
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIncorporation of a self-immolative spacer enables mechanically triggered dual payload release†
Yu-Ling
Tseng
 ,
Tian
Zeng
and
Maxwell J.
Robb
*
,
Tian
Zeng
and
Maxwell J.
Robb
*
Division of Chemistry and Chemical Engineering, California Institute of Technology, Pasadena, California 91125, USA. E-mail: mrobb@caltech.edu
First published on 20th December 2023
Abstract
Polymers that release functional small molecules in response to mechanical force are promising materials for a variety of applications including drug delivery, catalysis, and sensing. While many different mechanophores have been developed that enable the triggered release of a variety of small molecule payloads, most mechanophores are limited to one specific payload molecule. Here, we leverage the unique fragmentation of a 5-aryloxy-substituted 2-furylcarbinol derivative to design a novel mechanophore capable of the mechanically triggered release of two distinct cargo molecules. Critical to the mechanophore design is the incorporation of a self-immolative spacer to facilitate the release of a second payload. By varying the relative positions of each cargo molecule conjugated to the mechanophore, dual payload release occurs either concurrently or sequentially, demonstrating the ability to fine-tune the release profiles.
Introduction
Polymers that release functional small molecules in response to mechanical force are promising for a range of applications including catalysis, sensing, and drug delivery.1–3 In polymer mechanochemistry, small molecule release is accomplished by using mechanical force to drive productive chemical transformations in stress-responsive molecules termed mechanophores, which are incorporated into polymer chains to achieve force transduction.4–6 Mechanical force is an appealing external stimulus that is routinely applied using a variety of methods including solution-phase ultrasonication, or tension and compression in polymeric materials, which afford spatiotemporal control over mechanophore activation.7 In the context of molecular release, a recently reported method for activating mechanochemical reactions using biocompatible focused ultrasound under physiological conditions presents exciting opportunities for biomedical applications including drug delivery.8 These myriad techniques for external mechanochemical activation coupled with the excellent selectivity and control afforded by molecular structure design have attracted significant interest in the development of mechanophores for small molecule release.9 For example, gem-dichlorocyclopropane mechanophores have been designed to release HCl.10,11 A flex activation strategy has been devised for the liberation of a small molecule furan from an oxanorbornadiene mechanophore12 and the release of catalytically active N-heterocyclic carbenes from carbodiimide adducts.13 Mechanophores have also been developed for the release of carbon monoxide,14,15 metal ions,16–18 and other specific small molecules.19,20The development of mechanophores that release multiple different payloads, however, is limited. Herrmann and Göstl have developed a mechanophore platform based on a judiciously designed disulfide motif with cargo molecules attached via β-carbonate linkages.21 Mechanical cleavage of the disulfide bond initiates a 5-exo-trig cyclization with concomitant cargo release. The mechanophore can be assembled in a convergent fashion such that a distinct cargo molecule is appended via each carbonate linkage on either side of the mechanically active disulfide bond.22 Research has leveraged the bifunctional character of this platform to trigger the concurrent release of a drug molecule and a fluorescent reporter, enabling indirect real-time tracking of drug distribution in vitro following ex situ mechanochemical activation. Combination chemotherapy is another possible application of dual payload release, in which the simultaneous delivery of two or more chemotherapeutic molecules elicits a synergistic therapeutic effect.23 Despite the advantageous simplicity of the disulfide mechanophore platform, its susceptibility to activation via chemical or enzymatic reduction or thiol exchange reactions limits mechanochemical selectivity and payload scope has largely been limited to alcohols and phenols.24
