
a,
Frédéric
Avenier
c,
Gonzalo
Jiménez-Osés
bd and
Josiel B.
Domingos
*a
aLaboratory of Biomimetic Catalysis (LaCBio), Department of Chemistry, Federal University of Santa Catarina (UFSC), Campus Trindade, Florianópolis, 88040-900, SC, Brazil. E-mail: josiel.domingos@ufsc.br
bCenter for Cooperative Research in Biosciences (CIC BioGUNE), Basque Research and Technology Alliance (BRTA), Bizkaia Technology Park, Building 800, Derio, 48160, Spain
cInstitut de Chimie Moléculaire et des Matériaux d’Orsay (UMR 8182), Université Paris Saclay, 9140, Orsay Cedex, France
dIkerbasque, Basque Foundation for Science, 48013, Bilbao, Spain
First published on 16th February 2024
Abstract
The palladium-mediated uncaging reaction of allene substrates remains a promising yet often overlooked strategy in the realm of bioorthogonal chemistry. This method exhibits high kinetic rates, rivaling those of the widely employed allylic and propargylic protecting groups. In this study, we investigate into the mechanistic aspects of the C–O bond-cleavage deallenylation reaction, examining how chloride levels influence the kinetics when triggered by Pd(II) complexes. Focusing on the deallenylation of 1,2-allenyl protected 4-methylumbelliferone promoted by Allyl2Pd2Cl2, our findings reveal that reaction rates are higher in environments with lower chloride concentrations, mirroring intracellular conditions, compared to elevated chloride concentrations typical of extracellular conditions. Through kinetic and spectroscopic experiments, combined with DFT calculations, we uncover a detailed mechanism that identifies AllylPd(H2O)2 as the predominant active species. These insights provide the basis for the design of π-allylpalladium catalysts suited for selective uncaging within specific cellular environments, potentially enhancing targeted therapeutic applications.
Introduction
Bioorthogonal uncaging reactions driven by transition metal ions represent an emerging and powerful approach for the activation of caged molecules within living systems.1–6 Operating in both extra- and intracellular environments, metal complexes and nanoparticles have demonstrated the ability to selectively catalyze C–O or C–N bond cleavage reactions without disruption to native biological processes.7,8 More recently we demonstrated that also some C–C bonds could be cleaved by transition metal ions in vivo.9 The efficiency of the uncaging process, determined by the paired caging group and catalyst, is significantly influenced by the functional group's affinity for the metal ion and the ion's capacity to promote the bond breaking of the corresponding functional group. The choice of metal ion, its oxidation state, and the nature of the ligand, considering the substrate's characteristics, are pivotal for achieving success in gain-of-function studies of proteins,10–12 prodrug activation,13–23 and cell surface engineering.24,25In the development of metal catalytic systems, a thorough understanding of the mechanism, including the medium's influence on the reaction, is indispensable.26 A significant portion of research in bioorthogonal chemistry employs biological aqueous buffers for catalytic studies. Prominent among these are phosphate-buffered saline (PBS), Earl's balanced salt solution (EBSS), Hank's balanced salt solution (HBSS), and minimum essential medium (MEM). These isotonic solutions are formulated to maintain osmolarity and pH in biological contexts. Notably, chloride ions, present in concentrations exceeding 100 mmol L−1, are the principal components in these buffers. Given their electronic properties and prevalence, chloride ions are uniquely positioned in biological environments to effectively coordinate with transition metal centers. Their presence can substantially influence whether a metal catalyst undergoes successful activation through ligand exchange with solvents or substrates. However, a noteworthy detail, often overlooked during the optimization phase of new transition metal bioorthogonal catalytic system development, is the variability in chloride ion concentrations, ranging from 4–30 mmol L−1 intracellularly to approximately 100–140 mmol L−1 extracellularly. While research on the role of chloride ions in the mechanism of palladium-catalyzed reactions has historically focused on oxidative additions from Pd(0) in organic solvents,27,28 there is a distinct scarcity of systematic studies that explore their effects in uncaging reactions promoted by Pd in aqueous environments.
We thus hypothesize that the difference in the concentration of chloride ions inside and outside of the cells may directly affect the structure of the transition metals complexes, by ligand exchange processes, and thus tune the catalytic activity of the metal ion. This potential alteration, likely via ligand exchange processes, may consequently modulate the metal's catalytic activity. To substantiate our hypothesis, we have devised a mechanistic study for the uncaging reaction of an O-allenyl protected coumarin substrate, facilitated by palladium complexes (Scheme 1A). While the allenyl group has recently been demonstrated to be a highly potent bioorthogonal caging group, exhibiting sensitivity to a diverse range of transition metals,29,30 it remains notably underexplored in bioorthogonal uncaging reactions compared to allyl and propargyl groups. This underutilization likely stems from a lack of mechanistic insights and a comprehensive understanding of its reactivity. As such, our work focuses on assessing the deliberate impact of chloride ion concentration on the C–O bond cleavage rate and yield, harnessing kinetic, energetic, and structural analyses.
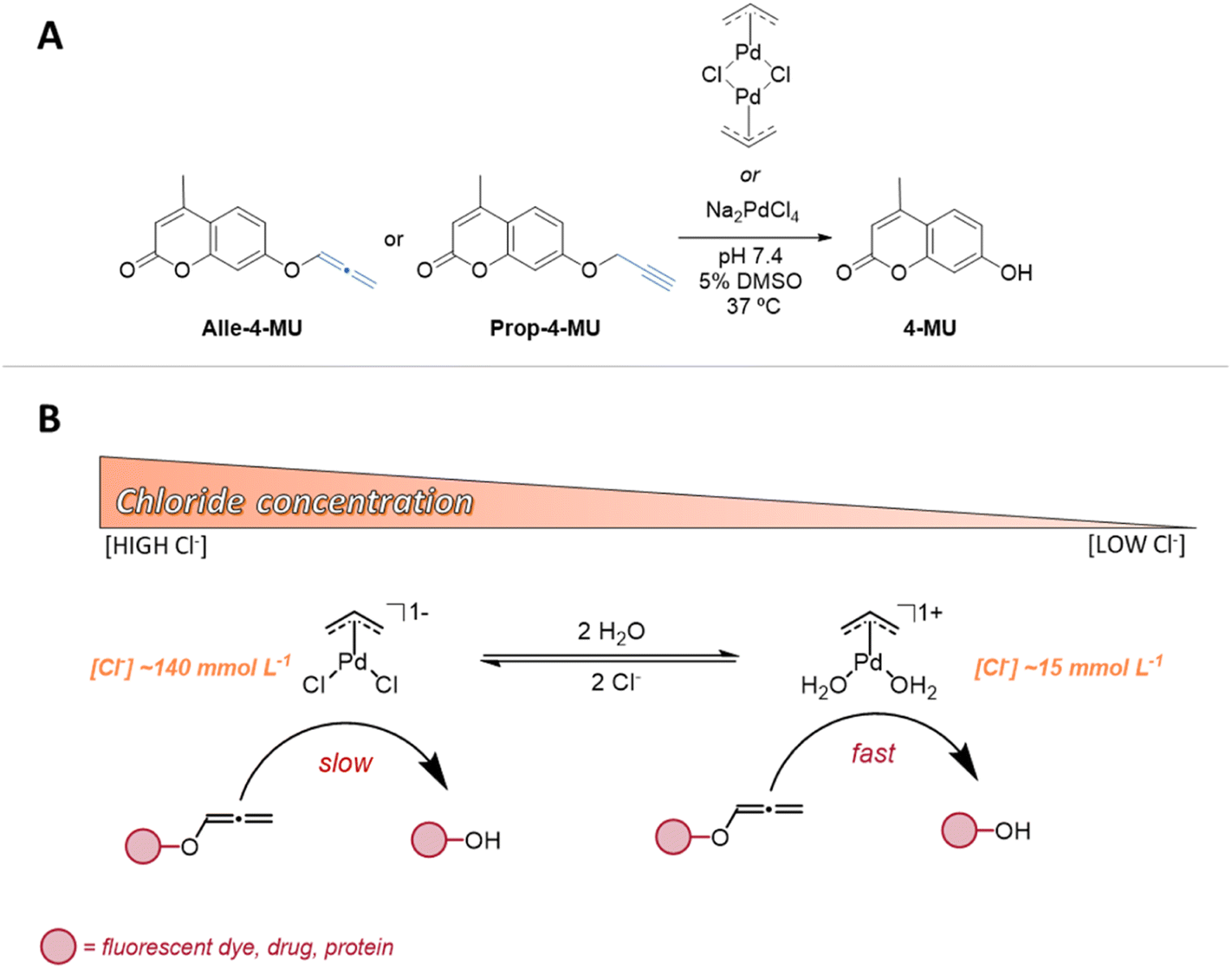 | ||
| Scheme 1 (A) Uncaging reaction of allenyl and propargyl protected 4-methylumbelliferone (4-MU) triggered by palladium(II) complexes. (B) Effect of chloride in the O-deallenylation uncaging reaction mediated by the Allyl2Pd2Cl2 catalyst. | ||
In alignment with our hypothesis, our findings indicate that differences in chloride ion concentration indeed directly affect the structure of the Allyl2Pd2Cl2 catalyst, thus tuning the catalytic activity of the metal through ligand exchange processes (Scheme 1B). This study aims to shed light on these specific interactions and contribute to the broader understanding of bioorthogonal uncaging reactions by metal complexes.
Results and discussion
In the initial phase of our mechanistic study, we set out to determine the kinetics of the uncaging reaction, specifically, the C–O bond-cleavage of the 1,2-allenyl protected 4-methylumbelliferone (Alle-4-MU). This reaction was triggered by two distinct Pd(II) complexes: sodium tetrachloropalladate salt (Na2PdCl4) and the π-allyl dimer (Allyl2Pd2Cl2), as depicted in Scheme 1. While the primary focus of this study centers on the uncaging of Alle-4-MU, we incorporated the widely recognized propargyl caged substrate (Prop-4-MU) for comparison purposes. Crucially, in all experiments, substoichiometric quantities of metal catalysts were employed. This approach not only aligns with standard protocols for in-depth catalytic mechanistic studies31,32 but also positions the study well for subsequent biological applications. It's worth noting that the majority of reported uncaging reaction studies deviate from this approach, often using greater amounts of the metal systems. This divergence can obscure the understanding of the catalytic mechanism. Monitoring of the reactions was performed in an aqueous phosphate-buffered medium (pH 7.4) using UV-vis spectroscopy, wherein the appearance of the phenolate product (4-MU) was detected at a wavelength of 364 nm (Fig. S1†).Our preliminary kinetic evaluations were conducted using both substrates, with phosphate-buffered saline (PBS, [Cl−] = 140 mmol L−1) as the reaction medium and Na2PdCl4 as the catalyst. As illustrated in the kinetic profiles (Fig. S2 and S3†), the reaction involving the substrate Alle-4-MU exhibited a half-life (t1/2) of approximately 1.8 min. This is significantly shorter than the half-life observed for the Prop-4-MU substrate (t1/2 = 19 minutes). This highlights the inherently superior reactivity of the allenyl caging group. In every instance, two turnovers for product formation were noted. Under these conditions, the reaction with Alle-4-MU demonstrated a biphasic kinetic profile, suggesting two parallel reactions, each yielding the same product but at different rate constants. A comparable kinetic pattern was previously identified in an O-depropargylation reaction catalyzed by Na2PdCl4.33 That study postulated that the initial phase of the kinetic profile represented a swift reaction concluding after two turnovers, attributable to product inhibition of the PdCl42− complex. In contrast, the subsequent phase was slower, yielding the same product, and was catalyzed by Pd(0) nanoparticles generated during the first phase. For O-deallenylation, the catalyst also undergoes modification after the first turnover, leading to a shift in the mechanism, but there was no indication that Pd(0)-NPs were being formed, suggesting that the participation of a Pd(II) species is most likely. Through deconvolution of these biphasic kinetics profiles, we were able to discern the two pseudo first-order rate constants (Fig. S4 and S5†). Notably, the initial phase is approximately 20 times faster than the second one.
Contrasting the reactivity of the bioorthogonal reactant pairs, the second-order rate constants (k2) were determined (entries 1–4, Table 1, and Fig. S6 and S7†). These results clearly highlighted the superior reactivity of the allenyl substrate, especially in the initial phase of the reaction, with rate constants approximately 30 times higher than those observed in the depropargylation reaction, considering the initial phase of the biphasic kinetics. When utilizing the Allyl2Pd2Cl2 catalyst, which is often preferred in bioorthogonal reactions due to its efficacy and biocompatibility, the deallenylation reaction remains faster and yields better results than the depropargylation reaction in the PBS medium (entries 7–10, Table 1, Fig. S8 and S9†). However, its reactivity is not as pronounced as when the Na2PdCl4 salt is applied.
| Entry | Catalyst | Substrate | [NaCl] (mmol L−1) | k 2 (L mol−1 s−1) | [Product]b (%) |
|---|---|---|---|---|---|
| a Aqueous phosphate-buffered media (ionic strength (I) = 140 mmol L−1 (NaClO4)). b Maximum product conversion in 180 min of reaction with 25 mol% of Pd. c For the initial phase. d Second phase of the biphasic kinetics. | |||||
| 1 | Na2PdCl4 | Prop | 140 | 3.4 | 49 |
| 2 | Na2PdCl4 | Alle | 140 | 104.1,c 2.0d | 50 |
| 3 | Na2PdCl4 | Prop | 0 | 3.5 | 48 |
| 4 | Na2PdCl4 | Alle | 0 | 15.2 | 57 |
| 5 | Allyl2Pd2Cl2 | Alle | 0 | 88.9 | 84 |
| 6 | Allyl2Pd2Cl2 | Alle | 15 | 19.8 | 71 |
| 7 | Allyl2Pd2Cl2 | Alle | 140 | 2.0 | 52 |
| 8 | Allyl2Pd2Cl2 | Prop | 0 | 56.0 | 37 |
| 9 | Allyl2Pd2Cl2 | Prop | 15 | 4.5 | 39 |
| 10 | Allyl2Pd2Cl2 | Prop | 140 | 0.4 | 15 |
We subsequently examined the impact of chloride concentration on the kinetics of the uncaging reaction for both substrates. Comparisons were made between the reactions in PBS ([Cl−] = 140 mmol L−1) and PB ([Cl−] = 0 mmol L−1), at pH 7.4. Using Na2PdCl4 as the catalyst and the Prop-4-MU substrate, a minimal difference was noted (entries 1 and 3, Table 1, and Fig. S10†). For the Alle-4-MU substrate, the biphasic kinetic profile in PBS transitioned to a monophasic profile in PB (entries 2 and 4, Table 1 and Fig. S11†), with k2 about 7 times larger than in the slow phase in PBS. When the Allyl2Pd2Cl2 catalyst was employed, there was a pronounced difference in the kinetics (Fig. S12 and S13†). In the chloride-free buffer (PB), both reactions were markedly faster and achieved higher yields, particularly for the deallenylation reaction (entries 5–10, Table 1).
A paramount criterion for a bioorthogonal reaction is rapid kinetics, ideally exhibiting second-order rate constants greater than 10 L mol−1 s−1.26 Remarkably, the reaction of Alle-4-MU, catalyzed by Allyl2Pd2Cl2 in PB, demonstrated an outstanding second-order rate constant of 88.9 L mol−1 s−1, achieving an 84% product conversion (entry 5, Table 1, and Fig. S13 and S14†). This rate is comparable to those seen in other fast bioorthogonal reactions, such as cycloaddition reactions like SPAAC, SPANC, photoclick 1,3-dipolar, CuAAC, and IEDDA.34
In the PBS medium, the deallenylation reaction exhibited significantly reduced reactivity, highlighting the pivotal role of the chloride ion in this process. Consequently, we investigated the effect of varying chloride concentrations. We systematically adjusted this concentration by incorporating different amounts of NaCl into the buffer solution. As illustrated in Fig. 1, even minor changes in chloride concentration markedly influenced the deallenylation reaction rates when promoted by Allyl2Pd2Cl2. Comparable with deallenylation, chloride variation showed the same trend in the rate of depropargylation reaction mediated by Allyl2Pd2Cl2 (Fig. S17†), however with lower yields and rates (entries 8–10, Table 1).
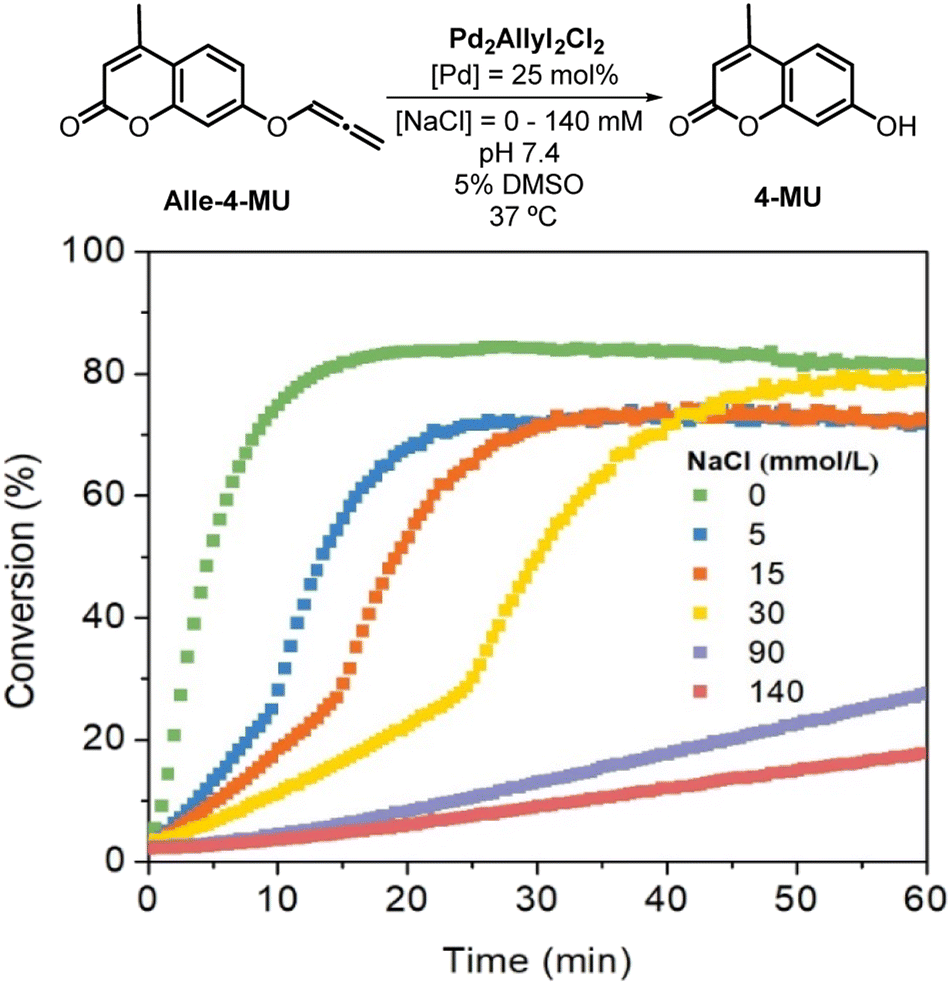 | ||
| Fig. 1 Uncaging reaction of Alle-4-MU promoted by Allyl2Pd2Cl2 in phosphate buffer with different NaCl concentrations (25 mol% of Pd, 37 °C, pH 7.4), ionic strength (I = 140 mmol L−1 (NaClO4)). | ||
To comprehensively elucidate the role of chloride in these reactions, we explored deeper into mechanistic details. The second step of our mechanistic study aimed at characterizing the nature of the active catalytic species. Initially, we probed the impact of Pd oxidation state [Pd(II) vs. Pd(0)] on the reaction kinetics of both substrates. This was done by incorporating 10 equiv. of sodium ascorbate (SA) or 2.5 equiv. of EDTA into the reaction medium, with a palladium concentration of 25 mol%. SA is frequently employed as a reducing agent to generate Pd(0)-NPs from Na2PdCl4; upon its addition, the solution swiftly transitions to a light grey hue. Notably, this phenomenon was absent when using the Allyl2Pd2Cl2 complex. Neither the solution color nor the UV-vis spectrum of the complex was influenced by the presence of SA, suggesting that Allyl2Pd2Cl2 is not reduced by SA. After allowing SA to incubate in the reaction mixture for 10 min, the substrate was introduced to initiate the reaction. This pre-incubation period was essential to ensure the complete reduction of Pd(II) to Pd(0). Intriguingly, slight product formation was noted after 90 min of reaction with Na2PdCl4 (Fig. S18 and S19†). This suggests that the in situ formed Pd(0) is not particularly active in the deallenylation reaction. In contrast, when using the Allyl2Pd2Cl2 complex, the addition of SA brought about negligible changes to the kinetic profiles (Fig. S20–S23†). This validates that the catalyst remained largely unaffected by SA under the given reaction conditions.
Furthermore, introducing EDTA into the medium markedly decelerated both reactions (Fig. S24–S27†). Given that EDTA is a chelating agent, it readily forms stable complexes with Pd(II). This complexation potentially hinders substrate coordination and subsequent catalysis.35 These findings robustly advocate for the involvement of a Pd(II) catalytically active species in both the deallenylation and depropargylation reactions. Moving forward, our investigations focused solely on the Allyl2Pd2Cl2 catalyst, as Na2PdCl4 has shown greater vulnerability to the reductive conditions inherent within cellular environments.36
Earlier spectroscopic and potentiometric investigations on Allyl2Pd2Cl2 in aqueous media revealed an equilibrium among various monomeric π-AllylPd species, contingent upon both [Cl−] and pH levels. In environments with high chloride ion concentrations, the solution predominantly comprises AllylPdCl2. Conversely, in the absence of chloride and at neutral pH, AllylPd(H2O)2 emerges as the principal form.37,38
In alignment with this observation, the ligand exchange between chloride and water was followed by UV-vis spectral scanning kinetics (Fig. S28†) with a pre-formed AllylPd(H2O)2 species in a PB medium with the addition of NaClaq (140 mmol L−1) at 5 min. The disappearance of the AllylPd(H2O)2 and appearance of chloride species [AllylPdCl(H2O) and AllylPdCl2] occur at 275 and 360 nm, respectively. The first-order kinetics showed that the ligand exchange possesses a reaction half-life of 42 s, demonstrating that the ligand exchange is a fast process and indicating that the choice of starting species may not influence the catalytic process due to their rapid interconversion. To further evaluate the stability of the π-allyl ligand, we conducted a 1H NMR analysis on Allyl2Pd2Cl2 in D2O. The spectrum predominantly displayed the signals associated with the η3-allylic protons. These signals remained consistent throughout a 140 min kinetic assessment, indicating that no significant modifications, such as hydration, occurred (Fig. S29†). When sodium chloride was introduced at a concentration of 140 mmol L−1, the only discernible alteration in the 1H NMR signals was a reduction in the coupling constants (Fig. S30†). This is likely attributable to shifts in the electronic charge distribution within the solution. We also conducted Synchrotron X-ray Absorption Spectroscopy (XAS) analysis to assess the presence of Pd species in the solution. Three samples of Allyl2Pd2Cl2 were analyzed, both in PB and PBS buffers, as well as in the solid state. The XANES spectra in Fig. S31† clearly indicate that the oxidation state of Pd has not been affected by the composition of the buffers. Furthermore, the EXAFS analysis reveals that in the solid form and in PBS, only Pd–Cl and Pd–C bonds are observed, with coordination numbers NPd–C and NPd–Cl of 2 and 3, respectively, in both cases (Fig. S32a and Table S1†). Conversely, in the PB solution, a signal is observed for the Pd–O bond (NPd–O = 1.09, NPd–C = 2.5 and NPd–Cl = 2.0). These XAS analyses have confirmed, albeit under different conditions compared to the kinetic experiments, that chloride ions are more readily replaced by water molecules at lower chloride concentrations.
The collected evidence regarding the nature of the palladium's active catalytic species is concisely illustrated in Scheme 2. Within aqueous solutions, Allyl2Pd2Cl2 predominantly exists as three primary π-AllylPd species: the dichloride, the mono-aquo, and the diaquo forms. All these species potentially participate in the uncaging reactions. Given the chloride concentration used in the kinetics (Fig. 1) and by referencing the equilibrium constants K1 and K2 from the literature under analogous conditions,37 we calculated the proportion of each of the three π-AllylPd species present in solution (Table 2).
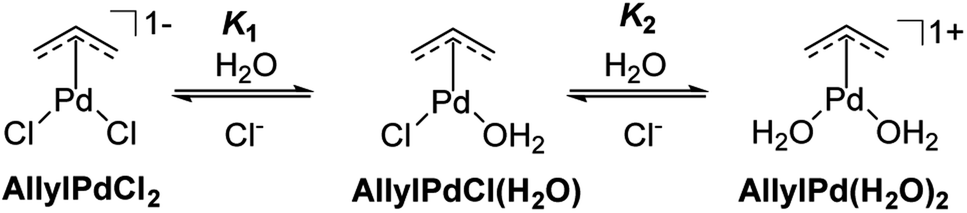 | ||
| Scheme 2 π-AllylPd species in equilibrium derived from Allyl2Pd2Cl2 in aqueous solution. | ||
| Entry | [Cl−] (mmol L−1) | AllylPdCl2 (%) | AllylPdCl(H2O) (%) | AllylPd(H2O)2 (%) |
|---|---|---|---|---|
| a The equilibrium constants used for the calculations are the ones reported in the literature: K1 = 2.5 × 10−2. K2 = 5.0 × 10−3 (25 °C, I = 100 mmol L−1 (NaClO4)).37 | ||||
| 1 | 0 | 0 | 0.5 | 99.5 |
| 2 | 5 | 9.0 | 45.2 | 45.8 |
| 3 | 15 | 31.0 | 51.6 | 17.4 |
| 4 | 30 | 50.4 | 42.0 | 7.6 |
| 5 | 90 | 77.2 | 21.5 | 1.3 |
| 6 | 140 | 84.4 | 15.1 | 0.5 |
From the accumulated data, including those presented in Tables 1 and 2, a distinct trend emerges: the rate of the deallenylation reaction using the Allyl2Pd2Cl2 complex increases as the chloride concentration decreases. This increase is attributable to the growing concentration of a more active species, AllylPd(H2O)2. At a chloride concentration of 140 mmol L−1 (resembling extracellular conditions), the proportion of AllylPd(H2O)2 is merely 0.5%, corresponding to a k2 value of 2.0 L mol−1 s−1. In contrast, at 15 mmol L−1 of chloride (emulating intracellular conditions), the proportion of AllylPd(H2O)2 rises to 17.4% and is associated with a k2 value of 19.8 L mol−1 s−1. At 0 mmol L−1 of chloride (with 99.5% of AllylPd(H2O)2), the k2 value reaches 88.9 L mol−1 s−1. This pattern suggests that the reactivity of the bioorthogonal reactant pairs is directly impacted by the concentration of the diaquo form of the π-AllylPd catalyst. Extrapolating these findings to biological contexts, one could reasonably infer that this deallenylation reaction mediated by the Allyl2Pd2Cl2 reaction would be more effective within the intracellular environment than in the extracellular milieu, with the diaquo form emerging as the most active catalytic species. Previous studies demonstrate that the Allyl2Pd2Cl2 catalyst enters cells effectively, facilitating the uncaging of allenyl and propargyl groups.11,31 Despite the similar transformation of the drug/catalyst cisplatin (Pt(NH3)2Cl2) inside the cell, where it transitions into its more active aquo forms, specifically [Pt(NH3)2Cl(H2O)]1+ and [Pt(NH3)2(H2O)2]2+,39 there is no straightforward explanation for why the aquo forms would also be more active than the dichloride Pd species in the uncaging reactions presented here.
To further substantiate our findings, we investigated the deallenylation reaction using ESI-MS analysis in both positive and negative ion modes (Fig. S33–S36†). This reaction was conducted in the presence of 12.5 mol% of Pd2Ally2Cl2 (equivalent to 25 mol% of Pd) in water at 37 °C. Three aliquots were sampled at distinct time points: immediately after mixing the reagents (0 min), and then at 15 and 30 min into the reaction.
In the positive ion mode at the beginning of the reaction, the predominant peak was identified as a cluster comprising two substrate molecules and a sodium ion (m/z 451.1), which diminished rapidly thereafter. Several π-AllylPd species were discerned in this mode. The major species include: [AllyPd]+ (m/z 146.9), [AllylPd-DMSO]+ (m/z 224.9), [AllylPd-(Alle-4-MU)]+ (m/z 361.0), and [AllylPd-(Alle-4-MU)2]+ (m/z 575.1), all of which exhibited high intensities. In the negative ion mode, the predominant peak was attributed to the product [4-MU]− (m/z = 174.7). The array of species detected in the solution can be attributed to a combination of factors. These encompass solvent–ligand exchange dynamics in the solution and the ion-molecule as well as ion–ion interactions occurring therein. Additionally, the analysis underscores the high stability of the [π-AllylPd]+ species and its aptitude for effectively coordinating to substrate molecules. The species identified via ESI-MS, in conjunction with our kinetic and additives introduction study findings, suggest a mechanism that initiates with the substrate coordinating to the AllylPd(H2O) species. This is followed by hydration of the allene moiety, culminating in hydrolysis to yield the phenol product, thereby regenerating the diaquo form of the π-AllylPd catalyst.
The deallylation reaction mechanism was computationally explored using quantum mechanics (Fig. 2 and S37†). The initial step involves the coordination of Alle-4-MU (abbreviated as A) to the diaquo η3-allylpalladium(II) (AllylPd(H2O)2) catalyst via the inner π-bond of the allene (B), accompanied by the release of a water molecule. Notably, this complex exhibits low reactivity, given that no transition structure was identified for the direct water molecule attack on the inner carbon atom of the allene. Moreover, high activation barriers were computed for water molecule attacks on both the central (ΔG‡ ≈ 39 kcal mol−1) and terminal (ΔG‡ approximates to 32 or 34 kcal mol−1) carbon atoms of the allene B (Fig. S38†). These pathways, however, are dead-end pathways. Conversely, while the σ-coordination of Pd to the allene's central carbon atom (C) is thermodynamically less favored (ΔGσ–π is roughly 17 kcal mol−1), it results in a more reactive configuration. As such, a water molecule attacking the allene's inner carbon atom [TS1, with ΔG‡ about 31 kcal mol−1 as the rate-determining step (RDS)] evolves into intermediate D. This intermediate undergoes rapid intramolecular proton transfer (E*), leading to the barrierless decaging of 4-MU (TS2*) and subsequent discharge of σ-complex F. Notably, only high-energy geometries could be fully optimized for these species (E* and TS2*), as relaxed scans along the breaking C–O bond from lower-energy conformations always show downhill profiles (Fig. S39†). Remarkably, π-complex G exhibits greater stability than σ-complex F (ΔGσ–π approximates to 9 kcal mol−1). A ligand exchange involving this species and a water molecule regenerates the AllylPd(H2O)2 catalyst, releasing hydroxyallene (H) in the process. Ultimately, this hydroxyallene tautomerizes to acrolein, serving as the thermodynamic driver for the entire mechanism (ΔG is about −24 kcal mol−1). It's crucial to underline that this mechanism effectively includes the hydrolysis of acrolein's carbonyl group while masked as an enol, that is, a protected hydroxyallene.
In exploring the reaction profile with the dichloride η3-allylpalladium(II) (AllylPdCl2) as a catalyst, the energy was calculated to be approximately 7 kcal mol−1 higher (Fig. 2 and S40†). This translates to a notably slower reaction rate. One possible explanation for this outcome is the neutral nature of complex C′, which stands in contrast to the positively charged C (+1). Consequently, C exhibits greater electrophilicity. Moreover, the water molecule coordinated to Pd aids in stabilizing both ROH and/or H2O species present in the transition structures and intermediates. These computational insights align with experimental observations, suggesting that in media with elevated chloride concentrations, the catalyst's activity diminishes.
The decaging mechanism for propargyl derivatives was similarly assessed using Prop-4-MU (Aalkyne) (Fig. S41 and S42†). For this mechanism, water is able to react with the π-complex Balkyne, targeting the terminal carbon atom (TS1alkyne). Notably, this attack presents a higher activation barrier (ΔG‡ ≈ 34 kcal mol−1) compared to its allene counterpart. Following this, the hydrated intermediate, Dalkyne, can directly undergo concomitant intramolecular proton transfer and decaging of 4-MU with a comparable activation energy (TS2alkyne, ΔG‡ ≈ 35 kcal mol−1), culminating in the release of the σ-complex F. Consequently, the resulting products from the alkyne derivative decaging align with those discerned from the allene derivative, specifically, 4-MU and acrolein. However, the kinetics of the former are anticipated to be slower, given the approximately 4–5 kcal mol−1 higher activation energy of the calculated reaction pathway.
The theoretical results agree with the experimentally determined activation parameters for the deallylation reaction in a chloride-free medium (PB), using 12.5 mol% of Pd2Ally2Cl2 (25 mol% of Pd), as observed from the Eyring plot (Fig. S43†). The derived experimental values for the enthalpy of activation (ΔH‡ = 8.18 kcal mol−1) and the entropy of activation (ΔS‡ = −35.30 cal mol−1 K−1) are consistent with our proposed mechanism. Specifically, these values hint at the formation of a single bond during the hydration step, which aligns with the rate-determining step depicted in Fig. 2 (TS1). Furthermore, we scrutinized the impact of ionic strength on the reaction kinetics, as detailed in Fig. S44,† by plotting the logarithm of rate constants against the square root of ionic strength. The observed primary salt effect suggests the formation of a transition state carrying a unit charge.
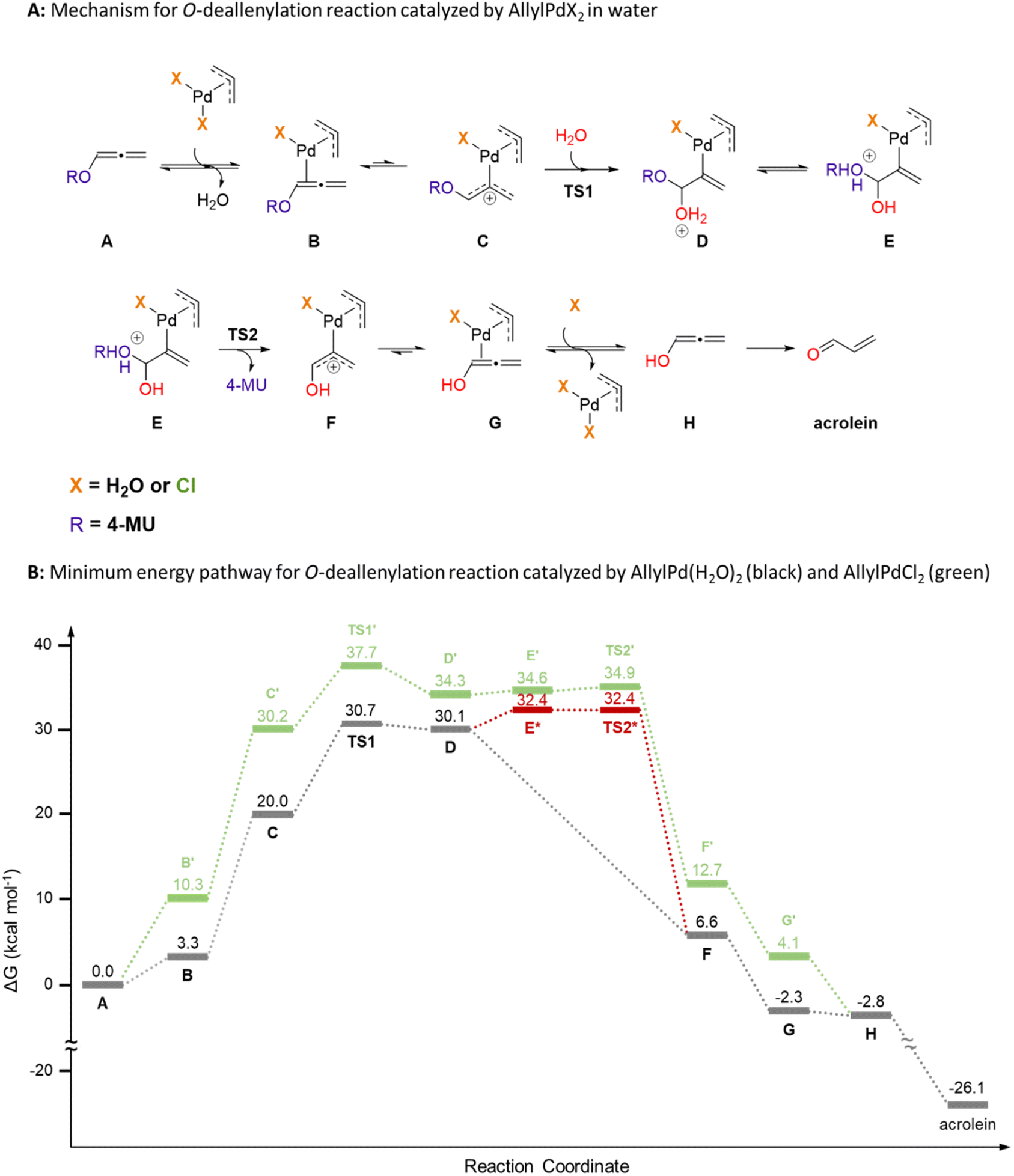 | ||
| Fig. 2 Proposed mechanism and minimum energy pathway (MEP) calculated with PCM(H2O)/M06-2X/6-311+G(d,p)+SDD(Pd) for the deallenylation reaction of Alle-4-MU (abbreviated as A). (A) Mechanism for the deallenylation reaction catalyzed by η3-allylpalladium(II) complexes in water. (B) Minimum energy pathway for the deallenylation reaction mediated by AllylPd(H2O)2 (black) and AllylPdCl2 (green). For the stationary points E* and TS2* (in red) only high-energy conformations could be fully optimized. Therefore, the MEP is proposed to proceed downhill to decaged product F after tautomerization of intermediate D. | ||
Conclusions
We have demonstrated that the Allyl2Pd2Cl2 complex acts as an efficient catalyst for the O-deallenylation reaction under biocompatible conditions, exhibiting second-order kinetics with reaction rates reaching up to 89 L mol−1 s−1. This rate shows significant sensitivity to chloride ion concentrations, increasing as the chloride concentration decreases. This phenomenon is likely due to the higher concentration of the more catalytically active diaquo form at lower chloride concentrations. Theoretical modeling suggests that the aquo intermediate involved in the rate-determining step is more electrophilic and more stabilized than its chloride-containing counterpart. These findings are crucial for biological applications, given the typical chloride concentration range of 4 to 140 mmol L−1 in cellular environments. Our insights facilitate the design of novel Pd–allyl catalysts specifically tailored for the selective uncaging of drugs or proteins within distinct cellular settings. For example, designing Pd–allyl complexes that exhibit slower ligand exchange with water could optimize targeted delivery in the extracellular matrix. Conversely, utilizing Pd–allyl complexes with varied substituents on the allyl group might increase the deallenylation reaction rate in intracellular environments. Although the precise catalytic behavior of these reactions in biological media, such as cell cultures and animal models, has yet to be fully determined, our current knowledge on the mechanism of action of these common palladium complexes lays the groundwork for developing highly efficient biorthogonal catalysts.Data availability
We have provided all relevant data in the ESI† of the paper.Author contributions
G. M. D. F. conceptualized the study, conducted experiments, and wrote the original draft of the manuscript. E. L. analyzed the data and contributed to the manuscript writing. A. L. S. J. performed some kinetic experiments. B. L. A. conducted XAS data analysis. C. D. N. and G. J.-O. carried out the computational studies and contributed to the manuscript writing. F. A. analyzed the data. J. B. D. conceived and supervised the study, analyzed the data, and contributed to the writing of the original draft.Conflicts of interest
The authors declare no competing interests.Acknowledgements
We are grateful to the Brazilian National Council for Scientific and Technological Development (CNPq) for the fellowships awarded to GMDF, EL, ALSJ, and JBD, as well as financial support (process 408534/2022-2). We also extend our gratitude to the Agencia Estatal Investigacion of Spain (AEI) for grants PID2021-125946OB-I00 to GJO and CEX2021-001136-S to CIC bioGUNE. Additionally, the authors are thankful to CAPES PrInt Call – Program for Institutional Internationalization for supporting JBD and FA (process 88887.310560/2018-00). This research used facilities of the Brazilian Synchrotron Light Laboratory (LNLS). The EMA beamline staff is acknowledged for the assistance during the experiments under the proposal 20221845. The authors also thank LABIME-UFSC for help with ESI-MS analysis.Notes and references
- J. Wang, X. Wang, X. Fan and P. R. Chen, ACS Cent. Sci., 2021, 7, 929–943 CrossRef CAS PubMed.
- M. O. N. Van De L’Isle, M. C. Ortega-Liebana and A. Unciti-Broceta, Curr. Opin. Chem. Biol., 2021, 61, 32–42 CrossRef PubMed.
- A. Seoane and J. L. Mascareñas, Eur. J. Org Chem., 2022, 2022, e202200118 CrossRef CAS PubMed.
- C.-M. Hirschbiegel, X. Zhang, R. Huang, Y. A. Cicek, S. Fedeli and V. M. Rotello, Adv. Drug Delivery Rev., 2023, 195, 114730 CrossRef CAS PubMed.
- A. Sathyan, L. Deng, T. Loman and A. R. A. Palmans, Catal. Today, 2023, 418, 114116 CrossRef CAS.
- H. Madec, F. Figueiredo, K. Cariou, S. Roland, M. Sollogoub and G. Gasser, Chem. Sci., 2023, 14, 409–442 RSC.
- E. Latocheski, G. M. D. Forno, T. M. Ferreira, B. L. Oliveira, G. J. L. Bernardes and J. B. Domingos, Chem. Soc. Rev., 2020, 49, 7710–7729 RSC.
- S. L. Scinto, D. A. Bilodeau, R. Hincapie, W. Lee, S. S. Nguyen, M. Xu, C. W. am Ende, M. G. Finn, K. Lang, Q. Lin, J. P. Pezacki, J. A. Prescher, M. S. Robillard and J. M. Fox, Nat. Rev. Methods Primers, 2021, 1, 1–23 CrossRef.
- G. M. Dal Forno, E. Latocheski, A. Beatriz Machado, J. Becher, L. Dunsmore, A. L. St. John, B. L. Oliveira, C. D. Navo, G. Jiménez-Osés, R. Fior, J. B. Domingos and G. J. L. Bernardes, J. Am. Chem. Soc., 2023, 145, 10790–10799 CrossRef CAS PubMed.
- H. Ai, J. W. Lee and P. G. Schultz, Chem. Commun., 2010, 46, 5506–5508 RSC.
- J. Li, J. Yu, J. Zhao, J. Wang, S. Zheng, S. Lin, L. Chen, M. Yang, S. Jia, X. Zhang and P. R. Chen, Nat. Chem., 2014, 6, 352–361 CrossRef CAS PubMed.
- X. Wang, Y. Liu, X. Fan, J. Wang, W. S. C. Ngai, H. Zhang, J. Li, G. Zhang, J. Lin and P. R. Chen, J. Am. Chem. Soc., 2019, 141, 17133–17141 CrossRef CAS PubMed.
- J. T. Weiss, J. C. Dawson, C. Fraser, W. Rybski, C. Torres-Sánchez, M. Bradley, E. E. Patton, N. O. Carragher and A. Unciti-Broceta, J. Med. Chem., 2014, 57, 5395–5404 CrossRef CAS PubMed.
- T. Völker, F. Dempwolff, P. L. Graumann and E. Meggers, Angew. Chem., Int. Ed., 2014, 53, 10536–10540 CrossRef PubMed.
- J. T. Weiss, J. C. Dawson, K. G. Macleod, W. Rybski, C. Fraser, C. Torres-Sánchez, E. E. Patton, M. Bradley, N. O. Carragher and A. Unciti-Broceta, Nat. Commun., 2014, 5, 3277 CrossRef PubMed.
- J. T. Weiss, N. O. Carragher and A. Unciti-Broceta, Sci. Rep., 2015, 5, 9329 CrossRef CAS PubMed.
- G. Y. Tonga, Y. Jeong, B. Duncan, T. Mizuhara, R. Mout, R. Das, S. T. Kim, Y.-C. Yeh, B. Yan, S. Hou and V. M. Rotello, Nat. Chem., 2015, 7, 597–603 CrossRef CAS PubMed.
- M. Tomás-Gamasa, M. Martínez-Calvo, J. R. Couceiro and J. L. Mascareñas, Nat. Commun., 2016, 7, 12538 CrossRef PubMed.
- B. Rubio-Ruiz, J. T. Weiss and A. Unciti-Broceta, J. Med. Chem., 2016, 59, 9974–9980 CrossRef CAS PubMed.
- M. A. Miller, B. Askevold, H. Mikula, R. H. Kohler, D. Pirovich and R. Weissleder, Nat. Commun., 2017, 8, 15906 CrossRef CAS PubMed.
- A. M. Pérez-López, B. Rubio-Ruiz, V. Sebastián, L. Hamilton, C. Adam, T. L. Bray, S. Irusta, P. M. Brennan, G. C. Lloyd-Jones, D. Sieger, J. Santamaría and A. Unciti-Broceta, Angew. Chem., Int. Ed., 2017, 56, 12548–12552 CrossRef PubMed.
- B. J. Stenton, B. L. Oliveira, M. J. Matos, L. Sinatra and G. J. L. Bernardes, Chem. Sci., 2018, 9, 4185–4189 RSC.
- M. Sancho-Albero, B. Rubio-Ruiz, A. M. Pérez-López, V. Sebastián, P. Martín-Duque, M. Arruebo, J. Santamaría and A. Unciti-Broceta, Nat. Catal., 2019, 2, 864–872 CrossRef CAS PubMed.
- J. Wang, B. Cheng, J. Li, Z. Zhang, W. Hong, X. Chen and P. R. Chen, Angew. Chem., Int. Ed., 2015, 54, 5364–5368 CrossRef CAS PubMed.
- M. Szponarski, F. Schwizer, T. R. Ward and K. Gademann, Commun. Chem., 2018, 1, 1–10 CrossRef CAS.
- E. M. Sletten and C. R. Bertozzi, Acc. Chem. Res., 2011, 44, 666–676 CrossRef CAS PubMed.
- K. Fagnou and M. Lautens, Angew. Chem., Int. Ed., 2002, 41, 26–47 CrossRef CAS.
- A. Jutand, Appl. Organomet. Chem., 2004, 18, 574–582 CrossRef CAS.
- N. Krause and A. S. Hashmi, Modern Allene Chemistry, Wiley-VCH, 2004 Search PubMed.
- B. Alcaide and P. Almendros, Chem. Soc. Rev., 2014, 43, 2886–2887 RSC.
- J. Wang, S. Zheng, Y. Liu, Z. Zhang, Z. Lin, J. Li, G. Zhang, X. Wang, J. Li and P. R. Chen, J. Am. Chem. Soc., 2016, 138, 15118–15121 CrossRef CAS PubMed.
- M. Martínez-Calvo, J. R. Couceiro, P. Destito, J. Rodríguez, J. Mosquera and J. L. Mascareñas, ACS Catal., 2018, 8, 6055–6061 CrossRef PubMed.
- S. E. Coelho, F. S. S. Schneider, D. C. de Oliveira, G. L. Tripodi, M. N. Eberlin, G. F. Caramori, B. de Souza and J. B. Domingos, ACS Catal., 2019, 3792–3799 CrossRef CAS.
- B. L. Oliveira, Z. Guo and G. J. L. Bernardes, Chem. Soc. Rev., 2017, 46, 4895–4950 RSC.
- B. L. Oliveira, B. J. Stenton, V. B. Unnikrishnan, C. R. de Almeida, J. Conde, M. Negrão, F. S. S. Schneider, C. Cordeiro, M. G. Ferreira, G. F. Caramori, J. B. Domingos, R. Fior and G. J. L. Bernardes, J. Am. Chem. Soc., 2020, 142, 10869–10880 CrossRef CAS PubMed.
- J. Konč, V. Sabatino, E. Jiménez-Moreno, E. Latocheski, L. R. Pérez, J. Day, J. B. Domingos and G. J. L. Bernardes, Angew. Chem., Int. Ed., 2022, 61, e202113519 CrossRef PubMed.
- L. A. Katsman, M. N. Vargaftik, A. P. Belov and Ya. K. Syrkin, Bull. Acad. Sci. USSR, 1971, 20, 1005–1007 CrossRef.
- N. G. Satsko, A. P. Belov, I. I. Moiseev and Y. K. Syrkin, Bull. Acad. Sci. USSR, Div. Chem. Sci., 1971, 20, 2463–2465 CrossRef.
- D. Wang and S. J. Lippard, Nat. Rev. Drug Discovery, 2005, 4, 307–320 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc06408e |
| This journal is © The Royal Society of Chemistry 2024 |