
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnravelling a bench-stable zinc-amide compound as highly active multitasking catalyst for radical-mediated selective alk(en)ylation of unactivated carbocycles under mild conditions†
Sangita
Sahoo
,
Subarna
Manna
and
Arnab
Rit
 *
*
Department of Chemistry, Indian Institute of Technology Madras, Chennai 600036, India. E-mail: arnabrit@iitm.ac.in
First published on 24th February 2024
Abstract
The direct functionalization of unactivated organic moieties via C–C bond formation has long fascinated synthetic chemists. Although base-metal systems are steadily emerging in this area, achieving multitasking activity in a single catalyst to execute several such functionalizations under mild conditions is challenging. To address this, we herein report an effective protocol for the selective C-alk(en)ylation of indene/fluorene with alcohol as a green alkylating agent employing a naturally abundant and eco-friendly zinc-derived compound, for the first time. Notably, this study unveils the unique potential of a bench-stable Zn compound bearing an amidated imidazolium salt towards C–C bond-forming reactions utilizing an array of alcohols, ranging from aliphatic to aromatic and, attractively, even secondary alcohols. Moreover, this readily scalable protocol, which proceeds via an underdeveloped radical-mediated borrowing hydrogen protocol (an aldehyde is generated from an alcohol, and subsequent condensation with indene/fluorene provides the corresponding alkenylated products) established based on a range of control experiments, works effortlessly under mild conditions using a low catalyst loading. Notably, this approach affords remarkable selectivity towards alkylated or alkenylated products with a high level of functional group tolerance and chemoselectivity. Crucially, the catalytic activity of these Zn compounds can be attributed to their hydrogen atom transfer (HAT) capability, while their selectivity towards different products can be understood in terms of employed reaction conditions. Lastly, the synthetic utility of obtained products was showcased by their late-stage functionalization to access unsymmetrical 9,9-disubstituted fluorenes, which are potentially useful for various optoelectronic applications.
Introduction
Carbon–carbon bond forming reactions have garnered a great deal of attention in the field of synthetic chemistry due to their diverse reactivity profiles and paramount importance in medicinal chemistry, agrochemicals, and natural product synthesis.1 In this context, direct alkylation of an activated organic moiety at the α-carbon of an electron withdrawing group may be easily achieved,2 but remains challenging for compounds possessing a methylene functionality, such as fluorene and indene (Scheme 1a).3 Additionally, the widespread applications of C-alkylated carbocycles (indene/fluorene) as useful compounds in synthetic chemistry, polymer chemistry, and the field of organic electronics, has continued to motivate researchers to develop sustainable C–C bond-forming reactions.4 Traditionally, these reactions use highly genotoxic alkylating agents, e.g. alkyl halides, nitro-alkanes, and sulfonate esters and suffer from chemoselectivity issues due to the formation of over-alkylated by-products and hazardous waste.5a,b Further, benzofulvenes can be obtained via the condensation of indene/fluorene with aldehydes.5c In this regard, the use of alcohols as benign alkylating agents has drawn considerable attention, owing to their high abundance and relatively low toxicity, which makes the process simple, safe, and sustainable, as it generates water as the sole byproduct.6a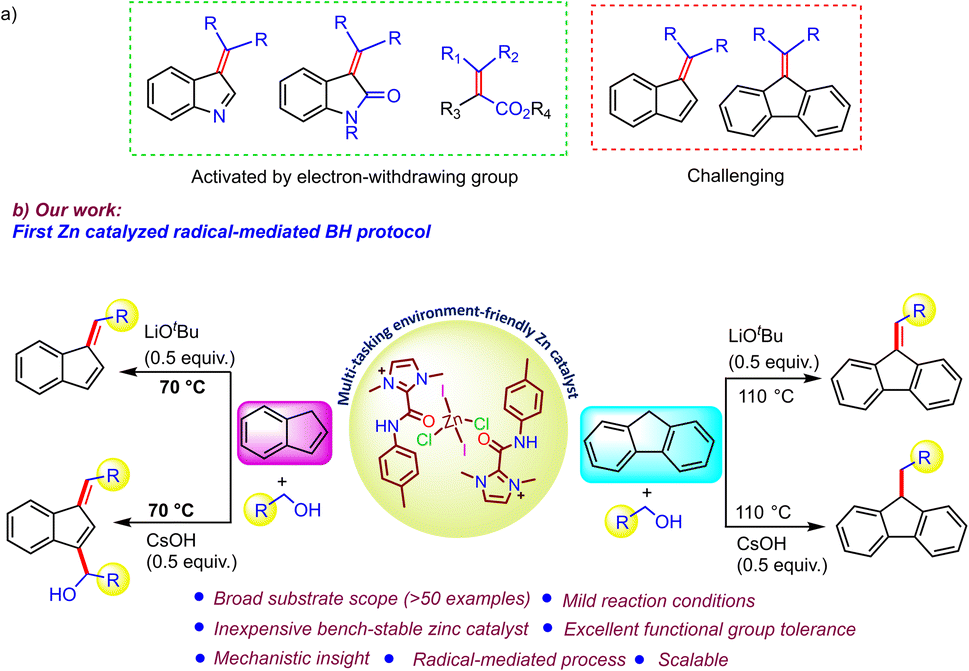 | ||
| Scheme 1 Overview of C-alk(en)ylation and our present study. | ||
Based on this, a novel catalytic methodology has emerged, commonly known as the “Borrowing Hydrogen/Hydrogen Autotransfer (BH/HA)” pathway, wherein an alcohol is dehydrogenated to form a more reactive carbonyl compound, followed by a condensation and hydrogenation process to give the alkylation of various organic moieties.1a,6b–d In this direction, Ding et al. described the facile C3-alkylation of indenes using a spirocyclic IrIII–NHC pincer complex in 2022.7a However, only two examples based on 3d-transition-metal-catalyzed approaches for the C-alk(en)ylation of indene and fluorene with alcohol under a BH protocol have been reported so far.7b,c In 2021, Srimani and coworkers reported an NNS-ligand-supported manganese complex for the C1-alk(en)ylation of fluorene/indene,7b and later, Adhikari et al. utilized a nickel azo phenolate complex for fluorene alkylation.7c Although these base-metal-mediated processes are promising, it is necessary to develop other non-precious-metal-catalyzed effective BH protocols for the selective alk(en)ylation of indene/fluorene for diverse substrates that could be carried out under mild conditions. Along this line, zinc, as an inexpensive, highly abundant, and environmentally friendly metal with Lewis acid character,8a–e could be an attractive choice for the borrowing hydrogen protocol but has been scarcely explored as a catalyst.8f In particular, there are no reports of the functionalization of unactivated molecules like fluorene/indene. Thus, it would be appropriate to study the activity of Zn-based catalysts in the borrowing hydrogen process.
Furthermore, in contrast to the conventional metal-hydride pathway,1a,6b,c a radical-mediated BH protocol is beginning to develop as an exciting and highly attractive new approach to access a wide range of structural motifs.9a–c Due to the advantage of mild reaction conditions arising from the high reactivity of free radicals, the use of radicals generated from alcohols as active intermediates as an alternative route to the typical metal-hydride-promoted BH protocol is of great interest to synthetic chemists. Therefore, it is essential to rationally develop a catalyst that can effectively cleave methylene C–H bonds in alcohols for the generation of C-centered radicals for coupling with the substrates under consideration. Consequently, research has focused on exploiting redox-active ligands that can unlock such synthetic pathways, which have previously been considered to be highly challenging or unfeasible. The redox-active nature of secondary amide moieties has been known for a long time, as they easily generate amidyl radicals with high reactivity.10 In light of the above principle, we herein disclose a redox-active bench-stable C2-amidated imidazolium salt that can store and deliver electrons as required with the help of the imidazolium moiety in the backbone. Accordingly, a well-defined C2-amidated imidazolium-salt-supported zinc compound has been developed as a very effective catalyst for various types of selective C-alk(en)ylations of indenes/fluorenes utilizing various alcohols. The salient features of our current catalytic protocol are as follows: (1) a radical-mediated effective pathway that operates under mild conditions without the need for any external stimuli; (2) the unique nature of the catalyst, in which the Zn(II) center acts as a template to hold two amide N–H moieties together for facile intramolecular electron transfer to generate a reactive radical intermediate; (3) changing the product selectivity by easy tuning of the reaction conditions, showing the multi-tasking activity of the present zinc catalyst; (4) wide substrate scope (in terms of alcohol, including aliphatic and secondary alcohols, and indene/fluorene) with a high level of functional group tolerance; (5) detailed mechanistic understanding based on several control experiments; (6) easy access to various important structural motifs for optoelectronic devices via post-modification of the obtained products.
Results and discussion
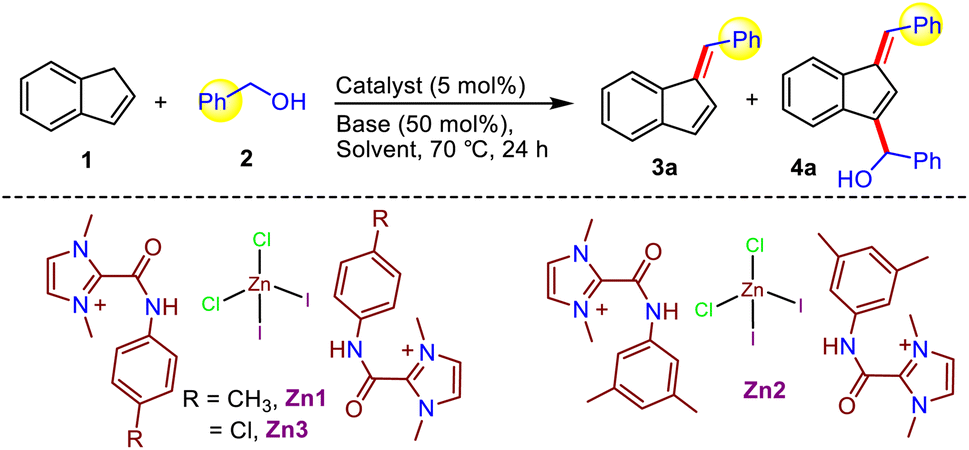
We first synthesized various C2-amidated azolium salts [L1-3]I following a previously reported protocol.11 Subsequently, Zn compounds (Zn1-3) were prepared by reacting corresponding amidated salts [L1-3]I with anhydrous ZnCl2 in methanol and isolated as air- and moisture-stable white solids in 79–87% yields (Scheme 2). These compounds were fully characterized using NMR and IR spectroscopy, as well as single-crystal X-ray crystallography for Zn1. The 1H NMR spectra of compounds Zn1-3 display the signals of the characteristic amide N–H protons in the region of 11.24–11.96 ppm; they are shifted slightly downfield compared to the peaks of corresponding amidated imidazolium salts, and the presence of an amide moiety was supported by IR studies (see ESI†). Further, the diamagnetic nature of the compound Zn1 due to the presence of the Zn2+ ion was confirmed by molar magnetic susceptibility calculation (χm = 0) following the Evans method (see ESI†). | ||
| Scheme 2 (a) Synthesis of compounds Zn1–3. (b) Molecular structure of compound Zn1 with ellipsoids at the 50% probability level; all hydrogen atoms are omitted for clarity except for the N–H proton, and the N-methyl group is shown as a capped stick model. | ||
Most amide–metal complexes show one of the following four types of coordination: chelating, monodentate O-or N-binding, or bridging (Fig. 1).12 However, for the compounds Zn1-3, halide-coordinated tetrahedral zinc ion is situated between two cationic amidated imidazolium moieties.
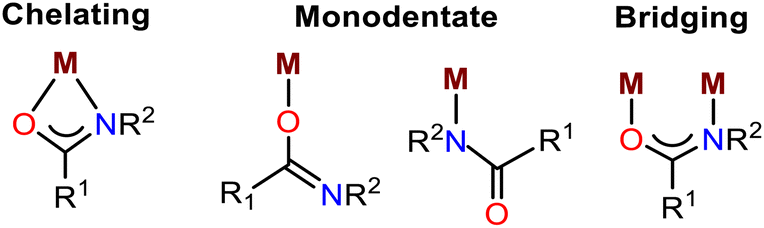 | ||
| Fig. 1 Possible metal (M) coordination modes of an amide ligand. | ||
With the unusual compounds Zn1-3 in hand, we next focused on exploring the efficacy of these amide-derived Zn-based systems in catalyzing organic transformations, especially C–C bond formation utilizing a lignocellulose-derived alcohol as a green alkylating agent. Accordingly, at the outset, we optimized the reaction conditions for our target functionalization of indene with benzyl alcohol employing in situ generated compound Zn1 (Table 1). Gratifyingly, the alkylation of indene with benzyl alcohol using C2-amidated azolium salt [L1]I (10 mol%) along with LiOtBu (0.5 equiv.) and ZnBr2 (5 mol%) in benzene at 70 °C for 24 h provided the C1-alkenylated product in 61% yield instead of an alkylated product (entry 1). To further improve the yield, the use of various zinc salts, such as ZnCl2, Zn(OAc)2, and Zn(OTf)2, were examined while keeping all other parameters unaltered; among them, ZnCl2 was found to be the best for the present indene alkenylation (68%, entry 2–4). With this understanding, we next studied the activity of the isolated compounds Zn1–3 featuring a ZnCl2I2 moiety. It is worth noting that the isolated compound Zn1 having an amide N-p-tolyl group displayed better activity (83%) than the corresponding in situ generated compound and the related catalysts Zn2-3 with either an electron donating (N-3,5-Me2-Ph) or an electron-withdrawing (N-4-Cl-Ph) amide substituent (entry 2 vs. 5–7). Hence, further optimization studies were performed using the isolated compound. Optimization of the reaction conditions revealed that other solvents, such as DCE, DMF, DMSO, acetonitrile, and dioxane, are not suitable (entry 5 vs. 10 and 18–21, Table S1†), and that LiOtBu is the best base. Further screening at different temperatures established that 70 °C is the most effective for this protocol. Next, only a trace amount of product formation was detected in the absence of either the catalyst or the base, implying that the catalyst and base are required here (entry 14–15). Conversely, after thorough optimization studies, we learned that 4a, which is doubly functionalized at the C1- and C3-position of indene, could be obtained as the major product (71% yield) from the same reaction by employing 2.2 equiv. benzyl alcohol and CsOH instead of LiOtBu at 70 °C (entry 12). It is important to note that the formation of such doubly functionalized indenes utilizing alcohol as an alkylating agent has not been described before. Crucially, the nature of the base and specific ratio of indene to alcohol are critical to achieve the desired selectivity under milder reaction conditions. To the best of our knowledge, at present, only one report is available for indene C1-alkenylation using a Mn-NNS complex as a catalyst; however, it requires a significantly higher temperature (110 °C) along with a stoichiometric amount of base (NaOH, 1 equiv.).7b Compared to this report, our eco-friendly Zn-based methodology works under much milder conditions (70 °C and 0.5 equiv. of base), and more importantly, this is the first report on radical-mediated indene alkenylation in which either C1-alkenylated or a doubly functionalized (C1- and C3-position) indene derivatives could be selectively obtained from the same starting material simply by tuning the reaction conditions (base and alcohol equiv., entry 5 vs. 12).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | Base | Indene![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :alcohol :alcohol |
Solvent | Yield (%) | |
| 3a | 4a | |||||
| a Reaction conditions: indene (0.5 mmol), benzyl alcohol (0.55–1.1 mmol), catalyst (5 mol%), base (0.25 mmol), solvent (2 mL), 70 °C, 24 h. | ||||||
| 1 | [L1]I + ZnBr2 | LiOtBu | 1:1.1 |
Benzene | 61 | — |
| 2 | [L1]I + ZnCl2 | LiOtBu | 1:1.1 |
Benzene | 68 | — |
| 3 | [L1]I + Zn(OAc)2 | LiOtBu | 1:1.1 |
Benzene | 29 | — |
| 4 | [L1]I + Zn(OTf)2 | LiOtBu | 1:1.1 |
Benzene | 35 | — |
| 5 | Zn1 | LiO t Bu |
1:1.1 |
Benzene | 83 | — |
| 6 | Zn2 | LiOtBu | 1:1.1 |
Benzene | 59 | — |
| 7 | Zn3 | LiOtBu | 1:1.1 |
Benzene | 67 | — |
| 8 | Zn1 | NaOtBu | 1:1.1 |
Benzene | 37 | 15 |
| 9 | Zn1 | CsOH | 1:1:1 |
Benzene | 12 | 43 |
| 10 | Zn1 | LiOtBu | 1:1.1 |
DCE | 53 | — |
| 11 | Zn1 | LiOtBu | 1:1.1 |
Toluene | 41 | — |
| 12 | Zn1 | CsOH |
1:2.2 |
Benzene | 10 | 71 |
| 13 | Zn1 | LiOtBu | 1:2.2 |
Benzene | 80 | — |
| 14 | — | LiOtBu | 1:1.1 |
Benzene | <10 | — |
| 15 | Zn1 | — | 1:1.1 |
Benzene | Trace | — |
| 16 | ZnCl2 | LiOtBu | 1:1.1 |
Benzene | Trace | — |
| 17 | [L1]I | LiOtBu | 1:1.1 |
Benzene | 30 | — |
The optimized protocol was then employed towards the C1-alkenylation of indene with diverse sterically and electronically distinct primary alcohols to determine the scope of the present protocol (Table 2). First, electron-rich and -deficient aromatic alcohols possessing an array of substituents at p- and m-positions delivered desired C1-alkenylated products (3a–i, 3j–k) in good-to-excellent yields. Further, sterically congested o-substituted alcohols (2l–m) were found to be compatible to deliver the respective alkenylated products in good yields of 59–61%. Intriguingly, even the very electron-poor p-NO2 and –CN substituted alcohols 2f–g delivered the corresponding alkenylated products (3f–g) in 63–67% yields without affecting these readily reducible functional groups, although a slightly longer duration was required. Following that, alcohols with more-electron-donating substituents (e.g., iPr and even NMe2, 2h–i) were also noted to be active. Moreover, this protocol is highly tolerant to halogen substituents, furnishing the respective products (2d–e, 2j–m) with no sign of dehalogenation, and further, polyaromatic substrates such as alpha- and beta-naphthalen-1-yl-methanol were equally competent (∼80% yields, 3n–o).
| a Reaction conditions: indene (0.5 mmol), alcohol (0.55 mmol), Zn1 (5 mol%), and LiOtBu (0.25 mmol) in benzene at 70 °C for 24 h. b36 h. |
|---|

|
Pleasingly, the efficiency of this reaction was not suppressed even for heteroaromatic substrates such as 2-pyridyl methanol (2q), which are well-known to cause catalyst poisoning via their chelation. Interestingly, cinnamyl alcohol alkenylated indene as well to afford the product 3r in 77% yield, and notably, the internal olefinic double bond was completely intact. It is noteworthy that overall, the present protocol offers diverse scope for a wide variety of alcohols under mild reaction conditions with wide-ranging functional group compatibility that includes alkene, –NO2, –CN, alkoxy, and halide substituents, thus providing possible sites for post-synthetic modifications of the obtained product. Gratifyingly, substituted indenes (1t–w) also smoothly furnished the alkenylated products (3t–w) in moderate-to-good yields. It is worth mentioning that aliphatic alcohols are also compatible and delivered the coveted alkenylated products in good yields (3v–w, 65–69%). Subsequently, we explored the scope for the double-functionalization of indene (Table 3) under the optimized conditions developed in Table 1. Alcohols containing electron-donating (–Me or –OMe) and -withdrawing (–Br or –Cl) functional groups at the ortho- and para-positions of the phenyl ring afforded the corresponding products in good yields (4b–e); however, the presence of an ortho-substituent resulted in the product being obtained in relatively lower yield. Next, heteroaromatic alcohols were also found to be suitable (4f–g, 68–70% yields). Interestingly, substituted indene also provided the corresponding product 4h in 72% yield.
| a Reaction conditions: indene (0.5 mmol), alcohol (1.1 mmol), Zn1 (5 mol%), and CsOH (0.25 mmol) in benzene at 70 °C, 24 h. |
|---|

|
Methylidenefluorenes have emerged as versatile synthetic intermediates in many organic materials and are widely used in optoelectronic devices.7b Conventional methods for the synthesis of these compounds mostly rely on Wittig or Peterson olefination, which generates toxic wastes.13a,b Additionally, catalytic strategies utilizing expensive transition metals in combination with sensitive/toxic phosphine ligands have been developed over the years. For example, Gevorgyan and coworkers developed a palladium-catalyzed intramolecular hydroarylation of o-alkynyl biaryls using a phosphine ligand to synthesize these molecules.13c Along the same line, in 2014, Jin et al. utilized a Pd-catalyzed annulation of bis-biaryl alkynes in the presence of MnO2 as an oxidant and PivOH as an additive (10 mol%) to attain the same.13d However, non-precious-metal-based protocols are scarce. Therefore, the dehydrogenative construction of methylidene fluorenes from fluorene and inexpensive green alcohols catalyzed by an eco-friendly and bench-stable catalyst is highly alluring.
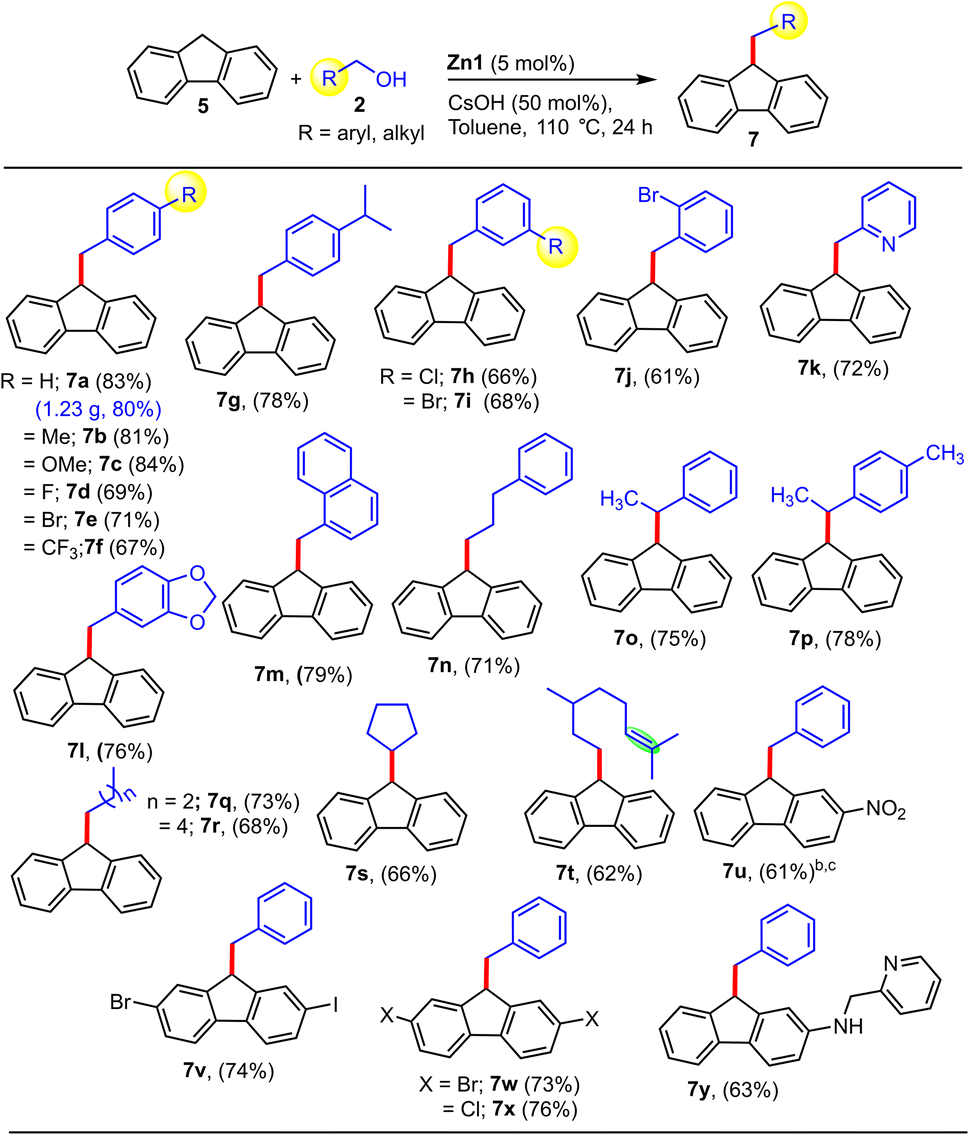
Thus, after the success in the alkenylation and double-functionalization of indene, we expanded the suitability of the present protocol towards the alkenylation of fluorene (Table S2†). After comprehensive optimization studies, we found that by employing the catalyst Zn1, LiOtBu, and 1.1 equiv. of alcohol w.r.t. fluorene at 110 °C for 24 h in toluene, the alkenylated product (6a) could be selectively obtained in a high yield of 78% (Table S2,† entry 2). Various reaction parameters were subsequently screened, and to our delight, by changing the base to CsOH and increasing the amount of alcohol to 2.2 equiv., we could selectively obtain the alkylated product 7a as the major product in excellent yield (83%, Table S2,† entry 5). The use of in situ generated catalyst under the same reaction conditions provided the product 7a in relatively lower yield (Table S2,† entry 7).
In essence, we could achieve the selective formation of the alkenylated and alkylated products from fluorene and alcohol in high yields utilizing the same Zn catalyst system via fine-tuning of the reaction conditions. Under the optimized conditions, we then explored the scope of alcohols for the fluorene alkenylation protocol (Table S2†). Alcohols with various substituents at the o-, m-, and p-positions all reacted readily to afford the desired products (6b–f). Of note, this protocol is equally effective for electron-donating and -withdrawing groups. Heteroaromatic and polyaromatic alcohols were also competent in furnishing the products 6g–h in 69–71% yields. Gratifyingly, halogen-substituted fluorenes also delivered the desired products 6i–k, which can be functionalized at the C–X (X = halide) bonds as required in good yield (71–75%), implying the good compatibility of fluorene derivatives in this reaction. Satisfyingly, aliphatic alcohols 2l–m also produced the alkenylated products 6l–m in good yields.
After this, we focused on the alkylation of fluorene (Table 5) using the optimized conditions (entry 5, Table S2†). Diverse benzyl alcohol derivatives featuring various m- and p-Ph substituents could be utilized to afford the coveted alkylated products in excellent yields (7a–7g, 7h–7i). Notably, derivatives with the strong electron-withdrawing p-CF3 or p-F substituents also furnished the respective products in good yields (7d, 7f). Further, ether-functionalized, polyaromatic, and hetero-aromatic alcohols such as 2-pyridyl methanol (no sign of catalyst deactivation was observed in the presence of this chelating substrate) were found to be effective. Aliphatic alcohols and secondary alcohols are described as significantly more challenging to use as an alkyl source than benzyl alcohol derivatives. Encouragingly, secondary aromatic and aliphatic alcohols were also successful substrates, furnishing the desired alkylated products (7o–p, 7s) in good yields (66–78%). It is worth noting that Gnanaprakasam et al. reported fluorene alkylation with secondary alcohols employing [Ru(p-cymene)Cl2]2 as a catalyst at 140 °C with a higher base loading (2 equiv.) for 48 h, and even under these rigorous conditions, the majority of secondary alcohols yielded alkenylated rather than the alkylated products,14 whereas our non-precious-metal Zn-based catalyst system can provide the alkylated products under much milder conditions. Promisingly, aliphatic alcohols (cyclic and acyclic, including secondary ones) delivered the desired alkylated products in good yields (7q–t, 62–73%), and intriguingly, when terpenols like citronellol were used as an alkylating agent, the respective product (7t) was obtained without affecting the alkene moiety, making it a chemoselective protocol. Interestingly, fluorene derivatives with strong electron-withdrawing groups, e.g. NO2-substituted fluorene (5u), as well as a heterocyclic derivative of fluorene (5y), were also found to be compatible to provide the desired alkylated products (7u and 7y) in good yields (61–63%). Impressively, halogen-substituted fluorenes (6v–x) are also compatible with the present methodology, providing the alkylated products in good yields of 73–76%. It should be noted that such halo-substituted compounds could be exploited as a key intermediate for the synthesis of various optoelectronic materials via standard C–X coupling reactions. In particular, in the case of halo-substituted product 7v, due to the presence of two different halogen atoms (Br vs. I), selectivity can be easily controlled during further transformations to other valuable organic compounds.
Next, to assess the suitability of present protocol in competitive reaction, we first examined the reaction of an equimolar mixture of benzyl alcohol and 1-phenyl ethanol with 9H-fluorene, which revealed that benzyl alcohol is more reactive than secondary alcohol (Scheme S2a†), which is in line with previous observations.7c A similar competition between benzyl alcohols substituted with an electron-donating or an electron-withdrawing group substituted indicated the greater reactivity of the electron-rich alcohol towards alkylation, providing the corresponding compound 7b in 76% (Scheme S2b†). This might be due to better stabilization of the reactive intermediate by the electron-donating substituent on the phenyl ring. Along the same line, we next attempted the alkenylation of indene with an equimolar mixture of benzyl alcohols substituted with an electron-donating or electron-withdrawing group, and the electron-rich alcohol was again noted to be more reactive than that the electron-poor alcohol (Scheme S2c†).
To evaluate the synthetic utility of obtained 9-monoalkylated fluorenes, we next focused on functionalizing some of them to obtain potentially useful unsymmetrically 9,9-disubstituted fluorenes, which can serve as monomers for the synthesis of function-oriented polyfluorenes and find valuable applications in various fields such as photovoltaic cells, electroluminescent displays, field-effect transistors, plastic lasers, and optical sensors. Accordingly, some unsymmetrically 9,9-disubstituted fluorene-based monomers were readily synthesized (8a–c) starting from the selected alkylated products, 7a and 7w (Scheme 3).5b It is worth noting that such unsymmetrical 9,9-difunctionalized fluorenes are difficult to obtain following reported classical methods due to the lack of selectivity towards mono-alkylated species for further functionalization. Furthermore, the reported synthesis of symmetrical compounds, e.g.8d, from 2,7-dibromofluorene requires a greater amount of benzyl bromide (7 equiv.) and higher temperature (110 °C), while the same compound can be easily synthesized from 7w, which can be obtained using a lower temperature (80 °C) and amount of benzyl bromide using our strategy.
 | ||
| Scheme 3 General transformations of 9-monoalkylated fluorenes into various important functionalized 9,9-dialkylated fluorene derivatives. | ||
Mechanistic studies
After the success of these catalytic reactions utilizing a simple and bench-stable Zn compound (Zn1) as a multi-tasking catalyst, we focused on understanding the nature of the active catalyst and mechanistic pathway for this protocol. Transition-metal-promoted borrowing hydrogen protocols typically proceed via a putative metal-hydride intermediate that has been well-established in the literature. However, recently, a radical pathway without the involvement of a metal-hydride intermediate has also been documented for an azo-phenolate ligand supported Ni complex.7c To determine whether the present protocol follows a typical metal-hydride pathway or a radical one, at the outset, radical scavengers such as BHT, galvinoxyl or CuCl2 were added to our standard catalytic reaction between fluorene and 4-methylbenzyl alcohol.15 The product formation (7b) was completely inhibited in the presence of galvinoxyl and CuCl2, and a drastic decrease in the formation of 7b was noted for BHT (Scheme 4a), establishing that the reaction advances through a radical-mediated pathway. Further, the inhibition of the reaction in the presence of CuCl2 suggests that a single-electron transfer (SET) process is in operation.15a,b Next, to understand the nature of the radical generated from the substrate during the catalytic cycle, we studied various reaction mixtures via HRMS, and to our delight, the investigation of a standard reaction to generate 7b in the presence of BHT revealed the formation of BHT-trapped alcohol (HRMS at m/z 341.2470, Scheme 4a(i), Fig. S15†). Similarly, in a stoichiometric reaction between benzyl alcohol and BHT in the presence of Zn1 and CsOH, we detected the analogous BHT-trapped ketyl radical (Scheme 4a(ii), HRMS at m/z 327.2435, Fig. S16†). All these studies establish the generation of a ketyl radical from the alcohol during the catalytic cycle. Further, radical reactions generally follow one of the two pathways: (a) two-electron hydride transfer or (b) one-electron HAT. To distinguish between these two pathways, the dehydrogenation of cyclobutanol was conducted under the standard conditions, and 1H NMR confirmed that cyclobutanone was not formed, indicating that a one-electron HAT pathway rather than a two-electron hydride pathway is involved in the mechanistic cycle (Scheme 4b, Fig. S17†).16 | ||
| Scheme 4 Control experiments for establishing a radical pathway. | ||
With the understanding that this was a ketyl-radical-mediated process, we next tried to determine the exact role of our catalyst system in this process, Accordingly, Zn1 was reacted with the base LiOtBu (1 equiv.), at room temperature, which generated a yellow solution (Scheme 5). Spectral analyses indicated the possible formation of an intermediate like Zn1a: (a) The absence of the N–H peak (which was present in Zn1, δ = 11.96 ppm in 1H NMR) indicated that the N–H proton might be rapidly shuttled between the amidated imidazolium salts, and the formation of free tBuOH (δ = 1.10 ppm) was observed, along with broadening of peaks in the 1H NMR spectrum compared to those of compound Zn1 (Fig. S21†). (b) The EPR (room temperature, Scheme 5d) spectrum shows a clear triplet isotropic pattern with a hyperfine coupling constant of 16 G at giso = 1.9737, indicating the formation of an N-centered amidyl radical (Scheme 5b). We believe that the presence of an amide N-p-tolyl group helps to stabilize the amidyl radical, and thus, could be readily detected by EPR measurement at room temperature; however, the formation of a C-centered amidyl radical during the catalytic cycle cannot be ruled out completely, although we could not detect it, given the shorter lifetime of such radicals.17 Intriguingly, when [L1]I alone was treated with LiOtBu (1 equiv.), no EPR signal could be detected at either room temperature or 77 K (Scheme 5c), suggesting that [L1]I alone cannot effectively stabilize the respective radical. To further strengthen the above hypothesis, we investigated the electrochemical behavior of compound Zn1. It is to be noted that with Zn(II) being a redox-inactive center, redox responses are expected because of the amidated salt-centered redox activity only.8c The employed amidated imidazolium salt [L1]I has two major sites for redox reactions: an NH group attached to the p-tolyl moiety and a keto group attached to the imidazolium-C2 position. To comprehend this, first we have checked the CV of [L1]I, which displayed two sequential single-electron oxidations at −0.086 and 0.269 V, while one single-step reduction at −0.139 V was noted. However, after the formation of compound Zn1, two redox couple peaks were observed at −0.016/0.005 and 0.252/0.267 V (Scheme 5e), which were due to redox reactions at amide –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and N–H moieties. All above studies demonstrate that the amidated imidazolium salt (L1) plays an active role in the catalytic reaction, while Zn possibly acts as a template in Zn1.
O and N–H moieties. All above studies demonstrate that the amidated imidazolium salt (L1) plays an active role in the catalytic reaction, while Zn possibly acts as a template in Zn1.
 | ||
| Scheme 5 (a) Reaction of [L1]I with base. (b) Reaction of Zn1 with base. (c and d) X-band EPR spectra of reactions (a) and (b). (e) Cyclic voltammogram of Zn1 in acetonitrile using glassy carbon as the working electrode, platinum wire as an auxiliary electrode, and saturated Ag/Ag+ as a reference electrode (referenced to Fc/Fc+, E1/2 = 0.22 V). Scan rate = 100 mV s−1. | ||
Moreover, the effects of different substituents on our amidated imidazolium salt on the activity of the different catalysts employed here can be easily understood based on the calculated bond dissociation energy (BDE) of the respective N–H bond, as EPR studies clearly established the formation of an N-centered amidyl radical in this process. Accordingly, the BDE values of the N–H bond in Zn1 and Zn2 were estimated by calculating reaction enthalpies at 298 K for the HAT reaction using an unsubstituted aminyl radical (˙NH2) as a reference and found to be 96.5 and 99.8 kcal mol−1, respectively, implying the better potential of Zn1 as a HAT catalyst, which is reflected in their catalytic performances (Table 1).10c The classical methods for generating amidyl radicals require a strong oxidant as well as external stimuli such as heat, light, or cathodic current, as in the case of Hofmann–Löffler–Freytag reaction, because of the high BDE of N–H bonds.18 However, our Zn-based catalyst system can generate respective N-centered radicals at ambient temperature and even without the need for any photochemical conditions. Thus, as a HAT catalyst, our zinc compound is undoubtedly more attractive, but no such system has previously been reported to the best of our knowledge. It is worth mentioning here that the only reported catalyst system for indene C1-alkenylation utilizing an alcohol, a Mn-NNS complex, follows a standard metal hydride pathway that requires a significantly higher temperature (110 °C) and a stoichiometric amount of base (NaOH, 1 equiv.).7b In comparison, our Zn-based radical pathway uses much milder conditions, providing substantial advantages over other catalyst systems for similar borrowing hydrogen protocols.
Further, to determine the various reaction intermediates, the progress of a model alkylation reaction of fluorene with benzyl alcohol was monitored using in situ1H NMR spectroscopy (Fig. S19†), which revealed a gradual conversion to aldehyde; the same was also observed for an independent benzyl alcohol oxidation reaction via GC-MS (Scheme 6a), along with the formation of a mixture of the alkenylated (6a) and alkylated fluorene product (7a). Simultaneous formation of both these products was supported by 1H NMR resonances at 7.00 ppm and 4.01 ppm corresponding to C9CH and C9–CH moieties, respectively. Further, as time elapsed, the amount of alkylated product (7a) continued to increase at the expense of the alkenylated product, supporting the intermediacy of the alkenylated species (6a) during the formation of 7a. A similar result was observed in the case of indene; initially, the alkenylated indene product was formed, and as the time progressed, the double-functionalization product was gradually produced (Fig. S20†). Next, a comparable yield (81–83%) of alkenylated fluorene (6a) was found to be obtained in reactions between fluorene and aldehyde using CsOH in either the presence or absence of catalyst (Scheme 6b), which suggests that the formation of 6a can be mediated by base alone, and that the aldehyde is a possible intermediate that is generated by alcohol dehydrogenation.
 | ||
| Scheme 6 Detection of catalytic intermediate. | ||
To understand the role of the base, we performed the reaction between alkenylated fluorene (6a) and benzyl alcohol under standard conditions with two different bases (LiOtBu and CsOH). Interestingly, no hydrogenation occurred with LiOtBu, while 79% alkylated product (7a) was obtained using CsOH (Scheme 6c), which is in line with our experimental observations (Table S1†). Similarly, in the case of indene, no double-functionalization product was observed in the presence of LiOtBu, while 66% alkylated product (4a) was obtained using CsOH (Scheme 6f). These results suggest that 3a and 6a are the possible intermediates; this was also suggested by in situ NMR studies of the functionalization of indene and fluorene, respectively. To understand the roles of the different bases, the alkenylation of fluorene was carried out under the standard conditions of Table 4 in the presence of benzophenone, which acts as a good electron acceptor, and the reaction was noted to be faster (a similar yield of the product 6a was noted even for a lower reaction duration of 12 h, Scheme S3†). Intriguingly, when the alkylation of fluorene was carried out under the standard conditions of Table 5 in the presence of benzophenone, the reaction was halted at the alkenylated product (Scheme S4†). These results clearly indicate that when LiOtBu is used, HOtBu or its derivatives accepts the electrons from the catalytic intermediate, and thus, the reaction stops at the alkenylation stage. This is not the case for the CsOH-based protocol, in which electrons are transferred from Zn1c to the alkenylated species (6a) to generate the alkylated product 7a. Additionally, when we performed the alkenylation reaction of fluorene (under the standard conditions of Table 4) in the presence of 0.5 equiv. of water, 17% of the alkylated product (7a) was indeed observed along with the expected 64% of the alkenylated product (6a) (Scheme 6g). When the same experiment was carried out using 2.2 equiv. of alcohol, a slight increase in the formation of alkylated products was observed (23%). These results suggest that the generated water in the reaction medium definitely plays some role in facilitating the alkylation process. Although the impact of the cations present in the base is not clear yet, we can surmise that the size of the metal cation of the employed base might also affect the catalytic process (entry 5 vs. 8, Table 1) in accordance with a previous report.19
| a Reaction conditions: fluorene (0.5 mmol), alcohol (0.55 mmol), Zn1 (5 mol%), and LiOtBu (0.25 mmol) in toluene at 110 °C for 24 h. |
|---|

|
| a Reaction conditions: fluorene (0.5 mmol), alcohol (1.1 mmol), Zn1 (5 mol%), and CsOH (0.25 mmol) in toluene at 110 °C for 24 h. b36 h. c130 °C, NMR conversion. |
|---|
|
Further, when the same experiment was conducted using deuterated benzyl alcohol-d2, the alkylated product 7a–d1 was obtained with 71% deuterium incorporation at the alcohol β-position (less sterically encumbered position), whereas 21% deuterium incorporation was detected at the C9-position of fluorene via1H NMR (Scheme 6d, Fig. S18†), establishing that the deuterium/hydrogen from the employed alcohol is transferred to the final alkylated products.7c Next, a Hg-dropping experiment confirmed the homogeneous nature of the catalyst (Scheme 6e).20 Finally, kinetic analysis of the present Zn-catalyzed alkylation of fluorene using 4-methylbenzyl alcohol by varying the catalyst loading (3–6 mol%) as well as the amount of alcohol (1.4–2.6 equiv.) revealed that the reaction is pseudo-first-order with respect to the catalyst Zn1 as well as the alcohol (Fig. 2).
 | ||
| Fig. 2 (a) and (d): Kinetic time profile of fluorene alkylation; growth of the alkylated product as monitored using UV-vis spectroscopy. Reaction conditions: fluorene (0.5 mmol), 4-methylbenzyl alcohol (1.1 mmol for (a) and variable amount of alcohol for (d)), Zn1 (variable catalyst loading for (a) and 5 mol% for (d)), CsOH (0.25 mmol), toluene, 110 °C. Corresponding rate constant calculations with regard to the catalyst (b and c) and alcohol (e and f). | ||
Taking all mechanistic studies into consideration, a plausible mechanistic pathway is depicted in Scheme 7. First, the catalytically active radical species Zn1a is generated from the reaction between Zn1 and the base following either a deprotonation/single-electron transfer sequence or a concerted proton-coupled electron transfer (PCET).10a Subsequently, Zn1a in the presence of benzyl alcohol produces a species like I, followed by the abstraction of the benzylic proton, resulting in the formation of a ketyl radical as in intermediate Zn1bvia a HAT process. Intermediate Zn1b next undergoes single-electron transfer between the ketyl radical and electrophilic amide CO moiety to generate C-centered amidyl radical anion species Zn1c.21 Subsequently, the release of the desired aldehyde (confirmed by 1H NMR spectroscopy, δ = 10.00 ppm, Fig. S22†) followed by its coupling with indene/fluorene produces the alkenylated product with the regeneration of Zn1. Further, in the case of CsOH, transfer of electrons from Zn1c to the generated alkenylated species 6, followed by H-atom transfer yields the alkylated product 7 and regenerates Zn1.
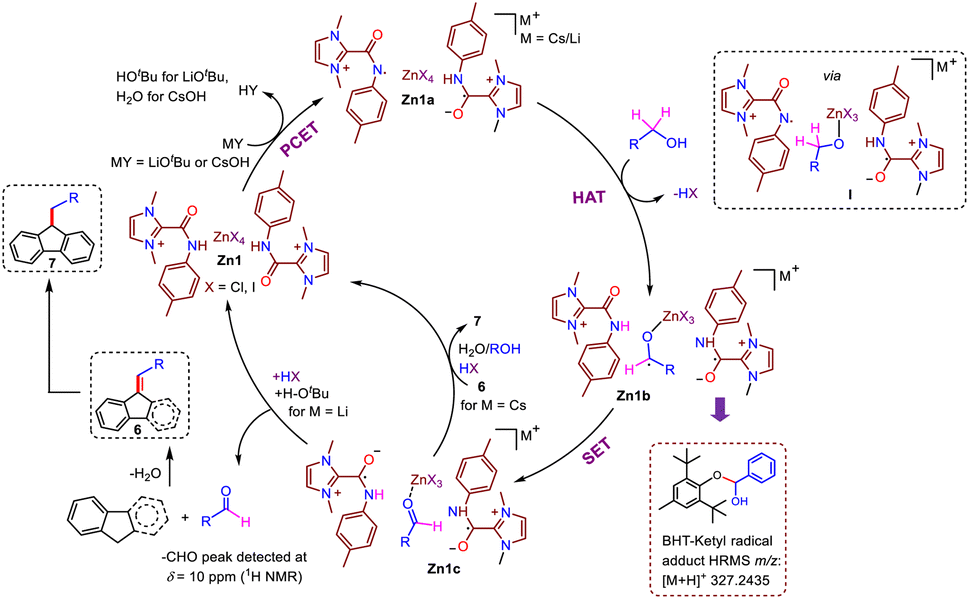 | ||
| Scheme 7 Plausible mechanistic pathway. | ||
Conclusion
In summary, we have developed an efficient strategy for the selective alkylation and alkenylation of fluorene and indene derivatives under mild conditions via a borrowing hydrogen pathway using a redox-active amidated imidazolium salt supported Zn compound, wherein the Zn(II) center acts as a template to hold two amide N–H moieties together for facile intramolecular electron transfer to generate a reactive catalytic radical intermediate. It is worth mentioning that this is the first time that the use of eco-friendly zinc to catalyze the diverse functionalization of indene/fluorene derivatives utilizing alcohol has been achieved, to the best of our knowledge. This robust protocol offers several advantages, such as low catalyst loading (with the catalyst displaying multitasking activity), milder reaction conditions, broad substrate scope (compatible with a range of alcohols, ranging from aliphatic to aromatic, and encouragingly, secondary alcohols) with a high level of functional group tolerance, and chemoselectivity. The post-modification of the products and gram-scale reaction authenticate the utility of the developed methodology. Further, detailed mechanistic studies including theoretical calculations established a reaction pathway that proceeds via the generation of a ketyl radical formed through a HAT process, which helps in understanding the reactivity trends of this catalyst system. Intriguingly, this unique catalytic system effectively generates aldehyde from alcohol via a radical-mediated pathway, followed by condensation with indene/fluorene to form the alkenylated products. We believe that this new redox-active amide system containing an imidazolium unit in the backbone, used in combination with an eco-friendly and earth-abundant Zn center to generate reactive radical species, represents a strong candidate for developing new synthetic methodologies and will certainly attract more research attention in the near future.Data availability
Experimental data including experimental procedures and characterization data; the NMR and ESI-MS spectra of the new compounds; detailed computational studies are available in the ESI.†Author contributions
A. R. conceived and designed the project. S. S. and S. M. performed the experiments. S. S. and A. R. wrote the manuscript and all authors approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are thankful to SERB (grant no CRG/2020/000780) for the financial support. S. S. and S. M. thank CSIR, India for their PhD fellowships. We acknowledge instrumental facilities at the Department of Chemistry and SAIF, IIT Madras. Dr K.R.R.’s lab is acknowledged for CV measurements, and we thank Dr Joyram Guin for the helpful discussion.References
- For selected references, see: (a) A. Corma, J. Navas and M. J. Sabater, Chem. Rev., 2018, 118, 1410–1459 CrossRef CAS PubMed; (b) B. Rao and R. Kinjo, Chem.–Asian J., 2018, 13, 1279–1292 CrossRef CAS PubMed; (c) Y. Obora, ACS Catal., 2014, 4, 3972–3981 CrossRef CAS; (d) C. Gunanathan and D. Milstein, Science, 2013, 341, 1229712 CrossRef PubMed.
- For selected references, see: (a) F. Bartoccini, M. Retini and G. Piersanti, Tetrahedron Lett., 2020, 61, 151875 CrossRef CAS; (b) S. Chakraborty, P. Daw, Y. B. David and D. Milstein, ACS Catal., 2018, 8, 10300–10305 CrossRef CAS PubMed; (c) N. Deibl and R. Kempe, J. Am. Chem. Soc., 2016, 138, 10786–10789 CrossRef CAS PubMed.
- For selected references, see: (a) B. Blank and R. Kempe, J. Am. Chem. Soc., 2010, 132, 924–925 CrossRef CAS PubMed; (b) K. D. Nguyen, B. Y. Park, T. Luong, H. Sato, V. J. Garza and M. J. Krische, Science, 2016, 354, 300 CrossRef CAS PubMed; (c) D.-W. Lee, K.-H. Kwon and C. S. Yi, Science, 2011, 333, 1613–1616 CrossRef CAS PubMed.
- For selected references, see: (a) A. J. Liedtke, B. C. Crews, C. M. Daniel, A. L. Blobaum, P. J. Kingsley, K. Ghebreselasie and L. J. Marnett, J. Med. Chem., 2012, 55, 2287–2300 CrossRef CAS PubMed; (b) L.-H. Xie, C.-R. Yin, W.-Y. Lai, Q.-L. Fan and W. Huang, Prog. Polym. Sci., 2012, 37, 1192–1264 CrossRef CAS; (c) A. C. Grimsdale, K. L. Chan, R. E. Martin, P. G. Jokisz and A. B. Holmes, Chem. Rev., 2009, 109, 897–1091 CrossRef CAS PubMed.
- For selected references, see: (a) H. H. Seltzman, C. Shiner, E. E. Hirt, A. F. Gilliam, B. F. Thomas, R. Maitra, R. Snyder, S. L. Black, P. R. Patel, Y. Mulpuri and I. Spigelmen, J. Med. Chem., 2016, 59, 7525–7543 CrossRef CAS PubMed; (b) R. K. Kadu, P. B. Thakur and V. R. Patil, Polym. Bull., 2019, 76, 595–613 CrossRef CAS; (c) E. Ghera and V. Sprinzak, J. Am. Chem. Soc., 1960, 82, 4945–4952 CrossRef CAS.
- For selected references, see: (a) G. Guillena, D. J. Ramón and M. Yus, Angew. Chem., Int. Ed., 2007, 46, 2358–2364 CrossRef CAS PubMed; (b) S. P. Shan, X. Xiaoke, B. Gnanaprakasam, T. T. Dang, B. Ramalingam, H. V. Huynh and A. M. Seayad, RSC Adv., 2015, 5, 4434–4442 RSC; (c) J. Campos, Phys. Sci. Rev., 2018, 3, 20170017 Search PubMed; (d) A. E. Owen, A. Preiss, A. McLuskie, C. Gao, G. Peters, M. Bühl and A. Kumar, ACS Catal., 2022, 12, 6923–6933 CrossRef CAS; (e) M. Kaur, N. U. D. Reshi, K. Patra, A. Bhattacherya, S. Kunnikuruvan and J. K. Bera, Chem.–Eur. J., 2021, 27, 10737–10748 CrossRef CAS PubMed.
- For selected references, see: (a) K.-L. Dai, Q.-L. Chen, W.-P. Xie, K. Lu, Z.-B. Yan, M. Peng, C.-K. Li, Y.-Q. Tu and T.-M. Ding, Angew. Chem., Int. Ed., 2022, 61, e202206446 CrossRef CAS PubMed; (b) A. Mondal, R. Sharma, D. Pal and D. Srimani, Chem. Commun., 2021, 57, 10363–10366 RSC; (c) A. Biswas, A. K. Bains and D. Adhikari, Catal. Sci. Technol., 2022, 12, 4211–4216 RSC.
- For selected references, see: (a) S. Mandal, S. Mandal and K. Geetharani, Chem.–Asian J., 2019, 14, 4553–4556 CrossRef CAS PubMed; (b) F. Ritter, T. P. Spaniol, I. Douair, L. Maron and J. Okuda, Angew. Chem., Int. Ed., 2020, 59, 23335–23342 CrossRef CAS PubMed; (c) X. Zhang, G.-P. Lu, K. Wang, Y. Lin, P. Wang and W. Yi, Nano Res., 2022, 15, 1874–1881 CrossRef CAS; (d) N. Hidalgo, C. Romero-Pérez, C. Maya, I. Fernández and J. Campos, Organometallics, 2021, 40, 1113–1119 CrossRef CAS PubMed; (e) A. Rit, A. Zanardi, T. P. Spaniol, L. Maron and J. Okuda, Angew. Chem., Int. Ed., 2014, 53, 13273–13277 CrossRef CAS PubMed; (f) V. Sankar, M. Kathiresan, B. Sivakumar and S. Mannathan, Adv. Synth. Catal., 2020, 362, 4409–4414 CrossRef CAS.
- For selected references, see: (a) A. K. Bains, A. Kundu and D. Adhikari, ChemCatChem, 2023, 15, e2023005 CrossRef; (b) C. Pratley, S. Fenner and J. A. Murphy, Chem. Rev., 2022, 122, 8181–8260 CrossRef CAS PubMed; (c) M. D. Kärkäs, ACS Catal., 2017, 7, 4999–5022 CrossRef.
- For selected references, see: (a) K. Kwon, R. T. Simons, M. Nandakumar and J. L. Roizen, Chem. Rev., 2022, 122, 2353–2428 CrossRef CAS PubMed; (b) H. Chen and S. Yu, Org. Biomol. Chem., 2020, 18, 4519–4532 RSC; (c) K. Ohmatsu, R. Suzuki, Y. Furukawa, M. Sato and T. Ooi, ACS Catal., 2020, 10, 2627–2632 CrossRef CAS.
- S. Bauri, A. Ramachandran and A. Rit, Chem.–Asian J., 2023, 18, e202201301 CrossRef CAS PubMed.
- A. V. Lee and L. L. Schafer, Eur. J. Inorg. Chem., 2007, 2243–2255 CAS.
- For selected references, see: (a) M. Banerjee, S. J. Emond, S. V. Lindeman and R. Rathore, J. Org. Chem., 2007, 72, 8054–8061 CrossRef CAS PubMed; (b) Y. Ooyama, R. Sagisaka, T. Enoki, N. Tsunoji and J. Ohshita, New J. Chem., 2018, 42, 13339–13350 RSC; (c) N. Chemyak and V. Gevorgyan, J. Am. Chem. Soc., 2008, 130, 5636–5637 CrossRef PubMed; (d) J. Zhao, N. Asao, Y. Yamamoto and T. Jin, J. Am. Chem. Soc., 2014, 136, 9540–9543 CrossRef CAS PubMed.
- M. A. Shaikh, S. G. Agalave, A. S. Ubale and B. Gnanaprakasam, J. Org. Chem., 2020, 85, 2277–2290 CrossRef CAS PubMed.
- For selected references, see: (a) V. K. Pandey, S. Sahoo and A. Rit, Chem. Commun., 2022, 58, 5514–5517 RSC; (b) V. K. Pandey, C. S. Tiwari and A. Rit, Org. Lett., 2021, 23, 1681–1686 CrossRef CAS PubMed; (c) Y. Yamamoto, S. Onuki, M. Yumoto and N. Asao, J. Am. Chem. Soc., 1994, 116, 421–422 CrossRef CAS.
- For a selected reference, see: R. Ray, S. Chandra, D. Maiti and G. K. Lahiri, Chem.–Eur. J., 2016, 22, 8814–8822 CrossRef CAS PubMed.
- J. Hioe, D. Šakić, V. Vrcek and H. Zipse, Org. Biomol. Chem., 2015, 13, 157–169 RSC.
- K. Xu, J. Yang, H. Qin and F. Liu, Eur. J. Org Chem., 2023, 26, e202300543 CrossRef CAS.
- R. R. Putta, S. Chun, S. B. Lee, J. Hong, S. H. Choi, D.-C. Oh and S. Hong, J. Org. Chem., 2022, 87, 16378–16389 CrossRef CAS PubMed.
- V. Artero and M. Fontecave, Chem. Soc. Rev., 2013, 42, 2338–2356 RSC.
- For selected references, see: (a) K. Singh, A. Kundu and D. Adhikari, ACS Catal., 2022, 12, 13075–13107 CrossRef CAS; (b) A. K. Bains, A. Kundu, S. Yadav and D. Adhikari, ACS Catal., 2019, 9, 9051–9059 CrossRef CAS; (c) A. Banik, P. Datta and S. K. Mandal, Org. Lett., 2023, 25, 1305–1309 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, supplementary figures, characterization data, NMR spectra, DFT details. CCDC 2284848. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc06334h |
| This journal is © The Royal Society of Chemistry 2024 |