Over the past several years, our group has developed a versatile mechanophore platform enabling the mechanically triggered release of functionally diverse small molecules that relies on masked 2-furylcarbinol derivatives.25–28 Upon mechanochemical activation, the furan–maleimide mechanophore undergoes a formal retro-Diels–Alder reaction to reveal a thermally unstable 2-furylcarbinol derivative, which then spontaneously decomposes to release its covalently bound cargo. This general mechanophore design exhibits excellent selectivity and is capable of efficiently releasing a wide range of molecular payloads.26 In 2021, we described a mechanophore in which an aryloxy substituent is installed at the 5-position of the furan scaffold, serving both as an electron donating group to enhance molecular release and the position of polymer attachment (Scheme 1a).27 The synthesis of this aryloxy-substituted mechanophore is efficient and scalable, while maintaining competency for the release of phenols and even more challenging arylamine payloads on reasonable timescales. Furthermore, we discovered a unique fragmentation pathway for the 5-aryloxy-substituted 2-furylcarbinol derivative that resulted not only in the release of the intended cargo molecule attached via a carbonate or carbamate linker, but also cleavage of the aryloxy group from the furan. We reasoned that attack of the intermediate furfuryl cation by methanol present in solution generated an ortho ester analogue that subsequently collapsed to expel the tyrosol derivative. We further hypothesized that this secondary fragmentation process could potentially be exploited to achieve dual cargo release.29,30
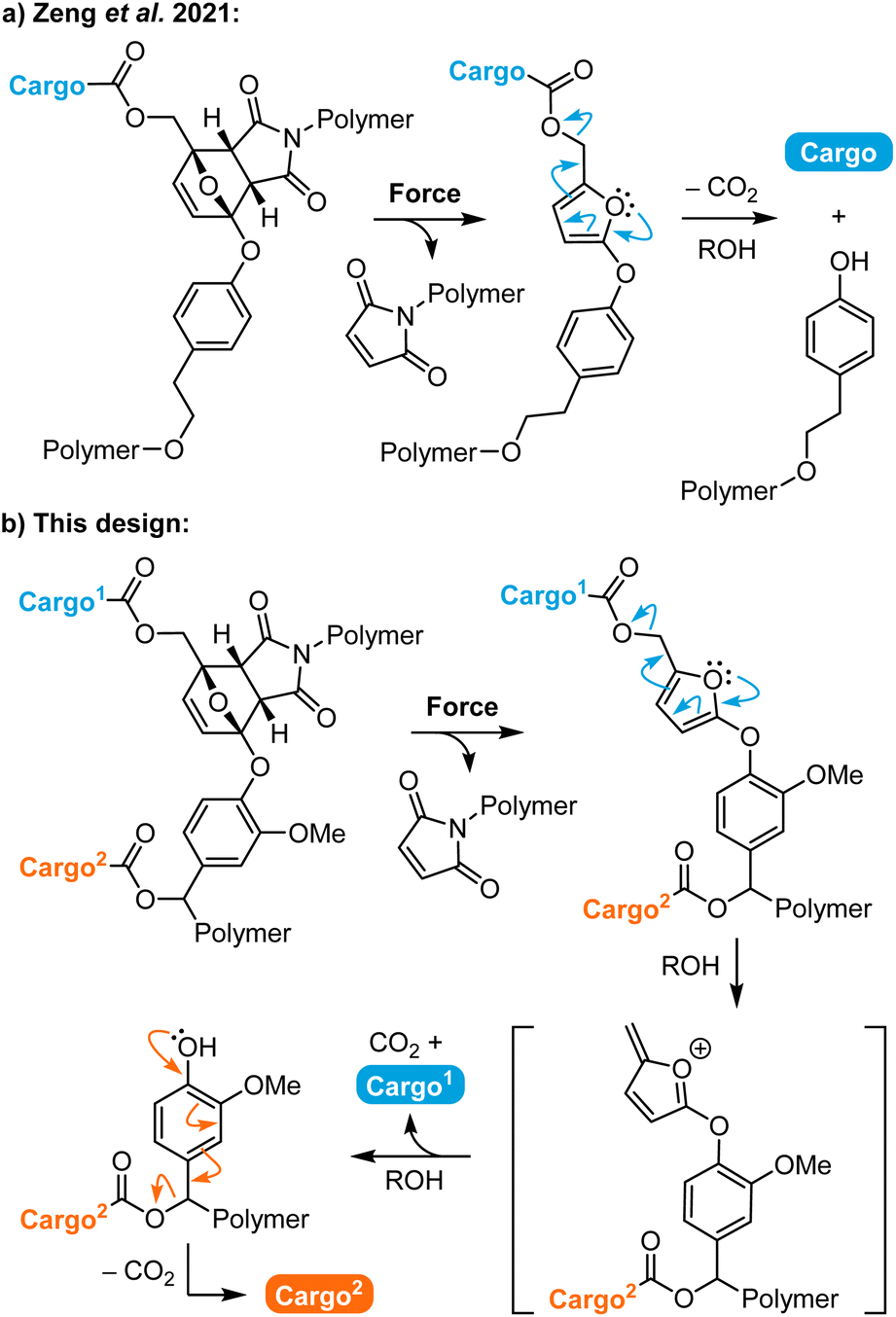 | ||
| Scheme 1 (a) Mechanically triggered small molecule release is accompanied by a secondary fragmentation pathway resulting in expulsion of a tyrosol-functionalized polymer. (b) Incorporation of a self-immolative spacer enables mechanically triggered dual payload release via a multistep retro-Diels–Alder/fragmentation–decarboxylation cascade. | ||
Here, we leverage the unique fragmentation of this 5-aryloxy-substituted 2-furylcarbinol scaffold by incorporating a self-immolative spacer that enables the mechanically triggered release of two distinct cargo molecules from a single mechanophore (Scheme 1b). Upon mechanochemical activation, the furan–maleimide Diels–Alder adduct is expected to undergo a formal retro-[4 + 2] cycloaddition reaction to reveal an unstable 2-furylcarbinol derivative, which decomposes via a furfuryl cation intermediate according to the arrow pushing mechanism shown in Scheme 1 to release the first covalently bound cargo molecule. Further fragmentation unmasks the judiciously designed phenolic spacer, which self-immolates in a 1,6-elimination process and another decarboxylation event to release the second cargo molecule. We characterize the reactivity of this novel mechanophore design through small molecule model experiments, and demonstrate mechanically triggered dual cargo release from polymers upon ultrasound-induced mechanochemical activation. Furthermore, we show that the release profiles for the two different payloads can be substantially modulated by varying their relative attachment position on the mechanophore.
Results and discussion
We first investigated the effect of the self-immolative spacer on the decomposition of 5-aryloxy-substituted furan model compound 1 toward dual cargo release. The model compound incorporates an aryloxy substituent derived from vanillin that is capable of a 1,6-elimination mechanism upon expulsion from the furan.31 For comparison to our prior studies,26,27 we chose to study the release of fluorogenic aminocoumarin (ArNH22) and hydroxycoumarin (ArOH 3) as model payloads, which were connected via carbamate and carbonate linkages, respectively (Fig. 1, see the ESI† for synthetic details). Preliminary experiments performed on a simpler 5-aryloxy-substituted furfuryl carbamate compound indicated that both fragmentation processes illustrated in Scheme 1b occur in close succession.27 Furthermore, the release of phenols via 1,6-elimination of similar self-immolative linkers is expected to be faster than the release of an arylamine via decomposition of the furfuryl carbamate.27,32 Therefore we anticipated that the release of ArNH22 and ArOH 3 would occur on similar timescales, with initial decomposition of the 2-furylcarbinol derivative and the release of ArNH22 being rate determining in this case.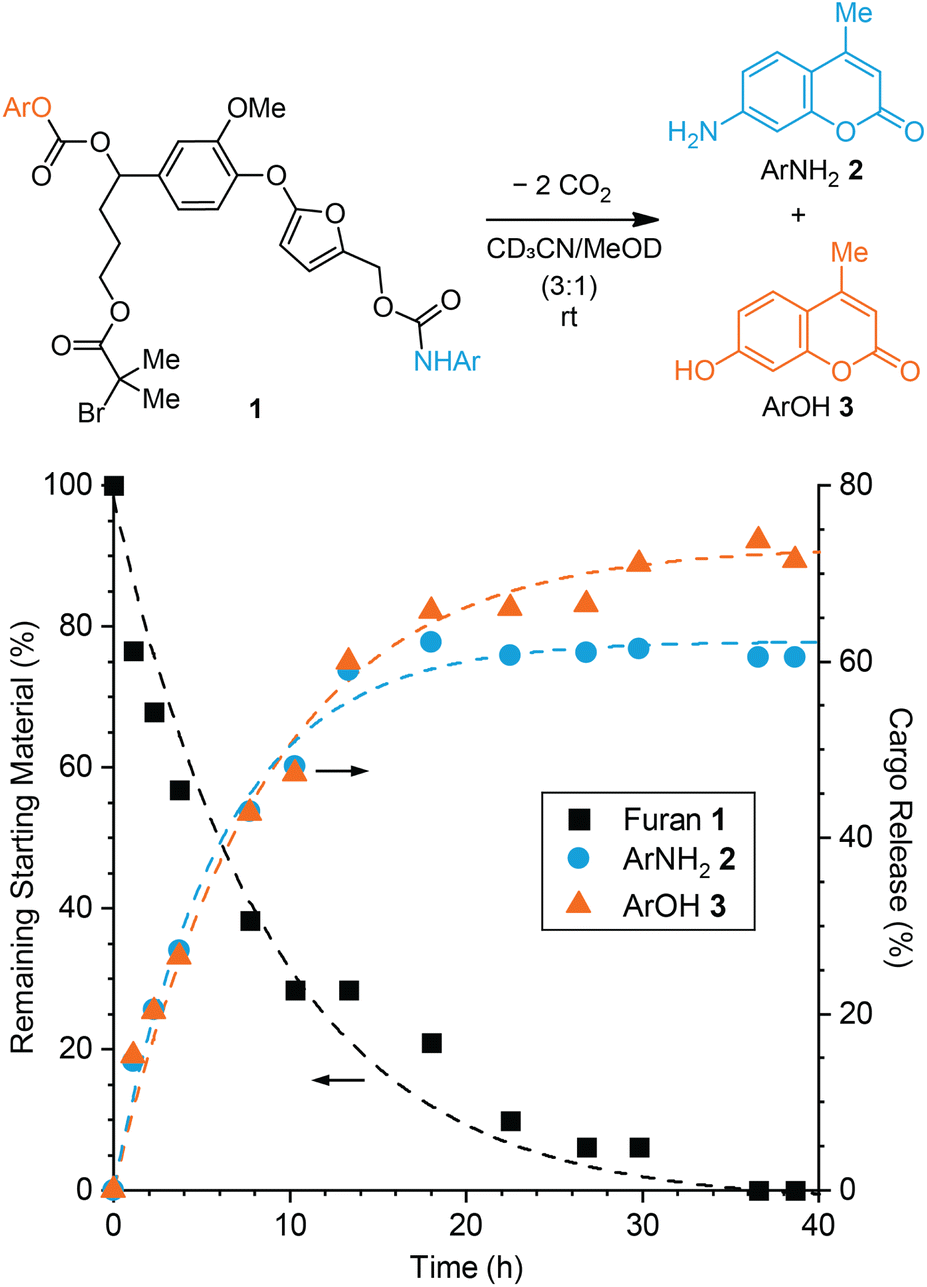 | ||
Fig. 1 Reaction of model compound 1 in polar protic solvent at room temperature produces ArNH22 and ArOH 3via a dual fragmentation–decarboxylation cascade. Time-dependent reaction profiles for the conversion of furan 1 and the generation of 2 and 3 characterized by 1H NMR spectroscopy (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 MeCN-d3/MeOD, [1]0 = 29 mM). A rebound product is also formed under these conditions accounting for an additional ∼32% release of ArNH22 as a result of the relatively high substrate concentration (see ESI† for details). :1 MeCN-d3/MeOD, [1]0 = 29 mM). A rebound product is also formed under these conditions accounting for an additional ∼32% release of ArNH22 as a result of the relatively high substrate concentration (see ESI† for details). | ||
Furfuryl carbamate 1 is transiently stable in aprotic solvents like chloroform and acetonitrile; however, the addition of methanol into an acetonitrile solution of the small molecule initiates fragmentation at room temperature resulting in the release of both payloads.25 To characterize the kinetics of cargo release, model compound 1 was dissolved in CD3CN followed by addition of CD3OD to afford a 29 mM solution (3:1 v/v, respectively) and the reaction was monitored by 1H NMR spectroscopy at room temperature (Fig. 1 and Fig. S1†). Model compound 1 was fully converted to products in 40 h, liberating ArNH22 in ∼61% yield and ArOH 3 in ∼72% yield. The release of ArNH22 and ArOH 3 follows first order kinetics with half-lives of t1/2 = 4 and 6 h, respectively. Release kinetics are roughly 2-fold faster compared to our previously studied 5-aryloxy-substituted 2-furylcarbinol derivative, which we attribute to increased electron density on the aryloxy group due to the methoxy substituent. Another set of peaks was observed in the 1H NMR spectrum that corresponds to the formation of a minor product resulting from rebound of the liberated nucleophilic ArNH22 onto the quinone methide derived from the self-immolative linker (Fig. S2†). As expected,26 the formation of this rebound product is concentration dependent and is not observed in reactions with a lower substrate concentration of 20 μM (Fig. S3†), on the same order of the mechanophore concentration in typical mechanochemical activation experiments (vide infra). Taking the amount of rebound product into account, the total yield of ArNH22 is ∼93% in the experiment described by Fig. 1, illustrating the efficiency with which fragmentation of the furfuryl carbamate occurs. The slightly lower yield of ArOH 3 compared to ArNH22 suggests that incomplete fragmentation or other side reactions occur in the second step involving the self-immolative linker. Nevertheless, the relatively high release yields of both cargo molecules and the similar release kinetics of ArNH22 and ArOH 3 highlight the potential of this platform for mechanically triggered dual payload release.
Encouraged by the reactivity of the small molecule model compound incorporating a self-immolative linker, we next set out to investigate mechanically triggered dual cargo release from a masked 2-furylcarbinol mechanophore incorporated into a polymer. Density functional calculations using the constrained geometries simulate external force (CoGEF) technique33,34 predict the desired formal retro-[4 + 2] cycloaddition reaction of the target furan–maleimide adduct upon mechanical extension leading to the formation of the intended 5-aryloxy-substituted 2-furylcarbinol derivative (Fig. S4†). Therefore, the synthesis commenced from compound 4, which was prepared in three steps from vanillin in 50% overall yield (Scheme 2, see the ESI† for details). Next, deprotection and nucleophilic substitution with 5-bromofurfural afforded furaldehyde 5 in 74% yield over the two steps. Esterification of the primary alcohol proceeded via carbodiimide coupling with α-bromoisobutyric acid to produce 6 in 44% yield. Reduction of the aldehyde with sodium borohydride at −40 °C followed by a [4 + 2] cycloaddition reaction with a pre-functionalized maleimide dienophile at room temperature produced endo-Diels–Alder adduct 7 in 51% yield after isolation via silica gel chromatography. Finally, the two cargo molecules were installed in a two-step sequence, first by selective functionalization of the primary alcohol with a coumarin isocyanate followed by installation of the second cargo with a coumarin chloroformate to generate bis-initiator 8 in 48% yield, which was then employed in the controlled radical polymerization of methyl acrylate with Cu wire/Me6TREN in DMSO35 to afford PMA-1 (Mn = 106 kDa; Đ = 1.13) containing a chain-centered mechanophore. A similar synthetic procedure using N-methylmaleimide was used to prepare chain-end functional polymer PMA-Control (Mn = 98 kDa; Đ = 1.15, see the ESI† for details). The electron rich aryloxy substituent imparts substantial thermal stability to Diels–Alder adduct 7.27 Heating a solution of 7 in toluene-d8 at 70 °C for 5 and 24 h results in only ∼5 and 25% retro-Diels–Alder reaction, respectively, while no retro-Diels–Alder reaction was observed at room temperature over several weeks (Fig. S5 and S6†).
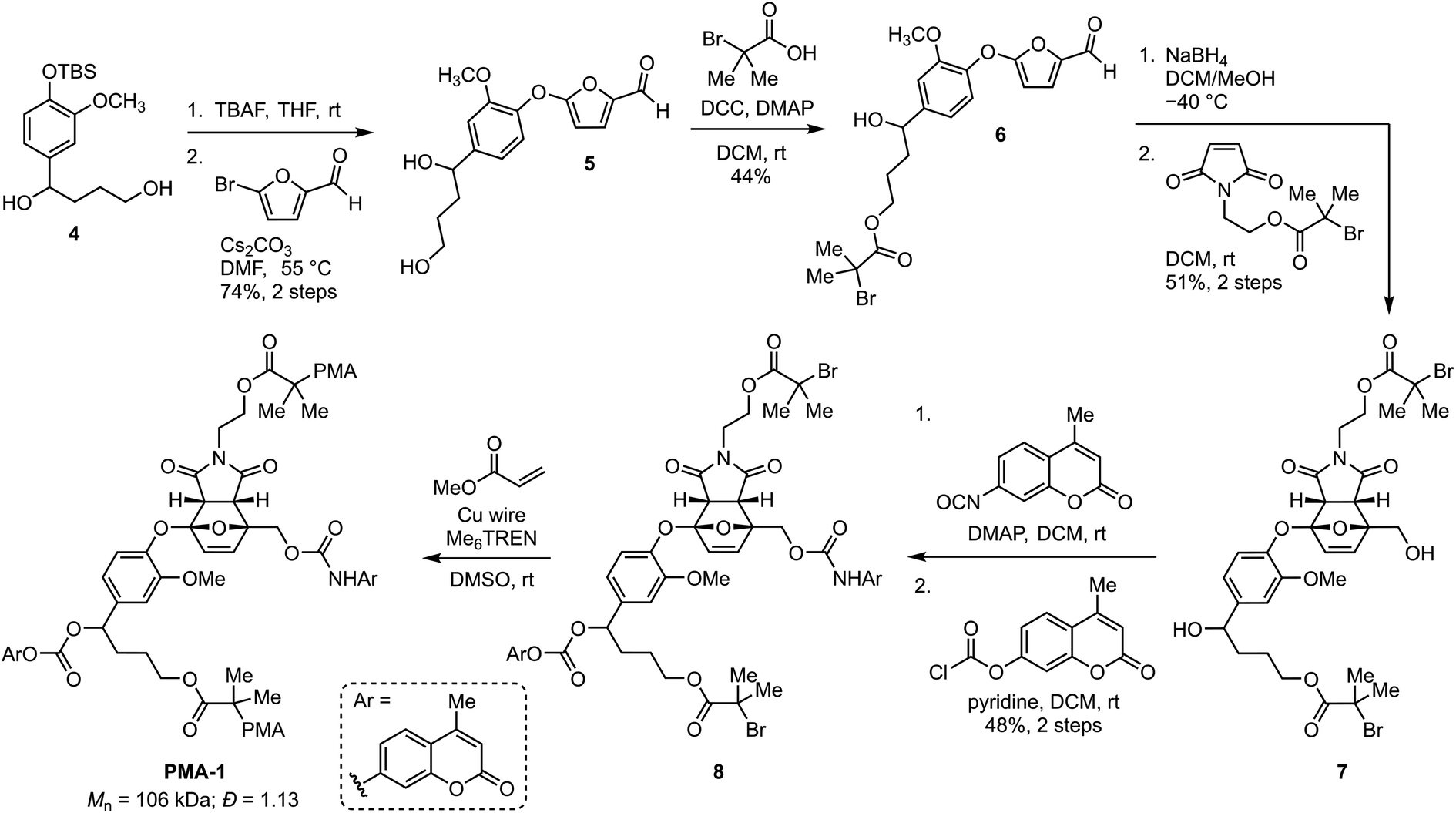 | ||
| Scheme 2 Synthesis of poly(methyl acrylate) (PMA) polymer PMA-1 containing a chain-centered mechanophore loaded with two distinct molecular payloads. | ||
The mechanochemical reactivity of the mechanophore in PMA-1 was evaluated using pulsed ultrasonication (1 s on/1 s off, 12–13 °C, 20 kHz, 30% amplitude, 16.4 W cm−2) and cargo release was characterized by high-performance liquid chromatography (HPLC). To compare the kinetics of payload release from PMA-1 with that from isolated small molecule model compound 1, a 2 mg mL−1 solution of PMA-1 in 3:1 MeCN/MeOH was subjected to ultrasonication for 60 min (sonication “on” time), and then cargo release was quantified at regular time intervals from aliquots of the solution incubated at room temperature (Fig. 2). We note that fluorescence spectroscopy can also be used to selectively quantify the formation of ArNH22 (but not ArOH 3), which provided similar results and additional confidence in the HPLC measurements (see the ESI† for additional details). The release of ArNH22 from the mechanically unmasked 2-furylcarbinol intermediate generated from PMA-1 occurs with a half-life of t1/2 = 11 h, which is slightly shorter than our previously reported substrate with a non-self-immolative tyrosol linker (t1/2 = 15 h),27 consistent with the more electron rich aryloxy group derived from vanillin. Notably, the release of the second payload, ArOH 3, occurs with a similar half-life of t1/2 = 13 h under these conditions. Normalized cargo release is plotted in Fig. 2 to highlight the relative release rates of each payload. These results are qualitatively consistent with the release kinetics measured for small molecule model compound 1 and demonstrate the rapid fragmentation–elimination–decarboxylation cascade of the self-immolative linker that follows initial furfuryl carbamate decomposition leading to the release of the first cargo, ArNH22 (see Fig. 1).
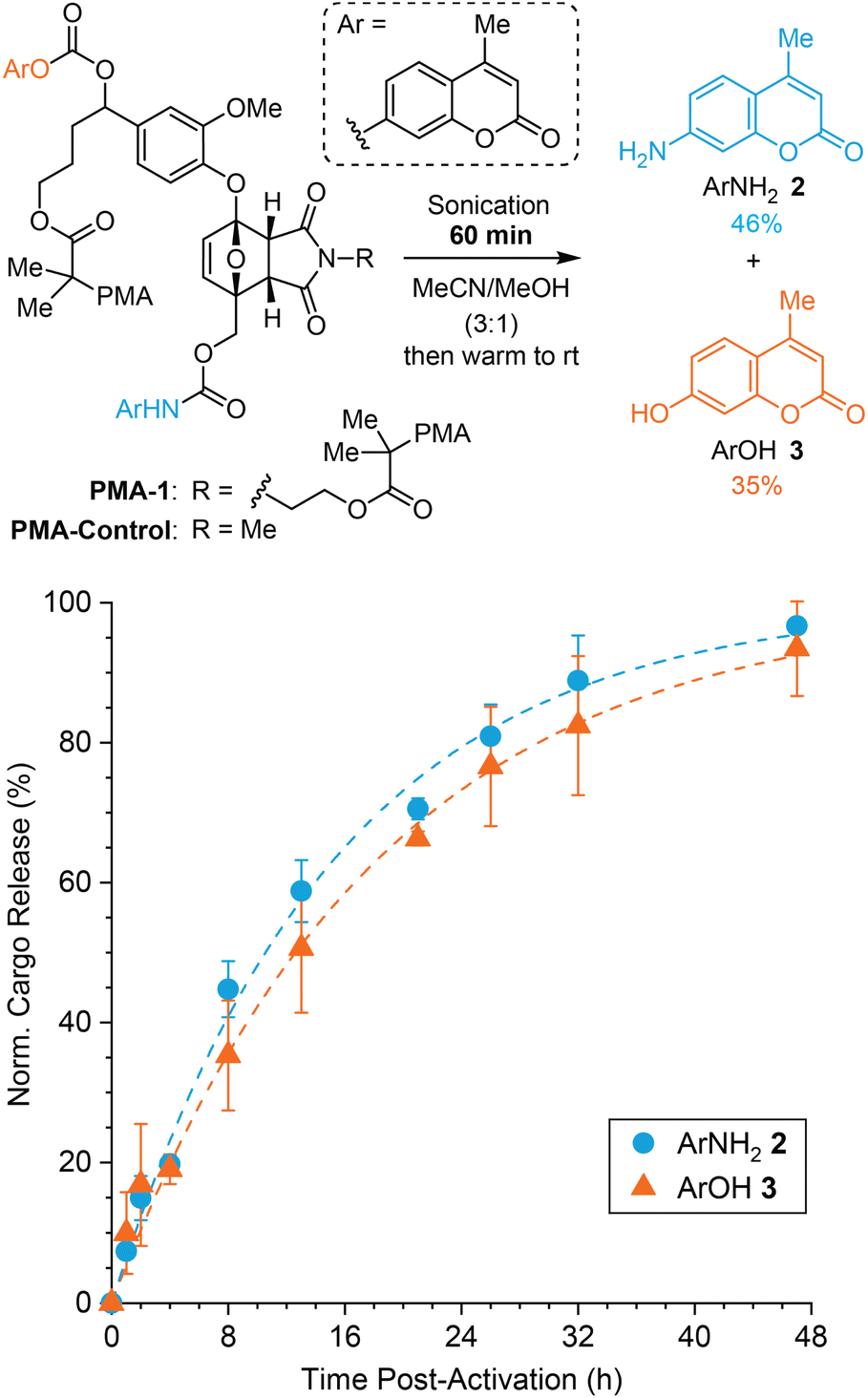 | ||
| Fig. 2 Mechanically triggered release of ArNH22 and ArOH 3 upon ultrasound-induced mechanochemical activation of PMA-1 (2 mg mL−1 in 3:1 MeCN/MeOH). Cargo release was measured at room temperature beginning immediately after sonication of PMA-1 for 60 min as characterized by quantitative HPLC. The dashed lines are fits to simple first order kinetics. The initial value was subtracted from each measurement and the data were normalized to the plateau value of the fitted curve to highlight the relative release rates of each payload. Error bars represent standard deviation from three replicate experiments. | ||
Increasing the duration of ultrasonication leads to a predictable increase in payload release from PMA-1 with increasing mechanophore conversion, achieving average yields of 64% for ArNH22 and 55% for ArOH 3 after 5 h of sonication (Fig. 3). Aliquots were periodically removed from the sonicated solution of PMA-1 over the course of the experiment and incubated at room temperature for 48 h to allow the complete fragmentation of the mechanically generated furan intermediate before quantifying cargo release by HPLC. Consistent with the experimental results for the decomposition of isolated small molecule 1, the amount of released ArNH22 is higher than ArOH 3 at every point in the reaction, again suggesting that the second fragmentation process involving the self-immolative linker is slightly less efficient than initial fragmentation of the mechanically unmasked 2-furylcarbinol derivative. Gel permeation chromatography was also used to determine changes in molar mass resulting from polymer chain scission. Rather than the typical bimodal evolution of molar mass that is characteristic of mid-chain scission,36 we observed a continuous shift in the average molar mass upon ultrasonication of PMA-1 (Fig. S7†). This trend is similar to results with our original aryloxy-substituted masked 2-furylcarbinol mechanophore,27 which we attribute to the relatively broad molar mass distribution (i.e., Đ > 1.1) that increases the competition between mechanophore activation and nonspecific chain scission and ultimately results in lower mechanophore activation efficiency.37,38 Importantly, negligible cargo release was observed upon ultrasonication of PMA-Control that incorporates the cargo-loaded mechanophore at the polymer chain end, which is not subjected to mechanical force, confirming the mechanical origin of payload release from PMA-1 (Fig. 3).
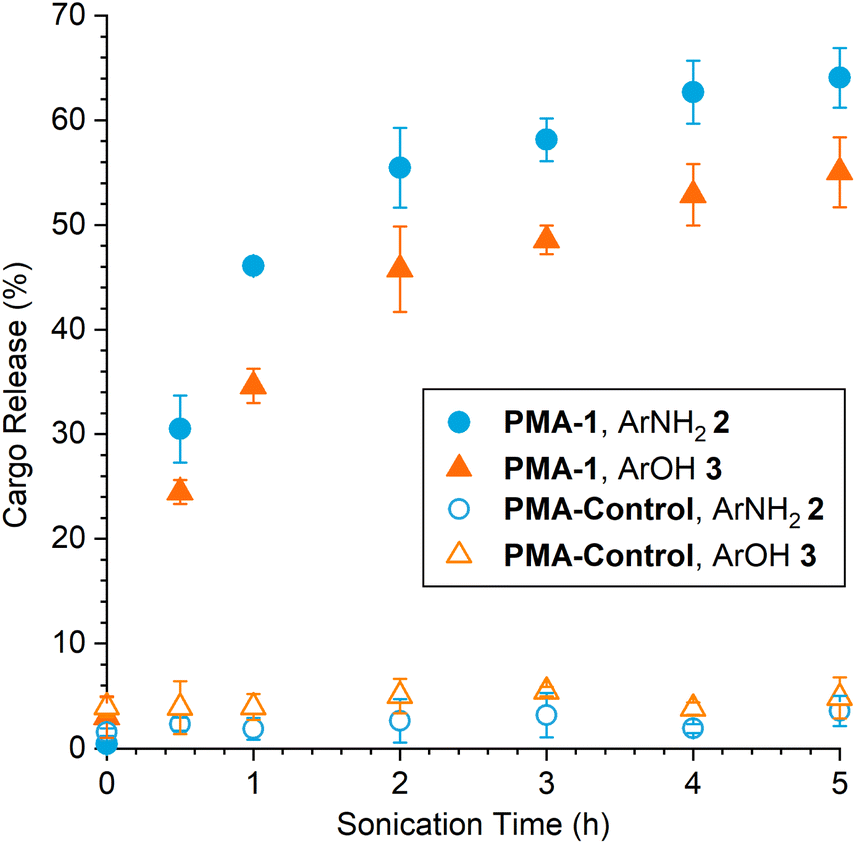 | ||
| Fig. 3 Mechanically triggered release of ArNH22 and ArOH 3 from PMA-1 and PMA-Control subjected to varying durations of sonication. Cargo release was quantified by HPLC after incubation at room temperature for 48 h following sonication. Error bars represent standard deviation from three replicate experiments. | ||
To further probe the dual release mechanism and also demonstrate the ability to systematically modulate the kinetics of payload release, we prepared polymer PMA-2 (Mn = 120 kDa, Đ = 1.35) incorporating a chain centered mechanophore in which the two cargo molecules, ArNH22 and ArOH 3, are conjugated at opposite positions compared to PMA-1 (Fig. 4). Based on the proposed fragmentation mechanism and the pronounced difference in reactivity between the furfuryl carbonate and furfuryl carbamate,26,32 we would expect that the release of ArOH 3 will occur rapidly, with much slower release of the second payload, ArNH22, conjugated to the self-immolative linker (Fig. S8†). A modified synthetic protocol compared to PMA-1 was required due to challenges forming the carbonate selectively at the primary alcohol on Diels–Alder adduct 7. Instead, ArNH22 was first installed via carbamate formation on the secondary alcohol of furfural derivative 6, which was then carried through a similar synthesis as the one illustrated in Scheme 2 (see the ESI† for details). To evaluate the kinetics of cargo release from PMA-2, a 2 mg mL−1 polymer solution in 3:1 MeCN/MeOH was subjected to similar ultrasonication conditions as above for 60 min and cargo release was subsequently monitored at room temperature using HPLC (Fig. 4). Again, normalized cargo release is plotted in Fig. 4 to highlight the relative rates of release of each payload. The non-normalized data are provided in Fig. S9.† As anticipated, the release of ArOH 3 reached a maximum prior to the first measurement and remained essentially constant over time, indicating that furfuryl carbonate decomposition occurs essentially immediately following mechanical activation of the mechanophore.27 In contrast, the release of the second cargo, ArNH22, occurred with a significantly longer half-life of t1/2 = 18 h. After 60 min of sonication, ArNH22 was released in 24% yield and ArOH 3 was released in 32% yield. Here, the even higher dispersity of PMA-2 compared to PMA-1 likely contributes to the diminished cargo release yields after the same extent of sonication, resulting from lower mechanophore conversion under similar conditions. We speculate that the higher dispersity may reflect the closer proximity of the carbamate group to the benzylic stereocenter on the bis-initiator. Similar to the results above, the release of the first cargo molecule from the initially generated furfuryl carbonate is again slightly more efficient than the release of the second cargo conjugated to the self-immolative linker. Notably, however, the dramatic difference in cargo release kinetics observed for PMA-2 compared to PMA-1 highlights the ability to fine-tune the relative release profiles based on the identity of the cargo and the attachment position on the mechanophore scaffold. Moreover, these results provide additional support for the stepwise fragmentation mechanism, indicating that expulsion of the self-immolative aryloxy group occurs after initial decomposition of the mechanically generated 2-furylcarbinol derivative. Importantly, negligible cargo release was observed after incubating PMA-2 in 3:1 MeCN/MeOH for 96 h at room temperature (Fig. S10†), fully consistent with control experiments demonstrating the stability of the primary carbonate under these conditions and supporting the mechanical origin of payload release.27 The ability to precisely control the relative release profiles of two different drug molecules, for example, is a unique attribute afforded by this mechanophore design, making it potentially suitable for applications in schedule-dependent combination therapies with enhanced therapeutic efficacy.39,40
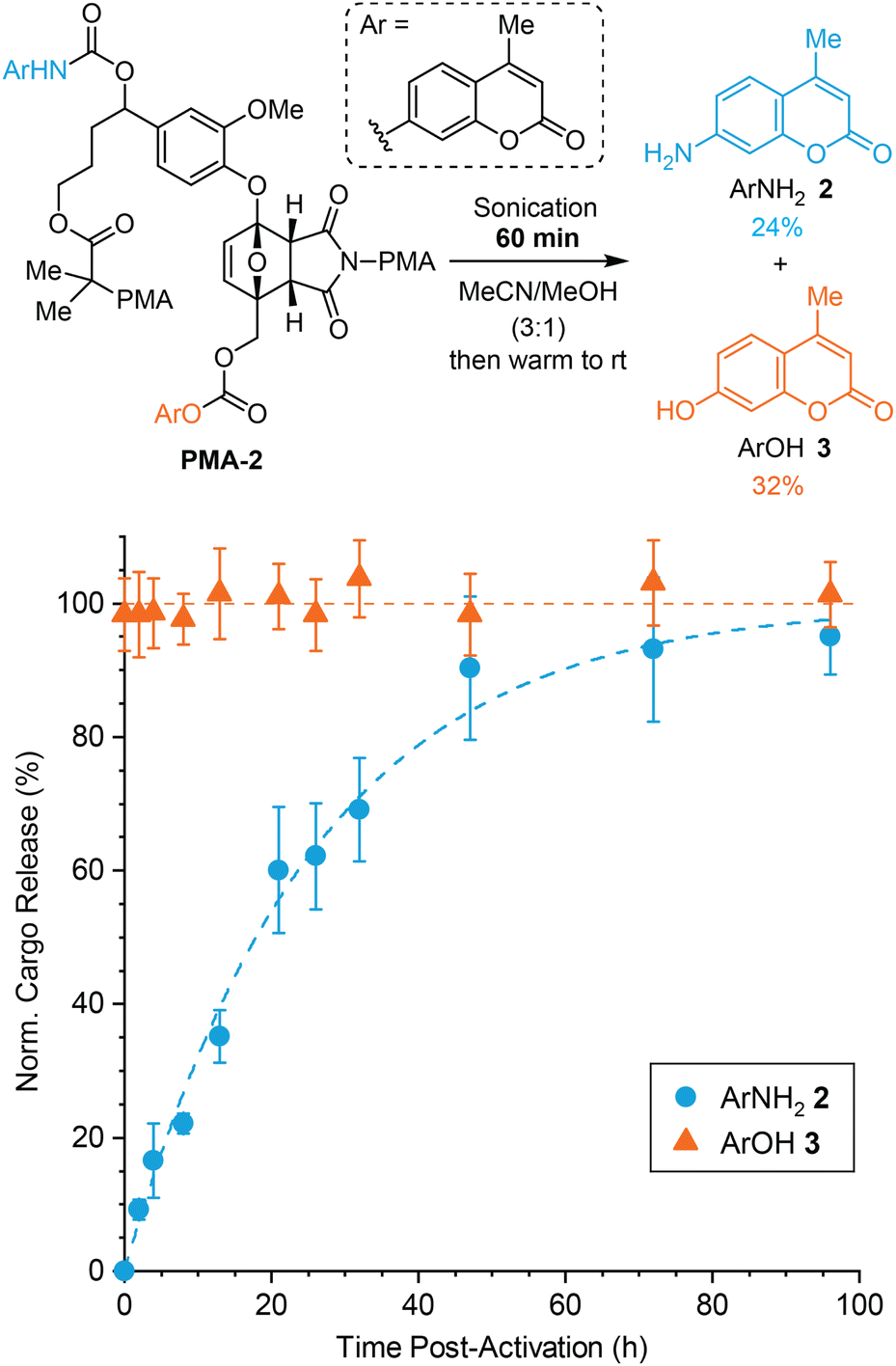 | ||
| Fig. 4 Mechanically triggered release of ArNH22 and ArOH 3 upon ultrasound-induced mechanochemical activation of PMA-2 (2 mg mL−1 in 3:1 MeCN/MeOH). Cargo release was measured at room temperature beginning immediately after sonication of PMA-2 for 60 min and characterized by quantitative HPLC. The data for ArNH22 were normalized to the plateau value of the fit to simple first order kinetics (dashed blue line), while the data for ArOH 3 was normalized to the average percent release across all measurements. Error bars represent standard deviation from three replicate experiments. | ||
Conclusions
We have designed a novel mechanophore based on a masked 5-aryloxy-substituted 2-furylcarbinol derivative incorporating a self-immolative spacer facilitating the mechanically triggered release of two distinct molecular payloads. The mechanophore design leverages a unique fragmentation pathway involving the initial decomposition of a mechanochemically unveiled 2-furylcarbinol derivative to release the first payload followed by a second fragmentation process resulting in expulsion of the aryloxy substituent. Installation of a judiciously designed aryloxy spacer capable of self-immolation via a 1,6-elimination mechanism enables the release of a second payload molecule from a single mechanophore. Notably, we demonstrate that the relative release rates of the two cargo molecules are varied over a wide range based on both the reactivity of the payload/linker group and the position of conjugation to the mechanophore. By simply switching the attachment positions of two model payloads, mechanically triggered release occurs either concurrently or sequentially, resulting in remarkably different release profiles. The selectivity, modularity, and tunable release kinetics afforded by this mechanophore design provide a promising platform for a variety of potential applications including real-time monitoring of drug distribution, combination chemotherapy, and even dual catalysis.Data availability
All data is available in the paper and/or ESI.†Author contributions
Y.-L. T., T. Z., and M. J. R. designed the research. Y.-L. T. performed the experiments. Y.-L. T., T. Z., and M. J. R. analyzed the data. Y.-L. T. and M. J. R. wrote the manuscript. M. J. R. provided guidance during all stages of the project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Arnold and Mabel Beckman Foundation through a Beckman Young Investigator Award and the National Institute of General Medical Sciences of the National Institutes of Health (R35GM150988) is gratefully acknowledged. We thank the Center for Catalysis and Chemical Synthesis of the Beckman Institute at Caltech for access to equipment. Y.-L. T. gratefully acknowledges the J. Yang & Family Foundation for a first year fellowship at Caltech. M. J. R. gratefully acknowledges the Alfred P. Sloan Foundation for a Sloan Research Fellowship and the Camille and Henry Dreyfus Foundation for a Camille Dreyfus Teacher-Scholar Award.References
- R. Groote, R. T. M. Jakobs and R. P. Sijbesma, Mechanocatalysis: forcing latent catalysts into action, Polym. Chem., 2013, 4, 4846–4859 RSC.
- M. A. Ghanem, A. Basu, R. Behrou, N. Boechler, A. J. Boydston, S. L. Craig, Y. Lin, B. E. Lynde, A. Nelson, H. Shen and D. W. Storti, The role of polymer mechanochemistry in responsive materials and additive manufacturing, Nat. Rev. Mater., 2021, 6, 84–98 CrossRef CAS.
- Y. Zhang, J. Yu, H. N. Bomba, Y. Zhu and Z. Gu, Mechanical Force-Triggered Drug Delivery, Chem. Rev., 2016, 116, 12536–12563 CrossRef CAS PubMed.
- M. M. Caruso, D. A. Davis, Q. Shen, S. A. Odom, N. R. Sottos, S. R. White and J. S. Moore, Mechanically-Induced Chemical Changes in Polymeric Materials, Chem. Rev., 2009, 109, 5755–5798 CrossRef CAS PubMed.
- M. K. Beyer and H. Clausen-Schaumann, Mechanochemistry: The Mechanical Activation of Covalent Bonds, Chem. Rev., 2005, 105, 2921–2948 CrossRef CAS PubMed.
- J. Li, C. Nagamani and J. S. Moore, Polymer Mechanochemistry: From Destructive to Productive, Acc. Chem. Res., 2015, 48, 2181–2190 CrossRef CAS PubMed.
- M. Stratigaki and R. Göstl, Methods for Exerting and Sensing Force in Polymer Materials Using Mechanophores, ChemPlusChem, 2020, 85, 1095–1103 CrossRef CAS PubMed.
- Y. Yao, M. E. McFadden, S. M. Luo, R. W. Barber, E. Kang, A. Bar-Zion, C. A. B. Smith, Z. Jin, M. Legendre, B. Ling, D. Malounda, A. Torres, T. Hamza, C. E. R. Edwards, M. G. Shapiro and M. J. Robb, Remote control of mechanochemical reactions under physiological conditions using biocompatible focused ultrasound, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2309822120 CrossRef CAS PubMed.
- B. A. Versaw, T. Zeng, X. Hu and M. J. Robb, Harnessing the Power of Force: Development of Mechanophores for Molecular Release, J. Am. Chem. Soc., 2021, 143, 21461–21473 CrossRef CAS PubMed.
- C. E. Diesendruck, B. D. Steinberg, N. Sugai, M. N. Silberstein, N. R. Sottos, S. R. White, P. V. Braun and J. S. Moore, Proton-Coupled Mechanochemical Transduction: A Mechanogenerated Acid, J. Am. Chem. Soc., 2012, 134, 12446–12449 CrossRef CAS PubMed.
- Y. Lin, T. B. Kouznetsova and S. L. Craig, A Latent Mechanoacid for Time-Stamped Mechanochromism and Chemical Signaling in Polymeric Materials, J. Am. Chem. Soc., 2020, 142, 99–103 CrossRef CAS PubMed.
- M. B. Larsen and A. J. Boydston, “Flex-Activated” Mechanophores: Using Polymer Mechanochemistry To Direct Bond Bending Activation, J. Am. Chem. Soc., 2013, 135, 8189–8192 CrossRef CAS PubMed.
- H. Shen, M. B. Larsen, A. G. Roessler, P. M. Zimmerman and A. J. Boydston, Mechanochemical Release of N-Heterocyclic Carbenes from Flex-Activated Mechanophores, Angew. Chem., Int. Ed., 2021, 60, 13559–13563 CrossRef CAS PubMed.
- Y. Sun, W. J. Neary, Z. P. Burke, H. Qian, L. Zhu and J. S. Moore, Mechanically Triggered Carbon Monoxide Release with Turn-On Aggregation-Induced Emission, J. Am. Chem. Soc., 2022, 144, 1125–1129 CrossRef CAS PubMed.
- S. Nijem, Y. Song, R. Schwarz and C. E. Diesendruck, Flex-activated CO Mechanochemical Production for Mechanical Damage Detection, Polym. Chem., 2022, 13, 3986–3990 RSC.
- M. Di Giannantonio, M. A. Ayer, E. Verde-Sesto, M. Lattuada, C. Weder and K. M. Fromm, Triggered Metal Ion Release and Oxidation: Ferrocene as a Mechanophore in Polymers, Angew. Chem., Int. Ed., 2018, 57, 11445–11450 CrossRef CAS PubMed.
- Y. Sha, Y. Zhang, E. Xu, Z. Wang, T. Zhu, S. L. Craig and C. Tang, Quantitative and Mechanistic Mechanochemistry in Ferrocene Dissociation, ACS Macro Lett., 2018, 1174–1179 CrossRef CAS PubMed.
- Y. Zhang, Z. Wang, T. B. Kouznetsova, Y. Sha, E. Xu, L. Shannahan, M. Fermen-Coker, Y. Lin, C. Tang and S. L. Craig, Distal conformational locks on ferrocene mechanophores guide reaction pathways for increased mechanochemical reactivity, Nat. Chem., 2021, 13, 56–62 CrossRef CAS PubMed.
- Y. Lu, H. Sugita, K. Mikami, D. Aoki and H. Otsuka, Mechanochemical Reactions of Bis(9-methylphenyl-9-fluorenyl) Peroxides and Their Applications in Cross-Linked Polymers, J. Am. Chem. Soc., 2021, 143, 17744–17750 CrossRef CAS PubMed.
- F. Yang, T. Geng, H. Shen, Y. Kou, G. Xiao, B. Zou and Y. Chen, Mechanochemical Release of Fluorophores from a “Flex-activated” Mechanophore, Angew. Chem., Int. Ed., 2023, e202308662 CAS.
- S. Huo, P. Zhao, Z. Shi, M. Zou, X. Yang, E. Warszawik, M. Loznik, R. Göstl and A. Herrmann, Mechanochemical bond scission for the activation of drugs, Nat. Chem., 2021, 13, 131–139 CrossRef CAS PubMed.
- Z. Shi, Q. Song, R. Göstl and A. Herrmann, Mechanochemical activation of disulfide-based multifunctional polymers for theranostic drug release, Chem. Sci., 2021, 12, 1668–1674 RSC.
- Q. Hu, W. Sun, C. Wang and Z. Gu, Recent advances of cocktail chemotherapy by combination drug delivery systems, Adv. Drug Delivery Rev., 2016, 98, 19–34 CrossRef CAS PubMed.
- Z. Shi, Q. Song, R. Göstl and A. Herrmann, The Mechanochemical Release of Naphthalimide Fluorophores from β-Carbonate and β-Carbamate Disulfide-Centered Polymers, CCS Chem., 2021, 3, 2333–2344 CrossRef CAS.
- X. Hu, T. Zeng, C. C. Husic and M. J. Robb, Mechanically Triggered Small Molecule Release from a Masked Furfuryl Carbonate, J. Am. Chem. Soc., 2019, 141, 15018–15023 CrossRef CAS PubMed.
- X. Hu, T. Zeng, C. C. Husic and M. J. Robb, Mechanically Triggered Release of Functionally Diverse Molecular Payloads from Masked 2-Furylcarbinol Derivatives, ACS Cent. Sci., 2021, 7, 1216–1224 CrossRef CAS PubMed.
- T. Zeng, X. Hu and M. J. Robb, 5-Aryloxy substitution enables efficient mechanically triggered release from a synthetically accessible masked 2-furylcarbinol mechanophore, Chem. Commun., 2021, 57, 11173–11176 RSC.
- C. C. Husic, X. Hu and M. J. Robb, Incorporation of a Tethered Alcohol Enables Efficient Mechanically Triggered Release in Aprotic Environments, ACS Macro Lett., 2022, 11, 948–953 CrossRef CAS PubMed.
- M. Shamis, H. N. Lode and D. Shabat, Bioactivation of Self-Immolative Dendritic Prodrugs by Catalytic Antibody 38C2, J. Am. Chem. Soc., 2004, 126, 1726–1731 CrossRef CAS PubMed.
- K. Haba, M. Popkov, M. Shamis, R. A. Lerner, C. F. Barbas III and D. Shabat, Single-Triggered Trimeric Prodrugs, Angew. Chem., Int. Ed., 2005, 44, 716–720 CrossRef CAS PubMed.
- M. Staderini, A. Gambardella, A. Lilienkampf and M. Bradley, A Tetrazine-Labile Vinyl Ether Benzyloxycarbonyl Protecting Group (VeZ): An Orthogonal Tool for Solid-Phase Peptide Chemistry, Org. Lett., 2018, 20, 3170–3173 CrossRef CAS PubMed.
- A. Alouane, R. Labruère, T. Le Saux, F. Schmidt and L. Jullien, Self-Immolative Spacers: Kinetic Aspects, Structure–Property Relationships, and Applications, Angew. Chem., Int. Ed., 2015, 54, 7492–7509 CrossRef CAS PubMed.
- M. K. Beyer, The mechanical strength of a covalent bond calculated by density functional theory, J. Chem. Phys., 2000, 112, 7307–7312 CrossRef CAS.
- I. M. Klein, C. C. Husic, D. P. Kovács, N. J. Choquette and M. J. Robb, Validation of the CoGEF Method as a Predictive Tool for Polymer Mechanochemistry, J. Am. Chem. Soc., 2020, 142, 16364–16381 CrossRef CAS PubMed.
- N. H. Nguyen, B. M. Rosen, G. Lligadas and V. Percec, Surface-Dependent Kinetics of Cu(0)-Wire-Catalyzed Single-Electron Transfer Living Radical Polymerization of Methyl Acrylate in DMSO at 25 °C, Macromolecules, 2009, 42, 2379–2386 CrossRef CAS.
- K. L. Berkowski, S. L. Potisek, C. R. Hickenboth and J. S. Moore, Ultrasound-Induced Site-Specific Cleavage of Azo-Functionalized Poly(ethylene glycol), Macromolecules, 2005, 38, 8975–8978 CrossRef CAS.
- Z. S. Kean, G. R. Gossweiler, T. B. Kouznetsova, G. B. Hewage and S. L. Craig, A Coumarin Dimer Probe of Mechanochemical Scission Efficiency in the Sonochemical Activation of Chain-Centered Mechanophore Polymers, Chem. Commun., 2015, 51, 9157–9160 RSC.
- A. C. Overholts and M. J. Robb, Examining the Impact of Relative Mechanophore Activity on the Selectivity of Ultrasound-Induced Mechanochemical Chain Scission, ACS Macro Lett., 2022, 733–738 CrossRef CAS PubMed.
- B. J. M. Braakhuis, V. W. T. R. van Haperen, M. J. P. Welters and G. J. Peters, Schedule-dependent therapeutic efficacy of the combination of gemcitabine and cisplatin in head and neck cancer xenografts, Eur. J. Cancer, 1995, 31, 2335–2340 CrossRef PubMed.
- M. J. Lee, A. S. Ye, A. K. Gardino, A. M. Heijink, P. K. Sorger, G. MacBeath and M. B. Yaffe, Sequential Application of Anticancer Drugs Enhances Cell Death by Rewiring Apoptotic Signaling Networks, Cell, 2012, 149, 780–794 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc06359c |
| This journal is © The Royal Society of Chemistry 2024 |