
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Excited-state antiaromaticity relief drives facile photoprotonation of carbons in aminobiphenyls†
Josip
Draženović‡
a,
Croix J.
Laconsay‡
 b,
Nađa
Došlić
*c,
Judy
I-Chia Wu
*b and
Nikola
Basarić
*a
b,
Nađa
Došlić
*c,
Judy
I-Chia Wu
*b and
Nikola
Basarić
*a
aDepartment of Organic Chemistry and Biochemistry, Ruđer Bošković Institute, Bijenička Cesta 54, 10000 Zagreb, Croatia. E-mail: nbasaric@irb.hr
bDepartment of Chemistry, University of Houston, Houston, TX 77204, USA. E-mail: jiwu@central.uh.edu
cDepartment of Physical Chemistry, Ruđer Bošković Institute, Bijenička Cesta 54, 10000 Zagreb, Croatia. E-mail: nadja.doslic@irb.hr
First published on 4th March 2024
Abstract
A combined computational and experimental study reveals that ortho-, meta- and para-aminobiphenyl isomers undergo distinctly different photochemical reactions involving proton transfer. Deuterium exchange experiments show that the ortho-isomer undergoes a facile photoprotonation at a carbon atom via excited-state intramolecular proton transfer (ESIPT). The meta-isomer undergoes water-assisted excited-state proton transfer (ESPT) and a photoredox reaction via proton-coupled electron transfer (PCET). The para-isomer undergoes a water-assisted ESPT reaction. All three reactions take place in the singlet excited-state, except for the photoredox process of the meta-isomer, which involves a triplet excited-state. Computations illustrate the important role of excited-state antiaromaticity relief in these photoreactions.
Introduction
Excited-state proton transfer reactions typically occur between a proton donor (e.g., NH or OH) and an electronegative heteroatom.1 Sometimes, however, the proton acceptor can be a carbon atom.2 Examples of facile photoprotonation in aromatic organic compounds by aqueous acids have been known since the mid-1980's.3–5 In 2002, Wan et al. reported the first example of direct photoprotonation of a carbon atom of an aromatic ring via excited-state intramolecular proton transfer (ESIPT).6,7 Irradiating 2-phenylphenol in D2O–CH3CN solution led to regioselective D-exchange at the 2′ and 4′ positions. Deuteration at the 2′ position was believed to proceed through ESIPT involving an initial hydrogen bonding interaction between the OH group of the phenol moiety and the π-system of the adjacent phenyl moiety. Upon irradiation, the phenolic OH proton shifts to the 2′ position of the unsubstituted phenyl ring. When the distance between the acidic and basic carbon site is far, regioselective D-exchange can occur by excited-state proton transfer (ESPT) involving a chain of protic solvents.8–14 Many examples of ESIPT and ESPT involving proton transfer, either from an OH group15–17 or an amine group18–20 to a basic carbon atom have been reported. These findings are at first sight somewhat surprising, since protonation of a carbon atom of an aromatic ring typically is a slow process in the ground state.21,22 Yet, why do some aromatic ring carbons gain basicity in the excited-state?Some of us recently related the phenomena of excited-state proton transfer in aromatic organic compounds to a reversal of aromaticity and antiaromaticity in the lowest ππ* excited-states.23o-Salicylic acid,24 for example, is 4n + 2 aromatic in the ground state, but shows enhanced antiaromatic character in the first 1ππ* state. Proton transfer from an OH group to the carbonyl site disrupts cyclic 4n + 2 conjugation in the six membered ring and this reaction alleviates excited-state antiaromaticity (Scheme 1a). The low barriers of many other excited state proton and electron transfer reactions, for example, ESIPT in phenol-benzoxazoles,25 the photoacidity of naphthols,26 and proton coupled electron transfer reactions in phenols27 have been attributed to the effects of excited-state antiaromaticity relief. The concepts of excited-state aromaticity and antiaromaticity were first explored by Dewar,28 Zimmerman,29–31 and later Dougherty32 to explain the mechanisms of pericyclic reactions. Later, Baird proposed, based on a set of perturbation molecular orbital theory analyses, that “the rules for ground state aromaticity are reversed in the first 3ππ* state: 4n rings display ‘aromatic’ character whereas 4n + 2 systems display ‘antiaromaticity’.33 Baird's rules apply also to the lowest ππ* singlet excited states of annulenes34–36 and has garnered considerable attention in many areas of applied organic photochemistry.37–40
 | ||
| Scheme 1 (a) Antiaromaticity relief in the ESIPT reaction of o-salicylic acid.23 (b) Antiaromaticity relief in the ESIPT reaction of o-aminobiphenyl. (c) Amino arene compounds investigated and carbon numberings for the phenyl and aniline rings. | ||
Here, we relate the potential for carbon atom photobasicity in aromatic ring to a reversal of aromaticity and antiaromaticity in the lowest ππ* excited states. In the ground state, protonating an aromatic ring and disrupting cyclic 4n + 2 π-conjugation is a slow process due to a loss of aromaticity. But in the lowest singlet or triplet excited state, these rings gain antiaromatic character and the ring carbon atoms display increased basicity, as protonation now affords a channel to alleviate excited-state antiaromaticity (Scheme 1b). To model the effects of excited-state antiaromaticity relief in the photoprotonation of aromatic ring carbons, here, we investigate the photophysics and photochemistry of a series of ortho-(1), meta-(2) and para-(3) aminobiphenyls (Scheme 1c). These chromophores can be found in many structures with important applications in supramolecular chemistry or material science.41 We now report a combined experimental and computational study, showing that the aromatic rings of aminobiphenyls can gain significant antiaromatic character in the singlet and/or triplet states, demonstrating that antiaromaticity relief is a major driving force for the photoreactivities of ortho-(1), meta-(2) and para-(3) aminobiphenyls.
Results and discussion
Photophysical properties
Absorption and emission spectra for the aminobiphenyls 1–3 were recorded in CH3CN (see Fig. S1–S3 in the ESI†). The absorption spectra of 1 and 2 show maxima at 305 nm, and that of 3 shows a maximum at 283 nm (Fig. 1, top). These absorption bands correspond to the S1 ← S0 transition and are characterized as ππ* states as confirmed by the symmetry of the computed orbitals. The higher singlet states of 1 (≈250 nm, shoulder), 2 (236 nm), and 3 (200 nm) indicate significant charge transfer character. The emission spectra of 1 and 2 nearly overlap and show maxima at 390 nm, while that of 3 shows a maximum at 361 nm (Fig. 1, bottom). Stokes' shifts of all three isomers range between 6600–7500 cm−1 (Table S1 in the ESI†). Quantum yields of fluorescence were measured by use of N-acetyl-tryptophanamide in water as a reference (Φf = 0.12).42 In accordance with an observed lower photoreactivity, 3 (Φf = 0.55) shows higher fluorescence compared to 1 (Φf = 0.21) and 2 (Φf = 0.26). | ||
| Fig. 1 Measured absorption (top) and normalized emission spectra (bottom) of 1–3 in CH3CN (λexc = 280 nm). | ||
Time-correlated single photon counting (TC-SPC) experiments were performed to measure the decay of fluorescence for 1–3 in CH3CN solutions (Fig. S7–S9 and Table S2 in the ESI†). The decay times do not fit to a single-exponential function. In addition to the major decay component (2.5–6.6 ns), a short decay component was detected (<100 ps); however, a precise measurement for the short decay component was not possible with the setup used. Dual fluorescence for aminonaphthalenes have been observed and attributed to emission from the La and Lb states.43 Our computations agree with this interpretation, since the excitation at 280 nm used for the SPC can give rise to the adiabatic transition to the S2 state. The other, less likely interpretation for dual emission would be that it originates from different vibrational levels of the excited states.44
Addition of a protic solvent (i.e., H2O) to CH3CN increases Φf for all three compounds (Fig. S4–S6 in the ESI†). We surmise that the presence of H2O may hinder vibrational modes that lead to a non-radiative decay from the S1 state, thereby enhancing fluorescence. Direct comparisons of the emission spectra for 1–3 in CH3CN–H2O and in CH3CN–D2O, reveal stronger fluorescence and a slower decay in D2O (Fig. 2, S10, S11 and Table S3 in the ESI†), especially for 1. This finding strongly indicates the involvement of H2O in blocking a non-radiative decay pathway from the singlet excited state; D2O shows slower decay kinetics due to a kinetic isotope effect. These findings are in line with the observed ESIPT reactivity of 1, which takes place only in the presence of H2O as a protic solvent.
 | ||
Fig. 2 Fluorescence spectra of 1 (λex = 280 nm) in CH3CN–H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) or CH3CN–D2O (1:1) (top) and the corresponding decays of fluorescence at 400 nm (bottom). Fitting data can be found in Table S3.† :1) or CH3CN–D2O (1:1) (top) and the corresponding decays of fluorescence at 400 nm (bottom). Fitting data can be found in Table S3.† | ||
Acid–base properties
UV-vis and fluorescence titrations in acidic media were performed to determine the pKa values and corresponding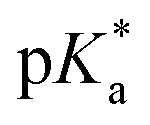 values in the S1 state for 1–3 (Table 1). Fig. 3 and 4 show the spectra for 3, and those of 1 and 2 are included in the ESI (Fig. S12–S18).† Increasing the acidity of the solution resulted in a hypochromic shift of the lowest-energy absorption band for 1–3 (i.e., due to protonation of the nitrogen atom) and a quenched fluorescence. The lower fluorescence quantum yields of 1–3 in acidic solutions are in line with an observed efficient photoreaction in acidic media. As shown in Table 1, compounds 1–3 display decreased basicity in the S1 state (lower
values in the S1 state for 1–3 (Table 1). Fig. 3 and 4 show the spectra for 3, and those of 1 and 2 are included in the ESI (Fig. S12–S18).† Increasing the acidity of the solution resulted in a hypochromic shift of the lowest-energy absorption band for 1–3 (i.e., due to protonation of the nitrogen atom) and a quenched fluorescence. The lower fluorescence quantum yields of 1–3 in acidic solutions are in line with an observed efficient photoreaction in acidic media. As shown in Table 1, compounds 1–3 display decreased basicity in the S1 state (lower 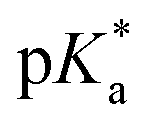 values). These results suggest that upon photoexcitation, electron density on the nitrogen atom is delocalized into the aromatic ring, and the nitrogen atom becomes less basic (see computed NPA charges for N in Fig. 6).
values). These results suggest that upon photoexcitation, electron density on the nitrogen atom is delocalized into the aromatic ring, and the nitrogen atom becomes less basic (see computed NPA charges for N in Fig. 6).
 values of 1–3 by UV-vis and fluorescence titrationa
values of 1–3 by UV-vis and fluorescence titrationa
| Compound | pKa (UV-vis) | pKa (fluorescence) |
|
|---|---|---|---|
|
a All titration experiments were conducted in CH3CN–H2O (1:4) at 25 °C.
|
|||
| 1 | 3.48 ± 0.02 | 3.27 ± 0.02 | 0.56 ± 0.04 |
| 2 | 3.94 ± 0.03 | 3.74 ± 0.06 | 0.15 ± 0.03 |
| 3 | 3.75 ± 0.01 | 3.80 ± 0.04 | 1.49 ± 0.02 |

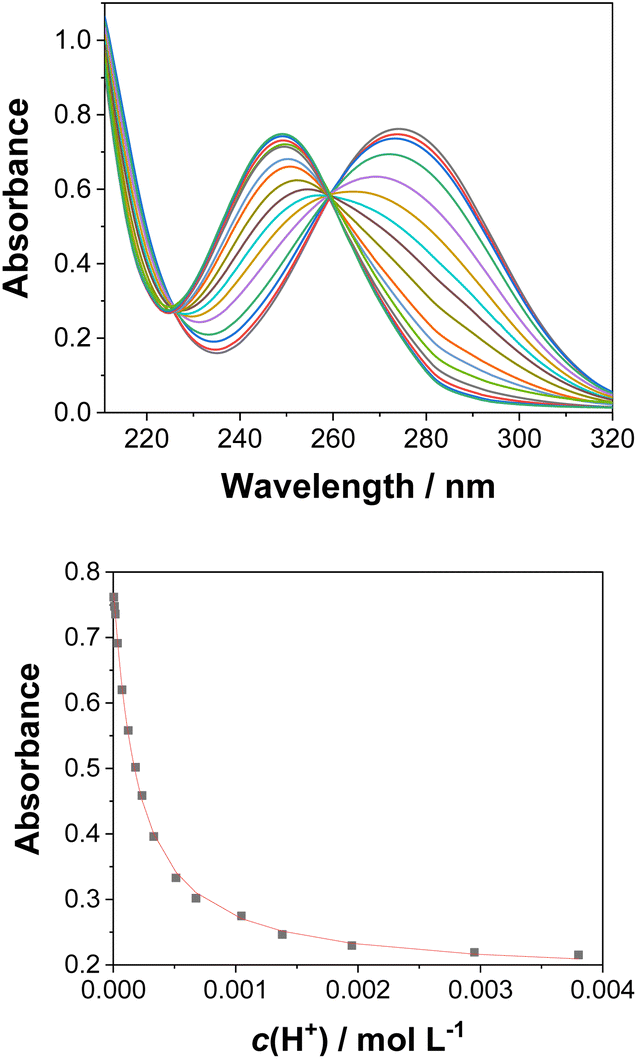 | ||
| Fig. 3 Absorption spectra of 3 (c0 = 3.67 × 10−5 M) in CH3CN–H2O (1:4) at different pH values (top), and dependence of the absorption at 274 nm on the pH (bottom). The black dots are experimental values and the red line corresponds to a model involving a one-step protonation equilibrium. | ||
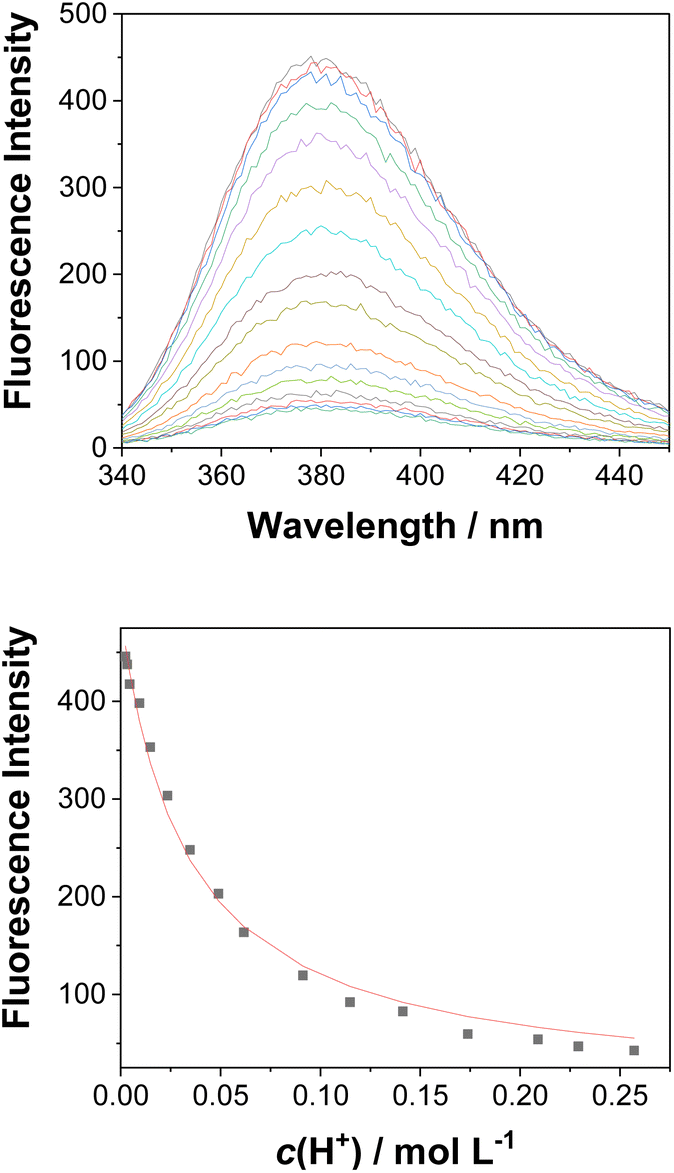 | ||
| Fig. 4 Fluorescence spectra (λex = 260 nm) of 3 (c0 = 4.51 × 10−6 M) in CH3CN–H2O (1:4) at different pH values (top), and dependence of the fluorescence intensity at 382 nm on the pH (bottom). The black dots are experimental values and the red line corresponds to a model involving a one-step protonation equilibrium. | ||
Irradiation experiments
Irradiating 1–3 in CH3CN–D2O (2:1) at pD = 2 at ≈300 nm led to regiospecific deuteration: specifically, at the C2′ position of the phenyl ring in 1 and 3, and the C4 position of the aniline ring in 2. Additionally, irradiating 2 gave a photoredox product 4 (vide infra). The acidity of the mixture was adjusted by addition of D2SO4 or a NaOD–D3PO4 buffer. The content of deuteration and position of the deuterium in molecules 1–3 was determined after aqueous workup from MS and 1H NMR, respectively. Experimental procedures and graphs showing dependence of D-exchange with the irradiation time, the D2O concentration, and the pH (pD) are included in the ESI (Fig. S19–S34).† Experiments for the naphthyl derivative of 1 under similar acidic conditions (pD = 2 in the presence of D2SO4) also showed efficient D-exchange.20
Irradiating 1 in CH3CN–D2O led to regiospecific D-exchange at the C2′position of the phenyl ring and yielded 1-D (Scheme 2). Deuteration depends on irradiation time (Fig. S19–S21 in the ESI†); 3 h irradiation gave almost complete D-exchange at the C2′ position, and upon longer irradiation, both ortho-H-atoms of the phenyl ring were replaced by D. A control experiment for 1 in CH3CN–D2O (2:1) at pD = 2 without irradiation showed no sign of D-exchange, confirming that D-exchange is a photochemical process. Moreover, the deuteration takes place over wide pH (pD) range with similar efficiency (see Fig. S23 in the ESI†). Most importantly, the D-exchange at pH ≈ 7 is not in agreement with a plausible mechanism that would involve electrophilic attack of H3O+ (D3O+), vide infra. A proposed reaction is shown in Scheme 2.6,20 Upon irradiation, 1 undergoes ESIPT giving aza-quinone methide 1-aQM, and the intermediate then re-aromatizes to 1-D. The last step is expected not to be pH- (pD-) dependent. The quantum yield for D-exchange in 1 (ΦR = 0.016 ± 0.004, Table S4†) in CH3CN–D2O (2:1, v/v, pD = 2) was determined by use of KI/KIO3 as an actinometer (Φ254 = 0.74).45,46 This value is lower compared to the quantum yield for D-exchange in 2-phenylphenol (Φ254 = 0.041 ± 0.004),6 as expected by the lower acidity of the NH2versus OH group.
 | ||
| Scheme 2 ESIPT reaction for D-exchange in 1. | ||
We found that the efficiency of D-exchange increases upon increased D2O content (between 0.1% to 1% D2O) and levels off at concentrations higher than 1% (see Fig. S22 in the ESI†). Interestingly, a higher D2O concentration does not lead to regioselective D-exchange at the more distant C4′ position. These results contrast with Wan's 2002 report on 2-phenylphenol, which showed both C2′ and C4′ deuterated photoproducts.6 It was found that, upon irradiating 2-phenylphenol, D-exchange at the C2′ position takes place via an ESIPT mechanism, whereas at higher D2O concentration, D-exchange at the C4′ position occurs via a H2O-mediated ESPT mechanism. Thus, deuteration of position C4′ may also take place in 1, but it is significantly less efficient compared to the ESIPT to position 2′. Note that we detected some tri-deuterated compound 1 after long irradiation (Fig. S19 in the ESI†), which may be due to the less efficient deuteration of the 4′ position, not detectable by 1H NMR (<5%).
To investigate whether D2O is required for D-exchange, we prepared a deuterated version of 1 (ND2, see the ESI† for details) and irradiated it in anhydrous CH3CN. No D-exchange occurred at the phenyl rings, suggesting that D2O is required for D-exchange to occur. To investigate whether an NH2 group is required for D-exchange, we prepared the corresponding dimethylated derivative 1(NMe2). Irradiated and non-irradiated samples of 1(NMe2) in CH3CN–D2O (2:1) both showed comparable minor D-exchange, more than ten times less efficient than for 1 (Table S7†), indicating that an NH2 group is necessary for D-exchange. To investigate whether photoinduced electrophilic attack of D3O+ to the carbon atom site contributes to D-exchange, we irradiated 1 in CH3CN–D2O in the presence of deuterated sodium phosphate buffer at different pD values (Table S6 and Fig S23 in the ESI†). D-exchange is low at pD < 3 and 1-D is observed at pD values 3–8, suggesting that an electrophilic D3O+ attack mechanism is unlikely (although it cannot be completely ruled out that some D-exchange may operate at very low pD values with much lower efficiency). These results obtained at pH values higher than the pKa also indicate that ESIPT involves proton transfer from an NH2 group (and not NH3+). Electrophilic attack of D3O+ to the deuterated carbon atom site (Scheme 3) is only competitive in very acidic solutions (i.e., when the concentration of D3O+ is high enough so that bimolecular reaction can compete with fast deactivation of 1 from S1: τ = 2.5 or 3.6 ns; Tables S2 and S3†).
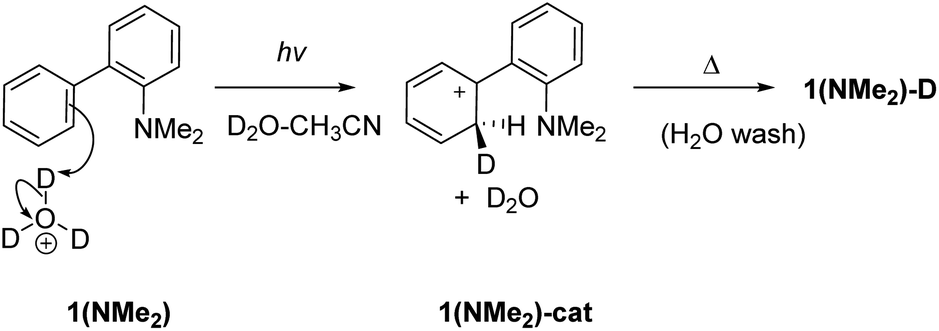 | ||
| Scheme 3 Photoinduced electrophilic attack of D3O+ at C2 for D-exchange in 1(NMe2). This mechanism is likely not important except for very acidic conditions. | ||
Compounds 2 and 3 cannot undergo ESIPT, since the acidic NH2 group is too far away from the neighboring phenyl ring. But water (D2O)-mediated excited-state proton transfer may give rise to D-exchange at distal sites.
Irradiating 2 in CH3CN–D2O gave two products, a compound 2-D resulting from regioselective D-exchange at the C4 position of the aniline ring, and a photoredox product 4 (Scheme 4). The photoproduct 4 could not be separated from the starting material. Attempts of HPLC separation provided their mixture or the starting compound 2. The structure of photoproduct 4 was therefore assigned based on NMR characterization of the mixture with 2 and MS analysis (see Fig. S24–S30 in the ESI†). The 15N–1H HMBC NMR correlation of the mixture clearly points to two different N-atoms (Fig. S29†). The structure for 4 was also confirmed by chemical synthesis. We performed a reduction of 3-nitrobiphenyl with Raney nickel and obtained the same mixture as the photolysis of 2 (see Fig. S31 in the ESI†). 2-D only is observed in acidic solution and may occur by electrophilic attack of D3O+ at the C4 position via water-assisted D-exchange. Computed partial charges for 2 support the observed regioselectivity, showing that in the S1 state, C4 is the most negatively charged ring carbon atom in the aniline ring (see Fig. 6, and discussion below).
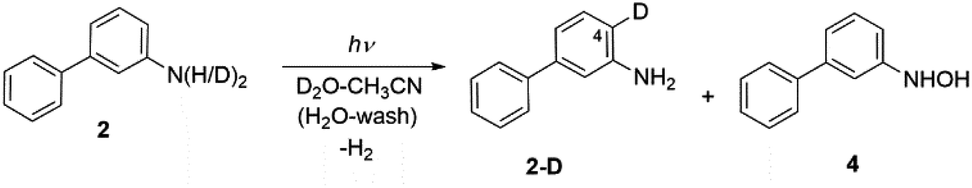 | ||
| Scheme 4 Excited-state proton transfer and photoredox reactions for 2. See Scheme 6 for the proposed photoredox mechanism for the formation of 4. | ||
Formation of a hydroxylamine photoproduct 4 suggests that photoexcitation of 2 triggers a photoredox process. To investigate how 4 was formed, compound 2 was irradiated in CH3CN–H2O, at different pH values and in the presence of different acids (H2SO4 and HCl). Product 4 was formed regardless of the pH values and the acid used, and these results suggest that an acidic media is not required for the observed photoredox process. We surmise that a proton-coupled electron transfer (PCET) reaction occurs between 2 and H2O, generating an OH radical and H atom (vide infra), which then forms 4 and releases H2. Although we did not detect the formation of H2, it is a probable product based on the proposed photoredox mechanism.
Irradiating 3 in CH3CN–D2O led to regioselective D-exchange at the C2′ position of the phenyl ring, giving 3-D (Scheme 5). D-exchange occurs only at very low pD values (see also Fig. S32–S34 in the ESI†) with a quantum yield of <10−4. The reaction takes place only in acidic solution and possibly proceeds through a water-assisted ESPT reaction involving D3O+. Computed partial charges for 3 support the observed regioselectivity, showing that in the S1 state, the C2′ and C4′ positions are most negatively charged ring carbon atoms in the phenyl ring (Fig. S52†). D-exchange at the C2′ position may be explained by a closer proximity to the NH2 group, making it a more accessible site via water-assisted ESPT. Note also that there are two C2′ positions but only one C4′ position, which may explain the observed protonation at C2′.
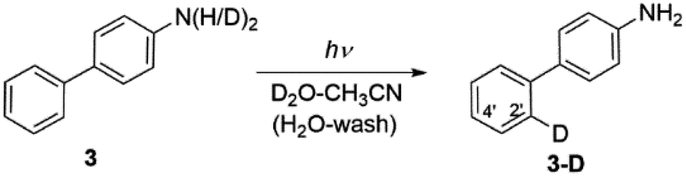 | ||
| Scheme 5 Excited-state proton transfer reaction for D-exchange in 3. | ||
Do these photoreactions happen in the singlet or triplet excited state? To investigate whether the ESPT of 1–3 and the photoredox of 2 occur from the singlet or triplet excited state, the experiments were repeated in CH3CN–D2O at pD = 2 and purged by N2 or O2. O2 strongly quenches triplet excited-states, and thus reactions that occur in the triplet state would be significantly quenched. For 1, we observed some quenching by O2 (Table S9 in the ESI†), suggesting that D-exchange may involve some triplet excited-state species. However, some quenching can occur in the S1 state. We expect that singlet–triplet energy transfer can give rise to singlet oxygen and produce the triplet excited state of 1. Note that the energy gap between the S1 and T1 state of 1 (E = 27.2 kcal mol−1) is within a reasonable range to populate 1O2 (E = 22.4 kcal mol−1). Spin is conserved in this process. D-exchange in 2 and 3 were not significantly quenched by O2, suggesting that these reactions do not involve triplet excited-states. The photoredox reaction of 2 giving hydroxylamine 4 was completely quenched by O2, indicating that the suggested PCET reaction for 2 involves a triplet excited state.
In summary, the three aminobiphenyl isomers 1–3 are likely undergoing three different photoreactions. The ortho-derivative, 1, undergoes ESIPT. We expect that the meta-derivative, 2, undergoes water-assisted ESPT and a photoredox reaction via PCET. We also expect that the para-derivative, 3, undergoes a water-assisted ESPT reaction. All of these reactions appear to take place in the singlet excited state, except for the photoredox process of 2, which involves a triplet excited state.
Laser flash photolysis
Laser flash photolysis experiments were carried out to probe for triplet excited states and reactive intermediates in the photoreactions of 1 and 2. All spectra and experimental details are included in the ESI (Fig. S35–S42†). In Ar-purged CH3CN and CH3CN–H2O solutions, a transient signal with a maximum of absorption at ≈350 nm was observed for 1 and 2 (Fig. 5). The transient decayed within nanoseconds (τ ≈ 100–300 ns) and was quenched by O2. | ||
| Fig. 5 Transient absorption spectra of 1 (c = 1.33 × 10−4 M), (top) and 2 (c = 3.68 × 10−5 M), (bottom) in Ar-purged (CH3CN–H2O) (4:1), A266 = 0.3, laser pulse 20 mJ. Insets: the decay of transient absorption at 360 nm for the Ar- and O2-purged solution. | ||
We assigned the transient signals to the triplet excited states of 1 and 2 by comparison to the transient absorption spectra of hydroxybiphenyls,47,48 and triplet excited states of aniline derivatives.49,501-aQM was not detected, possibly due to a short lifetime and the low efficiency of its formation. It is anticipated to have a short lifetime, similar to aza-QM, detected from 2-(2-aminophenyl)naphthalene,20 or similar QM derivatives formed from phenols.48 The radical cations of 1 and 2 and solvated electron also were not detected, which preclude a photoionisation mechanism. The tentative assignments of the transients observed in the LFP of 1 and 2 is provided in the ESI.†
Computed vertical excitations, charges, and nucleus-independent chemical shifts (NICS)
Computed vertical excitation energies for 1–3 at the ADC(2)/cc-pVDZ level are in good agreement with experimental UV-vis data (Tables S10–S12†). Plots of orbitals involved in the first three singlet excited states are included in Fig. S43–S45 of the ESI,† while the corresponding electron density difference plots are included in Fig. S46–S48.† All three compounds have ππ* character in the S1 state and show decreased electron density at the N atom and increased electron density in the phenyl ring in the S1 state. These findings are consistent with a red-shifted UV-vis spectra and quenched fluorescence at lower pH due to protonation of the N atom.Computed Natural Population Analysis (NPA) charges at the CPCM(CH3CN)-PBE0-D3/def2-TZVPP level for 1–3 in the S0 and S1 states are shown in Fig. S52 of the ESI,† and these results explain the regioselectivity of D-exchange in 1–3 (partial charges computed at the MP2/cc-pVDZ and ADC(2)/cc-pVDZ levels are included to Fig. S49–S51†). For 1 and 3, all carbon atoms of the phenyl ring (besides the linker carbons) become more negatively charged in the S1 state; this is consistent with an observed deuteration at the phenyl ring (i.e., Ring-1). In the S1 state of 1, C4′ of the phenyl ring bears the most negative charge, and the proximity of C2′ to the NH2 explains regioselective D-exchange via ESIPT at this position (Fig. S52†). In the S1 state of 3, C2′ and C4′ of the phenyl ring bear the most negative charge, yet the C2′ position is closer to the NH2 group and thus may be more accessible for water-assisted ESPT. For 2, all carbon atoms of the aniline ring (besides C3, which is attached to the NH2 group) become more negatively charged in the S1 state; this is consistent with an observed deuteration at the aniline ring (i.e., Ring-2). In the S1 state of 2, C4 of the aniline ring is the most negatively charged ring carbon atom, which supports the observed regioselective D-exchange at this position.
Computed nucleus-independent chemical shifts (NICS) at the CPCM(CH3CN)-PBE0-D3/def2-TZVPP level, based on the S0 and S1 state minimum geometries of 1–3, are shown in Fig. 6. NICS(1)zz values were computed at the phenyl (Ring-1) and aniline (Ring-2) ring centers. NICS for the S1 states were computed as triplet states based on geometries optimized in the S1 state with TD-CPCM(CH3CN)-PBE0-D3/def2-TZVPP. In the ground state, all three compounds show large negative NICS(1)zz values for Ring-1 and Ring-2, indicating strong aromatic character. In the S1 state, computed NICS for 1 and 3 show that Ring-1 is non-aromatic (i.e., NICS(1)zz values close to zero) and Ring-2 is antiaromatic (i.e., positive NICS(1)zz values).
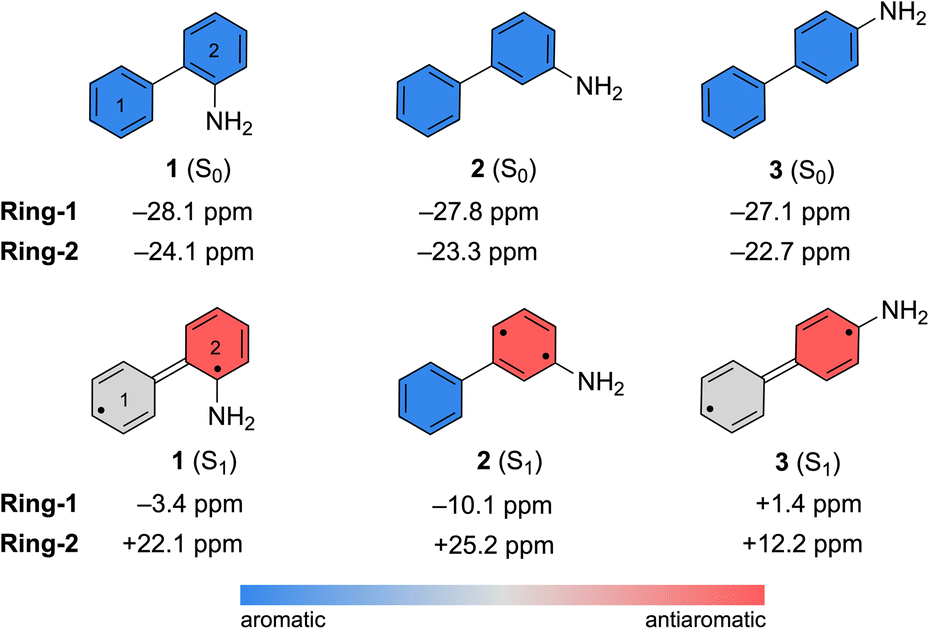 | ||
| Fig. 6 Computed NICS(1)zz values for the S0 and S1 state minimum geometries of 1–3. NICS(1)zz for the S1 state minimum geometries were computed as triplet states. | ||
In contrast, computed NICS(1)zz for the S1 state of 2 reveals a localized antiaromatic character in Ring-2, while Ring-1 retains aromaticity. Differences in the (anti) aromaticity patterns of 1–3 in the S1 state may have mechanistic consequences. We note that in 1 and 3, excited-state antiaromatic character of the aminobiphenyl compound is delocalized between Ring-1 and Ring-2. But in 2, it is localized in Ring-2. A higher photoreactivity of the aniline fragment in 2 may explain why it undergoes a photo-induced PCET reaction and uniquely deuterates at the aniline ring. In accordance, computed spin densities for the T1 state of 2 shows more spin in the aniline ring, while those of 1 and 3 display delocalized spins across the two rings (Fig. S53 in the ESI†).
Antiaromaticity relief in the ESIPT reaction of 1
A minimum energy pathway (MEP) was computed at the ADC(2)/cc-pVDZ level to investigate the ESIPT reaction of 1. Relevant structures (A–D) along the MEP are shown in Fig. 7a. Plots of the S1 state MEP and S0 state single-point energies at MP2/cc-pVDZ are shown in Fig. 7b. Structure A is the Franck–Condon point. Structure B is the lowest energy point on the MEP. Structure C is a transition state-like structure (i.e., proton is transferring—not necessarily a first-order saddle point) and the highest energy point after B. Structure D is a post-transition structure and the last point on the MEP considered. A few important observations emerge from Fig. 7: (1) the ESIPT reaction of 1 is a near-barrierless process in the S1 state. (2) As 1 relaxes from the Franck–Condon point, there is significant electron delocalization across the biphenyl linker (note shortened linker C–C bond length in B, as shown in Fig. 7a). (3) As D forms, the S1 and S0 state surfaces cross and a conical intersection brings the excited-state species back to the ground state. A computed MEP for 1 with H2O is included to Fig. S54, Tables S17 and S18 in the ESI,† showing a similar energy profile for ESIPT as that shown in Fig. 7. Cartesian coordinates of points A–D are included to Tables S13–16.†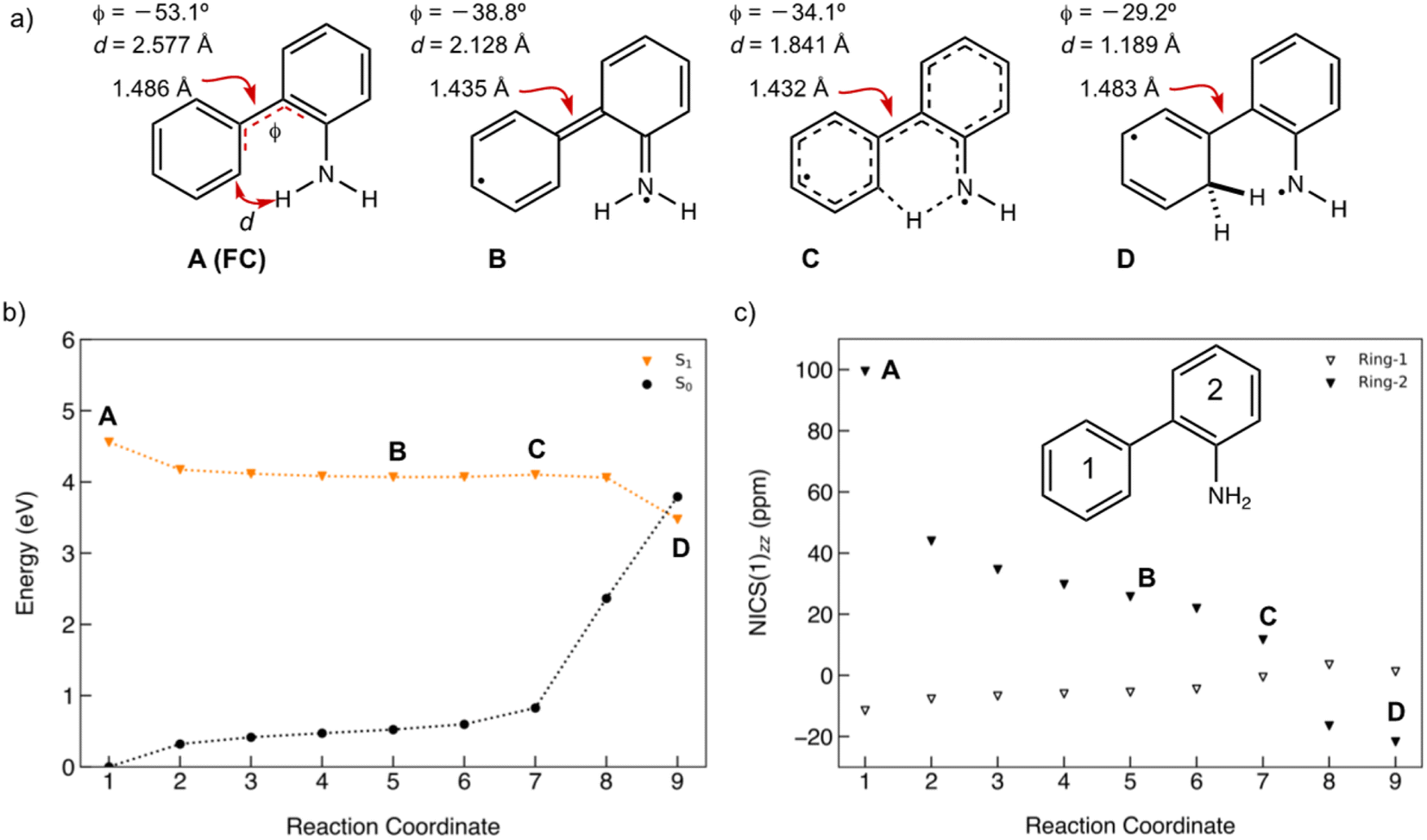 | ||
| Fig. 7 (a) Selected structures on the S1 state MEP for ESIPT in 1: the Franck–Condon point (A), the lowest energy point on the S1 state MEP (B), the highest energy point after (B) and (C), and a post-transition structure (D). d is the distance between the migrating H and the proton accepting carbon atom. Φ is the CCCC dihedral angle across the central linker CC bond. (b) A plot of the S1 state MEP computed at ADC(2)/cc-pVDZ for the ESIPT reaction of 1. Single-point S0 state energies were computed for each structure at MP2/cc-PVDZ. (c) NICS(1)zz values computed at CPCM(CH3CN)-PBE0-D3/def2-TZVPP for structures along the MEP (cf. orange curve in Fig. 7b). | ||
Nucleus-independent chemical shift tensor components of the principal axis perpendicular to the ring plane (NICS(1)zz)51–54 were computed at 1 Å above the centroids of Ring-1 (Fig. 7c, white triangles) and Ring-2 (Fig. 7c, black triangles) for each energy point along the MEP for the ESIPT reaction of 1.53 At A, Ring-1 (NICS(1)zz = −11.5 ppm) is aromatic and Ring-2 (+99.5 ppm) is strongly antiaromatic (see also computed spin density for A in Fig. S53†). From there, Ring-1 remains relatively non-aromatic throughout the ESIPT process (B to D) ranging from −5.5 ppm to +1.3 ppm. Yet, Ring-2 alleviates antiaromaticity as A (+99.5 ppm) relaxes to B (+25.8 ppm), and re-gains aromaticity as C (+11.7 ppm) evolves to D (−21.7 ppm). Changes in the geometries from A to D also are consistent with the effects of antiaromaticity relief and aromaticity gain in Ring-2 (Fig. 7a). From A to B, the linker C–C bond shortens (from 1.486 Å to 1.435 Å) and the dihedral angle of the biphenyl linker tends towards planarization (φ = −53.1° to φ = −38.8°, Fig. 7a), indicating increased quinoidal character in both rings in B. From C to D, the incipient C–H bond forms, and the linker C–C bond lengthens (from 1.432 Å to 1.483 Å) as the dihedral angle of the biphenyl linker twists even more towards planarity (from φ = −34.1° to φ = −29.2°), resulting in rearomatization of Ring-2 in D. NICS(1)zz values for all S1 state structures along the MEP were computed as triplet states at the CPCM(CH3CN)-PBE0-D3/def2-TZVPP level. In contrast to previous examples of ESPT reactions explored by us and by others,23,25,54 ESIPT in 1 is accompanied both by antiaromaticity relief (i.e., from relaxation of the Franck–Condon structure) and by aromaticity gain (i.e., proton transfer from the NH2 group C2′ in Ring-2).
Antiaromaticity relief in the PCET reaction of 2
To investigate the excited-state PCET reaction of 2, we approximated the MEP with a linearly interpolated pathway in internal coordinates computed at the ADC(2)/cc-pVDZ level. We expect this to be the first step in the photoredox reaction of 2 leading to 4. Upon photoexcitation of 2, the aniline moiety undergoes a PCET reaction, and the resulting H atom splits water to produce an OH radical, which then forms a bond with the N atom, producing 4 (Scheme 6). | ||
| Scheme 6 Proposed mechanism for the photoredox reaction of 2 leading to 4. | ||
A scheme of the MEP for the PCET reaction of 2, including only the S0, S1, and T1 states, is shown in Fig. 8b. Plots of MEPs at the ADC(2) level suggest that the S1 is populated, and an intersystem crossing from S1 to T2 followed by internal conversion to T1 is involved in the PCET process (see relevant structures (A–D) along the MEP of the PCET reaction in the S1 and T1 states are shown in Fig. 8a and a plot of the MEP is shown in Fig. 8b). Single-point energies at the MP2/cc-pVDZ level were computed for the S0 states along the MEP. Structure A corresponds to the Franck–Condon point. Structure B is the lowest energy point on the MEP and corresponds to the minimum energy structure of the S1 state. Structure C is a post-crossing point where the S1 and T1 state surface cross. At structure D, the T1 and S0 surface cross, and 2 can return to the ground state by intersystem crossing. A few important observations emerge from Fig. 8: (1) the PCET reaction of 2 is a non-adiabatic process. The reaction likely begins in the S1 state and crosses to a triplet state (a more elaborate energy plot including computed energies in the S2, T1, and T2 states is included in the ESI Fig. S55†). (2) As 2 relaxes from the Franck–Condon point, there is significant electron delocalization across the biphenyl linker (note shortened linker C–C bond length in B). (3) Computed Natural Transition Orbitals (NTO's) for structures A and B show ππ* character, while structures C and D show πσ* state character, suggesting that the PCET reaction is a non-adiabatic process involving electron transfer from the excited-state π-system followed by N–H bond breaking. (4) At structure D, the T1 and S0 state surfaces cross and intersystem crossing brings the excited-state species back to the ground state.
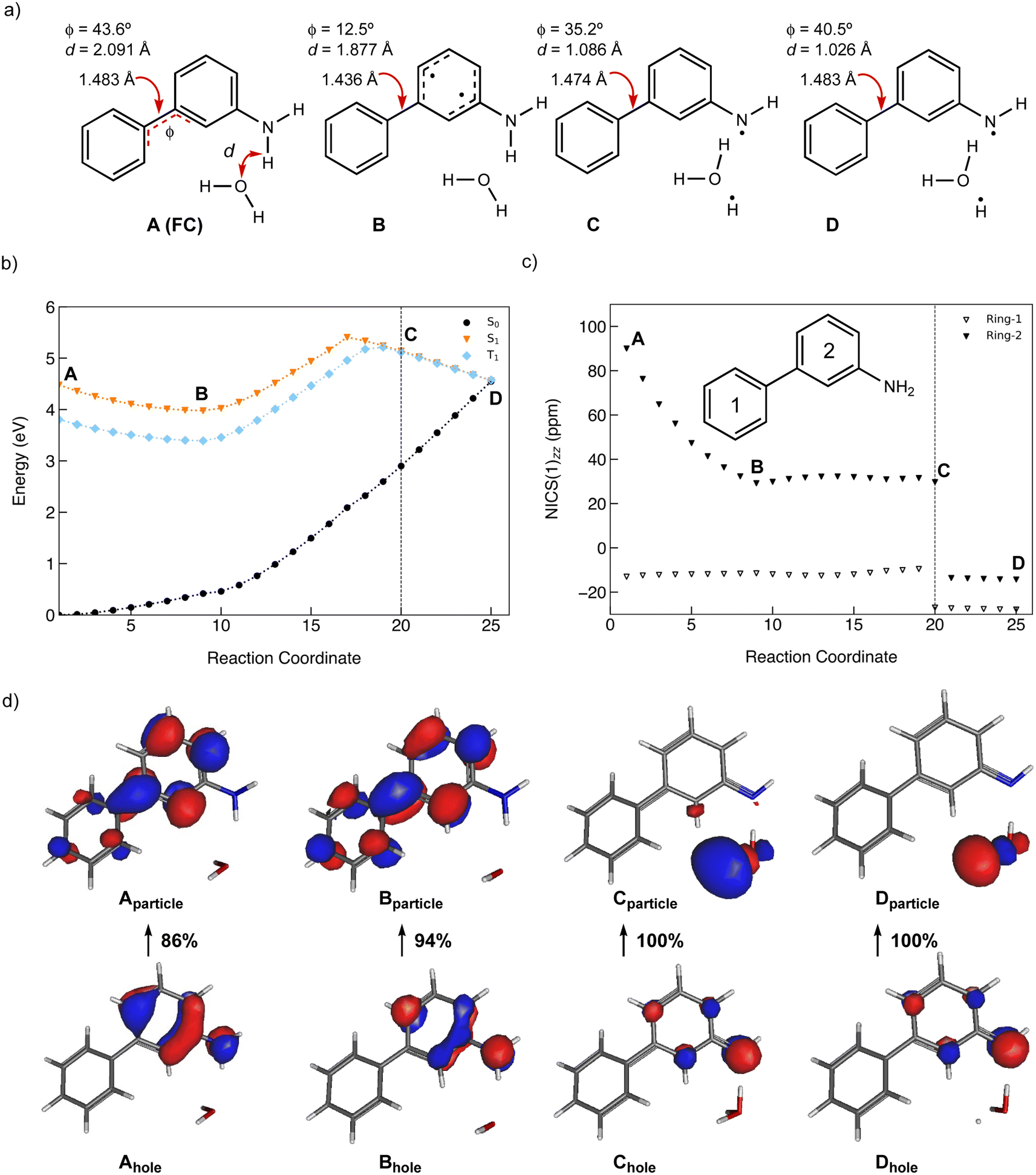 | ||
| Fig. 8 (a) Geometric features of the Franck–Condon state (A), the minimum of the S1 state the lowest energy point on the S1 state MEP (B), the highest energy point on the MEP (C), and a post-surface crossing structure (D). (b) Computed MEP plots at ADC(2)/cc-pVDZ for the PCET reaction of 2; the T1 and S0 state surfaces are computed single-point energies based on fully optimized S1 state geometries. d is the distance between the migrating H and the O atom on water. Φ is the CCCC dihedral angle across the central linker CC bond. (c) NICS(1)zz values computed at CPCM(CH3CN)-PBE0-D3/Def2-TZVPP for structures along the MEP for the PCET reaction of 2. (d) Dominant natural transition orbital (NTO) pairs for structure A–D. % values are the contributions of an NTO pair to the excited-state wavefunction (S1 for A and B, T1 for C and D). | ||
Changes in the geometry of 2 along the MEP for an excited-state PCET reaction show the effects of antiaromaticity relief. The C–C bond linker in structure A (1.483 Å) shortens as the structure relaxes from the Franck–Condon region to B (1.436 Å), and the dihedral angle of the two phenyl rings tends towards planarization (from φ = 43.6° to φ = 12.5°). Planarization allows the triplet spin to delocalize which alleviates excited-state antiaromaticity in Ring-2 (cf.Fig. 8c). Already in B, the forming O⋯H bond shortens from 2.091 Å (in A) to 1.877 Å.
Upon electron transfer from the aniline ring to the amine H atom, the geometry at C suggests the O–H bond is almost fully formed (d = 1.121 Å) and the C–C bond linker length (1.474 Å) and dihedral angle (35.2°) is returned to a Franck–Condon-like geometry. Computed T1 NICS(1)zz values for Ring-1 and Ring-2 along the MEP show that Ring-1 remains slightly aromatic during the PCET process (hovering around −12 ppm), and Ring-2 experiences a gradual decrease in antiaromaticity. At points 19 and 20, Ring-1 and Ring-2 undergo a drastic drop in NICS(1)zz magnitude, corroborating a PCET process that restores aromaticity at these points in rings 1 and 2, respectively.
Antiaromaticity relief in the water-assisted ESPT reaction of 3
Irradiating 3 in CH3CN–D2O showed selective D-exchange at the C2′ position (Scheme 5). To investigate the origin of this D-exchange, a MEP of 3 in a micro-solvated environment of five water molecules was computed on the S1 with ADC(2)/cc-pVDZ (Fig. 9a and b). Note that it is only a model, and in a real system more H-bonded H2O molecules are involved. Furthermore, such a relay processes are entropically unfavoured, and therefore, the process takes place with a low quantum yield, as observed in the experiment. Relevant structures (A–D) for the MEP of 3 are shown in Fig. 9a. The Franck–Condon structure (A) shows the hydrogen-bonded water chain connecting the ammonium moiety to the C2′ position. Intermediate structures, B and C, show two points along the MEP where a proton is being transferred intermolecularly to another H2O and C2′ carbon, respectively. Structure D is the point at which the S1 and S0 surfaces cross, and where the structure is brought back to the ground state via a conical intersection. Geometric signatures along the MEP show notable C–C bond contracting and lengthening in the linker C–C, denoted by the single-headed red arrow, along with trends towards planarity between Ring-1 and Ring-2 (φ tends toward 0° from A to C). The energy landscape near the Franck–Condon (FC) region, A, and point B is complicated by near-crossing of the S1 and S2 state surfaces, and thus the NICS(1)zz values for structures in this region cannot be properly described by the approach applied here. Optimized Cartesian coordinates for structures A–D are included to Tables S23–S26.†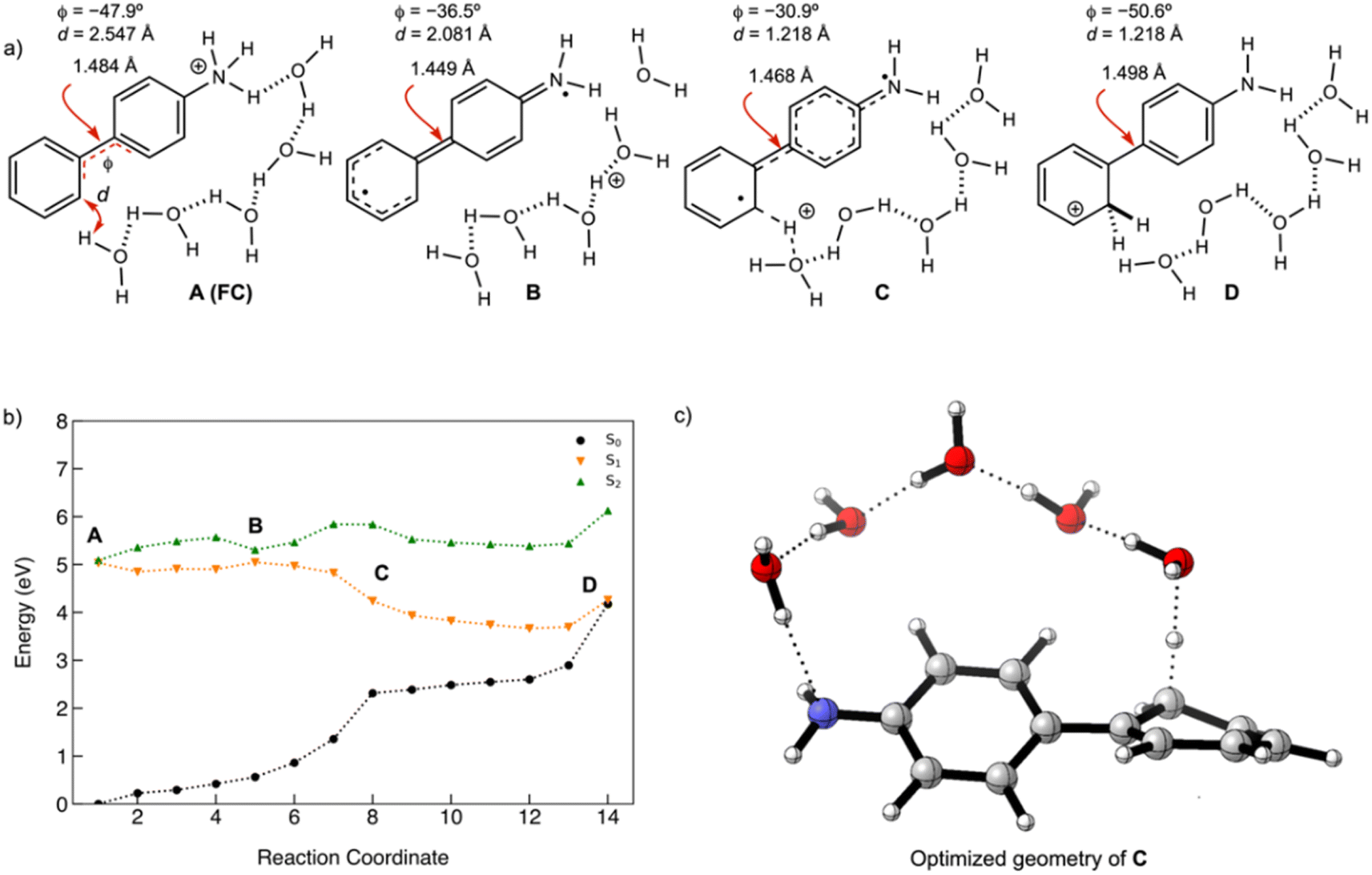 | ||
| Fig. 9 (a) Geometric features of the Franck–Condon state (A), S2–S1 near-crossing on MEP (B), ESPT to Ring-1 (C), and S1–S0 conical intersection (D). (b) Computed MEP at the ADC(2)/cc-pVDZ for the water-assisted ESPT reaction of 3 in the S1 state, and singlet-point energies in the S0 and S2 states. (c) Geometry for C showing proton transfer to the 2′ position in the benzene ring. | ||
Conclusions
Excited-state antiaromaticity relief can play important roles in many light-driven proton and electron transfer reactions. Here, we investigate and contrast the photoprotonation mechanism of three aminobiphenyl isomers. In all three cases, a proton formally migrates from an NH2 group to an aromatic ring carbon atom. All three isomers, ortho-, meta- and para-, can undergo excited-state proton transfer involving water, as documented by deuterium exchange experiments. In addition, the meta-isomer can undergo a photoredox reaction, involving proton-coupled electron transfer and water-splitting. We note that, in the meta-isomer, excited-state antiaromatic character is localized in the aniline moiety while the benzene moiety remains weakly aromatic (Fig. 6), and argue that strong antiaromaticity in the aniline ring may be responsible for driving the observed photoredox reaction.Experimental and computational methods
Preparative irradiation experiment in the presence of D2O
2-Aminobiphenyl (1) (100 mg, 0. 62 mmol) was dissolved in CH3CN–D2O (9:1, v/v, 100 mL) and transferred to a quartz Erlenmeyer flask with a rubber septum. The solution was purged with a stream of N2 for 30 min and irradiated in a Luzchem reactor equipped with 8 lamps (1 lamp ≈ 8 W) with the maximum output at 300 nm over 4 days. The flask equipped with a stirring bar was centered in the middle of the reactor, separated 15 cm from the lamps on each side. Information of the lamps irradiance can be found from the Luzchem supplier.55 After the irradiation, the solution was transferred to a separation funnel and water (100 mL) was added. An extraction with CH2Cl2 was carried out (3 × 75 mL). The extracts were dried over anhydrous MgSO4, filtered and the solvent was removed on a rotary evaporator. The residue was chromatographed on a column of silica gel by using CH2Cl2 as an eluent to remove high-weigh material formed in the photolysis. The residue (80 mg, 80%) was analysed by 1H NMR and MS. Since the 1H NMR indicated exchange of only one H by D, the sample was dissolved again in CH3CN–D2O (100 mL, 9:1), purged with N2 and irradiated in the Luzchem reactor over 4 days. After the same workup and chromatographic purification, 45 mg (45%) was obtained that was analyzed by 1H NMR and MS. Details of all photolyses conducted under different conditions for molecules 1–3 can be found in the ESI.†
2-Amino-2,2′-bisdeuteriobiphenyl (1-2D)
1H NMR (600 MHz, CD3OD): δ/ppm 7.43 (d, 2H, J = 7.8 Hz, H-3′), 7.33 (t, 1H, J = 7.8 Hz, H-4′), 7.10 (dt, J = 1.5 Hz, J = 7.9 Hz, H-4), 7.04 (dd, 1H, J = 1.5 Hz, J = 7.5 Hz, H-6), 6.83 (dd, 1H, J = 0.9 Hz, J = 7.9 Hz, H-3), 6.77 (dt, J = 1.5 Hz, J = 7.5 Hz, H-5). 13C {1H} NMR (150 MHz, CD3OD): δ/ppm 145.1, 141.1, 141.0, 131.2, 129.7, 129.4, 129.1 (t, J = 5.3 Hz), 128.0, 119.5, 117.1; MS (ESI+): m/z (%) 170 (0), 171 (23), 172 (100), 173 (13).Computational methods
Density functional theory (DFT) calculations were carried out in Gaussian 16 (revision C.01).56 Computed Natural Population Analysis (NPA) charges and dissected nucleus-independent chemical shifts, NICS(1)zz, were computed at the PBE0-D3/def2-TZVPP level of theory in a conductor-like polarizable continuum model (C-PCM).57–61 For each point of interest NICS(1)zz points were manually created using the Molecule software program. Bq atoms were placed 1 Å above each ring. Time-dependent density functional theory (TD-DFT) calculations were also computed in Gaussian 16 using the Tamm–Dancoff approximation. All Cartesian coordinates of structures optimized with DFT or TD-DFT are included in the ESI, Tables S28–S36.†Ground state geometries were optimized at MP2/aug-cc-pVDZ62 employing the resolution-of-identity (RI) approximation.63 Vertical excitation energies and oscillator strengths were computed using the algebraic diagrammatic construction to second order ADC(2) method64–66 as implemented in Turbomole 7.6.67 Calculations were performed with the cc-pVDZ and aug-cc-pVDZ basis sets both in the gas phase and in solution using the implicit solvation model COSMO68,69 with default parameters for acetonitrile (ACN). Excited states have been characterized in terms of natural transition orbitals70,71 computed by retaining only singly excited coefficients of the ADC(2) wave functions.72
Reaction pathways in the S1 state have been optimized using the double-ended reaction path optimization scheme woelfling73 of Turbomole. All reaction pathways start at the Franck–Condon geometry and end at the S1/S0 conical intersection (CI). CI geometries were optimized using the sequential penalty constrained optimization method of Levine et al.74 with default initial values of α = 0.025 Hartree and σ = 3.5.
Data availability
Supporting data are available in the main text and (ESI)† document, and are readily accessible at Public Documents_HrZZ-IP-2019-04-8008 at: https://mojoblak.irb.hr/s/PXPWDXZ2QCp3daC.Author contributions
Conceptualization: N. D., J. W., N. B.; data curation and formal analysis: J. D., C. L.; funding acquisition: N. D., J. W., N. B.; investigation: J. D., C. L., N. D.; methodology: C. L., N. D., J. W., N. B.; project administration and resources: N. D., J. W., N. B.; software: N. D., J. W.; supervision: N. D., J. W., N. B.; visualization: J. D., C. L., N. D., J. W., N. B.; writing: N. D., J. W., N. B.; review and editing: all. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by Croatian Science Foundation (HRZZ grant no. HRZZ-IP-2022-10-4658 and HRZZ-IP-2019-04-8008). NB and ND are grateful to the Adris foundation for supporting this work. JW thanks the National Institute of General Medical Sciences (NIGMS) of the National Institutes of Health (R35GM133548) and the Alfred P. Sloan Research Foundation (FG-2020-12811) for grant support. We acknowledge the use of the Sabine and Carya clusters and support from the Research Computing Data Core at the University of Houston.Notes and references
- Hydrogen-transfer reactions, ed. J. T. Hynes, J. P. Klinman, H.-H. Limbach and R. L. Schowen, Wiley-VCH, Weinheim, 2007 Search PubMed.
- P. Kalanderopoulos and K. Yates, Intramolecular Proton Transfer in Photohydration Reactions, J. Am. Chem. Soc., 1986, 108, 6290–6295 CrossRef CAS.
- H. Shizuka, Excited-State Proton-Transfer Reactions and Proton-Induced Quenching of Aromatic Compounds, Acc. Chem. Res., 1985, 18, 141–147 CrossRef CAS.
- N. Mathivanan, F. Cozens, R. A. McClelland and S. Steenken, Regioselective Photoprotonation of 1,3-Dimethoxybenzenes in 1,1,1,3,3,3-Hexafluoroisopropyl Alcohol. H-D Isotope-Exchange and Laser Flash Photolysis Studies, J. Am. Chem. Soc., 1992, 114, 2198–2203 CrossRef CAS.
- J. C. Roberts and J. A. Pincock, Methoxy-Substituted Stilbenes, Styrenes, and 1-Arylpropenes: Photophysical Properties and Photoadditions of Alcohols, J. Org. Chem., 2006, 71, 1480–1492 CrossRef CAS PubMed.
- M. Lukeman and P. Wan, A New Type of Excited-State Intramolecular Proton Transfer: Proton Transfer from Phenol OH to a Carbon Atom of an Aromatic Ring Observed for 2-Phenylphenol, J. Am. Chem. Soc., 2002, 124, 9458–9464 CrossRef CAS PubMed.
- S.-H. Xia, B.-B. Xie, Q. Fang, G. Cui and W. Thiel, Excited-state intramolecular proton transfer to carbon atoms: nonadiabatic surface-hopping dynamics simulations, Phys. Chem. Chem. Phys., 2015, 17, 9687–9697 RSC.
- M. Fischer and P. Wan, m-Quinone Methides from m-Hydroxy-1,1-Diaryl Alkenes via Excited-State (Formal) Intramolecular Proton Transfer Mediated by a Water Trimer, J. Am. Chem. Soc., 1998, 120, 2680–2681 CrossRef CAS.
- M. Fischer and P. Wan, Nonlinear Solvent Water Effects in the Excited-State (Formal) Intramolecular Proton Transfer (ESIPT) in m-Hydroxy-1,1-diaryl Alkenes: Efficient Formation of m-Quinone Methides, J. Am. Chem. Soc., 1999, 121, 4555–4562 CrossRef CAS.
- O. H. Kwon, Y.-S. Lee, H. J. Park, Y. Kim and D.-J. Jang, Asymmetric Double Proton Transfer of Excited 1:1 7-Azaindole/Alcohol Complexes with Anomalously Large and Temperature-Independent Kinetic Isotope Effects, Angew. Chem., Int. Ed., 2004, 43, 5792–5796 CrossRef CAS PubMed.
- O.-H. Kwon, Y.-S. Lee, B. K. Yoo and D. J. Jang, Excited-State Triple Proton Transfer of 7-Hydroxyquinoline along a Hydrogen-Bonded Alcohol Chain: Vibrationally Assisted Proton Tunneling, Angew. Chem., Int. Ed., 2006, 45, 415–419 CrossRef CAS PubMed.
- J. Ma, X. Zhang, N. Basarić and D. L. Phillips, Direct Observation of Photoinduced Ultrafast Generation of Singlet and Triplet Quinone Methides in Aqueous Solutions and Insight into the Roles of Acidic and Basic Sites in Quinone Methide Formation, J. Am. Chem. Soc., 2017, 139, 18349–18357 CrossRef CAS PubMed.
- X. Zhang, Y. Guo, E. Dallin, J. Ma, M. Dai and D. L. Phillips, Unusual Acetonitrile Adduct Formed via Photolysis of 4′-Chloro-2-Hydroxybiphenyl in Aqueous Solution, J. Org. Chem., 2020, 85, 11635–11640 CrossRef CAS PubMed.
- Y. Guo, X. Li, J. Ma and D. L. Phillips, Reaction Mechanisms of Photoinduced Quinone Methide Intermediates Formed via Excited-State Intramolecular Proton Transfer or Water-Assisted Excited-State Proton Transfer of 4-(2-Hydroxyphenyl)pyridine, J. Phys. Chem. Lett., 2021, 12, 11666–11672 CrossRef CAS PubMed.
- M. Lukeman and P. Wan, Excited-State Intramolecular Proton Transfer in o-Hydroxybiaryls: A New Route to Dihydroaromatic Compounds, J. Am. Chem. Soc., 2003, 125, 1164–1165 CrossRef CAS PubMed.
- M. Flegel, M. Lukeman, L. Huck and P. Wan, Photoaddition of Water and Alcohols to the Anthracene Moiety of 9-(2’-Hydroxyphenyl)anthracene via Formal Excited State Intramolecular Proton Transfer, J. Am. Chem. Soc., 2004, 126, 7890–7897 CrossRef CAS PubMed.
- N. Basarić, N. Došlić, I. Ivković, J.-H. Wang, M. Mališ and P. Wan, Very Efficient Generation of Quinone Methides through Excited State Intramolecular Proton Transfer to a Carbon Atom, Chem.–Eur. J., 2012, 18, 10617–10623 CrossRef PubMed.
- I. Saito, H. Sugiyama, A. Yamamoto, S. Muramatsu and T. Matsuura, Photochemical Hydrogen-Deuterium Exchange Reaction of Tryptophan. The Role in Nonradiative Decay of Singlet Tryptophan, J. Am. Chem. Soc., 1984, 106, 4286–4287 CrossRef CAS.
- H. Shizuka, L. M. Serizawa, T. Shimo, I. Saito and T. Matsuura, Fluorescence-Quenching Mechanism of Tryptophan. Remarkably Efficient Internal Proton-Induced Quenching and Charge-Transfer Quenching, J. Am. Chem. Soc., 1988, 110, 1930–1934 CrossRef CAS.
- J. Draženović, T. Rožić, N. Došlić and N. Basarić, Excited State Intramolecular Proton Transfer (ESIPT) from -NH2 to a Carbon Atom of Naphthyl Ring, J. Org. Chem., 2022, 87, 9148–9156 CrossRef PubMed.
- J. Wirz, Kinetics of proton transfer reactions involving carbon, Pure Appl. Chem., 1998, 70, 2221–2232 CrossRef CAS.
- A. P. Pelliccioli, P. Šebej and J. Wirz, Ketonization of enols in aqueous solution: is carbon protonation always rate-determining?, Photochem. Photobiol. Sci., 2012, 11, 967–971 CrossRef CAS PubMed.
- C.-H. Wu, L. J. Karas, H. Ottosson and J. I.-C. Wu, Excited-state proton transfer relieves antiaromaticity in molecules, Proc. Natl. Acad. Sci., 2019, 116, 20303–20308 CrossRef CAS PubMed.
- A. Weller, Innermolekularer Protonenübergang im angeregten Zustand, Z. Elektrochem., 1956, 60, 1144–1147 CrossRef CAS.
- B. J. Lampkin, Y. H. Nguyen, P. B. Karadakov and B. VanVeller, Demonstration of Baird's rule complementarity in the singlet state with implications for excited-state intramolecular proton transfer, Phys. Chem. Chem. Phys., 2019, 21, 11608–11614 RSC.
- Z. Wen, L. J. Karas, C.-H. Wu and J. I.-C. Wu, How does excited-state antiaromaticity affect the acidity strengths of photoacids?, Chem. Commun., 2020, 56, 8380–8383 RSC.
- L. J. Karas, C.-H. Wu and J. I.-C. Wu, Barrier-Lowering Effects of Baird Antiaromaticity in Photoinduced Proton-Coupled Electron Transfer (PCET) Reactions, J. Am. Chem. Soc., 2021, 143, 17970–17974 CrossRef CAS PubMed.
- M. J. S. Dewar, Aromaticity and Pericyclic Reactions, Angew. Chem., Int. Ed., 1971, 10, 761–776 CrossRef CAS.
- H. E. Zimmerman, On Molecular Orbital Correlation Diagrams, the Occurrence of Möbius Systems in Cyclization Reactions and Factors Controlling Ground- and Excited-State Reactions, J. Am. Chem. Soc., 1966, 88, 1564–1565 CrossRef CAS.
- H. E. Zimmerman, Molecular Orbital Correlation Diagrams, Möbius Systems and Factors Controlling Ground- and Excited-State Reactions. II, J. Am. Chem. Soc., 1966, 88, 1566–1567 CrossRef CAS.
- H. E. Zimmerman, Möbius-Hückel Concept in Organic Chemistry. Application of Organic Molecules and Reactions, Acc. Chem. Res., 1971, 4, 272–280 CrossRef CAS.
- R. C. Dougherty, Perturbation Molecular Orbital Treatment of Photochemical Reactivity. Nonconservation of Orbital Symmetry in Photochemical Pericyclic Reactions, J. Am. Chem. Soc., 1971, 93, 7187–7201 CrossRef CAS.
- N. C. Baird, Quantum Organic Photochemistry. II. Resonance and Aromaticity in the Lowest 3ππ* State of Cyclic Hydrocarbons, J. Am. Chem. Soc., 1972, 94, 4941 CrossRef CAS.
- F. Fratev, V. Monev and R. Janoschek, Ab initio Study of Cyclobutadiene in Excited States: Optimized Geometries, Electronic Transitions and Aromaticities, Tetrahedron, 1982, 38, 2929 CrossRef CAS.
- P. B. Karadakov, Ground- and excited-state aromaticity and antiaromaticity in benzene and cyclobutadiene, J. Phys. Chem. A, 2008, 112, 7303–7309 CrossRef CAS PubMed.
- P. B. Karadakov, Aromaticity and Antiaromaticity in the Low-Lying Electronic Ground States of Cyclooctatetraene, J. Phys. Chem. A, 2008, 112, 12707–12713 CrossRef CAS PubMed.
- L. J. Karas and J. I.-C. Wu, Baird's Rules at the Tipping Point, Nat. Chem., 2022, 14, 723–725 CrossRef CAS PubMed.
- M. Rosenberg, C. Dahlstrand, K. Kilså and H. Ottosson, Excited State Aromaticity and Antiaromaticity: Opportunities for Photophysical and Photochemical Rationalizations, Chem. Rev., 2014, 114, 5379–5425 CrossRef CAS PubMed.
- R. Papadakis and H. Ottosson, The Excited State Antiaromatic Benzene Ring: A Molecular Mr Hyde?, Chem. Soc. Rev., 2015, 44, 6472–6493 RSC.
- J. Yan, T. Slanina, J. Bergman and H. Ottosson, Photochemistry Driven by Excited-State Aromaticity Gain or Antiaromaticity Relief, Chem.–Eur. J., 2023, 29, e202203748 CrossRef CAS PubMed.
- V. Balzani, A. Credi and M. Venturi, Molecular Devices and Machines, Wiley, VCH, 2008 Search PubMed.
- N. Boens, W. Qin, N. Basarić, J. Hofkens, M. Ameloot, M. Pouget, J. P. Lefèvre, B. Valeur, E. Gratton, M. VandeVen, N. D. Silva, Y. Engelborghs, K. Willaert, A. Sillen, G. Rumbles, D. Phillips, A. J. W. G. Visser, A. Van Hoek, J. R. Lakowicz, H. Malak, I. Gryczynski, A. G. Szabo, D. T. Krajcarski, N. Tamai and A. Miura, Fluorescence Lifetime Standards for Time and Frequency Domain Fluorescence Spectroscopy, Anal. Chem., 2007, 79, 2137–2149 CrossRef CAS PubMed.
- S. R. Meech, D. V. O'Connor and D. Phillips, Photophysics of 1-Aminonaphthalenes, J. Chem. Soc., Faraday Trans. 2, 1983, 79, 1563–1584 RSC.
- K. E. Rieckhoff, E. R. Menzel and E. M. Voigt, Molecular Fluorescence from High Vibrational Levels of Excited Electronic States, Phys. Rev. Lett., 1972, 28, 261–263 CrossRef CAS.
- R. O. Rahn, Potassium Iodide as a Chemical Actinometer for 254 nm Radiation: Use of Iodate as an Electron Scavenger, Photochem. Photobiol., 1997, 66, 450–455 CrossRef CAS.
- S. Goldstein and J. Rabani, The Ferrioxalate and Iodide-Iodate Actinometers in the UV Region, J. Photochem. Photobiol., 2008, 193, 50–55 CrossRef CAS.
- N. Basarić, N. Cindro, D. Bobinac, L. Uzelac, K. Mlinarić-Majerski, M. Kralj and P. Wan, Zwitterionic biphenyl quinone methides in photodehydration reactions of 3-hydroxybiphenyl derivatives: laser flash photolysis and antiproliferation study, Photochem. Photobiol. Sci., 2012, 11, 381–396 CrossRef PubMed.
- J. Ma, X. Zhang, N. Basarić, P. Wan and D. L. Phillips, Observation of excited state proton transfer reactions in 2-phenylphenol and 2-phenyl-1-naphthol and formation of quinone methide species, Phys. Chem. Chem. Phys., 2015, 17, 9205–9211 RSC.
- I. Carmichael and G. L. Hug, Triplet-triplet absorption spectra of organic molecules in condensed phases, J. Phys. Chem. Ref. Data, 1986, 15, 1–250 CrossRef.
- A. P. Darmanyan, W. Lee and W. S. Jenks, Charge Transfer Interactions in the Generation of Singlet Oxygen O2(1Δg) by Strong Electron Donors, J. Phys. Chem. A, 1999, 103, 2705–2711 CrossRef CAS.
- P. von R. Schleyer, C. Maerker, A. Dransfeld, H. Jiao and N. J. R. van Eikema Hommes, Nucleus-Independent Chemical Shifts: A Simple and Efficient Aromaticity Probe, J. Am. Chem. Soc., 1996, 118, 6317–6318 CrossRef CAS PubMed.
- P. von R. Schleyer, H. Jiao, N. J. R. van Eikema Hommes, V. G. Malkin and O. L. Malkina, An Evaluation of the Aromaticity of Inorganic Rings: Refined Evidence from Magnetic Properties, J. Am. Chem. Soc., 1997, 119, 12669–12670 CrossRef CAS.
- P. von R. Schleyer, M. Manoharan, Z.-X. Wang, B. Kiran, H. Jiao, R. Puchta and N. J. R. van Eikema Hommes, Dissected Nucleus-Independent Chemical Shift Analysis of π-Aromaticity and Antiaromaticity, Org. Lett., 2001, 3, 2465–2468 CrossRef CAS PubMed.
- C. Corminboeuf, T. Heine, G. Seifert and P. von R. Schleyer, J. Weber. Induced Magnetic Fields in Aromatic [n]-Annulenes-Interpretation of NICS Tensor Components, Phys. Chem. Chem. Phys., 2004, 6, 273–276 RSC.
- For the output of the lamps see the catalogue of the Luzchem supplier at, https://www.luzchem.com/PartGroupList.php?part_ID=9, accessed on July 26, 2023.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision A.03, Gaussian, Inc., Willingford, CT, 2016 Search PubMed.
- C. Adamo and V. Barone, Toward Reliable Density Functional Methods without Adjustable Parameters: The PBE0 Model, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- V. Barone and M. Cossi, Quantum Calculation of Molecular Energies and Energy Gradients in Solution by a Conductor Solvent Model, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS.
- M. Cossi, N. Rega, G. Scalmani and V. Barone, Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model, J. Comput. Chem., 2003, 24, 669–681 CrossRef CAS PubMed.
- T. H. Dunning, Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS.
- F. Weigend and M. Häser, RI-MP2: first derivatives and global consistency, Theor. Chem. Acc., 1997, 97, 331–340 Search PubMed.
- J. Schirmer, Beyond the Random Phase Approximation: A New Approximation Scheme for the Polarization Propagator, Phys. Rev. A: At., Mol., Opt. Phys., 1982, 26, 2395–2416 CrossRef CAS.
- A. B. Trofimov and J. Schirmer, An Efficient Polarization Propagator Approach to Valence Electron Excitation Spectra, J. Phys. B: At., Mol. Opt. Phys., 1995, 28, 2299–2324 CrossRef CAS.
- A. Dreuw and M. Wormit, The Algebraic Diagrammatic Construction Scheme for the Polarization Propagator for the Calculation of Excited States, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2015, 5, 82–95 CAS.
- F. Furche, R. Ahlrichs, C. Hättig, W. Klopper, M. Sierka and F. Weigend, Turbomole, Wiley Interdiscip. Rev. Comput. Mol. Sci., 2014, 4, 91–100 CrossRef CAS.
- A. Klamt, Conductor-like Screening Model for Real Solvents: A New Approach to the Quantitative Calculation of Solvation Phenomena, J. Phys. Chem., 1995, 99, 2224–2235 CrossRef CAS.
- A. Klamt, V. Jonas, T. Bürger and J. C. W. Lohrenz, Refinement and Parametrization of COSMO-RS, J. Phys. Chem. A, 1998, 102, 5074–5085 CrossRef CAS.
- R. L. Martin, Natural transition orbitals, J. Chem. Phys., 2003, 118, 4775–4777 CrossRef CAS.
- I. Mayer, Using singular value decomposition for a compact presentation and improved interpretation of the CIS wave functions, Chem. Phys. Lett., 2007, 437, 284–286 CrossRef CAS.
- M. Sapunar, T. Piteša, D. Davidović and N. Došlić, Highly Efficient Algorithms for CIS Type Excited State Wave Function Overlaps, J. Chem. Theory Comput., 2019, 15, 3461–3469 CrossRef CAS PubMed.
- P. Plessow, Reaction Path Optimization without NEB Springs or Interpolation Algorithms, J. Chem. Theory Comput., 2013, 9, 1305 CrossRef CAS PubMed.
- B. G. Levine, J. D. Coe and T. J. Martínez, Optimizing conical intersections without derivative coupling vectors: application to multistate multireference second-order perturbation theory (MS-CASPT2), J. Phys. Chem. B, 2008, 112, 405–413 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: UV-vis and fluorescence data, detailed irradiation experimental procedures, LFP data, computational details, copies of 1H and 13C NMR spectra and coordinates of the optimized molecular structures. See DOI: https://doi.org/10.1039/d4sc00642a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |