
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProtein oxidation of fucose environments (POFE) reveals fucose–protein interactions†
Yixuan
Xie‡
ab,
Siyu
Chen‡
 a,
Michael Russelle
Alvarez
a,
Ying
Sheng
a,
Qiongyu
Li
a,
Emanual
Maverakis
c and
Carlito B.
Lebrilla
*ad
a,
Michael Russelle
Alvarez
a,
Ying
Sheng
a,
Qiongyu
Li
a,
Emanual
Maverakis
c and
Carlito B.
Lebrilla
*ad
aDepartment of Chemistry, University of California, Davis, Davis, California, USA. E-mail: cblebrilla@ucdavis.edu
bDepartment of Biochemistry and Molecular Biophysics, Washington University School of Medicine, St. Louis, Missouri 63110, USA
cDepartment of Dermatology, University of California, Davis, Sacramento, California, USA
dDepartment of Biochemistry, University of California, Davis, Davis, California, USA
First published on 11th March 2024
Abstract
Cell membrane glycoproteins are generally highly fucosylated and sialylated, and post-translational modifications play important roles in the proteins' functions of signaling, binding and cellular processing. For these reasons, methods for measuring sialic acid-mediated protein–protein interactions have been developed. However, determining the role of fucose in these interactions has been limited by technological barriers that have thus far hindered the ability to characterize and observe fucose-mediated protein–protein interactions. Herein, we describe a method to metabolically label mammalian cells with modified fucose, which incorporates a bioorthogonal group into cell membrane glycoproteins thereby enabling the characterization of cell-surface fucose interactome. Copper-catalyzed click chemistry was used to conjugate a proximity labeling probe, azido-FeBABE. Following the addition of hydrogen peroxide (H2O2), the fucose-azido-FeBABE catalyzed the formation of hydroxyl radicals, which in turn oxidized the amino acids in the proximity of the labeled fucose residue. The oxidized peptides were identified using liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS). Variations in degree of protein oxidation were obtained with different H2O2 reaction times yielding the acquisition of spatial information of the fucose-interacting proteins. In addition, specific glycoprotein–protein interactions were constructed for Galectin-3 (LEG3) and Galectin-3-binding protein (LG3BP) illustrating the further utility of the method. This method identifies new fucose binding partners thereby enhancing our understanding of the cell glycocalyx.
Introduction
The cell membrane is composed of a network of glycoconjugates, including glycoproteins, glycolipids, and glycoRNAs, that presents a dense matrix of carbohydrates involved in both cell–cell and cell–environment communications.1 This highly interactive glycan matrix, defined by extensive covalent and non-covalent interactions, is known as the glycocalyx. Among the relatively small number of monosaccharides that make up a diverse group of cell-membrane oligosaccharides, sialic acid and fucose play prominent roles and are involved in signaling, cell–cell interactions, and cell–microbe interactions.2–4 These saccharide residues are central to the ability of cell-surface oligosaccharides to interact with other biomolecules, creating interactive networks throughout the cell surface and extracellular space.When present, sialic acids most commonly modify the nonreducing termini of cell-surface glycans. Several approaches have been developed to investigate sialic acid-mediated interactions.5 However, far fewer analytical methods exist to characterize fucose–peptide interactions, due in part to the greater diversity of fucose linkages. Fucose–peptide interactions are typically studied using glycan arrays;6 however, this method provides only binding information with no spatial resolution for the chemical interactions. Another recently developed method to determine fucose-mediated interactions is FucoID, which utilizes in situ proximity-based transfer of fucosylated biotin using fucosyltransferases.7 This method utilizes fluorescence-based methods to detect the fucosylated biotin and is devoid of glycan structural information. Wibowo et al. synthesized photoactivatable glycopolymers to mimic cellular fucosylated glycoproteins, allowing the isolation of fucose-binding proteins through photo-cross-linking.8 While informative, this technique alone cannot elucidate the complex interactions of fucosylated glycoproteins on the cell membrane. More recently, Sun et al. integrated protein–protein cross-linking and enzymatic reactions to investigate cell-surface glycoprotein interactions.9 Similar crosslinking tools have also been developed in this laboratory for capturing sialylated glycoproteins and proximal proteins for mass spectrometry (MS) identification.10 This cross-linking method provides site-specific information about the interacting proteins; however, it may not be applied to fucose-associating proteins because fucose is labile during tandem mass spectrometry (MS/MS) analysis, making the detection of cross-linked products potentially unreliable.
In addition to the aforementioned techniques, proximity-based labelling strategies also have great potential for interrogating glycan–protein interactions.11,12 In our previous studies, we have employed an iron-based proximity tag, FeBABE, to identify the sialic acid environment on the cell surface.13 Similarly, Meyer et al. developed a photo labeling tool to reveal the sialylated glycoproteins in regulating cell-surface transporters.14 However, the same technique is difficult to apply for mapping fucose-associating proteins because the corresponding 6-azido-fucose (6AzFuc) cannot be efficiently incorporated into fucosylated glycans, while 6-alkynyl-fucose (6AlkFuc) blocks the activity of fucose transferases, which in turn alters the glycan profile.15,16 Of particular interest to the current study, Kizuka et al. reported a novel 7-alkynyl-fucose (7AlkFuc) probe, which can be metabolically incorporated into cell-surface fucosylated glycoproteins with low toxicity.17 We reasoned that an alkynyl group on fucose could be used as the handle for attaching the oxidative reagent FeBABE. The FeBABE catalyst motif when attached onto fucose residues of glycoproteins acts as a proximity-labeling probe allowing fucosylated glycoproteins and their binding partners to form stable complexes, followed by Fe-redox chemistry to oxidize the neighboring residues.18
Herein, we describe a proximity-based method to decipher the cellular fucosylated glycoprotein-associating environment in situ (Scheme 1). We termed the method “protein oxidation of fucose environments” (POFE). Briefly, a synthetic azido-FeBABE (AzFeBABE) probe was conjugated to the cell-surface fucosylated glycoproteins via copper-catalyzed azide–alkyne cycloaddition (CuAAC) click chemistry. Upon treatment with low-concentration hydrogen peroxide (H2O2), the incorporated iron catalyzed the generation of hydroxyl radicals, which oxidized amino acid residues residing in close proximity to the labeled fucose. The oxidized peptides were then identified using MS-based proteomic analysis. Furthermore, the hydroxyl radicals diffused to varying distances with different H2O2 incubation times, and thus, the spatial environment of fucose would be revealed by quantitative LC-MS/MS analysis.
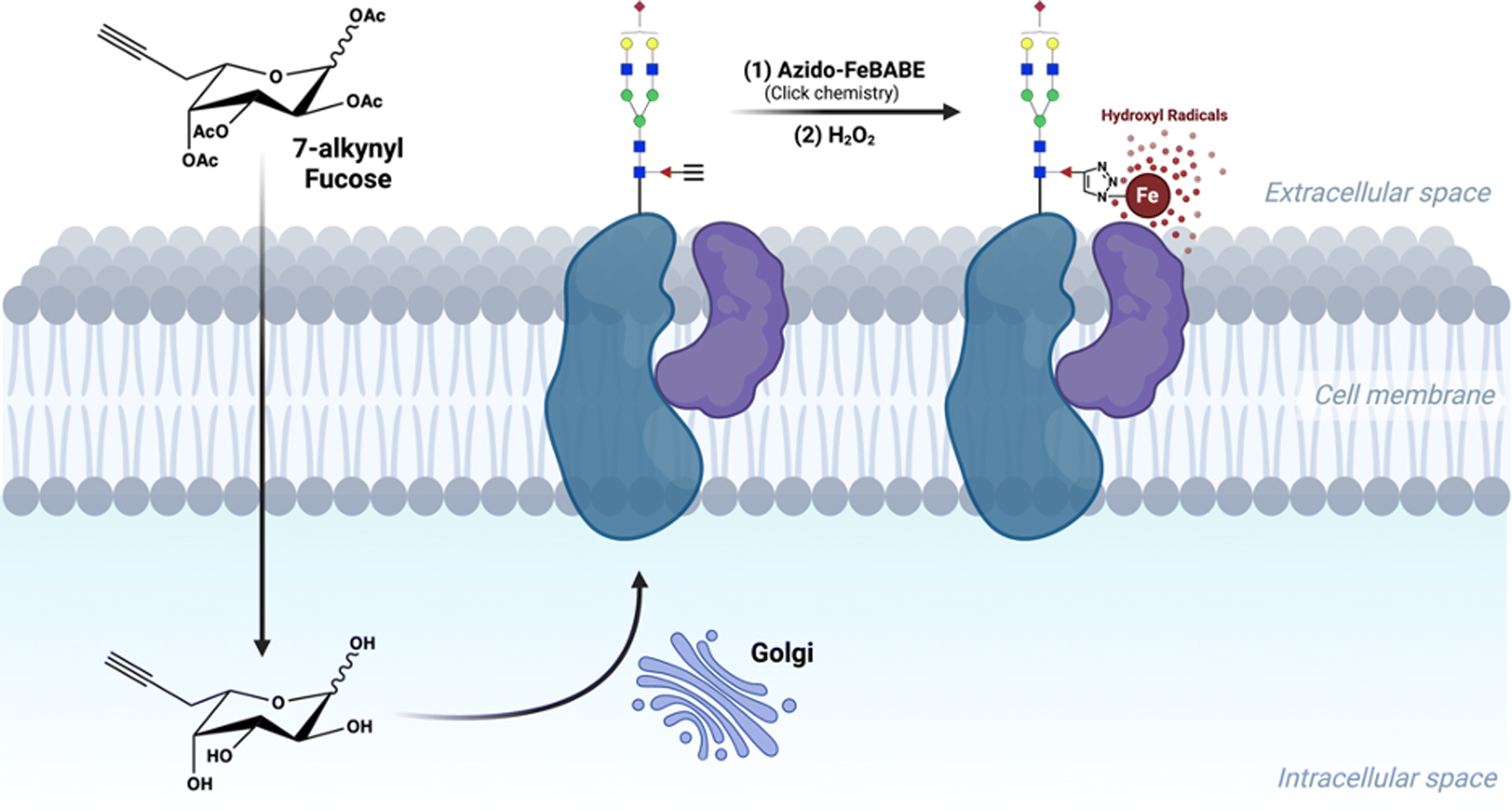 | ||
| Scheme 1 A representation of the 7-alkynyl fucose probe incorporated into fucosylated glycans on the cell surface, followed by the conjugation of the oxidative probe. Upon treatment with hydrogen peroxide, hydroxyl radicals were generated at the fucose site and oxidized the proteins in proximity. | ||
Results and discussion
Metabolic incorporation of fucose probes into cell-membrane glycoproteins
Several fucose bioorthogonal reporters containing azido or alkynyl groups have been proposed however the effect of the compounds on the glycomic profile remains relatively unknown.19 Monosaccharide derivatives with similar structures could result in significantly different incorporation efficiencies in the same cell line.20 Therefore, to assess the desired fucose probes for the PNT2 cell line, we tested three common fucose reporters 6AzFuc, 6AlkFuc, and 7AlkFuc. The incorporation efficiency of these fucose probes was determined through glycomic analysis using LC-MS/MS. This involved the release of the N-glycans followed by LC-MS/MS analysis yielding the N-glycomic profile of the cell membrane. Comparison of the extracted compound chromatogram (ECC) showed 7AlkFuc yielded the best incorporation among all three probes, modifying as much as 20% of fucose-containing glycans on PNT2 cells (Fig. 1a). 6AzFuc only resulted in 5% incorporation after three days of treatment, while 6AlkFuc was found to inhibit cell fucosylation in general. The extent of incorporation determined by the N-glycomic profile is consistent with previous incorporation rates determined by LC-MS/MS and gel-based methods.15,16 The structures of 7AlkFuc-modified glycans were further confirmed with tandem MS data (Fig. S1†). Consequently, 7AlkFuc was selected as the fucose bioorthogonal reporter in this study.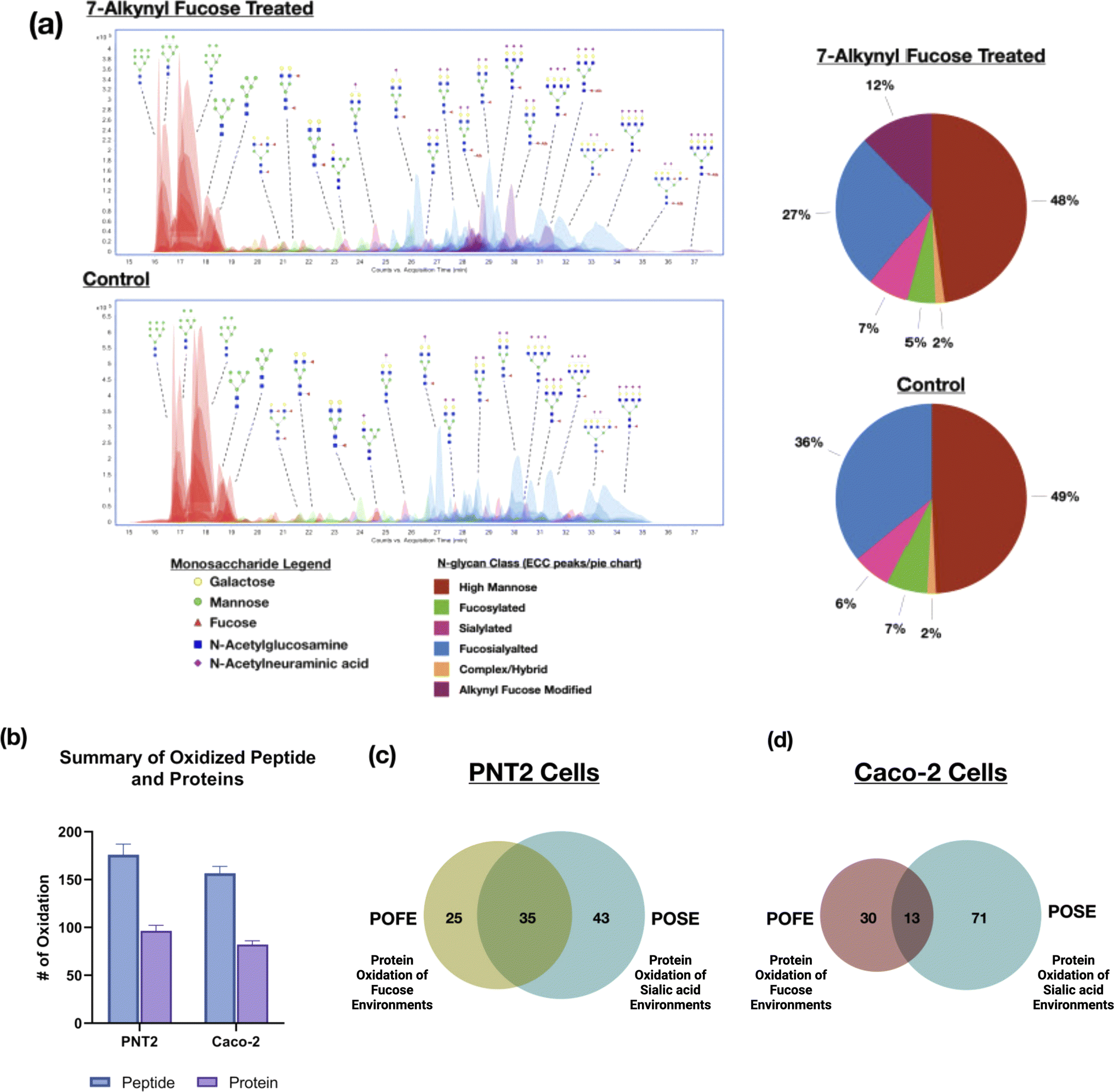 | ||
| Fig. 1 (a) N-glycomic profiles of 7-alkynyl fucose-treated and control PNT2 cells analyzed using HPLC-chip-QTOF MS. Over 200 glycans were identified in the chromatograms and were color-coded based on the glycan subtypes. Putative structures were annotated according to accurate masses and glycan compositions. (b) The numbers of oxidized proteins and peptides in PNT2 and Caco-2 cell lines generated using the POFE (protein oxidation of fucose environments) method. (c) The overlap of oxidized proteins identified from PNT2 cells using the POFE and POSE (protein oxidation of sialic acid environments) methods. (d) The overlap of oxidized proteins identified from Caco-2 cells using the POFE and POSE methods. | ||
To validate the incorporation and optimize the conditions of 7AlkFuc treatment for PNT2 cells, the alkynyl-modified glycoproteins were coupled with an azide-rhodamine reporter tag using CuAAC click chemistry and visualized by SDS-PAGE with in-gel fluorescence scanning. The signal increased with rising 7AlkFuc concentration and reached a plateau when the concentration reached 100 μM (Fig. S2†). Generally, with 100 μM treatment for 72 hours, the fucose bioorthogonal reporter was efficiently incorporated into cell fucosylated glycoproteins.
The localization of 7AlkFuc-containing glycoproteins was analyzed by labeling with coumarin azide and visualized using confocal microscopy. PNT2 cells were grown on a glass-bottom dish and treated with 7AlkFuc for three days. The cells were then incubated with 3-azido-7-hydroxycoumarin for 1 hour and labeled using CellMask™ Deep Red, a marker for the cell plasma membrane, for confocal imaging. The 7AlkFuc-treated cells showed a strong signal (red) after reaction with coumarin azide (Fig. S3†). This signal overlapped well with the membrane signal, indicating the incorporation of 7AlkFuc on the cell plasma membrane. In comparison, 6AlkFuc-treated cells displayed a much lower signal, consistent with the glycomic results obtained from LC-MS/MS analysis suggesting much lower incorporation (Fig. S4†). Notably, 6AzFuc was found mostly enriched inside the cells (Fig. S5†) as opposed to the membrane, which may be explained by its high incompatibility with being transferred onto cell proteins rendering the membrane incorporation pathway inefficient. This largely depends on the performance of glycosyltransferases in transporting the corresponding active unnatural sugar-nucleotides into glycoproteins. Some reporters such as 6AlkFuc can even induce unexpected inhibition of these carbohydrate-active enzymes.21,22
Interrogation of the glycan environment with fucose-centered probes and oxidative proteomic LC-MS/MS
To interrogate the fucose environment in the cell membrane, a metal ion-containing probe (FeBABE) was added through click chemistry to catalyze the production of localized hydroxide radicals. Quantitative proteomic analysis was performed to identify oxidized peptides while quantitating the extent of oxidation. Because peptide oxidation can happen during biological processes or common sample preparation workflows, we first determined the baseline of cellular background oxidation through two control experiments, one with H2O2 only (in the absence of the probe) and the other with AzFeBABE only (no H2O2 added) (Fig. S6†). Both experiments resulted in a similarly low level of background peptide oxidation and were further used as the baseline for the studies. The number of oxidized background proteins in this method was higher compared to the POSE (protein oxidation of sialic acid environments) method, which was likely due to the use of copper during the CuAAC reaction. Thus, to correct for oxidation caused by the CuAAC reaction, all controls and treatments were performed in copper-containing environments.We further validated the results of POFE by performing gene ontology analysis of the oxidized proteins and annotating them based on cellular components (ESI 1 and 2†).23 The majority of the oxidized proteins (128 proteins in PNT2 and Caco2) were designated as cell membrane components with a small fraction (12) of proteins in the extracellular space. Thus, nearly all the proteins were indeed found in the cell membrane or at least associated with the membrane. Two proteins – P29692 (EEF1D) and P61513 (RL37A) – were annotated as cytosolic. However, these proteins were oxidized after 15 minutes of incubation, at which point the hydroxyl radicals could have diffused into the cell.
In addition to PNT2 cells, we similarly tested the POFE method with Caco-2 cells. To identify the oxidized proteins with high confidence, we filtered the results generated by the software Byonic with the following criteria: Score > 300, DeltaMod score > 10.0, and log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ProbValue determined from the test > 2. We looked at oxidation modifications and intensities of peptides containing specific amino acid residues: +15.994915 to M, C, W, Y, F, H, L, I, R, V, T, P, K; +13.9793 to L, I, R, V, P, K; +31.9898 to M, W, Y, F, C; +47.9847 to C; −15.9772 to C; −23.0106 to H; −22.032 to H; −10.032 to H; −43.0534 to R; −30.0106 to D, E; and +47.9847 to C (ESI 3 and 4†).13,24 Furthermore, we manually annotated the spectra of peptides detected in the control, H2O2, and FeBABE–H2O2 experiments (Fig. S7†). The experiments yielded more than 150 oxidized peptides, corresponding to more than 80 proteins from each of the PNT2 and Caco-2 cells (Fig. 1b). Less protein oxidation was observed for Caco-2 cells due to the lower 7AlkFuc incorporation.25 Indeed, the efficiency of oxidation by the same probe on different cell lines can vary to a large extent. For example, PNT2 cells offered a much more effective incorporation for both ManNAz and 7AlkFuc than Caco-2 and similarly yielded more oxidation.26
ProbValue determined from the test > 2. We looked at oxidation modifications and intensities of peptides containing specific amino acid residues: +15.994915 to M, C, W, Y, F, H, L, I, R, V, T, P, K; +13.9793 to L, I, R, V, P, K; +31.9898 to M, W, Y, F, C; +47.9847 to C; −15.9772 to C; −23.0106 to H; −22.032 to H; −10.032 to H; −43.0534 to R; −30.0106 to D, E; and +47.9847 to C (ESI 3 and 4†).13,24 Furthermore, we manually annotated the spectra of peptides detected in the control, H2O2, and FeBABE–H2O2 experiments (Fig. S7†). The experiments yielded more than 150 oxidized peptides, corresponding to more than 80 proteins from each of the PNT2 and Caco-2 cells (Fig. 1b). Less protein oxidation was observed for Caco-2 cells due to the lower 7AlkFuc incorporation.25 Indeed, the efficiency of oxidation by the same probe on different cell lines can vary to a large extent. For example, PNT2 cells offered a much more effective incorporation for both ManNAz and 7AlkFuc than Caco-2 and similarly yielded more oxidation.26
The oxidized proteins using the fucose-centered probes were compared to our previous results based on sialic acid probes. For PNT2 cells, many of the glycans were sialofucosylated (containing both sialic and fucose residues). Indeed, comparison of the proteins oxidized with the fucose probe yielded a 60% overlap with the sialic acid probe (Fig. 1c). In contrast, comparison of the same fucose probe with different cell lines, Caco-2, yielded less than 30% similarities (Fig. 1d). The results can be rationalized by the differences in cell-surface glycosylation of the two cell lines. N-Glycan composition analyses using LC-MS yielded varying distributions of fucosylated-only, sialylated-only glycans, and sialofucosylated glycans. Specifically, PNT2 cells were mainly associated with sialofucosylated glycans (>36% total relative abundances) with only 7% of the glycans being fucosylated-only glycans (Fig. 1a). For Caco-2, we observed the reverse, with 40% of fucosylated-only glycans and 40% of sialofucosylated glycans. Consequently, the higher percentage of asialo-fucosylated glycans in Caco-2 cells resulted in a larger portion of unique proteins in the fucose environment that were not identified in the sialic acid environment.
Time-course functional analysis of proteins within the fucose environment
To probe the spatial distribution of proteins near fucose, we conducted multiple hydrogen peroxide treatments with varying reaction times, namely 5, 10, 15, and 30 minutes, and monitored the extent of oxidation using oxidative proteomics (Fig. S8†). The hydroxyl radicals produced by the probe diffuse to longer distances with extended reaction times providing spatial information relative to the fucose probe. The results were consistent with our previous work where we showed that the hydroxyl radicals produced by the probe diffused from the cell membrane towards intracellular proteins as a function of time.13 Previous single-particle tracking experiments of glycoproteins have suggested significant glycoprotein movement on the membrane as a result of protein diffusion.27–29 Thus, we can correlate both protein and hydroxyl radical diffusion by measuring the number of proteins oxidized at set time intervals. To compare the extent of protein oxidation as a function of incubation time, we compared the number of oxidized proteins at each time point with their identities and conducted a gene ontology analysis for Molecular Functions using STRING-DB.23 Based on these results, four spatial distributions were obtained based on the incubation times and the extent of oxidation of the proteins (Fig. S9†). We observed that for both PNT2 and Caco-2 lines, the total number of oxidized proteins increased as a function of incubation time (Fig. 2a).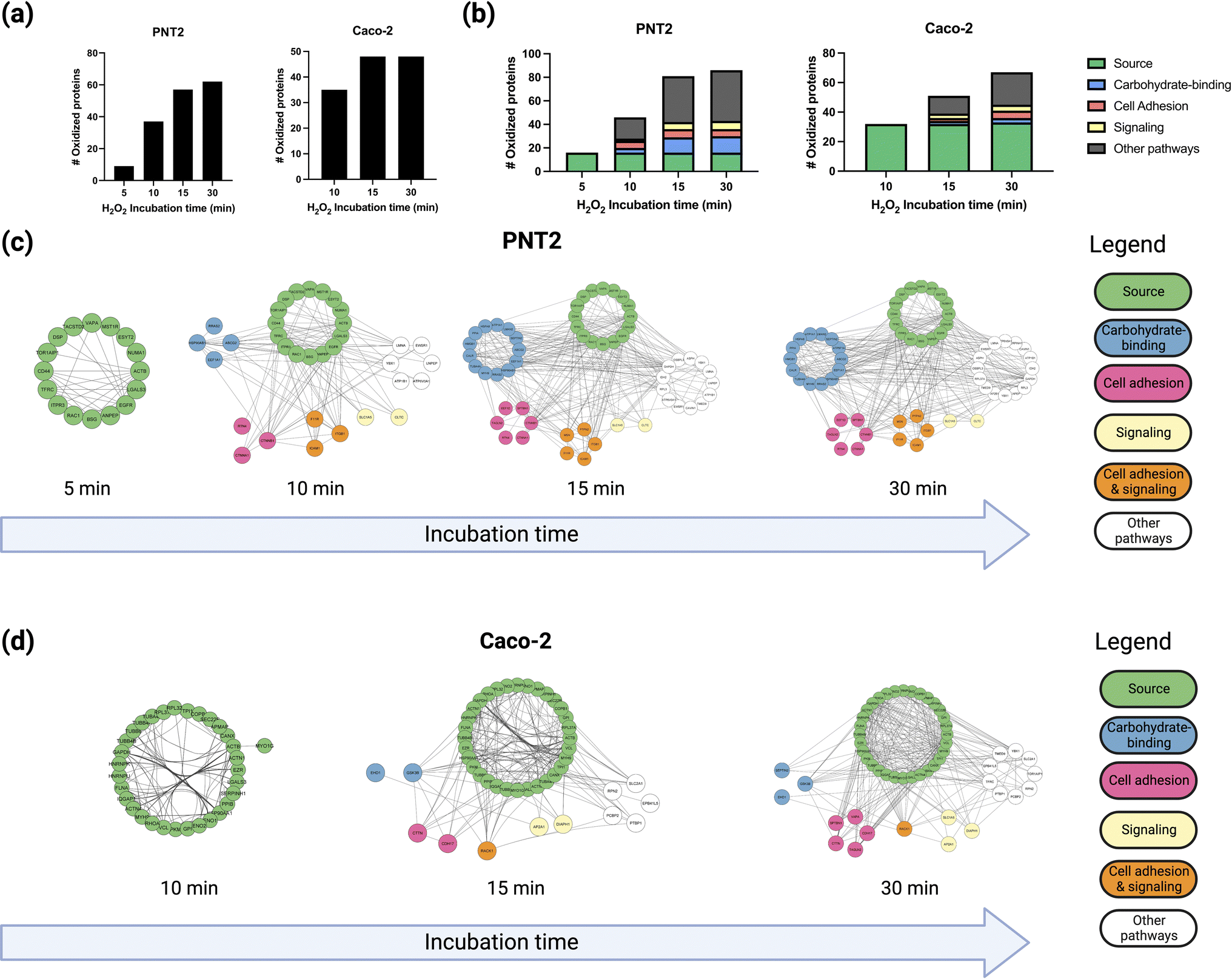 | ||
| Fig. 2 (a) The number of oxidized proteins in both PNT2 and Caco-2 cell lines increases with longer H2O2 incubation times. (b) Likewise, the number of proteins involved in carbohydrate-binding, cell adhesion, signaling, and other pathways increases with longer times. This translates to increasingly larger oxidation networks in both (c) PNT2 and (d) Caco-2 cells. Proteins in the oxidation networks are categorized and color-coded into different biological pathways based on being the source protein (green) or molecular functions: carbohydrate binding (blue), signaling (yellow), cell adhesion (pink), cell adhesion and signaling (orange), and other pathways (white). | ||
Gene ontology enrichment analyses for Molecular Function were conducted using STRING-DB, applying a 0.400 interaction score and 0.05 FDR value cutoff.23
Initially, a small group of proteins (16 proteins in total) were observed to be oxidized after 5 minutes of incubation, all of which were fucosylated according to a separate glycoproteomic analysis. The results suggested that the hydroxyl radicals were indeed generated near the fucose and in turn oxidized the fucosylated proteins themselves. These proteins included membrane glycoproteins such as BSG (basigin), CD44, and EGFR (epidermal growth factor receptor), which are well known to be present and abundant on the PNT2 cell membrane.30
After 10 minutes of reaction, additional glycoproteins and several non-glycosylated proteins such as CTNB1 (catenin beta-1) were oxidized. To understand further the nature of the protein complexes on the cell membrane, we annotated the functions of the proteins that were oxidized after each successive time-point (Fig. 2b). The interaction networks for both PNT2 (Fig. 2c and S10†) and Caoc-2 (Fig. 2d and S11†) grew larger with each succeeding time-point, with the number of proteins involved in carbohydrate-binding increasing considerably after 15 minutes of incubation. This result suggested that proteins involved in these pathways were being oxidized due to their interactions with the fucosylated glycoproteins. As shown in Fig. 3a and b (Fig. S12 and S13†), we evaluated the interactions of a specific glycoprotein, LEG3, and found a similar pattern from both PNT2 and Caco-2, in which there is an increase in carbohydrate-binding, cell adhesion, and signaling proteins after 15 minutes of incubation time. Collectively, these results suggest a time-dependent propagation of the oxidation reaction based on proximity to fucosylated source glycoproteins.
 | ||
| Fig. 3 In both (a) PNT2 and (b) Caco-2 cells, the oxidation network of LEG3 increases with increasing H2O2 incubation times. In PNT2, the proteins that are being oxidized by LGALS3 glycans are involved in carbohydrate-binding, cell adhesion, and signaling pathways while in Caco2, the proteins interacting with LGALS3 are involved in cell adhesion and other pathways. Proteins in the oxidation networks are categorized and color-coded into different biological pathways based on being the source protein (green) or molecular functions: carbohydrate binding (blue), signaling (yellow), cell adhesion (pink), cell adhesion and signaling (orange), and other pathways (white). | ||
We further annotated the oxidized proteins based on cellular component analysis. Cellular component analysis showed that all these proteins were found in the extracellular space or plasma membrane (Fig. S14a†). Additionally, we explored the membrane topology of these proteins using DeepTMHMM,31 and found that a large fraction of them contained transmembrane domains (Fig. S14b†). The results further confirm that these interactions are occurring on the plasma membrane and the highly interactive network in the cell glycocalyx. We combined the data from the first three layers, we constructed a highly interactive protein network through STRING analysis,23 and visualized the interactome using the Cytoscape software (Fig. S15†).32
After 30 minutes an additional 20 proteins were identified, however only four were annotated as plasma membrane proteins. The remainder were annotated as cytoplasm and nucleus proteins (data not shown). This was possibly due to the hydroxyl radicals diffusing across the membrane and labeling the intracellular proteins after 15 minutes.
In summary, more than 65% of the proteins interacting with the fucosylated glycans of the core proteins were annotated as binding proteins according to the Gene Ontology (GO) function annotation (Fig. S16a†). The binding functions were further sub-grouped into cell adhesion molecule binding, cadherin binding, protein-containing complex binding, and exogenous protein binding (Fig. S16b†). In addition, nearly 40% of the identified proteins were associated with cell adhesion molecule binding, consistent with the fact of fucose being highly involved in cell adhesion.32 In addition, dysregulation of fucose has been known to impair the attachment of colorectal cancer cells and affect key cell functions modulating tumor progression.33 Similarly, we found that the fucose-proximal proteins are enriched in proteins related to cell–cell adhesion. Also enriched was the biological function pathway of L1CAM (neural cell adhesion molecule L1) interactions. Indeed, it has been reported that core fucosylation impacted L1CAM proteolytic cleavage and the ability of L1CAM-supported melanoma metastasis.34 Overall, these results demonstrated that the current method can effectively map proteins within the surface fucosylated glycoprotein environment.
Modeling specific interactions of fucosylated glycans in cell membrane proteins
Identification of specific oxidation sites can provide direct experimental evidence for the interactions between individual proteins, revealing the potential interaction partners of surface glycoproteins. For example, LEG3 is known to interact with LG3BP, however the exact binding details are unknown.35 Both proteins are found in the PNT2 and Caco-2 cell lines. Glycoproteomic results show that LG3BP is fucosylated (Fig. S17†). The Phe190 residue in protein LEG3 was oxidized in both PNT2 and Caco-2 cells. The glycoproteomic profile mapped several glycosylation sites in LG3BP, including Asn69, Asn125, Asn192, Asn398, and Asn551. We then obtained the 3D structures of LEG3 and LG3BP from UniProt and PDB and performed protein–protein docking using HADDOCK.36 To identify the specific LG3BP N-glycosylation site responsible for the interactions, we specified interactions between LEG3–Phe190 (oxidized residue) and either LG3BP–Asn69/125/192/398/551. Modeling with HADDOCK predicted 115 possible structures with the highest-scoring and most statistically significant structure used for further analysis (Fig. S18†). To further validate the predicted complex structures of LEG3 and LG3BP, we manually removed sterically hindered structures, and then performed a distance filter between N-glycosite and oxidized residue (less than 20 Å, to account for the FeBABE probe). After validating the results, the complex structure between LEG3–Phe190 and LG3BP–Asn551 was identified to fit all these criteria, which suggests that the glycan in LG3BP–Asn551 is responsible for the oxidation of LEG3–Phe190.Afterwards, we incorporated N-glycan residues on LG3BP–Asn551 to determine the effect of specific N-glycan residues on the glycan–protein interactions. We modeled the following tetra-antennary N-glycans on Asn551: undecorated Hex7HexNAc6, core-fucosylated Hex7HexNAc6Fuc1, sialylated Hex7HexNAc6NeuAc1, and core-fucosylated Hex7HexNAc6Fuc1NeuAc1. After the equilibration, we monitored the frequency and types of contacts between the N-glycan and amino acid residues (Fig. 4a). First, we observed a drastically higher frequency of hydrogen-bonding interactions between the N-glycan and protein upon the addition of either a fucose (Fig. 4b) or a sialic acid (Fig. 4c) residue; the effect becomes more pronounced with the addition of both sialic acid and fucose (Fig. 4d). Based on the types of interactions, the sialic acid in Hex7HexNAc6NeuAc1 contributes additional hydrogen bonding and electrostatic interactions, specifically with a buried His217 residue in LEG3. On the other hand, fucose itself contributes to several hydrogen-bonding interactions. Furthermore, in the binding between LEG3 and LG3BP–Hex7HexNAc6 and Hex7HexNAc6NeuAc1, the galactose residues contribute the most hydrogen-bonding interactions, which is consistent with the previous results.37 In contrast, upon addition of core-fucose to the N-glycan, the N-acetylglucosamine residues then contribute the most hydrogen-bonding interactions. As shown in Fig. S19,† further comparison of the fucosylated N-glycan structures with undecorated N-glycan shows significantly different conformations (RMSD = 0.826, 309 atoms). This result suggests that the addition of core-fucose to the N-glycan changes the conformation of the N-glycan significantly, such that it exposes the core N-acetylglucosamine residues to more interactions, specifically with LEG3–Arg183.
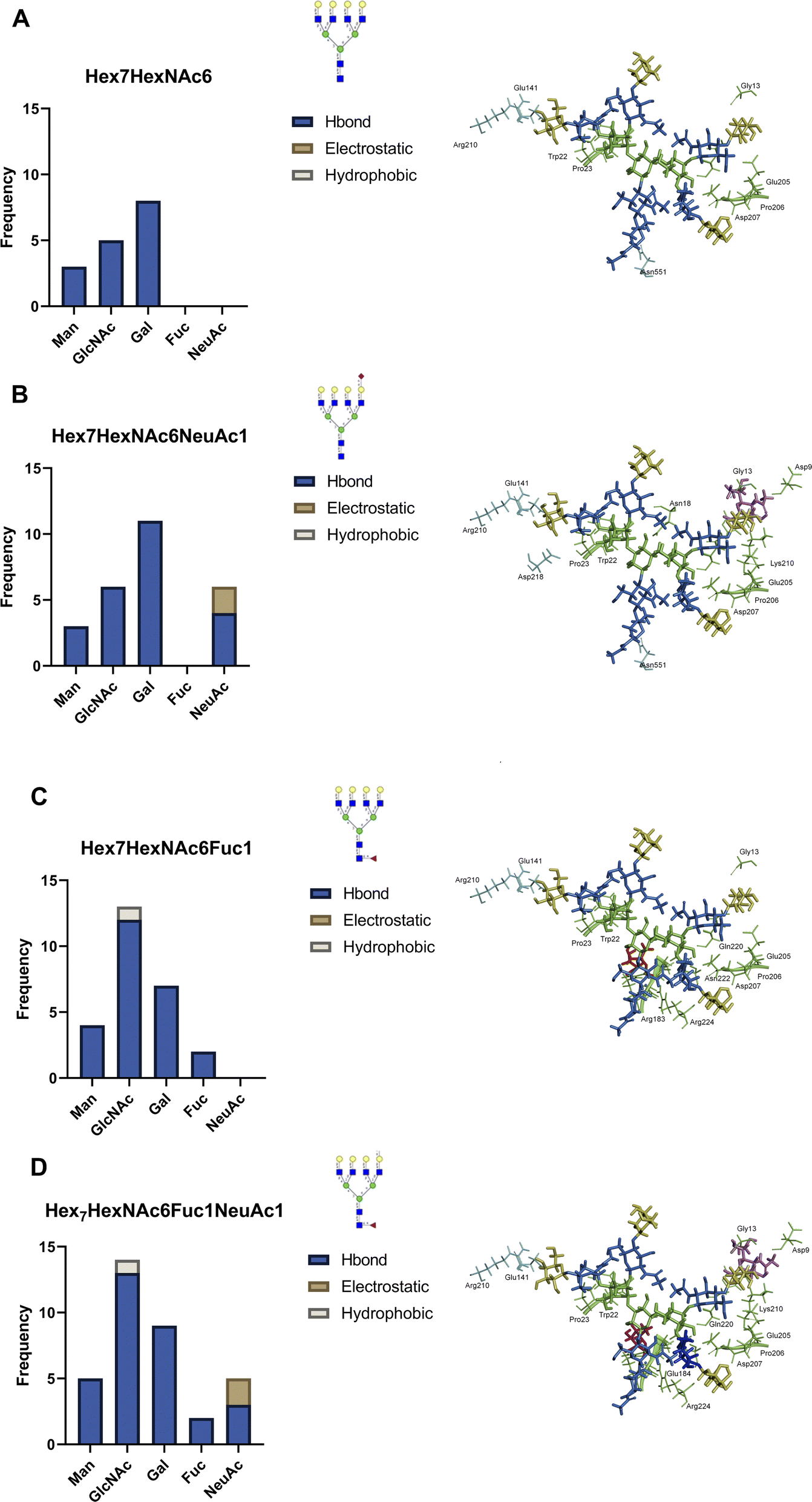 | ||
| Fig. 4 Modeled glycan–protein interactions between LEG3 and LG3BP–ASN511 N-glycan. The LG3BP–ASN511 glycosite was modeled with glycans (a) Hex7HexNAc6, (b) Hex7HexNAc6Fuc1, (c) Hex7HexNAc6NeuAc1, and (d) Hex7HexNAc6Fuc1NeuAc1. Frequency of protein interactions with glycan residues, including mannose (Man), N-acetylglucosamine (GlcNAc), galactose (Gal), fucose (Fuc), and sialic acid (NeuAc), was counted and classified as Hbond, electrostatic, or hydrophobic. | ||
When mapping these LG3BP–N-glycan and LEG3–amino acid residue interactions on the domain map of LEG3 (Fig. S20†), we observed several interesting features. Across the different N-glycan structures, several amino acid interactions are conserved: mannose interacting with LEG3–Gly13, Gly21, Trp22, Pro23, and galactose residues interacting with LEG3–Glu205, Pro206, Asp207, and Gln220. The addition of fucose in the N-glycan increases the number of interactions with the Galectin domain of LEG3, specifically with residues Arg183, Glu184, Glu185, Asn222, and Arg224. On the other hand, adding sialic acid to the N-glycan includes more interactions with Lys210, and specifically the electrostatic interaction with His217. Based on these results, it can be observed that the composition of the N-glycan in LG3BP drastically changes the molecular interactions with LEG3. Overall, by integrating the information from the POFE method with glycoproteomic analysis, specific fucose-mediated interactions can be investigated in detail.
Materials and methods
Materials
6-Azido-fucose (6AzFuc) and 6-alkynyl-fucose (6AlkFuc) were purchased from Carbosynth (San Diego, CA). 7-Alkynyl-fucose (7AlkFuc) was purchased from Vivitide (Gardner, MA). 1-(p-Bromoacetamidobenzyl)ethylenediamine-N,N,N′,N′-tetraacetic acid and iron(III) (FeBABE) were purchased from Dojindo (Rockville, MD). Sodium ascorbate, iodoacetamide (IAA), dithiothreitol (DTT), tris(2-carboxyethyl)phosphine (TCEP), 3-azido-7-hydroxycoumarin, tris(3-hydroxypropyltriazolylmethyl)amine (THPTA) and C18 SPE cartridges were purchased from Sigma-Aldrich (St. Louis, MO). Acetonitrile (ACN), formic acid, CellMask Deep Red plasma membrane stain, and porous graphitic carbon (PGC) SPE plates were purchased from Thermo Scientific (Waltham, MA). iSPE-HILIC cartridges were purchased from HILICON (Umea, Sweden).Synthesis of azido-functionalized p-bromoacetamidobenzyl–EDTA, iron(III) chelate (Az-FeBABE)
The azide group was introduced as previously described (Scheme 1).13 Briefly, the FeBABE was mixed with 1.5 equivalents of sodium azide at room temperature for 24 hours in the acetone/water in a ratio of 4:1, followed by argon flashing to remove the oxygen. The structure of the product was determined by LC-MS analysis in negative ionization mode.
Glycomic analysis with LC-MS/MS
The cell membrane fractions were resuspended with 100 μL of 5 mM DTT in 100 mM ammonium bicarbonate. The mixture was heated in boiling water for 3 minutes. The cleavage of N-glycans was performed by adding 2 μL of PNGase F followed by incubation in a 37 °C water bath overnight. The released N-glycans were separated using 200000×g for 30 minutes, and the supernatant was purified using porous graphitic carbon (PGC) on an SPE plate. The glycan samples were dried and reconstituted in 30 μL of nanopure water. The sample (5 μL) was injected and analyzed with an Agilent 6520 Accurate Mass Q-TOF LC/MS equipped with a PGC nano-chip (Agilent, CA). A binary gradient using solvent A with 3% (v/v) ACN and 0.1% (v/v) formic acid in water and solvent B with 90% (v/v) ACN and 1% (v/v) formic acid in water was applied to separate N-glycans at a 300 nL min−1 flow rate. The resulting chromatograms of glycans were extracted with the MassHunter Qualitative Analysis B08 software (Agilent, CA). N-Glycan compounds were identified using GlycoNote (https://github.com/MingqiLiu/GlycoNote), which contains the accurate mass and formula of human N-glycans, and the N-glycan structures were confirmed through tandem MS fragmentation.
Glycoproteomic analysis with LC-MS/MS
Proteins were denatured in 8 M urea at 55 °C, reduced with 10 mM TCEP, alkylated with 20 mM IAA, diluted to 1 M urea with 50 mM ammonium bicarbonate, and incubated with 2 μg of trypsin at 37 °C overnight. The resulting peptides were concentrated in vacuo. Glycopeptides were enriched by solid-phase extraction using ZIC-HILIC or iSPE®-HILIC cartridges (Nest Group, MA). The enriched products were eluted with a solution of 0.1% (v/v) trifluoroacetic acid in water and dried prior to mass spectrometric analysis.The dried peptides were reconstituted with 0.1% (v/v) FA in water and separated on an UltiMate WPS-3000RS nanoLC system using a C18 column (3 μm, 0.075 mm × 250 mm) coupled with a Fusion Lumos Orbitrap Mass Spectrometer (Thermo Scientific, CA). A binary gradient was applied using 0.1% (v/v) formic acid in (A) water and B 80% acetonitrile: 0–5 min, 4–4% B; 5–133 min, 4–32% B; 133–152 min, 32–48% B; 152–155 min, 48–100% B; 155–170 min, 100–100% B; 170–171 min, 100–4% B; 171–180 min, 4–4% B. The instrument was run in data-dependent mode with 1.8 kV spray voltage, 275 °C ion transfer capillary temperature, and the acquisition was performed with the full MS scanned from 700 to 2000 in positive ionization mode. Stepped higher-energy C-trap dissociation (HCD) at 30 ± 10% was applied to obtain tandem MS/MS spectra with m/z values starting from 120. Glycopeptide fragmentation spectra were annotated using Byonic software. Common modifications, including cysteine carbamidomethyl, methionine oxidation, asparagine deamidation, and glutamine deamidation, were assigned.
Dose-dependent labeling of glycoproteins
The PNT2 cells were treated with 7AlkFuc at different concentrations (0, 12.5, 25, 50, 100, and 200 μM), and then gel-based analysis was conducted following the previous protocol with some optimizations.38 Briefly, cell lysates from treated cells were diluted to 1 mg mL−1, and 12 μL of a freshly prepared click mixture containing 6 μL of 1.7 mM THPTA, 2 μL of 50 mM CuSO4, 2 μL of 1.25 mM rhodamine-azide, and 2 μL of freshly prepared 50 mM TCEP was added to 100 μL of the lysate. Upon addition of the click mixture, each sample was vortexed immediately and allowed to react at room temperature using the end-over-end mixer for 1 hour. The reaction was quenched with 14 μL of 4× sodium dodecyl sulfate (SDS) loading buffer. The proteins were resolved using precast 4 to 20% tris-glycine mini protein gel and visualized by in-gel fluorescence scanning using an LI-COR Imaging System (Lincoln, NE).Targeted oxidation of cell-membrane proteins
For oxidative mapping of the fucose environment on the cell surface, cells were subcultured in T75 flasks (three biological replicates for each treatment) and treated with regular media supplemented with 100 μM of 7AlkFuc for three days after reaching a 40% of confluency at 37 °C in a humidified incubator with 5% CO2. Then the cells were treated with a freshly prepared click reagent solution containing 250 μM THPTA, 50 μM CuSO4, 50 μM Az-FeBABE, and 2.5 mM sodium ascorbate. The click reaction was conducted for 10 minutes at 4 °C. Following the click reaction, the oxidation reaction was conducted by adding 100 μM H2O2 to produce localized oxidation at varying incubation times (5 to 30 minutes). The oxidation reaction was quenched with 10 mM methionine amide hydrochloride before the cells were rinsed and harvested in homogenization buffer containing 1:100 protease inhibitor, 0.25 M sucrose, and 20 mM HEPES–KOH (PH 7.4).
Cell membrane extraction
The procedures for cell membrane extraction were described following a previously established protocol.39 The harvested cells were lysed on ice with five alternating on and off pulses in 5 and 10 seconds intervals, respectively, using a probe sonicator (Qsonica, CT). Nuclear and mitochondria fractions and cellular debris were pelleted and removed by centrifugation at 2000×g for 10 min at 4 °C. The supernatants, containing the cell membrane fraction, were then ultra-centrifuged at 200000×g for 45 min at 4 °C to extract the cell membrane. The pellets were washed stepwise with 500 μL of 0.2 M Na2CO3 solution and 500 μL of water through the same ultra-centrifugation, to obtain the cell membrane fraction.
Proteomic analysis with LC-MS/MS
The extracted membrane pellets were reconstituted with 60 μL of 8 M urea and sonicated for 15 minutes. Then 2 μL of 100 mM TCEP was added to the samples and incubated for 30 minutes at room temperature to denature the proteins. Afterwards, 4 μL of 100 mM IAA was used to alkylate the free thiol groups in the dark for 20 minutes at room temperature. After diluting the urea and adjusting the pH of the samples by adding 420 μL of ammonium bicarbonate (NH4HCO3) solution, 2 μg of trypsin was added to the mixture and the digestion was conducted at 37 °C for 18 hours. The resulting peptides were desalted by solid-phase extraction with C18 cartridges and dried in vacuo.The peptide samples were reconstituted with 0.1% (v/v) FA in water and separated on an EASY-nanoLC system (Thermo Scientific, CA) using the Acclaim PepMap C18 column (3 μm, 0.075 mm × 250 mm) at a flow rate of 300 nL min−1. Water containing 0.1% formic acid and 80% acetonitrile containing 0.1% formic acid were used as solvents A and B, respectively. Peptides were resolved using the following binary gradient: 0–135 min, 2–25% B; 135–173 min, 25–40% B; 173–180 min, 40–100% B. The peptides were analyzed on a Q-Exactive Plus Orbitrap (Thermo Scientific, CA) with the full MS scanned from 200 to 2000 in positive ionization mode. The charge range of the precursor ions was 2 to 7 and the top 15 of them were selected for higher-energy collisional dissociation (HCD) fragmentation with stepped collision energy. Each selected precursor was excluded for 10 seconds after the fragmentation. The MS/MS spectra were collected for product ions with m/z over 130.
Proteomic data analysis and statistics
Acquired LC-MS/MS data were processed using Byonic software (Protein Metrics, CA) against the human protein database (UniProt UP000005640). The identified peptides from Byonic analysis were filtered based on a precursor and fragment mass accuracy of 20 and 10 ppm, respectively. Furthermore, a reverse sequence database was included in the Byonic search, in order to calculate protein FDR (cut-off = 1% FDR). Peptide IDs were filtered according to suggested Byonic cutoffs: Score > 300, logProb. > 2.0, and DeltaMod score > 10. Alkylation of cysteine with carbamidomethylation was assigned as a fixed modification. Deamidation of asparagine and glutamine, methylation of lysine and arginine, and acetylation of protein N-terminus were assigned as rare variable modifications. Oxidation modifications were selected as common variable modifications according to previous settings: +15.994915 to M, C, W, Y, F, H, L, I, R, V, T, P, K; +13.9793 to L, I, R, V, P, K; +31.9898 to M, W, Y, F, C; +47.9847 to C; −15.9772 to C; −23.0106 to H; −22.032 to H; −10.032 to H; −43.0534 to R; −30.0106 to D, E; and +47.9847 to C.24,40,41 After the identification, the peptides were quantified using the Byologic software (Protein Metrics, CA), by measuring the extracted ion chromatograms (XICs) of the oxidized peptides. Oxidized peptides were further annotated for pathway enrichment. Gene ontology enrichment analyses for Molecular Function were conducted using STRING-DB, applying a 0.400 interaction score and 0.05 FDR value cutoff.23
LEG3–LEG3BP glycoprotein modeling
From the oxidation experiment, the interaction between proteins Galectin-3 (LEG3, UniProt ID: P17931 (ref. 42)) and Galectin-3-binding protein (LG3BP, UniProt ID: Q08380 (ref. 43)) was identified. Specifically, the Phe190 residue in LEG3 was found to be oxidized in proximity of the glycans in LG3BP. LC-MS/MS glycoproteomic profiling showed several glycosylation sites in LG3BP, namely, Asn69, Asn125, Asn192, Asn398, and Asn551. The models of LEG3 and LG3BP were obtained from UniProt, and the complexes were constructed on HADDOCK,20 incorporating the interactions between LEG3–Phe190 and LG3BP–Asn69/Asn125/Asn192/Asn398/Asn551. After generating the structures, we selected the highest-scoring structure proposed by HADDOCK (Fig. S17†). We further validated by measuring the distance between LEG3–Phe190 and LG3BP–glycosylation sites while applying a distance filter of less than 20 Å to account for the size of the N-glycan and probe. Afterwards, tetra-antennary N-glycans Hex7HexNAc6, Hex7HexNAc6Fuc1, Hex7HexNAc6NeuAc1, and Hex7HexNAc6Fuc1NeuAc1 were modeled into sites LG3BP–Asn69/Asn125/Asn192/Asn398/Asn551 using CHARMM-GUI glycan modeler.44 After equilibration, the interactions between the LG3BP–glycan residues and LEG3 protein, specifically between LG3BP–glycan and LEG3–Phe190, were identified and quantified using BIOVIA Discovery Studio (Dassault Systemes).Conclusions
Characterization of glycan–protein interactions in situ has been challenging. In this study, we present the protein oxidation of fucose environment (POFE) method as a novel addition to the toolbox for cell-surface fucose interaction interrogation. The method can sketch the spatial protein environment of fucose by detecting proteins oxidized with localized hydroxyl radicals, as well as providing site-specific information of the oxidized proteins and fucosylated glycoproteins so that fucosylated glycan-mediated protein–protein interactions can be modeled. Critically, while abnormal glycosylation is often observed in cancer cells, the glycan interaction information offered by this method can be used to design innovative therapeutics.In the future, we expect that the glycan-based proximity labeling approach can be applied to other glycoforms after development of new carbohydrate reporters, and eventually, the combination of these techniques will provide a comprehensive view of the cellular glycocalyx environment and extend our knowledge in glycobiology.
Data availability
Raw and processed mass spectrometry data are freely available and can be found on the MassIVE repository (https://doi.org/10.25345/C52B8VP2W,MSV000093723).Author contributions
Y. X. and S. C. contributed equally to this work. Y. X. and S. C. designed and performed experiments, analyzed data, created the figures and wrote the manuscript. M. R. A. performed experiments, analyzed data, and wrote the manuscript. Y. S. performed experiments and analyzed data. Q. L. designed and performed experiments. E. M. analyzed data and wrote the manuscript. C. B. L. conceived the idea, supervised the study, and co-wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Research reported in this report was supported by General Medicine of the National Institutes of Health under the award numbers RO1GM049077 and RO1AG062240.Notes and references
- Essentials of Glycobiology, ed. A. Varki, R. D. Cummings, J. D. Esko, P. Stanley, G. W. Hart, M. Aebi, D. Mohnen, T. Kinoshita, N. H. Packer, J. H. Prestegard, R. L. Schnaar and P. H. Seeberger, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, 4th edn, 2022 Search PubMed.
- D. Park, N. Arabyan, C. C. Williams, T. Song, A. Mitra, B. C. Weimer, E. Maverakis and C. B. Lebrilla, Mol. Cell. Proteomics, 2016, 15, 3653–3664 CrossRef CAS PubMed.
- M. Schneider, E. Al-Shareffi and R. S. Haltiwanger, Glycobiology, 2017, 27, 601–618 CrossRef CAS PubMed.
- A. Varki, Nature, 2007, 446, 1023–1029 CrossRef CAS PubMed.
- J. E. Stencel-Baerenwald, K. Reiss, D. M. Reiter, T. Stehle and T. S. Dermody, Nat. Rev. Microbiol., 2014, 12, 739–749 CrossRef CAS.
- S. Serna, S. Yan, M. Martin-Lomas, I. B. H. Wilson and N.-C. Reichardt, J. Am. Chem. Soc., 2011, 133, 16495–16502 CrossRef CAS PubMed.
- Z. Liu, J. P. Li, M. Chen, M. Wu, Y. Shi, W. Li, J. R. Teijaro and P. Wu, Cell, 2020, 183, 1117–1133 CrossRef CAS PubMed.
- A. Wibowo, E. C. Peters and L. C. Hsieh-Wilson, J. Am. Chem. Soc., 2014, 136, 9528–9531 CrossRef CAS PubMed.
- F. Sun, S. Suttapitugsakul and R. Wu, Chem. Sci., 2021, 12, 2146–2155 RSC.
- Y. Xie, S. Chen, Q. Li, Y. Sheng, M. R. Alvarez, J. Reyes, G. Xu, K. Solakyildirim and C. B. Lebrilla, Chem. Sci., 2021, 12, 8767–8777 RSC.
- W. Qin, K. F. Cho, P. E. Cavanagh and A. Y. Ting, Nat. Methods, 2021, 18, 133–143 CrossRef CAS PubMed.
- A. E. Reeves and M. L. Huang, Curr. Opin. Chem. Biol., 2023, 72, 102233 CrossRef CAS PubMed.
- Q. Li, Y. Xie, G. Xu and C. B. Lebrilla, Chem. Sci., 2019, 10, 6199–6209 RSC.
- C. F. Meyer, C. P. Seath, S. D. Knutson, W. Lu, J. D. Rabinowitz and D. W. C. MacMillan, J. Am. Chem. Soc., 2022, 144, 23633–23641 CrossRef CAS PubMed.
- Y. Kizuka, S. Funayama, H. Shogomori, M. Nakano, K. Nakajima, R. Oka, S. Kitazume, Y. Yamaguchi, M. Sano, H. Korekane, T.-L. Hsu, H.-Y. Lee, C.-H. Wong and N. Taniguchi, Cell Chem. Biol., 2016, 23, 782–792 CrossRef CAS PubMed.
- Q. Zhou, Y. Xie, M. Lam and C. B. Lebrilla, Cells, 2021, 10, 2318 CrossRef CAS PubMed.
- Y. Kizuka, M. Nakano, Y. Yamaguchi, K. Nakajima, R. Oka, K. Sato, C.-T. Ren, T.-L. Hsu, C.-H. Wong and N. Taniguchi, Cell Chem. Biol., 2017, 24, 1467–1478 CrossRef CAS PubMed.
- S. M. Cheal, M. Ng, B. Barrios, Z. Miao, A. K. Kalani and C. F. Meares, Biochemistry, 2009, 48, 4577–4586 CrossRef CAS PubMed.
- D. Rabuka, S. C. Hubbard, S. T. Laughlin, S. P. Argade and C. R. Bertozzi, J. Am. Chem. Soc., 2006, 128, 12078–12079 CrossRef CAS PubMed.
- J. E. G. A. Dold and V. Wittmann, ChemBioChem, 2021, 22, 1243–1251 CrossRef CAS PubMed.
- S. Li, A. J. McCraw, R. A. Gardner, D. I. R. Spencer, S. N. Karagiannis and G. K. Wagner, Antibodies, 2021, 10, 44 CrossRef CAS PubMed.
- Y. Sheng, A. Vinjamuri, M. R. Alvarez, Y. Xie, M. McGrath, S. Chen, M. Barboza, M. Frieman and C. Lebrilla, Front. Mol. Biosci., 2022, 9, 799703 CrossRef CAS PubMed.
- D. Szklarczyk, A. L. Gable, D. Lyon, A. Junge, S. Wyder, J. Huerta-Cepas, M. Simonovic, N. T. Doncheva, J. H. Morris, P. Bork, L. J. Jensen and C. von Mering, Nucleic Acids Res., 2019, 47, D607–D613 CrossRef CAS PubMed.
- M. Bern, Y. J. Kil and C. Becker, Current Protocols in Bioinformatics, 2012 Search PubMed.
- C. Ma, H. Takeuchi, H. Hao, C. Yonekawa, K. Nakajima, M. Nagae, T. Okajima, R. S. Haltiwanger and Y. Kizuka, Int. J. Mol. Sci., 2020, 21, 6007 CrossRef CAS PubMed.
- D. Dayoung Park, G. Xu, M. Wong, C. Phoomak, M. Liu, N. E. Haigh, S. Wongkham, P. Yang, E. Maverakis and C. B. Lebrilla, Chem. Sci., 2018, 9, 6271–6285 RSC.
- Y. Cui, M. Yu, X. Yao, J. Xing, J. Lin and X. Li, Mol. Plant, 2018, 11, 1315–1327 CrossRef CAS PubMed.
- A. S. Hansen, M. Woringer, J. B. Grimm, L. D. Lavis, R. Tjian and X. Darzacq, eLife, 2018, 7, e33125 CrossRef.
- X. H. Vu, N. D. Dien, T. T. H. Pham, R. Jaffiol, C. Vézy, N. X. Ca and T. T. Trang, Biochim. Biophys. Acta, Biomembr., 2021, 1863, 183721 CrossRef CAS.
- G. D. Grass, L. B. Tolliver, M. Bratoeva and B. P. Toole, J. Biol. Chem., 2013, 288, 26089–26104 CrossRef CAS.
- J. Hallgren, K. Tsirigos, M. D. Pedersen, J. J. Almagro Armenteros, P. Marcatili, H. Nielsen, A. Krogh, O. Winther, bioRxiv, 2022, DOI:10.1101/2022.04.08.487609.
- P. Shannon, A. Markiel, O. Ozier, N. S. Baliga, J. T. Wang, D. Ramage, N. Amin, B. Schwikowski and T. Ideker, Genome Res., 2003, 13, 2498–2504 CrossRef CAS PubMed.
- A. Zipin, M. Israeli-Amit, T. Meshel, O. Sagi-Assif, I. Yron, V. Lifshitz, E. Bacharach, N. I. Smorodinsky, A. Many, P. A. Czernilofsky, D. L. Morton and I. P. Witz, Cancer Res., 2004, 64, 6571–6578 CrossRef CAS PubMed.
- P. Agrawal, B. Fontanals-Cirera, E. Sokolova, S. Jacob, C. A. Vaiana, D. Argibay, V. Davalos, M. McDermott, S. Nayak, F. Darvishian, M. Castillo, B. Ueberheide, I. Osman, D. Fenyö, L. K. Mahal and E. Hernando, Cancer Cell, 2017, 31, 804–819 CrossRef CAS PubMed.
- E. Joeh, A. E. Reeves, C. G. Parker and M. L. Huang, Curr. Protoc., 2021, 1, e104 CrossRef CAS PubMed.
- C. Dominguez, R. Boelens and A. M. J. J. Bonvin, J. Am. Chem. Soc., 2003, 125, 1731–1737 CrossRef CAS PubMed.
- J. Iwaki and J. Hirabayashi, Trends Glycosci. Glycotechnol., 2018, 30, SE137–SE153 CrossRef.
- Z. Lin, X. Wang, K. A. Bustin, K. Shishikura, N. R. McKnight, L. He, R. M. Suciu, K. Hu, X. Han, M. Ahmadi, E. J. Olson, W. H. Parsons and M. L. Matthews, ACS Cent. Sci., 2021, 7, 1524–1534 CrossRef CAS PubMed.
- Q. Li, Y. Xie, M. Wong, M. Barboza and C. B. Lebrilla, Nat. Protoc., 2020, 15, 2668–2704 CrossRef CAS PubMed.
- X. R. Liu, D. L. Rempel and M. L. Gross, Nat. Protoc., 2020, 15, 3942–3970 CrossRef CAS PubMed.
- O. Charvátová, B. L. Foley, M. W. Bern, J. S. Sharp, R. Orlando and R. J. Woods, J. Am. Soc. Mass Spectrom., 2008, 19, 1692–1705 CrossRef PubMed.
- J. Seetharaman, A. Kanigsberg, R. Slaaby, H. Leffler, S. H. Barondes and J. M. Rini, J. Biol. Chem., 1998, 273, 13047–13052 CrossRef CAS PubMed.
- Q08380 | SWISS-MODEL Repository, https://swissmodel.expasy.org/repository/uniprot/Q08380?csm=C488C2E99D77435B, accessed November 29, 2023.
- S.-J. Park, J. Lee, Y. Qi, N. R. Kern, H. S. Lee, S. Jo, I. Joung, K. Joo, J. Lee and W. Im, Glycobiology, 2019, 29, 320–331 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc06432h |
| ‡ Yixuan Xie and Siyu Chen contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |