
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHypergolic ionic liquids: to be or not to be?†
Souvick
Biswas
a,
Kazuumi
Fujioka
a,
Ivan
Antonov
a,
Grace L.
Rizzo
a,
Steven D.
Chambreau
 b,
Stefan
Schneider
c,
Rui
Sun
*a and
Ralf I.
Kaiser
*a
b,
Stefan
Schneider
c,
Rui
Sun
*a and
Ralf I.
Kaiser
*a
aDepartment of Chemistry, University of Hawai'i at Manoa, Honolulu, Hawaii 96822, USA. E-mail: ruisun@hawaii.edu; ralfk@hawaii.edu
bJacobs Technology, Inc., Edwards Air Force Base, California 93524, USA
cAir Force Research Laboratory, Edwards Air Force Base, California 93524, USA
First published on 18th December 2023
Abstract
Hypergolic ionic liquids (HIL) – ionic liquids which ignite spontaneously upon contact with an oxidizer – emerged as green space propellants. Exploiting the previously marked hypergolic [EMIM][CBH] – WFNA (1-ethyl-3-methylimidazolium cyanoborohydride – white fuming nitric acid) system as a benchmark, through the utilization of a novel chirped-pulse droplet-merging technique in an ultrasonic levitation environment and electronic structure calculations, this work deeply questions the hypergolicity of the [EMIM][CBH]-WFNA system. Molecular oxygen is critically required for the [EMIM][CBH]-WFNA system to ignite spontaneously. State-of-the-art electronic structure calculations identified the resonantly stabilized N-boryl-N-oxo-formamide [(H3B–N(O)–CHO)−; BOFA] radical anion as the key intermediate in driving the oxidation chemistry upon reaction with molecular oxygen of the ionic liquid. These findings challenge conventional wisdom of ‘well-established’ test protocols as indicators of the hypergolicity of ionic liquids thus necessitating truly oxygen-free experimental conditions to define the ignition delay upon mixing of the ionic liquid and the oxidizer and hence designating an ionic liquid as truly hypergolic at the molecular level.
Introduction
The very first synthesis of a hypergolic ionic liquid (HIL) by Schneider et al.1 – an ionic liquid such as 1-allyl-3-methylimidazolium dicyanamide and an oxidizer, which ignite spontaneously upon contact with each other – revolutionized the development of green bipropellants2 by replacing toxic and carcinogenic hydrazine (N2H4) and its derivatives such as methylhydrazine (H2NNH(CH3))3 with environment-friendly HILs. Upon contact with an oxidizer like white fuming nitric acid (WFNA, HNO3) or hydrogen peroxide (H2O2), HILs such as 1-allyl-3-methylimidazolium borohydride and trihexyltetradecyl-phosphonium borohydride revealed remarkably short ignition delays (IDs) defined by the time lag between the initial contact of the two components and the start of the ignition.4 This ID emerged as the key parameter to assess the performance of HILs with the goal to achieve IDs of less than 5 ms.5 These IDs have been determined by conventional drop test experiments, which allow a gravity-assisted descent of a (sub)milliliter sized droplet of the HIL into the reservoir of the oxidizer of a few to tens of milliliters; simultaneously a high-speed optical camera captures the images with milliseconds temporal resolution to extract the ID.1,4–18However, these drop test studies have come under harsh scrutiny contemplating that a vast majority of these experiments were conducted in air and hence in the presence of oxygen; further, the quantification of the amount of the reactant(s) has been challenging since the oxidizer represented the excess reagent thus prohibiting a molecular level understanding of the underlying ignition chemistry; catalytic surface effects from the vessel containing the oxidizer have not been taken into account either. Therefore, the lack of well-defined experimental parameters in these drop test studies can impose significant misinterpretations and deviations from genuine bipropellant-operated engines where both HIL and the oxidizer are injected as droplets into the combustion chamber.4,5,8,10–12,15,17–19
Here, we report on a novel protocol to systematically evaluate the ignition chemistry and hypergolicity of HILs exploiting 1-ethyl-3-methylimidazolium cyanoborohydride ([EMIM][CBH]) (Fig. 1) with white fuming nitric acid (WFNA, HNO3) as a benchmark. The experiments are carried out under controlled conditions in a closed process chamber by merging individually levitated droplets of the HIL with a droplet of the oxidizer (WFNA) through frequency chirped amplitude modulation of the carrier wave in an ultrasonic levitator apparatus20 (Fig. S1–S3, ESI, S1†). [EMIM][CBH] was selected as a prototype system of interest due to the high energy density of the boron-centered anion [BH3(CN)]−, thermal stability of the ionic liquid, and moisture insensitivity.21 This work explicitly avoids the shortcomings in conventional drop test studies and deeply questions traditional drop-test studies as a marker of hypergolicity. First, to acoustically levitate the droplets, a well-defined atmosphere was maintained through mixtures of argon (Ar) and molecular oxygen (O2) at distinct ratios from 0% to 60% oxygen. Second, the molar ratios of the reactants in the droplets were constrained and quantified (Table S1†). Third, surface effects in the oxidizer are eliminated since each droplet levitates freely instead in a conventional drop test setup where the oxidizer resides in a container. Finally, physico-chemical changes in the merging processes are traced through synchronized, temporally resolved high-speed optical and thermal cameras as well as ultraviolet-visible (UV-Vis) emission spectroscopy (Fig. S3 and S5†); these studies were merged with state-of-the-art electronic structure calculations to validate the experimental findings computationally (ESI, S2 and S6†).
 | ||
| Fig. 1 Measured ignition delays (IDs) for the reaction of [EMIM][CBH] (inset) with WFNA as a function of molecular oxygen (O2) contents inside the levitator process chamber. The errors in the ignition delays represent the 1 − σ errors in the averaged time differentials between individual merging and ignition frames. | ||
Using the [EMIM][CBH] – WFNA system as a benchmark, our studies provide compelling evidence of the critical need of molecular oxygen (O2) to initiate ignition; no ignition was noticed in the absence of molecular oxygen in the surrounding medium even at excess molar ratios of the oxidizer to the ionic liquid exceeding five. A remarkable shortening of the ignition delay from 15 ms to only 3 ms with increasing molecular oxygen contents was achieved (Fig. 1 and Table S2†). Boron dioxide (BO2) was identified as the key molecular species driving the ignition as evident from a bright green flame with maximum flame temperatures reaching about 3500 K. On a molecular level, using electronic structure calculations, the oxidation was determined to be driven by the reaction of the resonantly stabilized free N-boryl-N-oxo-formamide (BOFA) radical anion (HCON(O)BH3−) with molecular oxygen – opening up five highly exoergic (−183 to −421 kJ mol−1) channels including the gas phase formation of the boron dioxide (BO2) radical. These findings deeply question traditional textbook knowledge of drop test experiments as an indicator of ignition delays (IDs) and the fundamental classification of ionic liquids as hypergolic with substantial evidence presented here challenging the conventional wisdom that the [EMIM][CBH] – WFNA represents a hypergolic system.
Results
Visualization of merging events and ignition
The merging process of the droplets of [EMIM][CBH] and WFNA followed by the ignition event was monitored at distinct molecular oxygen – argon (Ar) ratios from 0% to 60% oxygen (O2) with droplets loaded in the lower and upper pressure node of the standing wave generated in the acoustic levitator, respectively (Movies S1–S13 and Fig. S6†). Series of snapshots captured by synchronized high-speed infrared (IR) and optical cameras for the ignition at 40% O2 – 60% Ar mixture are displayed in Fig. 2.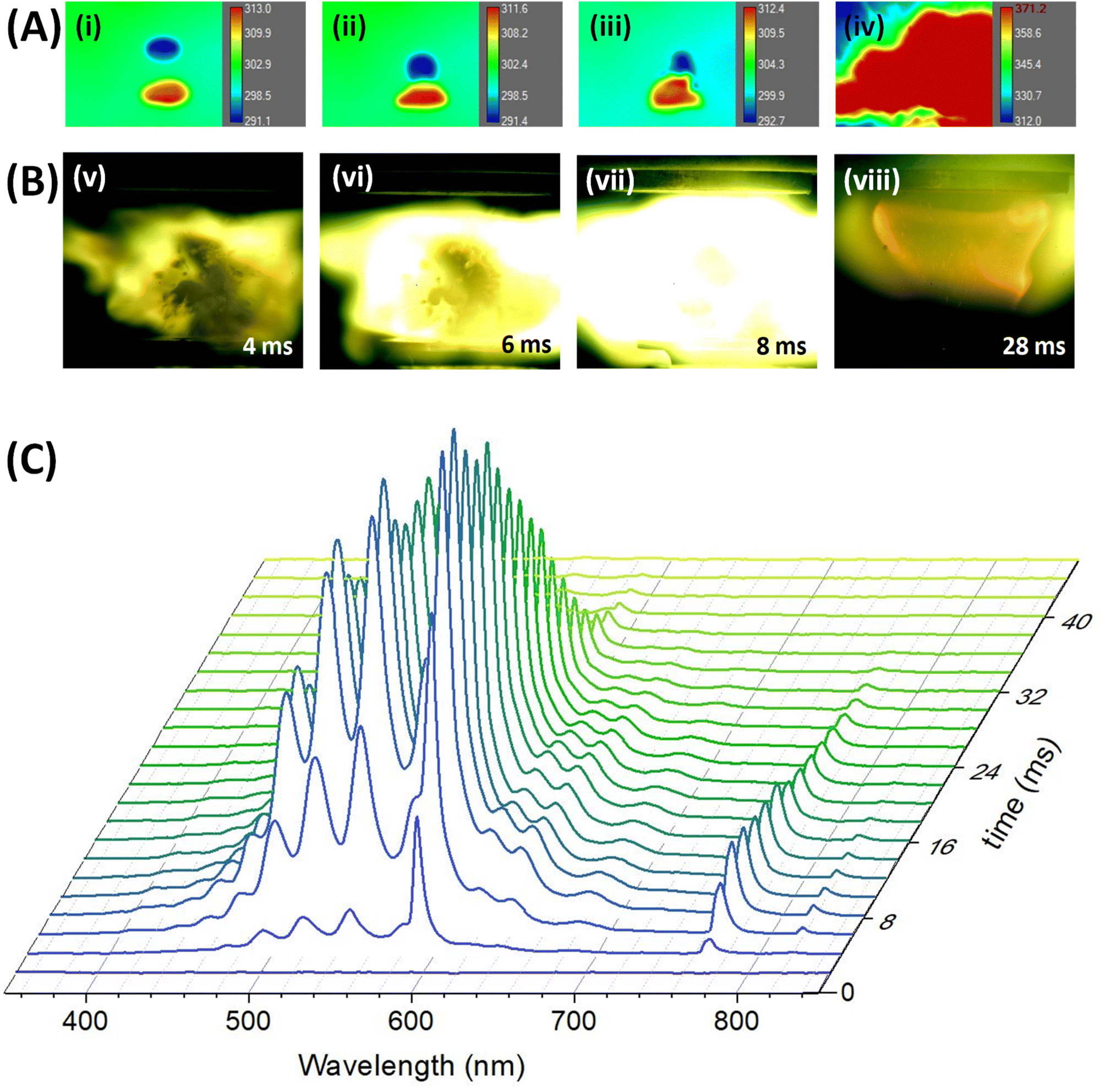 | ||
| Fig. 2 High-speed images captured by the synchronized (A) infrared camera and (B) optical camera to depict merging sequence between the levitating droplets of [EMIM][CBH] (bottom) and WFNA (top) followed by ignition. Frames correspond to the following: (i) pre-merging, (ii)-(iii) merging motion, (iv) merging, (v)–(viii) ignition. The infrared camera captures the merging instance, while the optical camera indicates the commencement of ignition. The progress of the ignition is traced spectroscopically by the (C) temporal evolution of the flame emission spectra. | ||
The IR images depict distinct stages comprising of (i) levitating reactant droplets (pre-merging), (ii)-(iii) merging motion upon application of the frequency-chirp pulse, and (iv) merging instance accompanied by temperature rises (exoergic); simultaneously, the ignition and post-ignition (combustion) events are captured by the optical camera operating at 1 kHz revealing bright greenish flames with yellowish tints. The IDs are determined from the time difference between the merging and the initiation of ignition (Fig. 1 and 2). As the molecular oxygen concentration rises, a significant reduction of the ID is observable. Quantitatively, the ID was reduced from 15 ± 7 ms (10% oxygen) to 3 ± 2 ms (60% oxygen); raising the oxygen content beyond this percentage did not reduce the ID further. Despite being previously designated as a HIL system, remarkably, the [EMIM][CBH] – WFNA droplets did not ignite in a pure argon atmosphere excluding molecular oxygen.
Emission spectra of the flame
To probe the underlying chemistry, the ignition process was also interrogated spectroscopically exploiting ultraviolet – visible (UV-Vis) emission spectroscopy in the 200 to 1100 nm spectral range; the time-resolved emission spectra are displayed for 40% O2 – 60% Ar mixture in Fig. 2(C), while emission traces at distinct molecular oxygen concentrations are presented in Fig. S7.† The changes in the overall emission spectral intensities with time indicate the commencement of the ignition, the spread of the flame to its maximum intensity, i.e. the combustion of the ionic liquid droplet, followed by diminishing to zero emission at the end of the oxidation process. This time sequence mirrors the recordings of the optical camera (Fig. 2(B)) and supports extended burn times of up to 100 ms as the molecular oxygen concentration rises to 60%. The stacked plots of the most intense flame emission spectra at distinct oxygen concentrations also reveal that the intensity of the emission is proportional to the percentage of molecular oxygen (Fig. 3(A) and S8†). The deconvolution of the emission traces (Fig. 3(B), Table S3 and Fig. S10†) reveals strong emission features of boron dioxide (BO2) radical (548.4 nm; A 2Πu–X 2Πg).22,23 | ||
| Fig. 3 (A) Stacked plot of the most intense spectra for the [EMIM][CBH]-WFNA ignition reactions at different molecular O2 concentrations. (B) Deconvoluted flame emission spectrum upon ignition in 40% O2/60% Ar atmosphere at 10 ms since merging. | ||
The detection of BO2 requires an oxidation of sp3 hybridized boron of the [CBH] anion; previous studies propose that this process likely involves boron-based transients, e.g. nitrato borane (H3B–O–NO2), boryl isocyanate (H3B–NCO), boryloxo aldehyde (H3B–O–CHO) and N-boryl-N-oxo-formamide (HCON(O)BH3−; BOFA).20 Boron monoxide (BO)24 could not be identified in the emission spectra presumably due to the intense emissions from boron dioxide. Traces of alkali metals present in any ionic liquid are also detectable via their characteristic atomic emission lines of sodium (Na) [589 nm, 2p63p 2P → 2p63 s 2S; 819 nm, 2p63d 2D → 2p63p 2P ] and potassium (K) [769 nm, 3p64p 2P → 3p64 s 2S].25 Temporal profiles of the dominating reaction intermediate BO2 for distinct initial concentrations of molecular oxygen reveal that the initial rate of appearance is much faster for 40% to 60% oxygen compared to lower concentrations, but BO2 survives much longer in the flame with time scale exceeding 50 ms (Fig. S9 and S11†). In contrast, for low initial oxygen concentrations (10–30%), BO2 is short-lived, revealing a sluggish initial growth; these findings suggest that the oxidation of the boron center of the [CBH] anion is facilitated by larger fractions of molecular oxygen. The dip in the BO2 profiles, for example at 18 ms in the case of 60% O2-containing atmosphere can be attributed to the transition from ignition at the diffusion layer of the ionic liquid droplet to the combustion process of the bulk ionic liquid droplet. The maximum temperatures were determined as 3512 K for 60% molecular oxygen, whereas at 20% oxygen, the peak temperature raised only to 2621 K (ESI, S5 and Fig. S9†).
Gas phase products
Finally, the [EMIM][CBH]-WFNA merging process was also accompanied by the production of gas phase products detected via FTIR spectroscopy (Fig. 4). In a pure argon inert gas atmosphere, characteristic features for nitrous oxide (N2O, 2270–2170 cm−1), hydrogen cyanide (HCN, 770–660 cm−1), and nitrogen dioxide (NO2, 1660–1560 cm−1) were identified (Fig. 4(A) and Table S4†) with major contribution of nitrogen dioxide (NO2) originating from the vapor of the reactant WFNA.20 Hydrogen cyanide (HCN), and nitrous oxide (N2O) are the result of the reaction of the [CBH] anion with WFNA, while the cation essentially stays intact; no carbon dioxide (CO2) was detected. However, in the presence of molecular oxygen, the aforementioned gas-phase products (HCN, N2O, NO2) were dominated by carbon dioxide (CO2); further, trace amounts of nitrous acid (HONO) and nitric oxide (NO) were identified.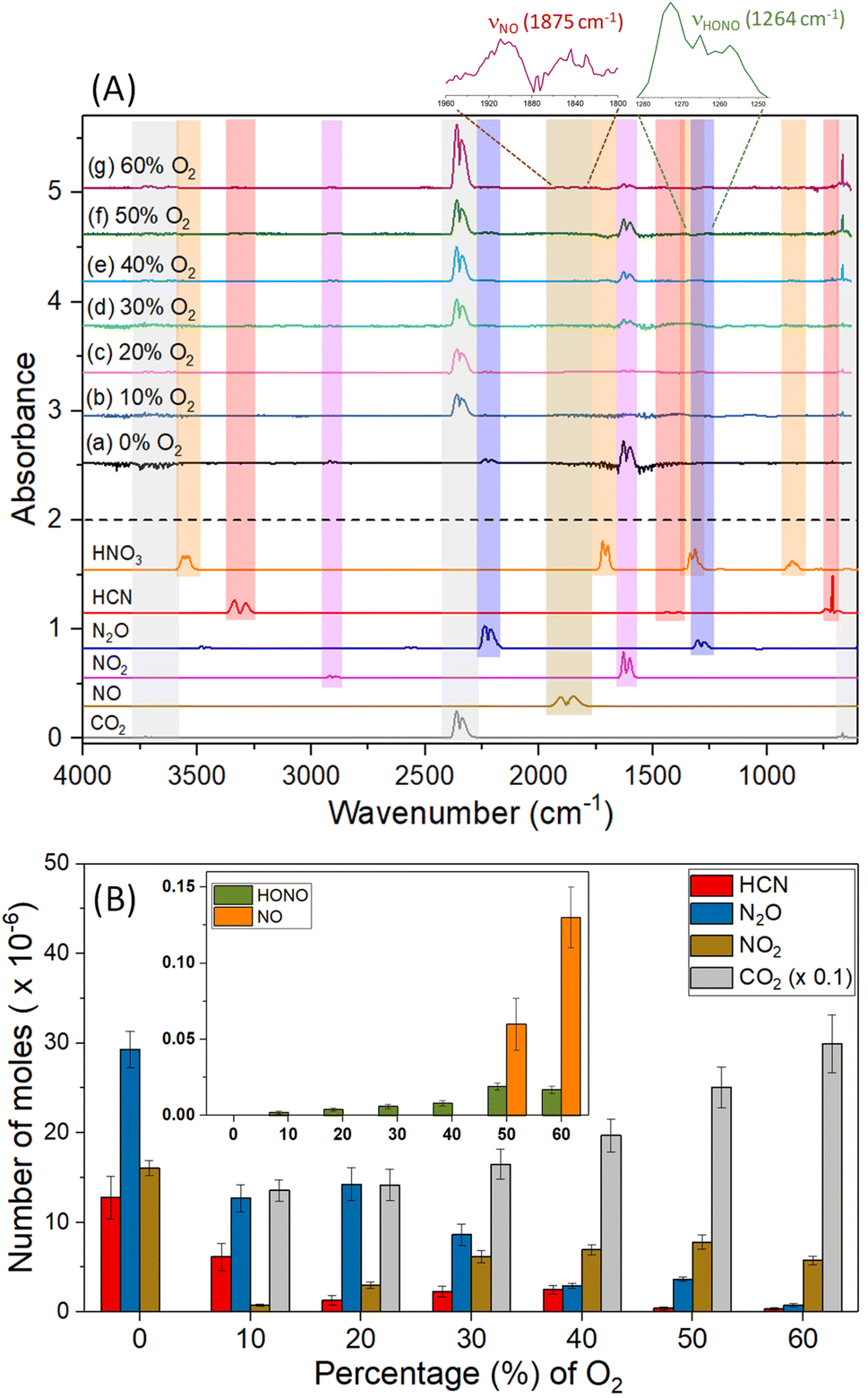 | ||
| Fig. 4 (A) FTIR spectra and (B) quantification of gas-phase products formed in the [EMIM][CBH]–WFNA reactions as a function of molecular O2 contents. | ||
The major source of carbon dioxide (CO2) can be linked to the combustion of the [EMIM] moiety holding six carbon atoms and percentage yield calculation assuming the cation as the primary source reveals that higher volume of molecular O2 assists the oxidation (Table S5†). The oxidation of hydrogen cyanide (HCN)26 is likely a minor channel supported by the finding that the amount of hydrogen cyanide (HCN) is significantly lowered with the increase of molecular oxygen, i.e. 12.8 ± 2.4 μmole versus 0.4 ± 0.1 μmole in 0% and 60% molecular oxygen, respectively. As we move from 10% to 60% molecular oxygen concentration, the yield of nitrous oxide (N2O) significantly decreases, which could be an outcome of the facilitated oxidation by the molecular oxygen to form nitrogen dioxide (NO2) and traces of nitric oxide (NO). The formation of these closed shell tri- and tetra-atomic products (HCN, N2O, NO2, HONO) is energetically favored as the mixing energy (Table S6†) between the liquid droplets can exceed the barrier to reaction of nitric acid (HNO3) and the [CBH] anion.20,27 Overall, since [EMIM][CBH] does not react with molecular oxygen, we have to conclude that reaction intermediates such as doublet radicals identified previously20 accessed during the reaction of the [CBH] anion with nitric acid (HNO3) are likely reactants with molecular oxygen.
Electronic structure calculation
To explore this hypothesis, we conducted extensive electronic structure calculations on the reactions of molecular oxygen with intermediates accessed in the [EMIM][CBH] – WFNA system at the CCSD(T)/cc-pVDZ//B3LYP/6-31G* level of theory (ESI, S2†). This study builds on previous unsupervised reaction pathway searches of the [CBH] – WFNA system and identified 3000+ critical points.20 In theory, molecular oxygen addition could take place at any position on the reaction surface given enough excess energy, but oxygen addition to the surfaces of the remaining closed-shell singlet reaction intermediates20 were found to be repulsive thus are not further considered.In contrast, reaction of molecular oxygen with the resonantly stabilized free radical (RSFR) N-boryl-N-oxo-formamide (BOFA) doublet radical anion (HCON(O)BH3−) (Fig. 5), in which the unpaired electron is delocalized over the two oxygen atoms (Fig. 5(A) and (B)), is feasible. The resonance of BOFA is well reflected by the shortened carbon–nitrogen bond of only 139 pm compared to 147 to 148 pm of classical carbon–nitrogen single bonds highlighting the partial double bond character of the carbon–nitrogen bond in BOFA. Likewise, the nitrogen–oxygen bond length of only 129 pm compared to typical single bonds of 136 pm also indicate a partial double bond character. Upon approaching BOFA, molecular oxygen forms a cyclic van-der-Waals complex i1, which is stabilized by 36 kJ mol−1 with respect to the separated reactants (Fig. 6). Considering the resonance stabilization, molecular oxygen abstracts a hydrogen atom via a low barrier of only 56 kJ mol−1 above the separated reactants rather than adding to the doublet radical site(s); this process forms the van-der-Waals complex i2, in which the hydroperoxyl radical (HOO) points its hydrogen atom to the nitrogen atom. The latter isomerized via a hydrogen migration from the hydroperoxyl radical to the oxygen atom of the N–O moiety and is accompanied by a B–O bond formation yielding the B–O–O functional group in i3. The i2 → i3 step in the shown PES involves the first covalent bond formation of molecular O2 with BOFA and it is associated with an exoergicity of 136 kJ mol−1. Hence, the origin of ignition in the oxidative environment can be chemically linked to this primary oxidation step of the boron center, and in the later stages (Fig. 6) of the reaction it eventually yields in boron dioxide (BO2), which is also experimentally detected. Next, the hydrogen atom at the N–O moiety in i3 shifts to the B–O–O moiety to i4. This intermediate can be formally seen as an insertion intermediate of molecular oxygen into one of the three boron–hydrogen bonds of BOFA (i1). This structure connects via concerted oxygen–oxygen bond cleavage, and hydroxyl migration to i5, which results in two hydroxy functional groups at the boron atom and also represents the global minimum (611 kJ mol−1) of the potential energy surface (PES). The latter can undergo a facile N–B bond rupture forming a van-der-Waals complex i8. It is important to note that commencing with intermediate i3, all transition states are located below the energy of the separated reactants and hence can be surmounted easily.
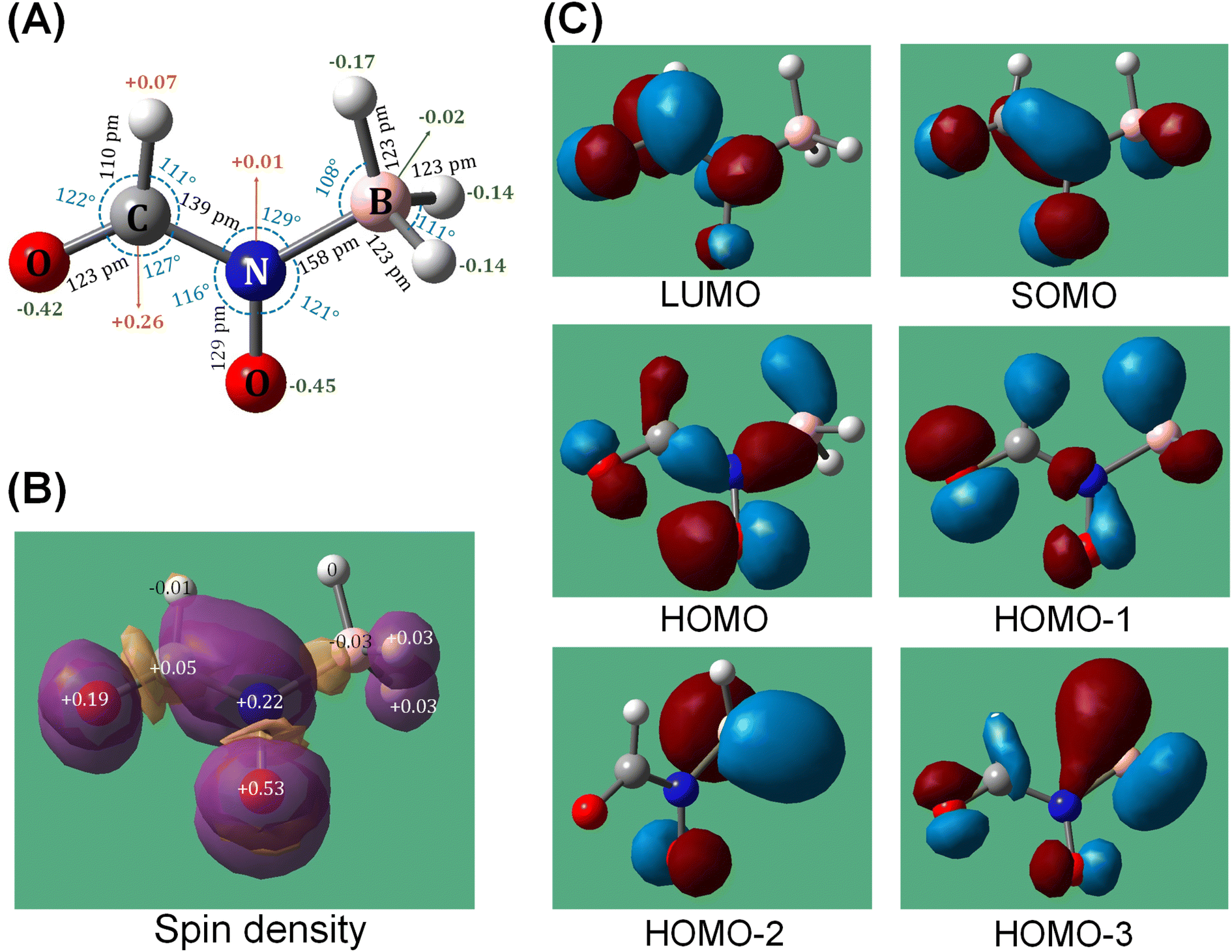 | ||
| Fig. 5 (A) Optimized structure with Mulliken charges, (B) spin densities on the individual atoms and (C) selected molecular orbitals of the intermediate- N-boryl-N-oxo-formamide [HCON(O)BH3−; BOFA] radical anion formed in the initial reaction steps of [EMIM][CBH] and WFNA. | ||
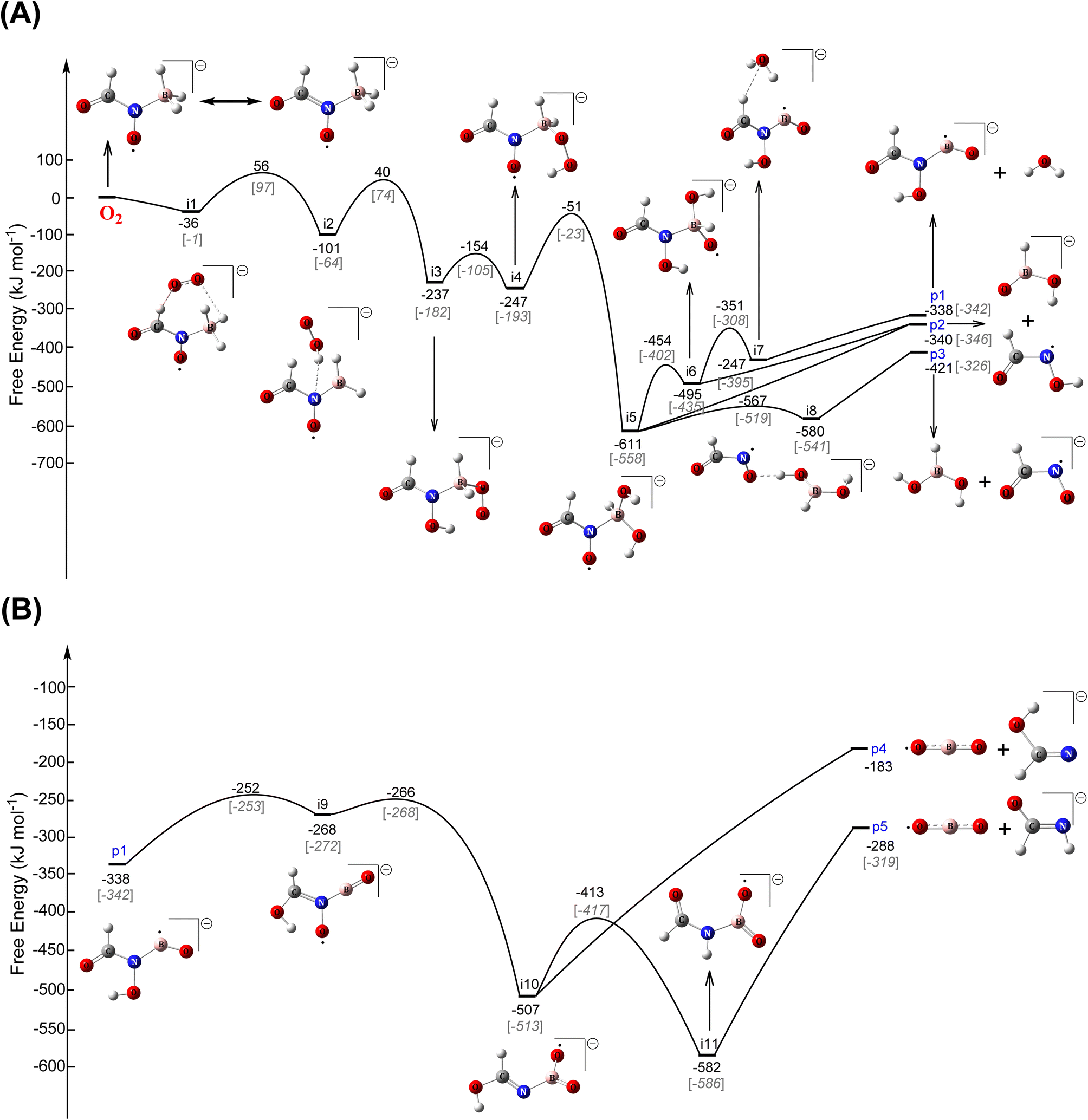 | ||
| Fig. 6 Potential energy surfaces (PES) for (A) the reaction of N-boryl-N-oxo-formamide [HCON(O)BH3−; BOFA] radical anion with molecular oxygen (O2), extending to (B) the formation of boron dioxide (BO2) radical. The energy values correspond to the free energies at 298.15 K (black) and 0 K in the parentheses (grey). | ||
Eventually, isomerization and decomposition of i5 and i8 opens three exoergic exit channels: N-hydroxy-formylboronamidate (HCON(OH)BO−) plus water (H2O) (p1), N-hydroxy-formamide (HCONOH) plus hydrogen boronate (HBO(OH)−) (p2), and boronic acid (BH(OH)2) plus N-oxo-formamidate (HCONO−) (p3). Among these products, the N-hydroxy-formylboronamidate (HCON(OH)BO−) radical anion was found to isomerize in four steps viai9 and i10 to reach eventually i11. The latter carries the O–B–O moiety and undergoes facile N–B bond rupture followed by the generation of the boron dioxide (BO2) radical and the formimidate (HCONH−) anion (p5) in an overall exoergic pathway. The formation of BO2 from i10 has been found to be relatively unfavorable yielding the co-product formimidate anion (HOCHN−) (p4), and the combined products are associated with an excess energy of 105 kJ mol−1 compared to p5. It is worth mentioning that these exoergic exit channels (p1–p5) combined contribute to the high ignition temperature ranges as recorded experimentally. Note that although the initial transition states from BOFA plus molecular oxygen (TS1) and from i2 to i3 (TS2) are located above the separated reactants, the actual formation of BOFA in the [EMIM][CBH] – WFNA system20 is exoergic by 15 kJ mol−1. Likewise, the experiments reveal a release of 3 to 12 kJ mol−1 of energy upon the initial mixing of the reactant droplets (Table S6†). Under thermal conditions and a Maxwell–Boltzmann distribution, 1% of the reactants can overcome the transition states TS1 and TS2 just initiating a vigorous oxidation [EMIM][CBH] – WFNA – molecular oxygen system. On the molecular level, these pathways involve the resonantly stabilized free N-boryl-N-oxo-formamide (BOFA) doublet radical anion (HCON(O)BH3−) as the key reactant. These findings provide persuasive computational evidence of the experimentally observed ‘need’ of molecular oxygen to engage an oxidation of [EMIM][CBH] with WFNA thus challenging the traditional picture of [EMIM][CBH] – WFNA as a hypergolic system.
Conclusion and outlook
A comprehensive experimental and computational study of the [EMIM][CBH]-WFNA system exploiting a novel chirped-pulse droplet-merging technique in a controlled environment deeply questions the hypergolicity of the [EMIM][CBH]-WFNA system even under oxidizer-rich conditions with moles of the oxidizer exceeding those of the ionic liquid by a factor of five. Molecular oxygen is critically required for the [EMIM][CBH]-WFNA system to ignite spontaneously with ignition delays as short as 3 ms for oxygen concentrations of 60%. Extensive electronic structure calculations match our experimental findings and identify the resonantly stabilized N-boryl-N-oxo-formamide (HCON(O)BH3−; BOFA) radical anion as a key intermediate in driving the overall oxidation chemistry of the ionic liquid. Although we note that BOFA might not be the only intermediate that molecular oxygen could add to in the [EMIM][CBH]-WFNA system, these findings deeply question the ‘well-established’ drop-test approach as an indicator of the hypergolicity of ionic liquids and necessitates truly oxygen-free experimental conditions as provided here to define the ignition delay upon mixing of the ionic liquid and the oxidizer and hence to label the ionic liquid as truly hypergolic. Finally, these findings imply that the addition of even small concentrations of molecular oxygen as an initiator in the rocket propulsion systems for [CBH]-based ionic liquids may critically boost the overall combustion performance while simultaneously aiming at the shortest ignition delays and reducing the amount of corrosive oxidizer (WFNA) required. Naturally, this work represents a very first step toward a systematic and novel experimental elucidation of the hypergolicity of ionic liquids. Further studies are aimed at exploring the non-boron ionic liquids, such as dicyanamide containing anion1 as well as at investigating the effects of distinct organic side groups in the cation, i.e. probing how the ignition delay is influenced by the organic substituent and hence elucidating structure – reactivity effects. Finally comparing the oxidizers, i.e., WFNA versus hydrogen peroxide (H2O2), is of interest thus providing a comprehensive framework of the true hypergolicity of ionic liquids on the molecular level.Data availability
The data that support the findings of this study are available in the article and ESI.† Additional data are available from the corresponding authors upon reasonable request.Author contributions
R. I. K. designed the experiments; S. B. carried out the experiments; S. B., I. A., and G. L. R. analyzed the data; S. D. C., and S. S. carried out the synthesis; K. F., and R. S. performed the theoretical calculations; S. B. wrote the original draft, S. B., R. S., and R. I. K. revised the draft and all of the authors discussed the data.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the Air Force Office of Scientific Research (AFOSR) (FA9550-21- 1-0377). The authors appreciate the Information Technology Service (ITS) from the University of Hawaii at Manoa for the computational resources.References
- S. Schneider, T. Hawkins, M. Rosander, G. Vaghjiani, S. Chambreau and G. Drake, Energy Fuels, 2008, 22, 2871–2872 CrossRef CAS.
- Q. Zhang and J. n. M. Shreeve, Chem.–Euro. J., 2013, 19, 15446–15451 CrossRef CAS PubMed.
- E. F. Rothgery, Kirk-Othmer Encyclopedia of Chemical Technology, 2004 Search PubMed.
- Q. Zhang and J. M. Shreeve, Chem. Rev., 2014, 114, 10527–10574 CrossRef CAS PubMed.
- Y. Zhang, H. Gao, Y. H. Joo and J. M. Shreeve, Angew. Chem., Int. Ed., 2011, 50, 9554–9662 CrossRef CAS PubMed.
- A. J. Alfano, J. D. Mills and G. L. Vaghjiani, Rev. Sci. Instrum., 2006, 77, 045109 CrossRef.
- S. D. Chambreau, S. Schneider, M. Rosander, T. Hawkins, C. J. Gallegos, M. F. Pastewait and G. L. Vaghjiani, J. Phys. Chem. A, 2008, 112, 7816–7824 CrossRef CAS PubMed.
- D. Chand, J. Zhang and J. n. M. Shreeve, Chem.–Euro. J., 2015, 21, 13297–13301 CrossRef CAS PubMed.
- H. Gao, Y.-H. Joo, B. Twamley, Z. Zhou and J. n. M. Shreeve, Angew. Chem., Int. Ed., 2009, 48, 2792–2795 CrossRef CAS PubMed.
- H. Gao and J. n. M. Shreeve, J. Mater. Chem., 2012, 22, 11022–11024 RSC.
- L. He, G.-H. Tao, D. A. Parrish and J. n. M. Shreeve, Chem.–Euro. J., 2010, 16, 5736–5743 CrossRef CAS PubMed.
- Y.-H. Joo, H. Gao, Y. Zhang and J. n. M. Shreeve, Inorg. Chem., 2010, 49, 3282–3288 CrossRef CAS PubMed.
- F. Lauck, J. Balkenhohl, M. Negri, D. Freudenmann and S. Schlechtriem, Combust. Flame, 2021, 226, 87–97 CrossRef CAS.
- S. Li, H. Gao and J. M. Shreeve, Angew. Chem., Int. Ed., 2014, 53, 2969–2972 CrossRef CAS PubMed.
- P. D. McCrary, P. A. Beasley, O. A. Cojocaru, S. Schneider, T. W. Hawkins, J. P. L. Perez, B. W. McMahon, M. Pfeil, J. A. Boatz, S. L. Anderson, S. F. Son and R. D. Rogers, Chem. Commun., 2012, 48, 4311–4313 RSC.
- S. Schneider, T. Hawkins, Y. Ahmed, M. Rosander, L. Hudgens and J. Mills, Angew. Chem., Int. Ed., 2011, 50, 5886–5888 CrossRef CAS PubMed.
- K. Wang, Y. Zhang, D. Chand, D. A. Parrish and J. n. M. Shreeve, Chem.–Euro. J., 2012, 18, 16931–16937 CrossRef CAS PubMed.
- Y. Zhang and J. n. M. Shreeve, Angew. Chem., Int. Ed., 2011, 50, 935–937 CrossRef CAS PubMed.
- M. Watanabe, M. L. Thomas, S. Zhang, K. Ueno, T. Yasuda and K. Dokko, Chem. Rev., 2017, 117, 7190–7239 CrossRef CAS PubMed.
- S. Biswas, I. Antonov, K. Fujioka, G. L. Rizzo, S. D. Chambreau, S. Schneider, R. Sun and R. I. Kaiser, Phys. Chem. Chem. Phys., 2023, 25, 6602–6625 RSC.
- Q. Zhang, P. Yin, J. Zhang and J. M. Shreeve, Chem.–Euro. J., 2014, 20, 6909–6914 CrossRef CAS PubMed.
- A. Fried and C. W. Mathews, Chem. Phys. Lett., 1977, 52, 363–367 CrossRef CAS.
- J. W. C. Johns, Can. J. Phys., 1961, 39, 1738–1768 CrossRef CAS.
- M. J. Baier, A. J. McDonald, K. A. Clements, C. S. Goldenstein and S. F. Son, Proc. Combust. Inst., 2021, 38, 4433–4440 CrossRef CAS.
- A. Kramida, Y. Ralchenko, J. Reader, and NIST ASD Team, NIST Atomic Spectra Database (version 5.10), National Institute of Standards and Technology, Gaithersburg, MD, 2022 Search PubMed.
- P. Dagaut, P. Glarborg and M. U. Alzueta, Prog. Energy Combust. Sci., 2008, 34, 1–46 CrossRef CAS.
- K. Fujioka, R. I. Kaiser and R. Sun, J. Phys. Chem. A, 2023, 127, 913–923 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and computational methods (Fig. S1–S5), droplet sizes of the reactants (Table S1), number of moles of the reactants with the varying molecular oxygen contents in the levitator process chamber along with measured ignition delays (Table S2), peak assignments for the deconvoluted flame emission spectra (Table S3), number of moles of the gaseous products formed (Table S4), percentage yield of carbon dioxide from the [EMIM]+ cation (Table S5), high-speed infrared and optical camera images depicting merging sequence and ignition at different fractions of molecular O2-contents (Fig. S6), temporal evolution of the flame emission spectra for the ignition upon varying molecular O2-contents (Fig. S7), flame emission intensities as a function of molecular oxygen contents (Fig. S8), temporal profiles of boron dioxide (BO2) and flame temperature during ignition at different molecular O2 volume fractions (Fig. S9), deconvolution of the flame emission spectra (Fig. S10), temporal profiles of all the intermediates during ignition (Fig. S11), flame temperature determination, Cartesian coordinates for the optimized structures including transition states in the potential energy surface and enthalpy changes during droplet merging (Table S6). Supplementary Movies S1 to S13. See DOI: https://doi.org/10.1039/d3sc05096c |
| This journal is © The Royal Society of Chemistry 2024 |