
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTertiary amide bond formation by an engineered asparaginyl ligase†
Simon J.
de Veer‡
 *,
Yan
Zhou‡
,
Thomas
Durek
,
David J.
Craik
* and
Fabian B. H.
Rehm
*
*,
Yan
Zhou‡
,
Thomas
Durek
,
David J.
Craik
* and
Fabian B. H.
Rehm
*
Institute for Molecular Bioscience, Australian Research Council Centre of Excellence for Innovations in Peptide and Protein Science, The University of Queensland, Brisbane, QLD 4072, Australia. E-mail: s.deveer@imb.uq.edu.au; d.craik@imb.uq.edu.au; fbhrehm@gmail.com
First published on 5th March 2024
Abstract
Transpeptidases are powerful tools for site-specific protein modification, enabling the production of tailored biologics to investigate protein function and aiding the development of next-generation therapeutics and diagnostics. Although protein labelling at the N- or C-terminus is readily accomplished using a range of established transpeptidases, these reactions are generally limited to forming products that are linked by a standard (secondary) amide bond. Here we show that, unlike other widely used transpeptidases, an engineered asparaginyl ligase is able to efficiently synthesise tertiary amide bonds by accepting diverse secondary amine nucleophiles. These reactions proceed efficiently under mild conditions (near-neutral pH) and allow the optimal recognition elements for asparaginyl ligases (P1 Asn and P2′′ Leu) to be preserved. Certain products, particularly proline-containing products, were found to be protected from recognition by the enzyme, allowing for straightforward sequential labelling of proteins. Additionally, incorporation of 4-azidoproline enables one-pot dual labelling directly at the ligation junction. These capabilities further expand the chemical diversity of asparaginyl ligase-catalysed reactions and provide an alternative approach for straightforward, successive modification of protein substrates.
Introduction
The use of transpeptidases for protein modification provides several advantages over chemical conjugation strategies, but perhaps the most appealing feature is their potential for exquisite site-specificity. As a result, several transpeptidases, particularly sortases, subtiligases and asparaginyl ligases, have gained widespread use.1 These enzymes typically catalyse the ligation of peptides or proteins via a reaction that involves nucleophilic attack by an N-terminal primary amine on a covalently linked enzyme-substrate intermediate. Although some transpeptidases are promiscuous with respect to the identity of the incoming N-terminal amine nucleophile, reactions involving secondary amines are rarely observed.2,3 Indeed, proline is reportedly not accepted as the N-terminal residue by a wide range of transpeptidases and amide ligases, despite their diverse specificities and catalytic mechanisms.4–6 Consequently, known transpeptidases are limited to generating products that are joined via a conventional (secondary) amide bond. However, transpeptidation reactions are often reversible and, if the recognition sequence for the enzyme is re-formed in the product, it can lead to re-recognition and proteolytic processing of the product.We hypothesised that ligating N-terminal secondary amines to form tertiary amide-bonded products might contribute to preventing product recognition. Such reactions have yet to be described for naturally occurring or engineered peptide ligases that are sufficiently versatile for practical use. Here, we show that the engineered asparaginyl ligase [C247A]OaAEP1 can efficiently form tertiary amide-bonded products and that several proline-containing products are subsequently resistant to significant recognition. We utilise separate aspects of the reaction involving proline-based nucleophiles to perform successive reactions on protein substrates and generate dual labelled products. We harness the disparity between acceptance of secondary amine nucleophiles but poor recognition of the resulting product to site-specifically label a protein at both termini using a single asparaginyl ligase. We also utilise the pyrrolidine ring of proline as a scaffold for displaying an additional chemical handle, which enables one-pot dual labelling directly at the ligation junction.
[C247A]OaAEP1 is a versatile ligase that has been used for a broad range of protein engineering and biotechnological applications.7–13 This enzyme acts on substrates bearing a tripeptide Asn–Gly–Leu (P1↓P1′–P2′) recognition motif, with cleavage of the Asn–Gly peptide bond generating a thioester-linked acyl-enzyme intermediate that is highly susceptible to nucleophilic attack. Conventional nucleophiles include peptides or proteins with an N-terminal Gly–Leu sequence, with the resulting transpeptidation reaction providing direct access to N- or C-terminally labelled proteins,9,13–16 head-to-tail cyclisation of peptides or proteins,14,17 or defined protein–protein fusions.11,15,18 However, these reactions typically reconstitute the Asn–Gly–Leu recognition sequence in the product, hence recognition and proteolytic processing of the product remains a possibility.
Previous efforts to avoid re-recognition events for asparaginyl ligases have focused on pH-dependent recognition of the P1 residue (Asp or Asn), which necessitates precise manipulation of the reaction pH between each labelling step,19 or varying the product P2′ residue to Val by incorporating GV nucleophiles that must be provided in substantial excess to generate the product.14 By contrast, altering P1′ in the formed product has not been explored – asparaginyl ligases have been reported to be tolerant of diverse amino acids at this position, with the one notable exception being proline.9
Results and discussion
To explore whether [C247A]OaAEP1 could catalyse the formation of tertiary amide-bonded products, we initially tested whether tetrapeptides bearing an N-terminal secondary amine could be ligated to a model peptide substrate. We first selected N-methylglycine and proline as examples of common, widely available secondary amino acids and assessed ligation of NMe-GLRL or PLRL to Ac-GWRNGLH in reactions comprising 100 nM [C247A]OaAEP1, 100 μM NGL peptide substrate and 200 μM NMe-GLRL or PLRL (2 molar equivalents) in 100 mM HEPES pH 7.5 (25 °C for 1 h). Reactions were quenched by adding 1% TFA, then analysed by reverse-phase HPLC (Fig. 1) and MALDI-TOF mass spectrometry (Fig. S1†). In ligation reactions using NMe-GLRL, only low levels of the tertiary amide-bonded product were observed (5% conversion, Fig. 1). However, ligation of PLRL was more effective (35% conversion, Fig. 1), with the level of product reaching approximately half that observed in reactions using an optimal peptide nucleophile GLRL under similar conditions (Fig. S2†). Interestingly, our finding that Pro is incorporated more efficiently than N-methylglycine in transpeptidation reactions parallels reported peptidyl transfer rates for Pro and other N-alkylamino acids during ribosomal synthesis, where Pro is incorporated more rapidly than N-methyl or N-butyl amino acids.20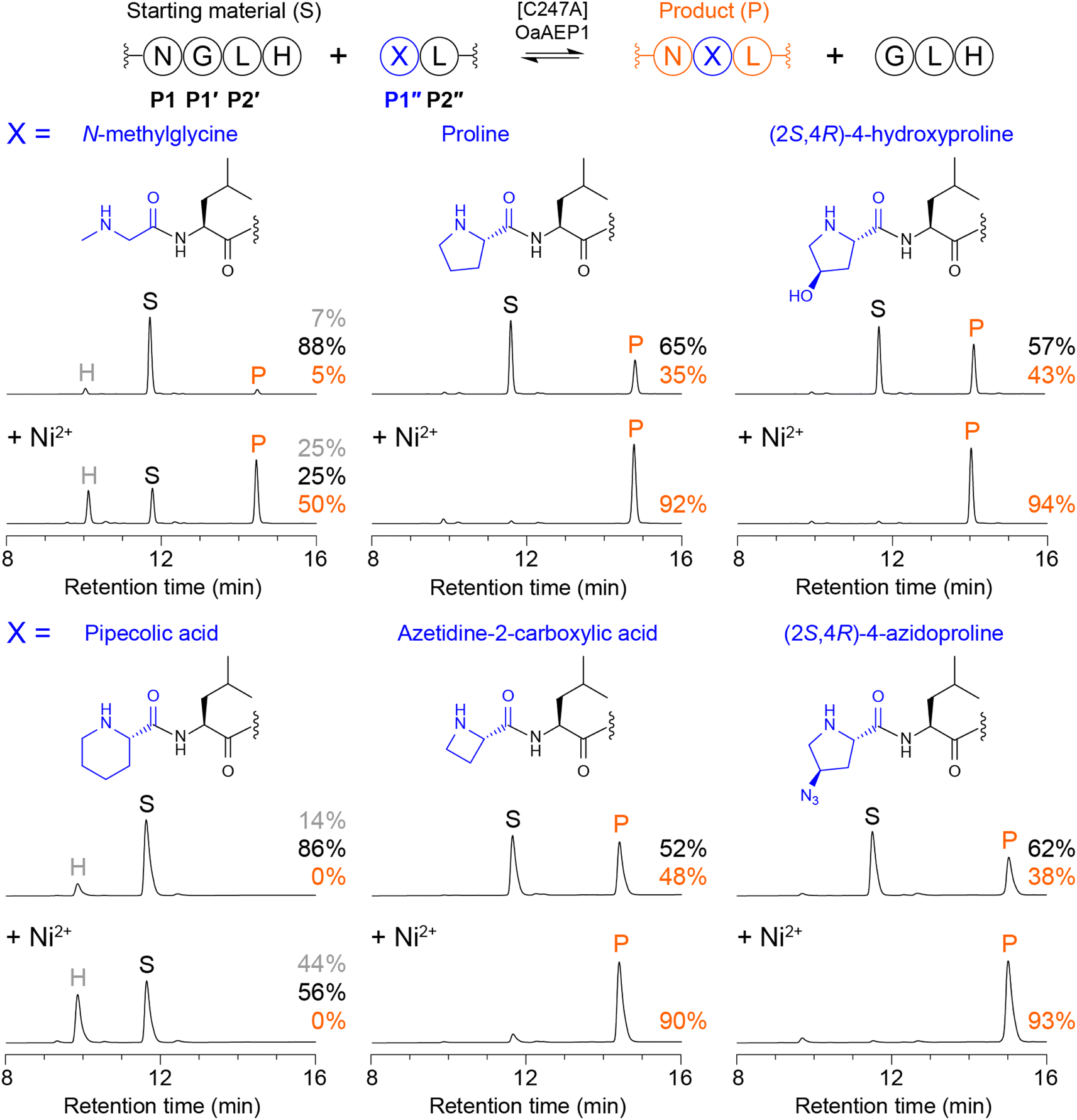 | ||
| Fig. 1 Enzymatic tertiary amide bond formation with model peptides. Reactions were run using 100 nM [C247A]OaAEP1, an NGLH-containing substrate (Ac-GWRNGLH, 100 μM), and tetrapeptides bearing an N-terminal secondary amine (XLRL where X = N-methylglycine, proline, (2S,4R)-4-hydroxyproline, pipecolic acid, azetidine-2-carboxylic acid or (2S,4R)-4-azidoproline, 200 μM, 2 equiv.) in 100 mM HEPES pH 7.5 (1 h at 25 °C). Shown are analytical reverse-phase HPLC traces (A280 nm). Peaks for starting material (S, black), product (P, orange), and hydrolysed substrate (H, grey) are indicated, as well as the % conversion to product. Spectra from MALDI-TOF MS are shown in Fig. S1.† Reactions labelled + Ni2+ (lower panels) were run with 300 μM NiCl2 included. Table S1 lists all calculated and observed peptide masses. | ||
To further explore the scope of [C247A]OaAEP1-catalysed tertiary amide bond formation, we tested a naturally occurring proline analogue (2S,4R)-4-hydroxyproline (Hyp), which is a key component of fibrillar collagen. In reactions using Hyp-LRL, we observed a further increase in the level of product formed (43% conversion, Fig. 1). We also examined varying the size of the pyrrolidine ring by screening six-membered (pipecolic acid, Pip) and four-membered (azetidine-2-carboxylic acid, Aze) cyclic amino acids. Ligation of Aze-LRL (48% conversion) was more efficient than PLRL but we did not detect product formation for Pip-LRL (Fig. 1), suggesting that the larger piperidine ring was too bulky. Additionally, we tested a proline analogue bearing a click handle, (2S,4R)-4-azidoproline (Azp), and found that Azp-LRL could be ligated to the NGL substrate (38% conversion) at a similar level to PLRL.
In the ligation reactions described above, cleavage of the NGL peptide substrate releases a GLH by-product that competes with the target peptide nucleophile. As the reaction proceeds and the concentration of substrate decreases, the concentration of GLH increases, eventually reaching an equilibrium where GLH is incorporated in favour of the target nucleophile. Anticipating that this effect was limiting the formation of tertiary amide-bonded products in our initial experiments, we turned to an approach for shifting the equilibrium in asparaginyl ligase-catalysed reactions to favour product formation. This method relies on quenching the released GLH tripeptide by supplementing the reaction with Ni2+, which tightly binds to N-terminal GLH motifs and weakens the nucleophilicity of the terminal amine.15 Such an approach enables efficient conjugation of otherwise less favoured nucleophiles.10,11
Repeating the earlier experiments in the presence of 300 μM NiCl2 (3 molar equivalents) generally led to improved formation of each tertiary amide-bonded product (Fig. 1). However, ligation of Pip-LRL remained unsuccessful, with quenching the released GLH peptide leading to increased levels of acyl donor peptide hydrolysis as a suitable peptide nucleophile was absent. Interestingly, we also observed that ligation at N-methylglycine remained less efficient than at Pro, Hyp, Aze or Azp. This effect could be partially overcome by increasing the concentration of NMe-GLRL to 800 μM (8 equiv.), which suppressed the competing hydrolysis reaction and enabled higher conversion to product (>80%, Fig. S3†). By contrast, supplementing the reaction with Ni2+ was sufficient to drive efficient formation of the tertiary amide-bonded product for PLRL, Hyp-LRL, Aze-LRL, and Azp-LRL at 200 μM peptide nucleophile (2 equiv.). These reactions reached ≥90% conversion to product (Fig. 1), which approaches the level of product formation for reactions using an optimal peptide nucleophile GLRL (Fig. S2†).
We subsequently performed time course experiments to compare the rate of product formation for 2 equiv. GLRL and a model secondary amine nucleophile PLRL in the presence or absence of Ni2+ (Fig. 2 and S4†). When Ni2+ was absent, reactions reached equilibrium within 30 min with higher levels of product formed for GLRL (65% conversion) than PLRL (34%). As noted above, this result reflects the greater capacity of GLRL, compared with PLRL, to compete with GLH released from the peptide substrate. However, when Ni2+ was added to quench the released GLH by-product, this advantage was nullified and reactions with GLRL and PLRL proceeded with similar kinetics.
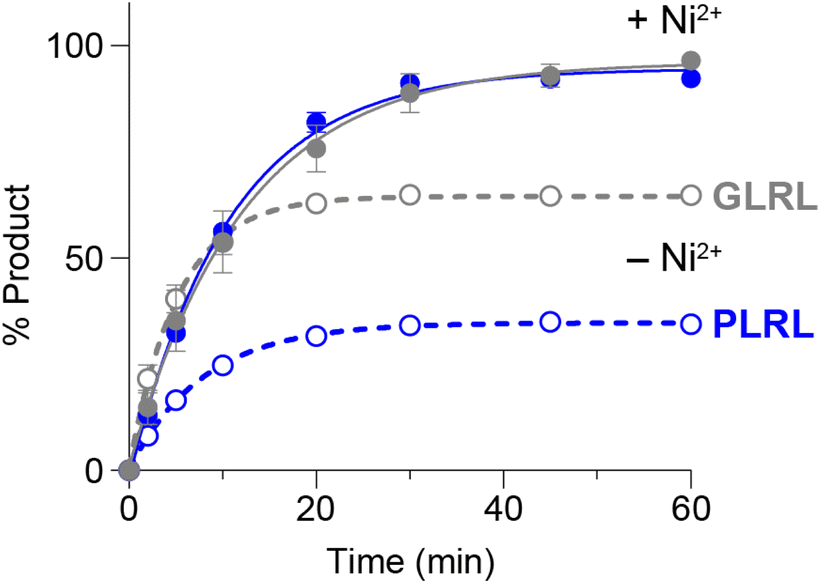 | ||
| Fig. 2 Time course for ligation of GLRL or PLRL to a model NGLH substrate. Reactions were run as described in Fig. 1 and analysed by reverse-phase HPLC (Fig. S4†). Data points represent the mean ± standard deviation (n = 3) for GLRL (grey) or PLRL (blue), in the presence (filled circles) or absence (open circles) of 300 μM NiCl2. Data were fit to one-phase association curves in GraphPad Prism. | ||
An additional effect of replacing Gly with Pro at P1′′ (incoming nucleophile) is that it introduces a chiral residue at the N-terminus. We tested whether D-Pro-LRL could also be ligated to the NGL peptide substrate, but observed high levels of acyl donor peptide hydrolysis and no detectable product formation (Fig. S3†). We also examined the effect of replacing L-Leu with D-Leu at P2′′ as we recently showed that this modification was well tolerated with P1′′ Gly.11 However, like D-Pro-LRL, P-D-Leu-RL was poorly incorporated and substantial levels of substrate hydrolysis were observed (Fig. S3†). These findings indicate that ligation reactions involving P1′′ Pro proceed under narrow stereochemical constraints.
Having shown that [C247A]OaAEP1 catalyses the formation of tertiary amide-bonded products, we next examined the degree to which these products are susceptible to re-recognition and proteolytic processing. We synthesised product mimetics for each secondary amino acid that could be ligated (Ac-GWRNXLH where X = NMe-Gly, Pro, Hyp, Aze or Azp) and performed reactions with 100 μM product mimetic, 200 μM GLRL (2 equiv.), 100 nM [C247A]OaAEP1, and 300 μM NiCl2 (25 °C for 18 h), as shown in Fig. 3. Of note, assessing recognition and processing of the product mimetics using a transpeptidation assay is more sensitive than monitoring peptide hydrolysis as GLRL is a substantially more effective nucleophile than water under these conditions.
 | ||
| Fig. 3 Recognition of product mimetics bearing a tertiary amide bond. Reactions were run using 100 nM [C247A]OaAEP1, NXLH-containing substrates (Ac-GWRNXLH where X = N-methylglycine, proline, (2S,4R)-4-hydroxyproline, azetidine-2-carboxylic acid, (2S,4R)-4-azidoproline or (2S,4S)-4-azidoproline, 100 μM), GLRL (200 μM, 2 equiv.) and NiCl2 (300 μM) in 100 mM HEPES pH 7.5 (18 h at 25 °C). Shown are analytical reverse-phase HPLC traces (A280 nm). Peaks for starting material (S*, black) and product (P*, orange) are indicated, as well as the % conversion to product. Spectra from MALDI-TOF MS are shown in Fig. S5.† Table S1† lists all calculated and observed peptide masses. | ||
Examining the processing of the five product mimetics revealed that each P1′ residue had different effects on recognition (Fig. 3). Compared to P1′ Gly in a conventional NGL substrate (>90% conversion within 1 h under similar conditions, Fig. S2†), addition of a methyl group (P1′ NMe-Gly) led to lower levels of substrate processing (56% conversion within 18 h, Fig. 3). However, P1′ Aze was more susceptible to processing, with conversion to product reaching near completion (95% conversion). By contrast, substrates with P1′ Pro or Hyp were largely resistant to processing (18% or 7% conversion to product, respectively). In general, proteolytic cleavage at Xaa-Pro sites is poorly favoured across a wide range of proteases, such that proteins with greater resistance to degradation can be engineered by introducing P1′ Pro at a susceptible cleavage site.21,22 Surprisingly, this effect was diminished in the product mimetic with P1′ Azp (73% conversion within 18 h). Previous studies on Azp derivatives have identified an azido gauche effect for (2S,4R)Azp and (2S,4S)Azp,23 and that each analogue has different impacts on peptide conformation.23,24 Having tested (2S,4R)Azp at P1′, we synthesised the corresponding peptide with (2S,4S)Azp and examined substrate processing. The (2S,4S)Azp substrate was highly resistant to processing (12% conversion within 18 h, Fig. 3), which is comparable to Pro and Hyp. We also synthesised (2S,4S)Azp-LRL and verified that this modification only affected product re-recognition but not product formation, which remained similar to (2S,4R)Azp-LRL (Fig. S6†).
These findings indicate that, in general, proline-containing products are poorly re-recognised and undergo limited enzymatic processing (Scheme 1). This effect is particularly evident in the divergent processing of substrates containing Pro or the related cyclic amino acid Aze, and appears to be modulated by additional steric or conformational factors, as noted for processing of Azp-containing products. Here, processing was heavily dependent on the stereochemistry of the azido substituent, such that (4R)Azp was susceptible to processing, whereas (4S)Azp was not.
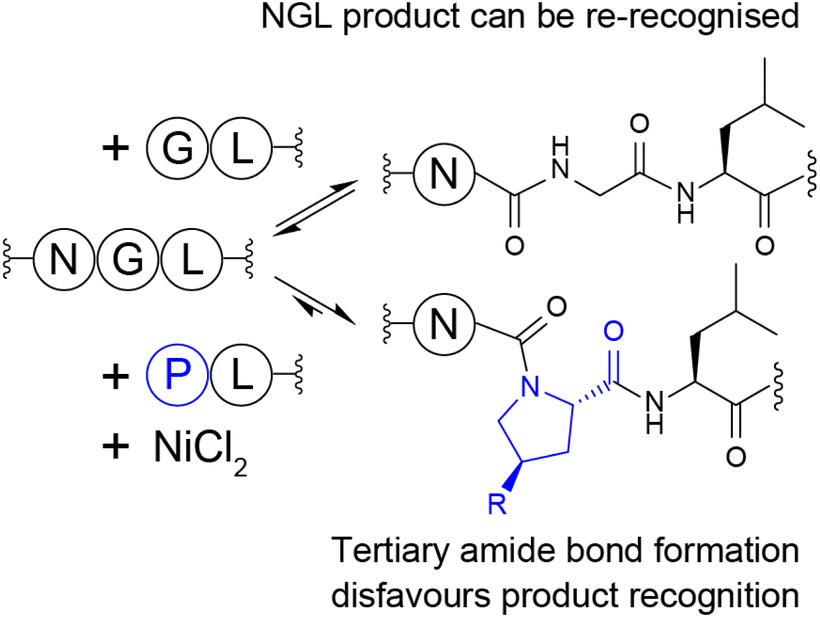 | ||
| Scheme 1 Tertiary amide bond formation by ligating proline (R = H), Hyp (R = OH) or (4S)Azp (R = N3) generates products that are poorly re-recognised, unlike reactions involving conventional GL nucleophiles, which re-form the recognition sequence in the product. | ||
To further assess the utility of asparaginyl ligase-catalysed tertiary amide bond formation, we explored whether the reaction could also be applied to protein substrates. Initially, we tested N-terminal labelling and synthesised a TAMRA-GRNGLH peptide substrate for conjugation to proteins bearing an N-terminal Pro–Leu motif. Such reactions require ligation of secondary amine nucleophiles from larger, more structured polypeptide chains compared with the synthetic tetrapeptides used in earlier experiments (Fig. 1). The model proteins we used for N-terminal labelling were small ubiquitin-like modifier (SUMO) and superfolder green fluorescent protein (sfGFP),25 which were each produced with an N-terminal PL extension. We observed that the TAMRA-peptide substrate was readily conjugated to both SUMO and sfGFP (Fig. 4A and B), demonstrating that tertiary amide bond formation can facilitate N-terminal protein labelling. Interestingly, labelling efficiency for sfGFP appeared to be slightly higher than SUMO (Fig. S7†), although both reactions reached ≥90% conversion within 4 h (Fig. 4A and B). Extending the reaction time to 18 h yielded near-complete conversion to product for both proteins (Fig. S7†).
 | ||
| Fig. 4 Protein labelling with tertiary amide bond formation. N-terminal labelling of (A) SUMO or (B) sfGFP25 with an N-terminal PL extension. Reactions contained 50 μM protein substrate, 500 μM TAMRA-GRNGLH, 2 mM NiCl2 and 200 nM [C247A]OaAEP1 in 100 mM HEPES pH 7 at 25 °C and were run for 4 h, then quenched via TFA addition (2%) prior to analysis by ESI-MS (see Fig. S7† for additional timepoints). Shown are reconstructed spectra with the observed substrate (S) and product (P) masses indicated. (C) C-terminal labelling of an MHCII-targeting nanobody (VHHMHCII)26 with a C-terminal NGLH-StrepTag extension. Reactions contained 50 μM protein substrate, 200 μM XLGK(biotin)RG (X = Pro, Hyp or Azp as indicated), 250 μM NiCl2 and 500 nM [C247A]OaAEP1 in 100 mM HEPES pH 7 at 25 °C and were run for 4 h, then quenched via TFA addition (2%) prior to analysis by ESI-MS (see Fig. S8† for additional timepoints). Shown are reconstructed spectra with the observed substrate (S) and product (P) masses indicated. For Azp-LGK(biotin)RG, the transpeptidation reaction (1) was followed by strain-promoted azide–alkyne cycloaddition (2) by adding 250 μM DBCO-AF488 and incubating for a further 4 h to generate the C-terminally dual labelled product in a one-pot reaction. In addition to ESI-MS, reaction products (1 and 2) were analysed by SDS-PAGE, with gels imaged after InstantBlue Coomassie staining (top panel) and by fluorescence scanning (lower panel). (D) C-terminal labelling of an MHCII-targeting nanobody (VHHMHCII)26 with a C-terminal NPLH-StrepTag extension. Reactions were conducted as described in (C) using PLGK(biotin)RG (see Fig. S9† for additional timepoints). Table S2† lists all calculated and observed protein masses. | ||
We also examined C-terminal labelling of a model protein, VHHMHCII, which is a single-domain antibody (nanobody) that recognises major histocompatibility complex class II (MHCII) molecules.26 This nanobody was recombinantly produced with a C-terminal extension comprising an NGLH recognition sequence followed by a StrepTag for affinity purification, and served as a substrate for ligation reactions using a biotinylated peptide, XLGK(biotin)RG where X = Pro, Hyp or Azp. Under mild reaction conditions (50 μM protein, 4 equiv. biotinylated peptide, 5 equiv. NiCl2 and 0.01 equiv. enzyme in 100 mM HEPES pH 7), we observed formation of each labelled product, with reactions reaching ≥90% conversion within 4 h (Fig. 4C). As observed for N-terminal labelling, conversion to product reached near-completion after extending the reaction time (Fig. S8†). For comparison, we also examined labelling of the nanobody substrate with GLGK(biotin)RG and found similar levels of product at each timepoint for the GL- and PL-peptides (Fig. S8†).
To assess whether the tertiary amide-bonded products formed in this context would also be resistant to re-recognition, we produced a nanobody where the AEP recognition sequence was mutated from NGLH to NPLH. In line with observations made using model peptides (Fig. 3), we found that the efficiency of labelling this protein substrate was substantially lower (5% conversion after 4 h, Fig. 4D; 7% after 18 h, Fig. S9†) under equivalent conditions.
We next turned to protein dual labelling and examined utilising the azide handle installed at the ligation junction upon conjugation of an Azp–Leu peptide. Having identified conditions for efficient C-terminal labelling using Azp-LGK(biotin)RG, we anticipated that introducing a second label via strain-promoted azide–alkyne cycloaddition27 could enable one-pot protein dual labelling directly at the ligation junction. After labelling VHHMHCII with Azp-LGK(biotin)RG to generate the single labelled product (1), we added a DBCO-functionalised fluorophore (DBCO-AF488, 250 μM) and observed quantitative conversion to the dual labelled product (2) within 4 h (Fig. 4C).
Given that reactions generating protected products have the potential to allow successive modifications to be carried out using the same enzyme, we next explored this possibility via tertiary amide bond formation. We examined dual labelling of CTC-445.2d, a bivalent de novo designed protein that binds to the SARS-CoV-2 spike protein at the interface that typically interacts with hACE2.28 The CTC-445.2d construct was extended at the C-terminus to include an NGLH recognition sequence and, at the N-terminus, we appended a pro-peptide bearing a GL motif that could be unmasked by TEV protease prior to the second reaction. We envisaged that generating a poorly favoured NPL recognition sequence in the product (Scheme 1) would enable subsequent reactions to be carried out without affecting the newly formed NPL site. To generate the dual labelled CTC-445.2d protein (Fig. 5), we first performed C-terminal labelling to conjugate PLGK(biotin)RG and form a tertiary amide bonded-product (1). This reaction reached >95% conversion within 4 h (Fig. S10†). After releasing the N-terminal pro-peptide (ENLYFQ) using TEV protease to reveal the downstream GL motif and removing excess peptide using a buffer exchange column, the second transpeptidation reaction was carried out to conjugate TAMRA-GRNGLH at the N-terminus and generate the site-specifically dual labelled CTC-445.2d protein (>95% conversion, Fig. 5).
 | ||
| Fig. 5 Site-specific, successive protein modification with tertiary amide bond formation. Dual labelling was conducted on the de novo designed, bivalent human angiotensin-converting enzyme 2 (hACE2) decoy CTC-445.2d.28 The protein substrate was extended C-terminally with an NGLH sequence and N-terminally with a TEV protease cleavage site (ENLYFQ) followed by a GL sequence. In the first step (1), the protein was C-terminally labelled with a PL-biotin peptide in a reaction comprising 50 μM protein, 500 μM PLGK(biotin)RG, 250 μM NiCl2 and 500 nM [C247A]OaAEP1 in 100 mM HEPES pH 7 for 4 h at 25 °C. After N-terminal deprotection using TEV protease and removal of excess peptide via a buffer exchange column, N-terminal labelling (2) was conducted on 50 μM protein, with 200 μM TAMRA-GRNGLH, 1 mM NiCl2 and 100 nM [C247A]OaAEP1 for 10 min in 100 mM HEPES pH 7 to yield the site-specifically dual labelled product. Shown are reconstructed ESI-MS spectra with the observed masses indicated. Higher resolution spectra for monitoring C-terminal labelling are shown in Fig. S10.† Table S2† lists all calculated and observed protein masses. | ||
Conclusions
In summary, we have demonstrated the capacity of the asparaginyl ligase [C247A]OaAEP1 to ligate N-terminal secondary amines on peptides and proteins. To our knowledge, these reactions represent the first examples of tertiary amide bond formation by a general-purpose transpeptidase. Importantly, we found that several of the resulting products were poor substrates for the enzyme, particularly products with an Asn–Pro, Asn–Hyp or Asn–(4S)Azp junction. Inefficient cleavage at Xaa–Pro sites is a relatively general phenomenon across a wide range of proteases – for transpeptidases, forming an Xaa–Pro junction contributes to suppressing product recognition events that would culminate in reaction reversal. Although such reversal can be overcome in standalone reactions by providing the peptide to be attached in excess,14 or by quenching the by-product released upon substrate cleavage to shift the equilibrium to favour product formation,15,16 these approaches still generate products that are susceptible to proteolytic processing, which is a drawback for carrying out several reactions sequentially. This challenge can be overcome by using enzymes with orthogonal substrate specificity,29 but here we demonstrate that tertiary amide bond formation can facilitate site-specific, successive protein modifications using a single transpeptidase.Unlike previously reported approaches, these ligation reactions are fully compatible with retaining the optimal recognition elements for asparaginyl ligases (P1 Asn and P2′′ Leu) and proceed efficiently under mild conditions (near-neutral pH). A further benefit of the approach is that the pyrrolidine ring of proline affords the opportunity to ligate nucleophiles bearing an additional chemical handle. We show that labelled peptides with an N-terminal Azp–Leu motif can be efficiently ligated to a protein substrate, which subsequently enables one-pot dual labelling directly at the ligation junction. This study broadens the known substrate scope of [C247A]OaAEP1-catalysed transpeptidation and we anticipate that such reactions will become increasingly valuable given the increasing complexity and diversity of engineered proteins and protein conjugates currently under development.
Data availability
All data are available in the article and ESI.† Further information is available from the authors upon reasonable request.Author contributions
S. J. D., D. J. C. and F. B. H. R. conceived the project. S. J. D., Y. Z. and F. B. H. R. performed the experiments. S. J. D., Y. Z., T. D., D. J. C. and F. B. H. R. analysed the data. S. J. D. and F. B. H. R. wrote the manuscript with input from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a grant from the Australian Research Council (ARC, DP200101299) and by access to the facilities of the ARC Centre of Excellence for Innovations in Peptide and Protein Science (CE200100012). D. J. C. is a National Health & Medical Research Council Leadership Fellow (2009564).References
- R. Pihl, Q. Zheng and Y. David, Nat. Rev. Chem, 2023, 7, 234–255 CrossRef CAS PubMed.
- A. M. Weeks and J. A. Wells, Nat. Chem. Biol., 2018, 14, 50–57 CrossRef CAS PubMed.
- H. K. Philpott, P. J. Thomas, D. Tew, D. E. Fuerst and S. L. Lovelock, Green Chem., 2018, 20, 3426–3431 RSC.
- M. Winn, M. Rowlinson, F. Wang, L. Bering, D. Francis, C. Levy and J. Micklefield, Nature, 2021, 593, 391–398 CrossRef CAS PubMed.
- T. Nuijens, A. Toplak, P. J. L. M. Quaedflieg, J. Drenth, B. Wu and D. B. Janssen, Adv. Synth. Catal., 2016, 358, 4041–4048 CrossRef CAS.
- G. K. Nguyen, S. Wang, Y. Qiu, X. Hemu, Y. Lian and J. P. Tam, Nat. Chem. Biol., 2014, 10, 732–738 CrossRef CAS PubMed.
- R. Yang, Y. H. Wong, G. K. T. Nguyen, J. P. Tam, J. Lescar and B. Wu, J. Am. Chem. Soc., 2017, 139, 5351–5358 CrossRef CAS PubMed.
- F. B. H. Rehm, T. J. Tyler, J. Xie, K. Yap, T. Durek and D. J. Craik, ChemBioChem, 2021, 22, 2079–2086 CrossRef CAS PubMed.
- T. M. S. Tang, D. Cardella, A. J. Lander, X. Li, J. S. Escudero, Y. H. Tsai and L. Y. P. Luk, Chem. Sci., 2020, 11, 5881–5888 RSC.
- F. B. H. Rehm, T. J. Tyler, K. Yap, S. J. de Veer, D. J. Craik and T. Durek, J. Am. Chem. Soc., 2021, 143, 19498–19504 CrossRef CAS PubMed.
- F. B. H. Rehm, T. J. Tyler, S. J. de Veer, D. J. Craik and T. Durek, Angew. Chem., Int. Ed., 2022, 61, e202116672 CrossRef CAS PubMed.
- M. Fottner, J. Heimgartner, M. Gantz, R. Muhlhofer, T. Nast-Kolb and K. Lang, J. Am. Chem. Soc., 2022, 144, 13118–13126 CrossRef CAS PubMed.
- Z. Lu, Y. Liu, Y. Deng, B. Jia, X. Ding, P. Zheng and Z. Li, Chem. Commun., 2022, 58, 8448–8451 RSC.
- F. B. H. Rehm, T. J. Harmand, K. Yap, T. Durek, D. J. Craik and H. L. Ploegh, J. Am. Chem. Soc., 2019, 141, 17388–17393 CrossRef CAS PubMed.
- F. B. H. Rehm, T. J. Tyler, K. Yap, T. Durek and D. J. Craik, Angew. Chem., Int. Ed., 2021, 60, 4004–4008 CrossRef CAS PubMed.
- Y. Xia, F. Li, X. Zhang, S. Balamkundu, F. Tang, S. Hu, J. Lescar, J. P. Tam and C. F. Liu, J. Am. Chem. Soc., 2023, 145, 6838–6844 CrossRef CAS PubMed.
- K. Yap, J. Du, F. Y. Looi, S. R. Tang, S. J. de Veer, A. R. Bony, F. B. H. Rehm, J. Xie, L. Y. Chan, C. K. Wang, D. J. Adams, L. H. L. Lua, T. Durek and D. J. Craik, Green Chem., 2020, 22, 5002–5016 RSC.
- Y. Deng, T. Wu, M. Wang, S. Shi, G. Yuan, X. Li, H. Chong, B. Wu and P. Zheng, Nat. Commun., 2019, 10, 2775 CrossRef PubMed.
- D. Zhang, Z. Wang, S. Hu, S. Balamkundu, J. To, X. Zhang, J. Lescar, J. P. Tam and C. F. Liu, J. Am. Chem. Soc., 2021, 143, 8704–8712 CrossRef CAS PubMed.
- M. Y. Pavlov, R. E. Watts, Z. Tan, V. W. Cornish, M. Ehrenberg and A. C. Forster, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 50–54 CrossRef CAS PubMed.
- L. G. Frenken, M. R. Egmond, A. M. Batenburg and C. T. Verrips, Protein Eng., 1993, 6, 637–642 CrossRef CAS PubMed.
- Y. Markert, J. Koditz, R. Ulbrich-Hofmann and U. Arnold, Protein Eng., 2003, 16, 1041–1046 CrossRef CAS PubMed.
- L. S. Sonntag, S. Schweizer, C. Ochsenfeld and H. Wennemers, J. Am. Chem. Soc., 2006, 128, 14697–14703 CrossRef CAS PubMed.
- M. Kumin, L. S. Sonntag and H. Wennemers, J. Am. Chem. Soc., 2007, 129, 466–467 CrossRef PubMed.
- J. D. Pedelacq, S. Cabantous, T. Tran, T. C. Terwilliger and G. S. Waldo, Nat. Biotechnol., 2006, 24, 79–88 CrossRef CAS PubMed.
- J. N. Duarte, J. J. Cragnolini, L. K. Swee, A. M. Bilate, J. Bader, J. R. Ingram, A. Rashidfarrokhi, T. Fang, A. Schiepers, L. Hanke and H. L. Ploegh, J. Immunol., 2016, 197, 4838–4847 CrossRef CAS PubMed.
- N. J. Agard, J. A. Prescher and C. R. Bertozzi, J. Am. Chem. Soc., 2004, 126, 15046–15047 CrossRef CAS PubMed.
- T. W. Linsky, R. Vergara, N. Codina, J. W. Nelson, M. J. Walker, W. Su, C. O. Barnes, T. Y. Hsiang, K. Esser-Nobis, K. Yu, Z. B. Reneer, Y. J. Hou, T. Priya, M. Mitsumoto, A. Pong, U. Y. Lau, M. L. Mason, J. Chen, A. Chen, T. Berrocal, H. Peng, N. S. Clairmont, J. Castellanos, Y. R. Lin, A. Josephson-Day, R. S. Baric, D. H. Fuller, C. D. Walkey, T. M. Ross, R. Swanson, P. J. Bjorkman, M. Gale Jr, L. M. Blancas-Mejia, H. L. Yen and D. A. Silva, Science, 2020, 370, 1208–1214 CrossRef CAS PubMed.
- T. J. Harmand, D. Bousbaine, A. Chan, X. Zhang, D. R. Liu, J. P. Tam and H. L. Ploegh, Bioconjugate Chem., 2018, 29, 3245–3249 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc06352f |
| ‡ Authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |